

Abstract
Abstract
Nanopore sequencing has emerged as a rapid and cost-efficient tool for diagnostic and epidemiological surveillance of SARS-CoV-2 during the COVID-19 pandemic. This study compared the results from sequencing the SARS-CoV-2 genome using R9 vs R10 flow cells and a Rapid Barcoding Kit (RBK) vs a Ligation Sequencing Kit (LSK). The R9 chemistry provided a lower error rate (3.5%) than R10 chemistry (7%). The SARS-CoV-2 genome includes few homopolymeric regions. Longest homopolymers were composed of 7 (TTTTTTT) and 6 (AAAAAA) nucleotides. The R10 chemistry resulted in a lower rate of deletions in thymine and adenine homopolymeric regions than the R9, at the expenses of a larger rate (~10%) of mismatches in these regions. The LSK had a larger yield than the RBK, and provided longer reads than the RBK. It also resulted in a larger percentage of aligned reads (99 vs 93%) and also in a complete consensus genome. The results from this study suggest that the LSK preparation library provided longer DNA fragments which contributed to a better assembly of the SARS-CoV-2, despite an impaired detection of variants in a R10 flow cell. Nanopore sequencing could be used in epidemiological surveillance of SARS-CoV-2.
Key points
• Sequencing SARS-CoV-2 genome is of great importance for the pandemic surveillance.
• Nanopore offers a low cost and accurate method to sequence SARS-CoV-2 genome.
• Ligation sequencing is preferred rather than the rapid kit using transposases.
Keywords: SARS-CoV-2, Nanopore, Sequencing, Flow cell, Genome assembly, COVID-19
Introduction
The human pathogen severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in Wuhan City (China) in early 2020 and causes the COVID-19 disease which is responsible for over one million deaths in less than one year since then. SARS-CoV-2 has spread over the world and has caused marked social, medical, and economical adaptations to the world. Complete genome sequences published in January 2020 (Wu et al. 2020) enabled the development of real-time reverse transcription–polymerase chain reaction (RT-PCR) assays for SARS-CoV-2 detection that have served as the diagnostic standard during the ongoing COVID-19 pandemic (van Kasteren et al. 2020). Sequencing the genome of the SARS-CoV-2 has provided relevant information on its mutation rate of the virus, its spreading dynamic, or its zoonotic origin (Boni et al. 2020). Genomic surveillance of SARS-CoV-2 is a key tool to know which lineages of the virus are circulating in each country, how often new sources of virus are introduced from other geographical areas, or as an indicator of the success of control measures, and how the virus evolves in response to interventions. Sequencing also provides invaluable insights when linked with detailed epidemiology data for epidemiological investigation of the evolution of the pandemic. All these aspects play a key role in surveillance of the pandemic. Joint efforts have contributed to create a nomenclature system for the different linages of the coronavirus (Rambaut et al. 2020).
Recent documentation of reinfections has been provided, demonstrating that different lineages of the SARS-CoV-2 can infect the same person (Tillett et al. 2020; To et al. 2020). Sequencing the genome of the coronavirus is necessary to confirm those reinfections and exclude medical relapses.
Fast and reliable sequencing of samples in hospitals is of main importance for this epidemiological surveillance. Oxford Nanopore Technologies (ONT, Oxford, UK) has developed several strategies for fast sequencing the SARS-CoV-2 genome that may be essential for quick diagnosis and monitoring the community transmission of the coronavirus.
The objective of this study was to evaluate the performance of different chemistry and sequencing strategies using ONT sequencing of the SARS-CoV-2 genome in terms of sequencing accuracy, detection of variants, and quality of the genome assembly.
Material and methods
Sample collection
A panel of clinical samples obtained during initial diagnostics in essential personnel from Madrid city hall services (police, firemen, emergency and health care workers, etc.) was included in this study. The sample with the lowest Ct value (19.24) from an in-house version of the recommended E-gene real-time RT-PCR (Corman et al. 2020) was selected for sequencing in this study. RNA was isolated anew from stored clinical samples using the IndiSpin® Pathogen kit (Indical Bioscience, Leipzig, Germany). Obtaining the cDNA and 400 bp amplicons was conducted using a protocol published by the ARTIC Network (Quick 2020), using primers from V2 (https://github.com/artic-network/artic-ncov2019/blob/master/primer_schemes/nCoV-2019/V2/nCoV-2019.tsv). The sample selected had the largest DNA concentration (15 ng/μl) measured using the Qubit fluorometer (ThermoFisher Scientific, 150 Waltham, MA, USA).
DNA sequencing
The MinION device was used for ONT sequencing. Two ONT kits were used to prepare the DNA library: The Rapid Barcoding kit (SQK-RBK004) and the Ligation Sequencing Kit (SQK-LSK109).
The first library was sequenced using a R9.4 flow cell by loading 175 ng onto the SpotON flow cell, according to the manufacturer’s instruction. The other library was sequenced using the R10 flow cell using the same amount of DNA. Both flow cells (FC) ran until exhaustion. Most of the reads were obtained during the first two hours of the run. The flow cells were controlled and monitored using the MinKNOW software (version 4.0.20, ONT).
Reads were basecalled using Guppy version 4.0.11 (community.nanoporetech.com), and the high accuracy version of the flip-flop algorithm.
Assembly
The SARS-CoV-2 virus from Wuhan strain Hu-1 genome (MN908947) was used as reference. The reads were aligned against SARS-CoV-2 genome using Minimap2 aligner (Li 2016), a general purpose alignment program to map DNA or long mRNA sequences against a reference database.
The total coverage of the genome for both sets of reads was calculated from the alignments using GenomeCoverageBed utility of the bedtools suite (Quinlan and Hall 2010), quantile-normalized and smoothed using a window width of 200 bp.
Variant calling
The variant calling genotyping from alignment files was performed using LoFreq (Wilm et al. 2012), VarScan (Koboldt et al. 2012) and Pilon (Walker et al. 2014). LoFreq is a fast and sensitive variant-caller for inferring single nucleotide variants (SNVs) and indels from Next Generation Sequencing data. It makes full use of base-call qualities and other sources of errors inherent in sequencing. VarScan employs a robust heuristic/statistic approach to call variants that meet desired thresholds for read depth, base quality, variant allele frequency, and statistical significance. This program needs to pre-process the alignment file to generate a mpileup format file. Pilon identifies small variants with high accuracy as compared to state-of-the-art tools and is unique in its ability to accurately identify large sequence variants including duplications and resolve large insertions. Deviations from the reference were analyzed.
De-novo assembly
The de novo assembly was performed using Canu (Koren et al. 2017). Canu is a fork of the celera assembler designed for high-noise single-molecule sequencing (such Oxford Nanopore MinION). In order to improve de genome assembly, we used Pilon to automatically improve draft assemblies. Pilon requires as input a FASTA file of the assembly along with the BAM files of reads aligned to the input FASTA file. Pilon uses read alignment analysis to identify inconsistencies between the input assembly and the evidence in the reads. The new assembled genomes were compared with reference genome using Gepard (Krumsiek et al. 2007) in order to thoroughly check the new assemblies.
Results
Quality check
After the quality control for ONT reads, a total of 16,991 (N50 = 409) and 9658 (N50 = 1059) good quality reads (Table 1) were retained from the R9 and R10 FC, respectively. The R9 yielded 6.48 Mb and the R10 7.73 Mb. ONT runs may yield much larger number of bases, however, in this case, the amount of DNA was limiting. The R9 yielded 90% of the reads within the first two hours, whereas 40% of the reads were sequenced during the same time in the R10 FC. The GC content distribution was computed from both runs using LongQC (Fukasawa et al. 2020). The SARS-CoV2 reference genome has a GC% of 37.97. Reads from R9 averaged a GC content of 39.99% (s.d. = 3.134), whereas GC content from R10 reads averaged 37.91% (s.d. = 3.30). Fig. 1 represents the GC fraction in reads obtained from each FC. The entire reads are expected to show sharper distribution, because they have smaller deviation due to longer sequences. The reads were chunked in 150 bp fragments, showing the same GC average contents although with slightly larger variability. This latter strategy is more robust to sequencing or sample differences, and this should be comparable to other data if the same target (biological replicates) is sequenced.
Table 1.
Minimap2 alignment summary results
| Flow cell and sequencing kit | Reads | Aligned reads | Unaligned reads | %aligned | Non-sense read fraction |
|---|---|---|---|---|---|
| R9-RBK004 | 16,991 | 15,827 | 1164 | 93.15 | 42% |
| R10-LSK009 | 9658 | 9548 | 110 | 98.86 | 18% |
Fig. 1.

GC content distribution from ONT sequences from (a) R9 set, (b) R10 flow-cells. The blue bars come from entire reads, and the red ones were computed from chunked (150 bp) subsequences
Alignment
Brief statistics of the alignments are shown in Table 1. A larger proportion (98.86%) of reads from R10 were aligned to the reference genome. Almost 7% of reads from R9 were not aligned to the reference genome vs only 1.1% from R10 reads. The alignments show a good quality of reads for the process. However, a larger amount of non-sense reads (Fukasawa et al. 2020) were detected from R9 FC. Nonsense reads are defined as unique reads that cannot be mapped onto sequences of any other molecules in the same library. This concept is similar to unmappable reads; however, mappability depends on references. According to Fukasawa et al. (2020), non-sense read fraction should be less than 30%. If the fraction of non-sense reads is a way high, it might indicate that either sequencing had some issues or simply coverage is insufficient.
Fig. 2 shows the mismatches per aligned read against the SARS-CoV2 reference genome. The mismatches distributions showed a mode of 3.5 and 7% of read length for R9 and R10, respectively. The R9 FC showed a lower rate of mismatches than R10, although it might be led by a shorter length of the reads, which is a consequence of the Rapid Barcoding kit (SQK-RBK004) used to prepare this library, as this kit requires a transposase fragmentation. However, the library loaded in the R10 FC was prepared using the Ligation Sequencing kit (SQK-LSK109) which does not fragment the DNA and is optimized for throughput. This might explain the slightly larger yield outcome from the library loaded in the R10 FC.
Fig. 2.
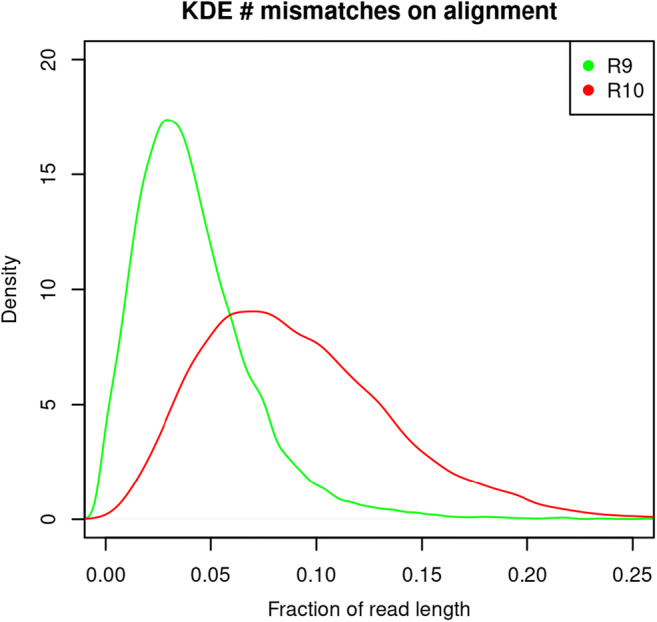
Fraction of mismatches per read against the SARS-CoV-2 reference genome
The read accuracy at the homopolymeric sites from each FC was evaluated. Homopolymeric sites in the SARS-CoV-2 reference genome were located with SeqKit (Shen et al. 2016). Longer homopolymeric regions in the SARS-Cov-2 reference genome were composed of thymine (7 nucleotides) and adenine (6 nucleotides), whereas guanine and cytosine homopolymers had a longer region of 3 nucleotides. Mismatches (Fig. 3A) and deletions (Fig. 3B) at homopolymer sites with length >2 were considered. The R9 chemistry produced a lower number of mismatches at the homopolymeric sites, but was more prone than R10 to produce deletions, mainly in thymine and adenine homopolymeric sites. The R10 chemistry produced a larger rate of mismatches, but a lower rate of deletions, although a rate >20% of deletions was observed at homopolymers <4 nucleotides. Both chemistries showed larger accuracy in adenine and thymine homopolymeric sites than in cytosine and guanine sites.
Fig. 3.

Read accuracy at homopolymeric sites in the SARS-CoV-2 genome for each FC type
Genome coverage
Fig. 4 shows the log2 normalized smoothed coverage plus 1 (to avoid zeroes) plot, generated using GNUPLOT (Williams and Kelley 2011). Most regions showed a coverage above 50×, and a coverage >200× was obtained for many regions regardless the FC type. Both FC types had lower coverage in the same regions. It denotes that the primers used produce a good coverage of the whole SARSCoV2 genome, although the 19 k–20 k region and the ending region showed a lower coverage than the rest of the genome.
Fig. 4.

Smoothed normalized coverage of reads by position from each type of FC (R9 and R10) against the SARS-CoV-2 genome (smoothing window width = 200 bp)
Variant calling
Indels and SNP (single nucleotide polymorphism) variants were identified using three softwares: LoFreq, Pilon, and VarScan. There was a large variability for the number of detected variants between different programs. LoFreq detected a larger number of SNP variants from R9 (25 vs 9), Pilon reported similar number of SNPs for both FC (6 vs 7), whereas VarScan detected larger number of SNPs from R10 (35 vs 12). Common SNPs detected in both FC were consistent, with 6–7 SNPs detected in common in both FC in all the analyses. Figs. 5 and 6 show the Venn diagrams of common SNPs by FC and software, respectively. The common SNP variants detected were in frequency >0.5, as shown in Fig. 7. Requiring a large frequency (>0.60) of the detected variants from nanopore long reads was consistent with the SNP variants detected from several software. Eight indels were reported in common from R9 and R10, however none of them were found with frequency >0.5 (Fig. 8). Forty-seven other indels were detected only from R9 FC, and 31 indels only from R10. The combination of these strategies (i.e. large variant frequency and selecting common variants reported from different software) seems to be a reliable strategy for variant calling from error prone long reads.
Fig. 5.

Common and unique SNPs (a) for R9 set, (b) for R10 set
Fig. 6.

Common and unique SNPs detected by (a) LoFreq, (b) Pilon, (c) VarScan
Fig. 7.

Allele frequencies of SNPs located in the SARS-CoV-2 genome detected by VarScan, LoFreq and Pilon
Fig. 8.

Allele frequencies of INDELs detected by VarScan located in the SARS-CoV-2 genome
De-novo assembly
Figs. 9 and 10 show the dotplot comparison between the SARS-CoV-2 reference genome and the assembly from R9 and R10 FC. Note that the R9 assembly is much shorter and sparse than the R10 assembly, with a larger number of contigs.
Fig. 9.

Dotplot comparison of SARS-CoV-2 reference (x-axis) vs. R9 assembly (y-axis)
Fig. 10.

Dotplot comparison of SARS-CoV-2 reference (x-axis) vs. R10 assembly (y-axis)
Discussion
A clinical sample with RT-qPCR Ct = 19.84 for SARS-CoV-2 was used in this study to amplify and sequence the coronavirus genome using nanopore long reads. Two chemistries and two different protocols were used in the study. A library prepared with the Rapid Barcoding kit (SQK-RBK004) was sequenced on an R9 FC, whereas a library prepared with the Ligation Sequencing kit (SQK-LSK109) was sequenced on an R10 FC.
Both runs yielded similar coverage of the SARS-CoV-2 reference genome. The R9 FC showed a smaller number of mismatches against the reference genome. The Rapid Barcoding kit resulted on shorter sequences as expected, as it uses a transposase fragmentation to insert the barcodes. The alignments showed a large number of variants but most of them in low frequency. Setting a threshold of 0.50 for the variant frequency led to a consistent number of variants per FC and library preparation kit of 6 to 7 variants, regardless of the software used. Despite of the larger number of mismatches from R10, the consensus sequence resulted in the same variants detected as from R9. Previous attempts to sequence the SARS-CoV-2 genome have provided accurate results for new variants discovery using rapid workflows based on the ARTIC protocol (Chan et al. 2020; Li et al. 2020; Moore et al. 2020). Other studies also showed amplicon sequencing using ONT flongle flow cells (Chan et al. 2020).
A de-novo assembly was attempted from both runs. In this case, the R10 FC yielded a more complete genome than the R9 run. We interpret that the larger size of the fragments and the larger number of Mb obtained, explained this better behavior, which is mainly due to the library preparation with the Ligation Sequencing Kit rather than to the R10 chemistry. Bull et al. (2020) achieved highly accurate consensus-level sequences, with SNVs detected at >99% sensitivity and >99% precision. Our results show that nanopore sequencing offers robust consensus sequence regardless of the chemistry used. This may help to optimize time and future protocols when sequencing SARS-CoV2 using ONT. Although the R10 FC is expected to achieve higher accuracy at homopolymeric regions than R9 FC, the errors are corrected in the consensus sequence. It must also be pointed out that a de novo assembly is not strictly necessary for genomic epidemiology surveillance. Mapping against a genome reference can be used instead, which would also facilitate the variant discovery.
All called mutations were already described in the GISAID (https://www.gisaid.org/) database by the date the sample was collected, except a mutation at position 21,727 with a C:T substitution on the S protein, which implies an amino-acid change S:P. This mutation was not observed further in the databases, hence we hypothesize that either this is a sequence error, or the transmission of this mutation was unsuccessful due to the strict lockdown imposed in Madrid from March to May 2020. The consensus sequences from both FC were introduced in the pangolin (v2.0) website tool (Rambaut et al. 2020). Both consensus reads belonged to the B1 lineage (Boni et al. 2020), which carries the D614G mutation. This variant is thought to have arisen in Italy at the beginning of the pandemic in Europe, and spread across Europe and later overseas. This mutation has been related with larger infectivity (Korber et al. 2020). This mutation has been previously detected in other studies in Spain (Díez-Fuertes et al. 2020).
It must be pointed out that an important limitation of this study is that each library preparation kit was only tested on one of the FC chemistry. It would have been informative to test the performance SQK-LSK109 on R9 and SQK-RBK004 on R10. This was not possible due to limited availability of DNA material. Nonetheless, we were able to extract some conclusions regarding the benefits and drawbacks from each chemistry and library preparation protocols regarding their convenience to sequence the SARS-CoV-2 genome. The results from this study suggest that the R10 chemistry does not improve the quality of the sequenced SARS-CoV-2 genome, and the Ligation Sequencing Kit is preferred for whole genome sequencing, as it yielded a higher sequencing depth and a much better genome assembly. This should be the kit of choice mainly at low initial DNA concentrations as in the case of this study. Nanopore offers a quicker turnaround genome sequencing for genomic epidemiology surveillance.
Acknowledgements
We are grateful to Madrid City Council for giving permission to use the samples in this particular study. We greatly thank all the technical and scientific personnel of INIA-CISA who worked voluntarily during the Spanish lockdown conducting mass diagnosis of samples from the essential operational sectors of Madrid City.
Author contribution
P.A.S., C.C.G., and J.F.P. extracted the RNA, and performed the cDNA transformation. O.G.R., M.G.R, and C.G. prepared the libraries and performed the cDNA sequencing. R.P.P. developed the computational pipelines for the assemblies and assisted on its analyses. O.G.R., J.F.P, and M.J.C. conceived the study and designed the experiments. O.G.R. wrote the manuscript. All authors helped in writing and configuring the last version of the manuscript.
Funding
This study has been partially funded by Madrid City Council under the contract service “Specific epidemiological and health studies of Covid-19 to know the prevalence of the disease in essential operational sectors”.
Data availability
The consensus sequence is available from GISAID (https://www.gisaid.org/) with accession number EPI_ISL_770129. Additional data are available from the authors upon reasonable request.
Declarations
Ethics approval and consent to participate
Samples were provided under a consent to participate statement.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Boni MF, Lemey P, Jiang X, Lam TTY, Perry BW, Castoe TA, Rambaut A, Robertson DL. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 2020;5:1408–1417. doi: 10.1038/s41564-020-0771-4. [DOI] [PubMed] [Google Scholar]
- Bull RA, Adikari TN, Ferguson JM, Hammond JM, Stevanovski I, Beukers AG, Naing Z, Yeang M, Verich A, Gamaarachchi H, Kim KW, Luciani F, Stelzer-braid S, Eden J, Rawlinson WD, Van Hal SJ, Deveson IW. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat Commun. 2020;11:6272. doi: 10.1038/s41467-020-20075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WS, Au CH, Lam HY, Wang CLN, Ho DNY, Lam YM, Chu DKW, Poon LLM, Chan TL, Zee JST, Ma ESK, Tang BSF. Evaluation on the use of nanopore sequencing for direct characterization of coronaviruses from respiratory specimens, and a study on emerging missense mutations in partial RdRP gene of SARS-CoV-2. Virol J. 2020;17:1–13. doi: 10.1186/s12985-020-01454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, Bleicker T, Brünink S, Schneider J, Schmidt ML, Mulders DG, Haagmans BL, van der Veer B, van den Brink S, Wijsman L, Goderski G, Romette J-L, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MP, Drosten C. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance. 2020;25(3):2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez-Fuertes F, Iglesias-Caballero M, García Pérez J, Monzón S, Jiménez P, Varona S, Cuesta I, Zaballos Á, Jiménez M, Checa L, Pozo F, Pérez-Olmeda M, Thomson MM, Alcamí J, Casas I. A founder effect led early SARS-COV-2 transmission in Spain. J Virol. 2020;95:e01583–e01520. doi: 10.1128/JVI.01583-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa Y, Ermini L, Wang H, Carty K, Cheung M-S. LongQC: a quality control tool for third generation sequencing long read data. G3: Genes|Genomes|Genetics. 2020;10(4):1193–1196. doi: 10.1534/g3.119.400864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, Angyal A, Brown RL, Carrilero L, Green LR, Groves DC, Johnson KJ, Keeley AJ, Lindsey BB, Parsons PJ, Raza M, Rowland-Jones S, Smith N, Tucker RM, Wang D, Wyles MD, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23(8):1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics. 2016;32:2103–2110. doi: 10.1093/bioinformatics/btw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang H, Mao L, Yu H, Yu X, Sun Z, Qian X, Cheng S, Chen S, Chen J, Pan J, Shi J, Wang X. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci Rep. 2020;10:1–10. doi: 10.1038/s41598-020-74656-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SC, Penrice-Randal R, Alruwaili M, Randle N, Armstrong S, Hartley C, Haldenby S, Dong X, Alrezaihi A, Almsaud M, Bentley E, Clark J, García-Dorival I, Gilmore P, Han X, Jones B, Luu L, Sharma P, Shawli G, Sun Y, Zhao Q, Pullan ST, Carter DP, Bewley K, Dunning J, Zhou EM, Solomon T, Beadsworth M, Cruise J, Crook DW, Matthews DA, Davidson AD, Mahmood Z, Aljabr W, Druce J, Vipond R, Ng L, Renia L, Openshaw PJM, Kenneth Baillie J, Carroll MW, Stewart J, Darby A, Semple M, Turtle L, Hiscox JA. Amplicon-based detection and sequencing of SARS-CoV-2 in nasopharyngeal swabs from patients with COVID-19 and identification of deletions in the viral genome that encode proteins involved in interferon antagonism. Viruses. 2020;12(10):1164. doi: 10.3390/v12101164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J (2020) nCoV-2019 sequencing protocol. In: 10.17504/protocols.io.bbmuik6w. 10.17504/protocols.io.bbmuik6w. Accessed 20 Sep 2020
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, du Plessis L, Pybus OG. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Le S, Li Y, Hu F, Zou Q. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS ONE. 2016;11(10):e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillett R, Sevinsky J, Hartley P, Kerwin H, Crawford N, Gorzalski A, Laverdure C, Verma S, Rossetto C, Jackson D, Farrell M, Van Hooser S, Pandori M (2020) Genomic evidence for a case of reinfection with SARS-CoV-2. Available at SSRN: 10.2139/ssrn.3680955
- To KK-W, Hung IF-N, Ip JD, Chu AW-H, Chan W-M, Tam AR, Fong CH, Yuan S, Tsoi H, Ng AC, Lee LL, Wan P, Tso E, To W-K, Tsang D, Chan K, Huang J, Kok K, Cheng VC-C, Yuen K-Y (2020) COVID-19 re-infection by a phylogenetically distinct SARS-coronavirus-2 strain confirmed by whole genome sequencing. Clin Infect Dis:ciaa 1275. 10.1093/cid/ciaa1275 [DOI] [PMC free article] [PubMed]
- van Kasteren PB, van der Veer B, van den Brink S, Wijsman L, de Jonge J, van den Brandt A, Molenkamp R, Reusken CBEM, Meijer A. Comparison of seven commercial RT-PCR diagnostic kits for COVID-19. J Clin Virol. 2020;128:104412. doi: 10.1016/j.jcv.2020.104412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Kelley C (2011) Gnuplot 4.5: an interactive plotting program. URL http://gnuplot.info. (Last accessed: 2021 February 14)
- Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The consensus sequence is available from GISAID (https://www.gisaid.org/) with accession number EPI_ISL_770129. Additional data are available from the authors upon reasonable request.