

Abstract
The emergence and global spread of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has resulted in an urgent need for evidence on medical interventions and outcomes of the resulting disease, coronavirus disease 2019 (COVID‐19). Although many randomized controlled trials (RCTs) evaluating treatments and vaccines for COVID‐19 are already in progress, the number of clinical questions of interest greatly outpaces the available resources to conduct RCTs. Therefore, there is growing interest in whether nonrandomized real‐world evidence (RWE) can be used to supplement RCT evidence and aid in clinical decision making, but concerns about nonrandomized RWE have been highlighted by a proliferation of RWE studies on medications and COVID‐19 outcomes with widely varying conclusions. The objective of this paper is to review some clinical questions of interest, potential data types, challenges, and merits of RWE in COVID‐19, resulting in recommendations for nonrandomized RWE designs and analyses based on established RWE principles.
There is now an urgent need for evidence to combat the emergence and global spread of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and the resulting disease, coronavirus disease 2019 (COVID‐19). Many randomized controlled trials (RCTs) evaluating treatments and vaccines for COVID‐19 are already in progress, 1 , 2 , 3 , 4 , 5 but the number of clinical questions of interest greatly outpaces the available resources to conduct RCTs. Patients and providers need evidence on vaccines, as well as medications used prior to, during, and after recovering from COVID‐19. Many medications and all vaccines for COVID‐19 may be granted emergency use authorizations (EAUs) or fast‐tracked for approval, resulting in widespread use before there is substantial evidence on safety or long‐term effects. This scenario can limit the conduct of additional RCTs, as some patients and providers may view use of the drugs as lacking clinical equipoise and therefore refuse randomization. Furthermore, randomized trials supporting the use of COVID‐19 treatments often enroll narrow patient populations. However, a much wider patient population is likely to receive these drugs in clinical practice, including patients who had long‐term mechanical ventilation during recovery and patients with long‐term COVID‐19 symptoms. RCTs also typically evaluate only one medication at a time, but patients hospitalized for COVID‐19 may receive multiple medications, and patients and providers need a better understanding of how these medications work when used together. For these reasons, there is growing interest in whether nonrandomized real‐world evidence (RWE) can be used to supplement RCT evidence and aid in clinical decision making.
RWE is evidence on the benefits and harms of treatments derived from real‐world data (RWD), routinely collected data relating to patient health status and/or delivery of health care. 6 , 7 Sources of RWD, such as health insurance claims, electronic health records (EHRs), and patient registries, are becoming increasingly consolidated, standardized, and accessible for research on therapeutics, and there is growing interest in using RWE to support decision making, both in the clinic and at regulatory and payer organizations. RWE studies relying on existing data can often be implemented more rapidly than RCTs, providing an important advantage in the context of a new and rapidly spreading disease with high morbidity and mortality. However, the importance of the clinical questions dictates that clinical decisions be made with high‐quality evidence, and concerns about the validity and reliability of RWE evaluating medical interventions remain. 8 , 9
Concerns about nonrandomized RWE were highlighted by the recent controversy over an RWE study evaluating hydroxychloroquine or chloroquine as treatment for patients hospitalized with COVID‐19. 10 Although now retracted, the study received considerable attention, as it claimed to show that patients in a large, multinational registry (Surgisphere) who received hydroxychloroquine or chloroquine, with or without a macrolide, had higher risks of both mortality and ventricular arrhythmia compared with patients who did not receive these drugs. On the basis of these findings, the hydroxychloroquine arm of the large World Health Organization SOLIDARITY trial was temporarily halted. 11 However, researchers soon raised serious concerns about the paper, including implausible numbers and no sharing of data or analytic code to support evidence from a previously unknown RWD source. 12 All authors of the RWE study not affiliated with Surgisphere ultimately rescinded their authorship, as they could “no longer vouch for the veracity of the primary data sources.” 13
Beyond the Surgisphere debacle, there has been a proliferation of RWE studies on medications and COVID‐19 outcomes with widely varying conclusions. 14 Although variation between RCT results is not infrequent either, some may argue that these discrepancies provide further evidence that RWE studies are of highly varying quality and cannot be relied on for decision making on medical interventions. Therefore, it is important to review the principles of high‐quality RWE design and analysis in the context of vaccines and medications for COVID‐19. The objective of this paper is to review several types of clinical questions of interest, potential data types, challenges, and merits of RWE in this context, resulting in recommendations for nonrandomized RWE designs and analyses that are more likely to succeed using established RWE principles.
MEDICATION EFFECTIVENESS IN AMBULATORY CARE SETTINGS
Much of the work on outpatient medications used for COVID‐19 has focused on how existing medications may lower disease severity among infected patients. For example, because of SARS‐CoV‐2’s interactions with angiotensin converting enzyme receptors, there have been a number of studies on how angiotensin converting enzyme inhibitor medications may impact the risk of a mild COVID‐19 case progressing to more serious illness among patients who were taking these drugs for their cardiovascular effects when they were diagnosed with COVID‐19. 15 , 16 , 17 There has also been considerable interest regarding whether hydroxychloroquine or other drugs could be prescribed as early treatment to prevent serious disease among patients testing positive for COVID‐19. When assessing study validity of the treatment effectiveness in this context it helps to differentiate between studies of treatments used for conditions other than COVID in patients who then happen to be infected vs. medications that are specifically prescribed after an infection in the hope to alleviate the presentation of COVID.
Data sources
The data available for the evaluation of outpatient medication effectiveness and safety in COVID‐19 will be familiar to many RWD researchers, including administrative claims and EHR data. Measurement of outpatient medication dispensings is typically well‐captured through pharmacy claims. 18 The primary outcomes of interest in studies of outpatient medications and COVID‐19 are hospitalization and death, which are typically well‐captured in common RWE data sources. An exception is out‐of‐hospital deaths, which are captured poorly in data sources that do not have linkage to administrative or other death records. Because insurance coverage for COVID‐19 testing is compulsory for symptomatic patients, administrative claims will record most tests, but tests paid for out‐of‐pocket will not be recorded. Test results are also not typically recorded in claims, but a positive result may be inferred from a test followed by an International Classification of Disease‐10 diagnosis code for confirmed COVID‐19 (U07.1).
In the United States, the use of open claims data—to date, not very commonly used in research—has also risen in prominence. 19 The administrative claims data typically used in pharmacoepidemiology are so‐called “closed claims,” (i.e., claims that have been submitted to an insurance company, adjudicated, and paid). They generally contain a full record of all care that an insurance company has covered, which is often the complete care that a patient has received. Closed claims also include an enrollment file, which gives month‐by‐month information on whether a patient has insurance coverage—and as such, sets an expectation for “observability,” or which claims a patient incurs that a researcher would expect to see. A major disadvantage of closed claims, however, is the time it takes for this adjudication to occur; most closed claims sources have a data lag of 3–6 months, and the Centers for Medicare and Medicaid Services (CMS) claims can lag from 6 to 9 months. In the study of COVID‐19, where treatment and disease patterns can change month‐by‐month, this lag time is for many questions unacceptable.
Open claims, on the other hand, are claims captured through practice management systems (the information systems that manage medical practices’ scheduling, billing, and other internal functions), “switches” or “clearinghouses” (the companies that route claims from healthcare providers to US insurers), or pharmacy benefit managers (companies that provide the link between pharmacies and insurance companies). These organizations receive claims information often within days of a patient’s medical or pharmacy encounter, and as such, open claims provide a near real‐time view of patient activity. Open claims include much of the same information content as closed claims—diagnosis codes, procedure codes, drug identifiers, and the like—but present several challenges that closed claims do not.
The first challenge with open claims is patient‐level completeness: as open claims are received largely from intermediaries between the provider of the medical service and the payer organization, and not all intermediaries will serve all payers, there may be some selectivity as to which providers’ data are included (Figure 1 ). However, if patients tend to consistently seek care from the same providers, then this challenge should be mitigated. This issue is similar in EHR data, but unlike with EHR data, an intermediary may serve a wider range of providers than an EHR will capture. In both cases, investigators must think about what proportion of outcomes are likely to be missed. A second aspect is claims‐level completeness, or the fraction of claims expected to be captured that are actually captured. Claims‐level completeness is time‐dependent. Open pharmacy claims are observable in near totality within several days. For medical and other claims, claims accrue over time with the majority expected to be available within 21 days.
Figure 1.

Patient representation in open vs. closed claims data.
A further challenge is the enrollment file. Although closed claims generally provide an enrollment file that can be used to establish a denominator, open claims are more like EHR data in that they do not carry enrollment information. As such, determination of the relevant denominator must be estimated through, for example, evidence of activity; that is, any patient on whom activity is observed within a specific timeframe is included in the denominator. To avoid biases due to under‐inclusion of healthy individuals, definitions of “activity” that are more sensitive vs. more specific are recommended. 20 For a particular patient, the eligible person‐time can be established similarly, where activity indicates that a patient’s information would be expected to be captured, and lack of activity causes that patient’s person‐time to be excluded.
Fully understanding the effectiveness and safety of an outpatient medication may require data beyond open and closed claims. In particular, laboratory data are useful to confirm COVID‐19 illness or infection, whereas hospital data can help understand outcomes as patients transition from outpatient illness to inpatient treatment. EHR data can provide needed clinical context on comorbidities, COVID severity, or other factors. As different providers will likely use different patient identifiers, generating cross‐dataset, linkable identifiers are critical. Companies like HealthVerity or IQVIA have technology to inspect identifiable information to generate a unique “token”; this token can then be shared without any identifiers and thus link patient data across. Using tokens can help create full, longitudinal records for patients across care settings and best characterize and contextualize the performance of medications, while preserving nonidentifiability of individual patients.
Study design considerations
Nonrandomized methods for examining the effectiveness and safety of medications used in an outpatient setting are well‐established. 21 , 22 , 23 , 24 The new‐user, active comparator cohort study design (Figure 2 ) has been repeatedly demonstrated as a critical tool in the RWE toolbox. It follows the target trial paradigm, anchoring measurement of covariates and follow‐up for outcomes to the “index date,” or date of initiation of exposure in each treatment group. 25 , 26 Using an active comparator medication with indications similar to the exposure of interest has been shown to reduce the impact of confounding by indication by balancing even some unmeasured patient characteristics. 27 This design may be appropriate when evaluating the effectiveness of early outpatient treatment on prevention of hospitalization or death in a population of patients with confirmed COVID‐19, but using it in the context of COVID‐19 can be challenging.
Figure 2.
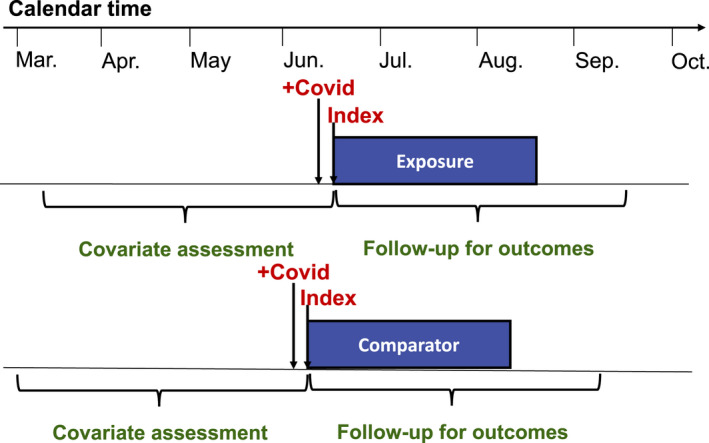
New user, active comparator, cohort study design in patients receiving early outpatient treatment for coronavirus disease 2019 (COVID‐19).
The rapid changes in COVID‐19 natural history and treatment decision making can create strong effects of calendar time. In particular, understanding of nonpharmacological treatments, such as prone positioning and use of supplemental oxygen, has improved rapidly, so that case fatality rates have gone down over time, independent of the medications used for treatments. Thus, it is important to ensure that any comparisons of medications control for calendar time so that patients in each treatment group have a similar distribution of the underlying risk of outcome. The need to control for calendar time also complicates selection of an appropriate comparator. Since the beginning of the pandemic, consensus on which medications, if any, may be effective in protecting against COVID‐19 illness in the outpatient setting has changed quickly. The novelty of the medications, or of the indication for existing drugs, in a space with few alternatives implies little choice of active comparators. For example, the rapid rise and fall of hydroxychloroquine prescribed to patients with COVID‐19 makes it both challenging to study as an exposure of interest as well as challenging to use as a comparator, because its time as a potential COVID‐19 treatment was limited. 28
Given the lack of a natural comparator in this context, investigators may instead consider comparing treated patients to patients receiving a positive COVID‐19 test but no treatment. Nonuser designs typically have stronger confounding by indication, because patients receiving any treatment are often different from those receiving no treatment at all. Identifying an index date for nonuser patients is also difficult, because there is no exposure for these patients on which to anchor index. In order to balance access to and utilization of the healthcare system between treatment groups, index dates may be instead anchored to a positive COVID‐19 test. Another common challenge that arises in nonuser designs is avoiding immortal time bias. A hallmark of potential immortal time bias is a design that first identifies all patients who use the exposure of interest at any time during the study period and subsequently identifies nonusers among the remaining patients by looking into the future using the existing data stream. 29 , 30 In this design, nonuser person time that occurs prior to a patient initiating the exposure of interest is necessarily immortal, and this person‐time is excluded from the nonuser group. Because there is no similar exclusion of immortal time in the exposure group, the nonuser group will appear worse than it should, creating bias (Figure 3 ). Mimicking a prospective randomized trial avoids this bias: patients should be selected for the nonuser group without regard to whether they go on to initiate the exposure of interest in the future, as in a risk‐set sampling design. 31
Figure 3.
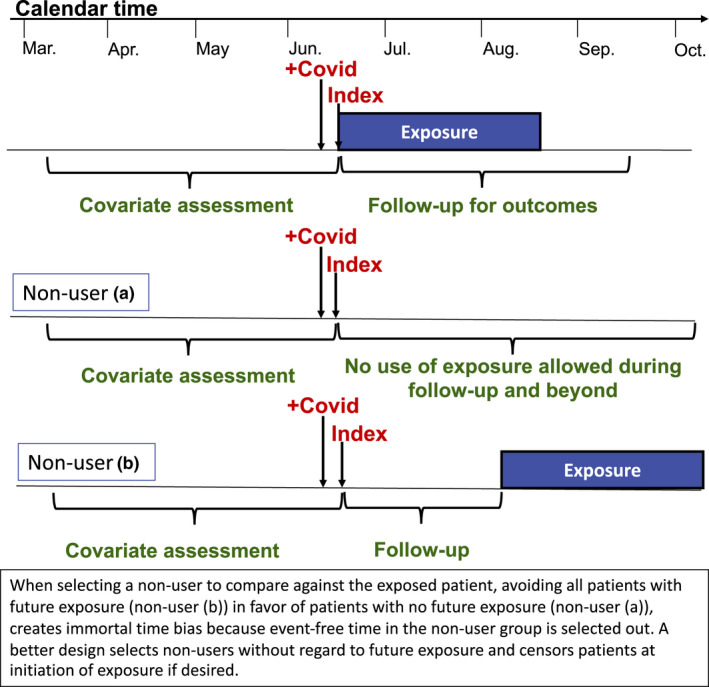
Potential study designs comparing patients receiving early outpatient treatment for coronavirus disease 2019 (COVID‐19) to nonusers.
Other considerations arise when evaluating the association of medications not specifically prescribed for treatment of COVID‐19 with severe disease or death among patients diagnosed with COVID‐19. In this context, a new‐user design will often not be possible, because patients may have been on the drugs of interest for months or years prior to their COVID‐19 diagnosis. Furthermore, outcomes on the medications prior to COVID‐19 infection is not of interest when studying the association with severe disease. Therefore, many such studies have identified a population of patients receiving a positive COVID‐19 test result and evaluated current use of medications at the time of the test. COVID‐19 outcomes can then be evaluated after the test (Figure 4 ). Because the medications under study are not prescribed specifically for treatment of COVID‐19, confounding by indication may be expected to be lower than in other studies evaluating medications and COVID‐19 outcomes. However, given the strong association between comorbidities and severe COVID‐19 disease, it is important to use an active comparator that is used in similar populations of patients with respect to rates of diabetes, cardiovascular disease, and other risk factors for severe COVID‐19 disease. In addition, it is important to verify that the comparator drug is used in patient populations with a similar distribution of race and socioeconomic status, given the increased risk of severe disease associated with Black and Hispanic patients and patients with lower socioeconomic status.
Figure 4.
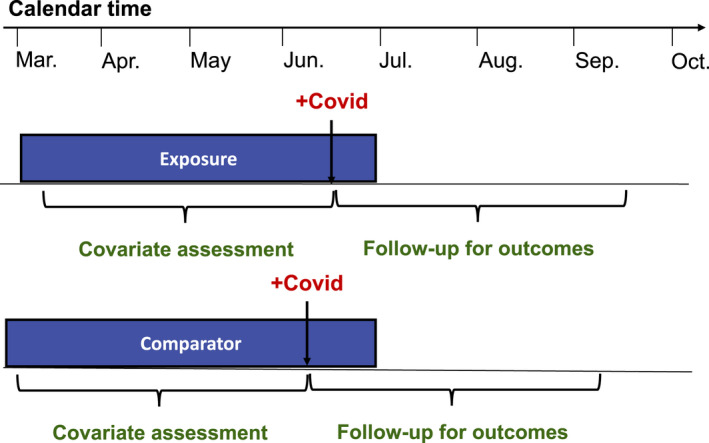
Prevalent user, active comparator, cohort study design evaluating the association of outpatient treatments not prescribed specifically for coronavirus disease 2019 (COVID‐19) with severe disease or death.
Some investigators may also consider the use of inpatient data sources, discussed in the next section, for evaluating the association between outpatient medications and severe disease by using records of outpatient medications collected at hospital admission for COVID‐19 and observing outcomes like death or intubation that occur during the hospitalization. However, this design is likely to suffer from severe selection bias, because patients must have COVID‐19 that is severe enough to warrant hospitalization to be included in the study. Patients who are on the drugs of interest that never present to the hospital are not included and the proportion of such patients may differ strongly between treatment groups.
TREATMENT EFFECTIVENESS IN HOSPITALIZED PATIENTS
For patients with mild COVID‐19 disease, supportive care has been the preferred management strategy 32 ; pharmacological treatment with possible antiviral effects has been primarily used for patients with moderate to severe disease requiring hospitalization. 33 , 34 Identifying treatments that can reduce morbidity and mortality in hospitalized patients is also one of the most urgent evidence needs related to COVID‐19. Therefore, the majority of comparative effectiveness research of COVID‐19 specific medications has been focused on evaluating treatment strategies to avoid death or intubation in patients hospitalized for COVID‐19.
Data sources
Typically, inpatient medical services are billed and reimbursed by the Diagnosis Related Group. Therefore, insurance claims data do not contain detailed information on the temporality of medication use in relation to other clinical events, which make it difficult to conduct claims data‐based research on inpatient medications. In contrast, EHR data of a care delivery system that provides inpatient services typically contain details of medication use, including timing, dose, and frequency, during hospitalization. EHR databases also provide rich time‐varying clinical data for patient phenotyping and confounding adjustments, such as vital signs, laboratory test results, smoking status, and body mass index. In addition, EHR data tend to have little time‐lag in data availability, which is an important advantage in studying a rapidly evolving disease like COVID‐19. For example, Geleris et al. used the EHR data of a large medical center in New York City to investigate the association between hydroxychloroquine use and intubation or death. Early in the pandemic, they were able to analyze 1,446 patients with a positive nasopharyngeal or oropharyngeal swab test result for the virus SARS‐CoV‐2 in the study EHR, and they published one of the earliest studies of hydroxychloroquine based on routine‐care data. 35
In the United States, another important data source for comparative effectiveness and safety research of inpatient pharmacotherapy is the Premier Healthcare Database (PHD), a repository of hospital utilization data from ~ 700 hospitals in all regions of the United States (covering 20–25% of all admissions in the United States). It includes data from hospital‐based encounters and provides information on patient demographics, hospital and physician characteristics, principal and secondary diagnoses, a date‐stamped log of all billed items (including medications, laboratory, and diagnostic services), and some cost data. 36 , 37 Researchers could potentially use this database to identify a large study population representative of hospitalized patients across the United States. For example, Shoaibi et al. compared adverse outcomes, including death and receiving intensive services, among patients hospitalized for COVID‐19 taking famotidine, proton pump inhibitors (PPIs), or hydroxychloroquine vs. famotidine nonusers. Using PHD, the authors identified 1,816 users of famotidine, 2,193 users of PPIs, 5,950 users of the hydroxychloroquine, and 26,820 non‐famotidine users. 38 Although it is substantially larger than other EHR‐based studies on the same topic, 39 PHD does not contain laboratory results to confirm COVID‐19 status, and the study cohort was defined as “condition, measurement or observation indicative of COVID‐19.” 38
In an inpatient setting, medication information can be generated at the following stages. (i) Electronic ordering data are produced when physicians prescribe a medication in the EHR system. What is prescribed is not necessarily taken by the patients because the medications can be ordered on an as‐needed basis, refused by the patients, or held by the provider based on prespecified holding parameters. (ii) Electronic medication administration data are generated when nurses scan patients’ identifying barcodes right before medication administration. (iii) Medication reconciliation is typically performed on admission (to compare the pre‐admission medication list with the admission orders) and at discharge (to compare the inpatient medications with the discharge orders). The pre‐admission medication list can be helpful in distinguishing new from prevalent users of the index drugs, which is important in avoiding common biases in drug effectiveness and safety research, such as depletion of susceptibles. 22 , 40 To optimize exposure assessment, researchers need to understand the type of medication data available in the study database and recognize its advantages and limitations. For example, Geleris et al. used electronic medication administration data from the EHR for their hydroxychloroquine study 35 and Shoaibi et al. used data generated by billed medication (corresponding to “administered medication”) in PHD for the study of famotidine vs. PPI. 38 Medication information based on inpatient administration data has little potential for drug exposure misclassification.
Clinical outcomes that are often of interest for COVID‐19 research include intensive care unit (ICU) admission, respiratory failure requiring mechanical ventilation, and inpatient mortality. These outcomes are typically well‐captured in the EHR or PHD. However, some nonICUs can be repurposed to serve as ICUs to manage patient surges during the COVID‐19 pandemic. So special attention is needed to check the validity and completeness of ICU status determination. For example, in the hydroxychloroquine study, Geleris et al. mentioned in the supplemental appendix, “Because of the dramatic increase in the need for ICU beds, multiple units that are not typically ICUs were converted to ICUs at various times as patient volumes increased, so identification of ICU patients was not possible.” 35 They thus used a composite of intubation or death as the primary end point.
To accurately assess patients “at risk to receive mechanical ventilation,” researchers will need information on code status because certain code status is not compatible with intubation (e.g., do‐not‐intubate or hospice care). As code status is a routine order during the admission process, it is often captured in EHR databases. Both the EHR and PHD are not an ideal data source for outcome ascertainment beyond the index admission. Relying on a single system EHR can lead to a substantial amount of misclassification for outcomes occurring after the index hospitalization due to out of system care, 41 but this bias can be potentially reduced by identifying a subcohort of higher data completeness with a validated algorithm. 42 On the other hand, PHD does not provide information after discharge and should only be used for research concerning inpatient events. 36
EHRs contain rich clinical information for confounding adjustment, patient phenotyping, and treatment heterogeneity evaluation. For example, in addition to demographic, comorbidity, and drug exposure variables, in the hydroxychloroquine analysis, Geleris et al. adjusted for vital signs, including heart rate, temperature, respiratory rate, blood pressure, and peripheral oxygen saturation, and a wide range of laboratory test results, including creatinine, D‐Dimer, ferritin, C‐reactive protein, lactate dehydrogenase, procalcitonin, and detailed oxygenation measured by the ratio of the partial pressure of arterial oxygen to the fraction of inspired oxygen (PaO2: FiO2). Body mass index and patient‐reported smoking status were also accounted for. In contrast, information on lifestyle factors, vital signs, and laboratory test results are not available in PHD. It is also important to note that PHD does not have comorbidity and medication use data before admission. For EHR‐based studies, some people may have longitudinal follow‐up within the study EHR (e.g., seeing the primary care physician in the system), but some may have incomplete data before admission (e.g., admitted via emergency department for the first time in the system). Consequently, misclassification of baseline covariates can be substantial for those who did not come to study EHR system for medical follow‐up. 41
For both EHR‐based and PHD‐based studies, researchers can use diagnoses recorded in the index admission to enhance identification of pre‐existing comorbidities, such as diabetes, hypertension, chronic obstructive pulmonary disease, congestive heart failure, and dementia, because the onset of these conditions likely precedes the hospitalization. On the other hand, one may not be able to distinguish with certainty whether an acute condition recorded as an inpatient diagnosis was an event that occurred during the index hospitalization or a relevant prior illness treated during the hospitalization, such as myocardial infarction, stroke, or venous thromboembolism, conditions that occur more commonly in patients with severe COVID‐19. 43 Researchers may need to apply additional algorithms or validations to accurately determine presence of these conditions based on the inpatient diagnosis.
Study design considerations
Most published studies of inpatient medications for COVID‐19 published so far have used a parallel‐group cohort design, similar to that described in the context of outpatient medications. However, within this general framework, there have been a wide variety of design choices, many known to result in major avoidable biases. For example, Geleris et al. set the study index to be 24 hours after hospital admission for all study patients and assessed exposure to hydroxychloroquine anytime from index through the end of follow‐up (Figure 5 ). Whereas this design allows investigators to capture outcome events occurring soon after hospital admission and allows for maximum capture of hydroxychloroquine exposure, it may suffer from immortal time bias, as patients who did not have hydroxychloroquine at baseline but received it later must have survived long enough to receive it, thereby giving the hydroxychloroquine group a mortality advantage compared with the nonuser group. This possibility was investigated in sensitivity analyses. Similar designs setting the index at or near hospital admission and assessing exposure during follow‐up for outcomes have been used in other studies of inpatient treatment of patients with COVID‐19. 10 , 44
Figure 5.
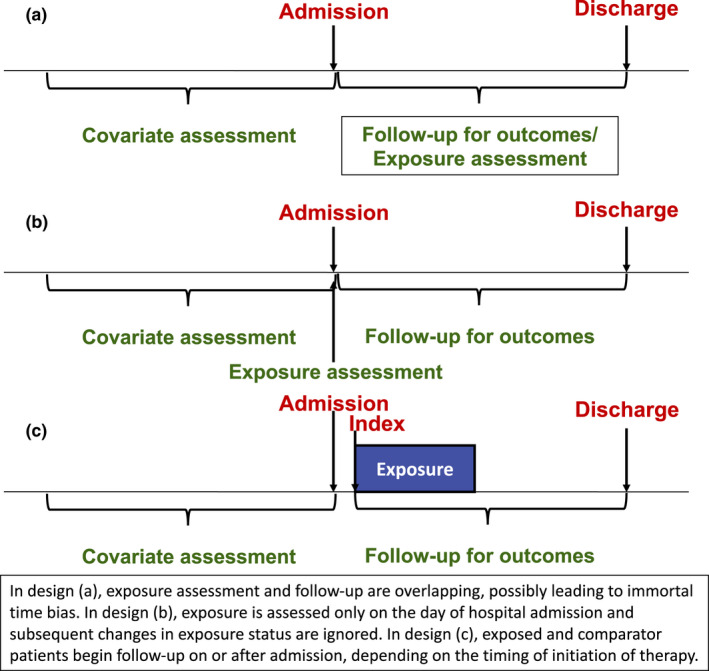
Potential study designs evaluating treatments for coronavirus disease 2019 (COVID‐19) in hospitalized patients.
The study of Shoaibi et al. instead classified patients by their medication use only on the index date, the day of admission. Follow‐up for outcomes began the day after admission, and changes to drug regimens during follow‐up were not considered. Although this design avoided concerns of immortal time bias, it also required a large database like PHD to ensure that there were sufficient numbers of patients receiving medications on the day of admission and would likely not be possible in a typical EHR database, depending on utilization patterns of the drugs under study. The design also cannot evaluate how changes in medication after the admission date impact study outcomes. This limitation may be especially impactful in a nonuser group, because many nonusers are likely to go on to initiate a study medication during subsequent days in the hospital.
In order to overcome these limitations, an alternative design could mirror the cohort design typically used for outpatient medications, where the index date is set to be the day when exposure is initiated. As in the study of outpatient medications, this design is relatively straightforward when using an active comparator, but selection of the index date is more complicated for a nonuser comparator. If users and nonusers are matched, then nonusers may be assigned the index date of their matched exposed patient, for example, day 2 after hospital admission. This design would require that measures of disease severity, such as oxygen saturation and levels of inflammatory markers, are available on the index date, because these measures are likely driving the decision to initiate treatment on that day. As in the study of outpatient medications, changes in underlying risk of death and severe disease from COVID‐19 over time require that patients being compared should have been hospitalized in a similar calendar period, so matching by month of hospitalization is also advisable.
At the initial peak of the pandemic in April 2020, many hospitals had a single treatment strategy and all patients received nearly identical treatment regimens, for example, hydroxychloroquine. However, not all hospitals used the same treatment strategy, and some implemented a time‐delay before starting similar strategies. Such treatment variation on the hospital level, but not patient level, could be exploited with instrumental variable analyses. If patients are quasi‐randomly sent to the closest hospital that can deal with COVID‐19, then the patient characteristics would be independent from the hospital’s treatment strategy, and, thus, valid inference may be achieved. 45 , 46
VACCINES
Operation Warp Speed (OWS), a partnership among several federal agencies and the biopharma industry, is intended to support promising vaccine candidates and approval or authorization by the US Food and Drug Administration (FDA) in order to make initial doses of COVID‐19 vaccine available by January 2021 and to produce and deliver 300 million doses of safe and effective vaccines by mid‐2021. OWS draws on experience with Zika and West African Ebola, but with unprecedented timelines. Although biopharma executes on clinical development plans, the government agency side of OWS leverages the US government to overcome hurdles that would otherwise hinder vaccine development or deployment. As a result, far less will be known about the long‐term safety of initially approved vaccines than is typically the case.
The first‐in‐human trials began in March 2020. In June, the FDA announced 50% effectiveness would be required for approval, and that an EUA may be considered after a median of 2 months following the completed vaccine regimen. Two companies (Moderna and Pfizer) recently reported better than 90% efficacy based on interim analyses and received US EUA in December 2020. 47 , 48 Although these trials are large (30,000 and 43,538 patients randomized in the Moderna and Pfizer trials, respectively), longer‐term safety will need to be monitored postapproval. The safety of COVID‐19 vaccines, like all vaccines, will be monitored using existing surveillance systems, but the FDA and other regulatory agencies are very likely to require postmarketing studies to further assess known or potential risks. A strong, principled, robust surveillance system will be essential to convince the public of the safety of the COVID‐19 vaccination program amid high vaccine hesitancy. 49 In addition, as more vaccine candidates are approved (as of January 2021 there were 66 in clinical trials in humans, 20 of which are in Phase 3), there may be a need for assessments of comparative effectiveness and safety, particularly as administration of some vaccines may be more difficult than others due to the need for multiple doses or cold‐chain distribution, thereby impacting effectiveness in routine care use.
Data sources
Several well‐known data sources exist for evaluating the safety of vaccines. 50 Passive vaccine surveillance and spontaneous reporting systems allow for an initial signal detection. In the United States, the Centers for Disease Control and Prevention (CDC) and the FDA use data from the Vaccine Adverse Event Reporting System (VAERS), a voluntary reporting system, to monitor safety and conduct descriptive analyses, disproportionality analysis, or observed‐to‐expected analyses. 51 , 52 Spontaneous reporting systems lack a denominator (e.g., total number of vaccinated individuals), precluding the ability to determine incidence of adverse events. In addition, due to the voluntary nature of safety reporting in VAERS, reporting rates may reflect reporting bias.
To confirm potential signals, post‐licensure phase IV surveillance studies have historically used a large distributed data network of linked healthcare claims and EHR databases for active safety surveillance. In 1990, the CDC, in collaboration with multiple managed care organizations, initiated the Vaccine Safety Datalink (VSD) project, linking vaccination data with medical outcome data, such as hospitalizations and outpatient visits. 53 , 54 The Post‐Licensure Rapid Immunization Safety Monitoring (PRISM) program, a part of the FDA’s Sentinel system, is another distributed data network consisting of vaccination data from immunization registries and several national health insurers that allows for prospective active safety monitoring of vaccination products. 55 , 56 Due to the large size of this data source, analysis of rare adverse events following immunizations can be conducted.
Recently, novel curated data sources have been developed for COVID‐19‐specific research purposes. Examples include COVID‐19 cohorts from HealthVerity, Health Catalyst, COTA Healthcare, and Hackensack Meridian Health’s Real world Evidence‐COVid‐RegistrY (RE‐COV‐RY), Optum COVID‐19 electronic health records, and IQVIA COVID Active Research Experience (CARE) Project registry. 57 , 58 , 59 , 60 , 61 In addition, the COVID‐19 Research Database, initiated by Datavant and a consortium of healthcare companies, includes de‐identified patient‐level data and is freely available to academic and scientific researchers. 62
A major hurdle in all of these data sources is accurate identification of administration of specific COVID‐19 vaccines. Use of healthcare claims or EHR data for vaccine studies typically relies on records of billing for the vaccine product. However, mass vaccination campaigns, such as the one expected for COVID‐19, often provide the vaccine free of charge, so that no claim for the vaccine is recorded. In addition, a lack of billing for the vaccine product makes it impossible to distinguish which specific vaccine is given when there are multiple vaccines available for a single disease. To avoid these problems, the American Medical Association recently announced an update to the Current Procedural Terminology code set that includes new codes for administration of specific vaccines: one for a 30 mcg dose and one for a 100 mcg dose, corresponding exactly to the Pfizer and Moderna vaccine candidates. 63 Implementation of these codes prior to approval or authorization of any COVID‐19 vaccine ensures that data on safety and comparative effectiveness can begin accruing with the first patients receiving the vaccines in routine care.
Study design considerations
RWE will be critical to timely safety surveillance and comparative effectiveness and safety studies, but mass vaccination campaigns will make typical postapproval comparative study designs especially challenging, as there are likely to be systematic differences among those vaccinated and those not vaccinated. Thus, monitoring of adverse event rates will be critical and inferential designs that do not require an unvaccinated comparator, such as the self‐controlled case series (SCCS) and self‐controlled risk interval (SCRI) designs will be needed. Comparative effectiveness analyses will benefit from the use of active comparators. Self‐controlled study designs, which are limited to people with the adverse event of interest (which can be seen as using only the “cases” from an underlying cohort study), leverage exposed and unexposed time within the same individual. Comparing these risk periods within the same individual avoids confounding by measured and unmeasured variables that do not change over the study period (see Table 1 ).
Table 1.
Self‐controlled case series vs. self‐controlled risk interval
| Self‐controlled case series (SCCS) | Self‐controlled risk interval (SCRI) | |
|---|---|---|
| Typical uses | Safety of drugs or vaccines on acute‐onset outcomes | Mainly used to study safety of vaccines on acute‐onset outcomes |
| Who’s included | Cases. Patients with the outcome during the study period. Never‐treated patients can be included to contribute to the estimation of time‐varying confounder effects | Exposed cases. Patients with the outcome and the exposure during the study period. |
| Study period | Typically calendar year(s) | Time immediately surrounding an exposure event. |
| Design | Incidence during risk interval compared to that during baseline time. All time that isn’t part of a risk interval (or a pre‐risk or washout interval, if used) is included as baseline time. | Incidence during risk interval compared to that during control interval. Control interval is typically short. |
| Advantage | Greater power than SCRI. All time outside risk periods (and washout + pre‐risk periods) is used as baseline time | Less susceptible to time‐varying confounding than SCCS |
| Disadvantage | More susceptible to time‐varying confounding than SCRI due to longer study period. Must be handled through adjustment. | Less power than SCCS |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The SCCS was developed to evaluate the impact of vaccines on acute adverse events but has been used in a range of vaccine and drug safety applications. Under this design, incidence rates are compared during risk intervals and baseline person‐time among individuals with the adverse event of interest. 64 , 65 , 66 , 67 The exposure intervals are defined within a specified study period, which may be defined by calendar time or by other time scales, such as age. A valid SCCS design requires that outcome events arise in a nonhomogeneous Poisson process, so risk of the event is changing over time, depending on a patient’s exposure status. If recurrences are correlated, an analysis of the first outcome only may be appropriate, provided the outcome is rare. SCCS designs also require that events do not influence subsequent exposures. Short‐term violations of this assumption, such as cases where the outcome is a short‐term indication or contraindication for exposure, can be handled by including a pre‐risk period; alternative solutions are available for cases where the outcome precludes any further exposure. Additionally, SCCS requires the outcome events do not influence the length of the observation period (e.g., by increasing the risk of mortality). It is important to note that, whereas the SCCS accounts for measured and unmeasured confounders that do not vary over time, time‐varying confounders must be adjusted for in the analysis.
The SCRI is a special case of the SCCS study design that has been widely used in vaccine safety. 68 , 69 Only vaccinated cases are included in the SCRI. The “risk interval” is defined following vaccination and one or more “control interval(s)” are defined either prevaccination or postvaccination (Figure 6 ). The control intervals are typically short in length and close in time to the risk interval. Like the SCCS, in the SCRI, the incidence rate of the event of interest in the risk interval is compared with the incidence rate in the control interval. The strengths and limitations of the SCRI are similar to the SCCS. The SCRI tends to be less susceptible to time‐varying confounders due to the shorter analysis period, but it has reduced statistical power due to the inclusion of less unexposed time. 70
Figure 6.
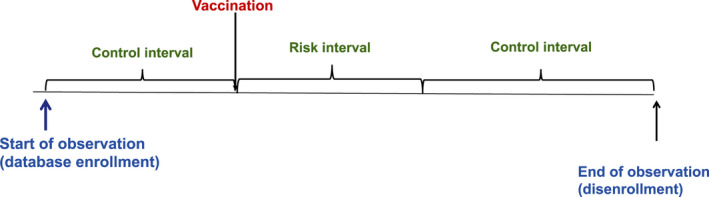
Self‐controlled risk interval design for assessment of vaccine safety.
APPROACHES TO STRENGTHEN THE CREDIBILITY OF RWE IN A MEDICAL CRISIS
The COVID‐19 pandemic has increased aspirations for the ability of RWE to provide badly needed evidence in a medical emergency. At the same time, the major policy and research impacts of poor‐quality RWE on COVID treatments have renewed criticisms that RWE cannot be trusted. Although this paper provides many suggestions on approaches to support methodological quality of RWE studies conducted to evaluate the safety and effectiveness of vaccines and medications for COVID, questions remain regarding how to strengthen the credibility of RWE in a pandemic and ensure that the RWE impacting decision making is of the highest quality possible.
The first step in evaluating the validity and credibility of RWE is understanding the RWD source from which it is derived. Although there are many well‐known and trusted RWD sources available for research, including the sources discussed in prior sections, the urgency of a pandemic may prompt investigators to explore new data sources. How can the medical and scientific communities quickly assess a new data source and come to understand its strengths and limitations? Any research purporting to use a new data source should provide ample description of data provenance, completeness, and aggregation practices. For example, if the data source relies on aggregation of medical records from multiple hospitals, as Surgisphere claimed, then at least a partial list of hospitals that are contributing records to the database should be provided, even if individual hospitals are anonymized in the data. Information on the completeness and precision of assessment of key confounding variables is also essential. There should also be some record of how EHR systems at different hospitals record different types of information. For example, what type of medication information is captured: medication dispensing, prescriptions written, medication reconciliation records, administration data, or only patient reports of medications used? If there is variability across hospitals in data capture, then that can be reported to help investigators understand the quality of measurement across the data. Other features of data aggregation and curation can also be reported, such as how often data are transferred to the database, what data checks are done by the data provider before transfer, and what checks are done by the data aggregator.
Assuming the data being used to produce RWE are credible and fit for use, then study design, analysis, and findings should be clearly reported to allow others to evaluate the study’s merits.
Due to the complexity of RWE derived from healthcare databases, it is often difficult to map simple text descriptions of study procedures provided in manuscripts into algorithms needed for extraction of study cohorts and definition of variables. 71 Space constraints further reduce the amount of detail reported in manuscripts. Therefore, providing detailed protocols that define all study details is preferable and likely to lead to a better understanding of design and analysis. There has also been considerable work on defining protocol templates that dictate clear and complete reporting of study details and promote study replicability, allowing important findings on COVID therapeutics to be quickly reproduced in the same database or in other databases.
Pre‐registration of such protocols further improves credibility of study findings. Particularly in the context of COVID, where evidence on treatments has both scientific and political ramifications, there may be additional concerns that RWE can be manipulated by conducting many RWD analyses and reporting only the small subset that produce a desired treatment effect. Publicly posting or registering a protocol prior to conducting analyses mitigates these concerns, because other investigators can verify that the reported analyses were prespecified and therefore not selected based on the favorability of their results.
Finally, detailed protocols and study reports must be reviewed by the scientific community in order to evaluate the scientific rigor of the design and whether the selected RWD source adequately measures the variables of interest, including all factors influencing the outcomes that might also affect the decision to use the specific treatments compared. This review will most often be facilitated through peer review of a manuscript submitted to a journal; however, the proliferation of published RWE studies of COVID‐19 treatments with major design flaws demonstrated that typical peer review may not be enough to ensure that only high‐quality studies are published. Evaluation of study design and analysis for RWE in COVID‐19 requires expertise not only in the relevant clinical areas but also in methodology for healthcare databases. When considering an RWE study for publication, journals should consider inviting at least one reviewer with a publication track record in using healthcare databases to assess the causal effects of treatments. Given its long history of using healthcare databases to assess the causal effects of treatments, the fields of pharmacoepidemiology or outcomes research can provide expertise on whether data are fit for use and whether the design avoids major biases and adequately addresses confounding.
CONCLUSION
Developing high‐quality nonrandomized studies from RWE databases is critical for supporting medical and public health decision making in the COVID‐19 pandemic. The large size of many healthcare databases can often answer questions on rare safety events of vaccines and treatments that cannot be answered in RCTs. In addition, RWE can contribute information on the comparative safety and effectiveness of medications and vaccines in routine care where complex and frail patients are receiving a rapidly increasing number of therapeutics. However, these advantages are accompanied by several potential disadvantages, including lack of controlled and standardized measurements and lack of baseline randomization to control confounding resulting from treatment selection. RWE is helpful for making prescribing decisions only after its limitations are thoroughly assessed and addressed.
Although this paper focused on the use of RWE to evaluate the causal effects of vaccines and treatments for COVID‐19, there are many other important uses of RWE related to COVID‐19. RWE has been the primary source of evidence describing patient symptoms and how patient characteristics influence risk of COVID‐19 morbidity and mortality. 72 , 73 , 74 , 75 RWE has also contributed to evaluations supporting the use of nonpharmacological interventions to reduce virus spread, such as mask wearing. 76 Future research may use RWE to evaluate the effectiveness of contact tracing and other public health measures; to assess the pandemic’s impact on other areas of health, such as rates of depression and suicide; and to measure the long‐term impacts of COVID‐19 infection.
FUNDING
This work was funded by the Division of Pharmacoepidemiology and Pharmacoeconomics, Department of Medicine, Brigham and Women’s Hospital and by Aetion, Inc. J.M.F. was supported by NHLBI grant R01HL141505.
CONFLICT OF INTEREST
Dr. Schneeweiss is consultant to WHISCON, LLC and to Aetion, Inc. of which he also owns equity. He is principal investigator of grants to the Brigham and Women’s Hospital from Bayer, Vertex, and Boehringer Ingelheim unrelated to the topic of this paper. Drs. Gatto and Rassen are employees of and have an ownership stake in Aetion, Inc. All other authors declared no competing interests for this work.
Acknowledgments
The authors thank Amanda Patrick and Pattra Mattox for editorial support and helpful comments.
References
- 1. Beigel, J.H. et al. Remdesivir for the treatment of Covid‐19 — preliminary report. N. Engl. J. Med. 383(10), 994 (2020). [DOI] [PubMed] [Google Scholar]
- 2. Brown, S.M. et al. Hydroxychloroquine versus azithromycin for hospitalized patients with suspected or confirmed COVID‐19 (HAHPS). Protocol for a pragmatic, open‐label, active comparator trial. Annals ATS. 17, 1008–1015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boulware, D.R. et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid‐19. N. Engl. J. Med. 383(6), 517–525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization . “Solidarity” clinical trial for COVID‐19 treatments <https://www.who.int/emergencies/diseases/novel‐coronavirus‐2019/global‐research‐on‐novel‐coronavirus‐2019‐ncov/solidarity‐clinical‐trial‐for‐covid‐19‐treatments>. Accessed September 2, 2020.
- 5. The RECOVERY Collaborative Group et al . Dexamethasone in hospitalized patients with Covid‐19 — preliminary report. N. Engl. J. Med 10.1056/NEJMoa2021436. [e‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonamici, S. H.R.34 ‐ 114th Congress (2015‐2016): 21st Century Cures Act. Published December 13, 2016 <https://www.congress.gov/bill/114th‐congress/house‐bill/34>. Accessed June 15, 2017.
- 7. US Food and Drug Administration . Prescription Drug User Fee Act (PDUFA) ‐ PDUFA VI: Fiscal Years 2018–2022 <https://www.fda.gov/forindustry/userfees/prescriptiondruguserfee/ucm446608.htm>. Accessed June 15, 2017.
- 8. Sherman, R.E. et al. Real‐world evidence—what is it and what can it tell us. N. Engl. J. Med. 375, 2293–2297 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Jarow, J.P. , LaVange, L. & Woodcock, J. Multidimensional evidence generation and FDA Regulatory Decision Making: defining and using “real‐world” data. JAMA 318, 703–704 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Mehra, M.R. , Desai, S.S. , Ruschitzka, F. & Patel, A.N. RETRACTED: Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID‐19: a multinational registry analysis. Lancet. 10.1016/S0140-6736(20)31180-6 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Mahase, E. Covid‐19: 146 researchers raise concerns over chloroquine study that halted WHO trial. BMJ. 369, m2197 (2020). [DOI] [PubMed] [Google Scholar]
- 12. Watson, J. et al. An open letter to Mehra et al and The Lancet. Zenodo <https://gonaturecom/3eRhiuo> (2020).
- 13. Mehra, M.R. , Ruschitzka, F. & Patel, A.N. Retraction—Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID‐19: a multinational registry analysis. Lancet. 395, 13–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Califf, R.M. , Hernandez, A.F. & Landray, M. Weighing the benefits and risks of proliferating observational treatment assessments: observational cacophony, randomized harmony. JAMA 324, 625 (2020). [DOI] [PubMed] [Google Scholar]
- 15. Khera, R. et al. Association of angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers with the risk of hospitalization and death in hypertensive patients with coronavirus disease‐19. medRxiv 10.1101/2020.05.17.20104943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schneeweiss, M.C. , Leonard, S. , Weckstein, A. , Schneeweiss, S. & Rassen, J. Renin‐angiotensin‐aldosterone‐system inhibitor use in patients with COVID‐19 infection and prevention of serious events: a cohort study in commercially insured patients in the US. medRxiv. (2020) https://www.medrxiv.org/content/10.1101/2020.07.22.20159855v1. [Google Scholar]
- 17. Lo, K.B. et al. Angiotensin converting enzyme inhibitors and angiotensin II receptor blockers and outcomes in patients with COVID‐19: a systematic review and meta‐analysis. Exp. Rev. Cardiovasc. Ther. 18, 919–930 (2020. [DOI] [PubMed] [Google Scholar]
- 18. Steiner, J.F. & Prochazka, A.V. The assessment of refill compliance using pharmacy records: methods, validity, and applications. J. Clin. Epidemiol. 50, 105–116 (1997). [DOI] [PubMed] [Google Scholar]
- 19. Lane, J.C.E. et al. Risk of hydroxychloroquine alone and in combination with azithromycin in the treatment of rheumatoid arthritis: a multinational, retrospective study. Lancet Rheumatol. 2, e698–e711 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rassen, J.A. , Bartels, D.B. , Schneeweiss, S. , Patrick, A.R. & Murk, W. Measuring prevalence and incidence of chronic conditions in claims and electronic health record databases. Clin. Epidemiol. 11, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneeweiss, S. A basic study design for expedited safety signal evaluation based on electronic healthcare data. Pharmacoepidemiol. Drug Saf. 19(8), 858–868 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson, E.S. et al. The incident user design in comparative effectiveness research. Pharmacoepidemiol. Drug Saf. 22(1), 1–6 (2013). [DOI] [PubMed] [Google Scholar]
- 23. Franklin, J.M. & Schneeweiss, S. When and how can real world data analyses substitute for randomized controlled trials? Real world evidence and RCTs. Clin. Pharmacol. Ther. 102, 924–933 (2017). [DOI] [PubMed] [Google Scholar]
- 24. Franklin, J.M. , Glynn, R.J. , Martin, D. & Schneeweiss, S. Evaluating the use of nonrandomized real‐world data analyses for regulatory decision making. Clin. Pharmacol. Ther. 105, 867–877 (2019). [DOI] [PubMed] [Google Scholar]
- 25. Hernán, M.A. et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology 19, 766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hernán, M.A. & Robins, J.M. Using big data to emulate a target trial when a randomized trial is not available. Am. J. Epidemiol. 183, 758–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patorno, E. et al. Claims‐based studies of oral glucose‐lowering medications can achieve balance in critical clinical variables only observed in electronic health records. Diabetes Obes. Metab. 20(4), 974–984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bull‐Otterson, L. et al. Hydroxychloroquine and chloroquine prescribing patterns by provider specialty following initial reports of potential benefit for COVID‐19 treatment ‐ United States, January‐June 2020. MMWR Morb. Mortal Wkly. Rep. 69, 1210–1215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suissa, S. Immortal time bias in observational studies of drug effects. Pharmacoepidemiol. Drug Saf. 16, 241–249 (2007). [DOI] [PubMed] [Google Scholar]
- 30. Suissa, S. Immortal time bias in pharmacoepidemiology. Am. J. Epidemiol. 167, 492–499 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Patorno, E. , Glynn, R.J. , Levin, R. , Lee, M.P. & Huybrechts, K.F. Benzodiazepines and risk of all cause mortality in adults: cohort study. BMJ 358, j2941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bajwah, S. et al. Managing the supportive care needs of those affected by COVID‐19. Eur. Respir. J. 55(4), 2000815 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanders, J.M. , Monogue, M.L. , Jodlowski, T.Z. & Cutrell, J.B. Pharmacologic treatments for coronavirus disease 2019 (COVID‐19): a review. JAMA 323, 1824–1836 (2020). [DOI] [PubMed] [Google Scholar]
- 34. Lin, K.J. et al. Pharmacotherapy for hospitalized patients with COVID‐19: treatment patterns by disease severity. Drugs 80, 1961–1972 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geleris, J. et al. Observational study of hydroxychloroquine in hospitalized patients with Covid‐19. N. Engl. J. Med. 382, 2411–2418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Premier Applied Sciences®, Premier Inc. Premier healthcare database: data that informs and performs <https://learn.premierinc.com/white‐papers/premier‐healthcare‐database‐whitepaper> (2020).
- 37. Schneeweiss, S. , Seeger, J.D. , Landon, J. & Walker, A.M. Aprotinin during coronary‐artery bypass grafting and risk of death. N. Engl. J. Med. 358, 771–783 (2008). [DOI] [PubMed] [Google Scholar]
- 38. Shoaibi, A. Comparative effectiveness of famotidine in hospitalized COVID‐19 patients. medRxiv (2020) https://www.medrxiv.org/content10.1101/2020.09.23.20199463v1 . [DOI] [PubMed] [Google Scholar]
- 39. Sethia, R. Efficacy of famotidine for COVID‐19: a systematic review and meta‐analysis. medRxiv 10.1101/2020.09.28.20203463. [DOI] [Google Scholar]
- 40. Schneeweiss, S. et al. Increasing levels of restriction in pharmacoepidemiologic database studies of elderly and comparison with randomized trial results. Med Care, 45(10 suppl), S131 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin, K.J. , Glynn, R.J. , Singer, D.E. , Murphy, S.N. , Lii, J. & Schneeweiss, S. Out‐of‐system care and recording of patient characteristics critical for comparative effectiveness research. Epidemiology 29, 356–363 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin, K.J. , Singer, D.E. , Glynn, R.J. , Murphy, S.N. , Lii, J. & Schneeweiss, S. Identifying patients with high data completeness to improve validity of comparative effectiveness research in electronic health records data. Clin. Pharmacol. Ther. 103, 899–905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klok, F. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19. Thromb. Res. 191, 145–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arshad, S. et al. Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalized with COVID‐19. Int. J. Infect. Dis. 97, 396–403 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McClellan, M. Does more intensive treatment of acute myocardial infarction in the elderly reduce mortality? Analysis using instrumental variables. JAMA 272, 859 (1994). [PubMed] [Google Scholar]
- 46. Brookhart, M.A. , Rassen, J.A. & Schneeweiss, S. Instrumental variable methods in comparative safety and effectiveness research. Pharmacoepidemiol. Drug Saf. 19, 537–554 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pfizer . Pfizer and BioNTech announce vaccine candidate against COVID‐19 achieved success in first interim analysis from phase 3 study <https://www.pfizer.com/news/press‐release/press‐release‐detail/pfizer‐and‐biontech‐announce‐vaccine‐candidate‐against> (2020).
- 48. Moderna . Moderna’s COVID‐19 vaccine candidate meets its primary efficacy endpoint in the first interim analysis of the Phase 3 COVE Study <https://investors.modernatx.com/news‐releases/news‐release‐details/modernas‐covid‐19‐vaccine‐candidate‐meets‐its‐primary‐efficacy> (2020).
- 49. Fisher, K.A. , Bloomstone, S.J. , Walder, J. , Crawford, S. , Fouayzi, H. & Mazor, K.M. Attitudes toward a potential SARS‐CoV‐2 vaccine: a survey of US adults. Ann. Intern. Med. 173(12), 964–973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen, R.T. Pharmacoepidemiologic studies of vaccine safety. In Phramacoepidemiology, 6th edn (eds. Strom, B.L. , Kimmel, S.E. & Hennessy, S. ). (John Wiley & Sons Ltd., New York, NY, 2020). [Google Scholar]
- 51. Shimabukuro, T.T. , Nguyen, M. , Martin, D. & DeStefano, F. Safety monitoring in the Vaccine Adverse Event Reporting System (VAERS). Vaccine 33, 4398–4405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mahaux, O. , Bauchau, V. & Van Holle, L. Pharmacoepidemiological considerations in observed‐to‐expected analyses for vaccines. Pharmacoepidemiol. Drug Saf. 25, 215–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen, R.T. et al. Vaccine Safety Datalink project: a new tool for improving vaccine safety monitoring in the United States. Pediatrics. 99, 765–773 (1997). [DOI] [PubMed] [Google Scholar]
- 54. DeStefano, F. & Vaccine Safety Datalink Research Group . The Vaccine Safety Datalink project. Pharmacoepidemiol. Drug Saf. 10, 403–406 (2001). [DOI] [PubMed] [Google Scholar]
- 55. Nguyen, M. , Ball, R. , Midthun, K. & Lieu, T.A. The Food and Drug Administration’s post‐licensure rapid immunization safety monitoring program: strengthening the federal vaccine safety enterprise. Pharmacoepidemiol Drug Saf. 21(suppl 1), 291–297 (2012). [DOI] [PubMed] [Google Scholar]
- 56. Baker, M.A. , Nguyen, M. , Cole, D.V. , Lee, G.M. & Lieu, T.A. Post‐licensure rapid immunization safety monitoring program (PRISM) data characterization. Vaccine 31(suppl 10), K98–K112 (2013). [DOI] [PubMed] [Google Scholar]
- 57. Health Catalyst . Covid‐19 health catalyst response https://www.healthcatalyst.com/covid‐19/. Accessed February 16, 2021.
- 58. COTA Team . COTA collaborates to understand COVID‐19 using real‐world evidence – new findings published https://cotahealthcare.com/cota‐collaborates‐to‐understand‐covid‐19‐new‐findings‐published/.
- 59. Hackensack Meridian Health . Hackensack Meridian Health DEVELOPS RE‐COV‐RY (Real world Evidence –COVid‐RegistrY) to study the effects of COVID‐19 and releases key findings on hypertension and ACE/ARB treatments (2020). https://www.hackensackmeridianhealth.org/press‐releases/2020/04/24/hackensack‐meridian‐health‐develops‐re‐cov‐ry‐real‐world‐evidence‐covid‐registry‐to‐study‐the‐effects‐of‐covid‐19‐and‐releases‐key‐findings‐on‐hypertension‐and‐ace‐arb‐treatments/.
- 60. Solow, B. Optum life sciences COVID‐19 resources. https://www.optum.com/business/resources/library/life‐sciences‐covid‐19‐resources.html
- 61. IQVIA . IQVIA COVID‐19 Active Research Experience (Care) Project https://www.helpstopcovid19.com.
- 62. COVID‐19 Research Database https://covid19researchdatabase.org.
- 63. Mills, R. AMA announces vaccine‐specific CPT codes for coronavirus immunizations <https://www.ama‐assn.org/press‐center/press‐releases/ama‐announces‐vaccine‐specific‐cpt‐codes‐coronavirus‐immunizations> (2020). Accessed November 16, 2020.
- 64. Farrington, C.P. Relative incidence estimation from case series for vaccine safety evaluation. Biometrics 51, 228–235 (1995). [PubMed] [Google Scholar]
- 65. Farrington, P. et al. A new method for active surveillance of adverse events from diphtheria/tetanus/pertussis and measles/mumps/rubella vaccines. Lancet 345, 567–569 (1995). [DOI] [PubMed] [Google Scholar]
- 66. Weldeselassie, Y.G. , Whitaker, H.J. & Farrington, C.P. Use of the self‐controlled case‐series method in vaccine safety studies: review and recommendations for best practice. Epidemiol. Infect. 139, 1805–1817 (2011). [DOI] [PubMed] [Google Scholar]
- 67. Whitaker, H.J. , Paddy Farrington, C. , Spiessens, B. & Musonda, P. Tutorial in biostatistics: the self‐controlled case series method. Stat. Med. 25, 1768–1797 (2006). [DOI] [PubMed] [Google Scholar]
- 68. Lee, G.M. et al. H1N1 and seasonal influenza vaccine safety in the vaccine safety datalink project. Am. J. Prev. Med. 41, 121–128 (2011). [DOI] [PubMed] [Google Scholar]
- 69. Tse, A. , Tseng, H.F. , Greene, S.K. , Vellozzi, C. , Lee, G.M. & V. S. D., Rapid Cycle Analysis Influenza Working Group . Signal identification and evaluation for risk of febrile seizures in children following trivalent inactivated influenza vaccine in the Vaccine Safety Datalink Project. 2010–2011. Vaccine. 30, 2024–2031 (2012). [DOI] [PubMed] [Google Scholar]
- 70. Li, R. , Stewart, B. & Weintraub, E. Evaluating efficiency and statistical power of self‐controlled case series and self‐controlled risk interval designs in vaccine safety. J. Biopharm. Stat. 26, 686–693 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang, S.V. et al. Reporting to improve reproducibility and facilitate validity assessment for healthcare database studies V1.0. Pharmacoepidemiol. Drug Saf. 26, 1018–1032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Petrilli, C.M. et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 369, m1966 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Suleyman, G. et al. Clinical characteristics and morbidity associated with coronavirus disease 2019 in a series of patients in metropolitan Detroit. JAMA Net. Open. 3, e2012270 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Redd, W.D. et al. Prevalence and characteristics of gastrointestinal symptoms in patients with severe acute respiratory syndrome coronavirus 2 infection in the United States: a multicenter cohort study. Gastroenterology 159, 765–767 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Killerby, M.E. et al. Characteristics associated with hospitalization among patients with COVID‐19—Metropolitan Atlanta, Georgia, March–April 2020. Morb. Mortal. Wkly. Rep. 69, 790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang, X. , Ferro, E.G. , Zhou, G. , Hashimoto, D. & Bhatt, D.L. Association between universal masking in a health care system and SARS‐CoV‐2 positivity among health care workers. JAMA 324 703–704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]