

Abstract
A new approach is suggested herein for the synthesis of pyrazole derivatives by reacting 4-nitrosemicarbazide with acetylacetone. Additional studies were done on the reaction of acetylacetone with semicarbazide and its derivatives (4-aminosemicarbazide, methylsemicarbazide, and dimethylsemicarbazide). The study on the reaction with acetylacetone resulted in monocyclic 3,5-dimethyl-N-nitropyrazole-1-carboxamide, monocyclic 5-hydroxy-3,5-dimethyl-2-pyrazoline, and bicyclic bis(3,5-dimethylpyrazole-1-carbonyl)hydrazine, and conditions for the formation of acetone semicarbazone were identified. The structures of the resultant compounds were validated by physicochemical analytical methods, including X-ray diffraction. The computer-aided screening in the PASS prediction software discovered a high biological activity of the newly obtained compounds.
Introduction
We have previously reported the synthesis of octaaza derivatives of mono- and tricyclic compounds such as 1,2,4,5,8,9,11,12-octaazacyclotetradeca-5,7,12,14-tetraene-3,10-dione and 2,3,4a,6,7,8a,9,10-octaaza-4,8-dioxo-3,4,4a,7,8,8a,9,9a,10,10a-decahydroanthracene by the reaction between 4-nitrosemicarbazide (1) and glyoxal.1 The known analogues, semicarbazide and 4-aminosemicarbazide (1,3-diaminourea and diaminourea), when reacted with glyoxal do not furnish anthracene derivatives under such conditions. 4-Nitrosemicarbazide (1) has been synthesized more recently starting from N,N′-dinitrourea2 and is a new reactant in organic chemistry and coordination compounds chemistry.3 Despite a few binding sites, the molecule of 4-nitrosemicarbazide exhibits regioselectivity toward monoaldehydes to yield the respective nitrosemicarbazones.4,5 4-Nitrosemicarbazide reacts with glyoxal (dialdehyde) and diacetyl (diacetone) to yield the respective linear glyoxal bis(nitrosemicarbazone) and diacetyl bis(nitrosemicarbazone).5 The resultant compounds can be used in further synthesis of novel cyclic nitro compounds6,7 and bioactive substances.8
In light of the aforesaid background, a logical question arises as to which compounds will be formed by the reaction between 4-nitrosemicarbazide and 1,3-dicarbonyl compounds since it is well known that the reaction of carbonic acid hydrazide derivatives (semicarbazide, 1,3-diaminourea, etc.) with 1,3-dicarbonyl compounds proceeds to give the respective hydrazones9−11 and pyrazoles.12−14 The pyrazole derivatives have been put to good use in practice because they possess diverse biological properties.15−18 By now, pyrazole has proved to be the most useable framework from among all the known diazoles for screening in the field of pharmaceutical and medicinal chemistry.15 More specifically, 3,5-dimethylpyrazole was found to exhibit antidiabetic16 and antipyretic17 actions, and the use of pyrazole derivatives as potent antibacterial drugs was described.18 The use of drugs in the pyrazolic series such as Analgin and Antipyrine has become common practice.
The studies in this field are being continued, and there are currently reports on the synthesis of other heterocyclic structures in similar reactions.19−23 That being said, the literature provides scarce data on the structure of intermediates in the synthesis of 3,5-dimethylpyrazoles. Among them, hydroxypyrazolines are still understudied because they were isolated in the pure form, only derivatives bearing the aryl radical which, as per X-ray diffraction (XRD) data, stabilizes the compound by hydrogen bonding between the hydroxyl and acetyl groups.24,25 The known hydroxy derivatives have already been proposed for use as antipyretics and analgesics in place of dipyrone,26 and they are also promising intermediates in the synthesis of medicinal drugs.
In the above context, it can be hypothesized that in case acetylacetone is used in condensation with 4-nitrosemicarbazide, the reaction is more likely to go in two directions, that is, to preserve the structure of compound 1 and furnish new linear or heterocyclic structures or to break down the structure of compound 1 and give the respective pyrazoles, or these two directions may take place at a time.
The achievement of the study objective would discover the regularities and direction of the chemical reactions between acetylacetone and semicarbazide derivatives leading to new linear compounds or heterocycles of the pyrazole framework starting from 4-nitrosemicarbazide depending on the chain length of diketones.
Results and Discussion
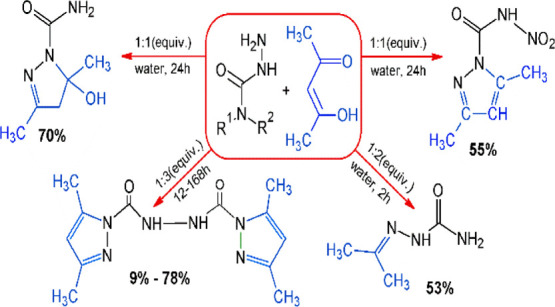
Linear glyoxal bis(nitrosemicarbazones) are quite easily obtainable through the condensation of 4-nitrosemicarbazide (1) with glyoxal at room temperature.4,5 In this regard, the reaction between 4-nitrosemicarbazide (1) and acetylacetone (2) was expected to give the respective hydrazone (3) (Scheme 1).
Scheme 1. Hypothesized Scheme for the Reaction between 4-Nitrosemicarbazide (1) and Acetylacetone.

However, the very initial experiments with acetylacetone demonstrated that when the starting reactants were mixed, no precipitation typical of hydrazones occurred. The UV spectrum at 300–320 nm had no absorption peak typical of bis-nitrosemicarbazones of dicarbonyl compounds.5 During the reaction, a bathochromic displacement of the maximum absorption band was observed from 256 nm typical of starting compound 1 to 276 nm. Besides, the generated spectrum had another absorption peak at 214 nm, which can be attributed to 3,5-dimethylpyrazole (5) because a characteristic absorption peak at 215 nm was observed for the standard sample (pyrazole 5) (Figure 1).
Figure 1.
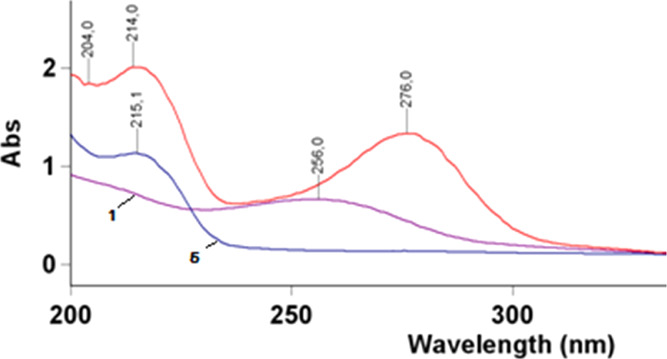
Electronic absorption spectra of starting compound 1, pyrazole 5, and the reaction product.
It can thus be assumed from the acquired UV spectrum that the reaction mixture consisted of mixed compounds, that is, the condensation product and pyrazole 5.
NMR spectroscopy discovered that the mixed compounds were composed of 85% dimethylpyrazole (5) and 15% 3,5-dimethyl-N-nitropyrazole-1-carboxamide (4). Two nonequivalent methyl groups detected by 1H and 13C NMR spectroscopy were referred to compound 4. A weak interaction between hydrogen atoms of one of the methyl groups (δ = 2.58 ppm) and a nitrogen atom (δ = 250 ppm) was found by 2D 1H 15N-multiple bond correlation (see the Supporting Information for the1H,15N-HMBC spectrum of 4). This nitrogen can be identified as an amide nitrogen with an additional acceptor group (a nitro group in this case).
The analysis of the NMR spectra of the resultant compound showed no signals (chemical shifts) relating to the −CH2– group and showed the asymmetric nature of the resultant compound, indicating that no structures of bis-hydrazones 2 and 3 were present.
The IR spectrum of compound 4 had an absorption band near 1666 cm–1, which can be ascribed to the associated band shape of amide I, ν(C=O). The spectrum had an absorption band around 3204 cm–1 typical of the associated NH bond and absorption bands representative of the nitramide group at 1594 and 1284 cm–1.27 3,5-Dimethyl-N-nitropyrazole-1-carboxamide (compound 4) is mentioned only in the PubChem database (CID 23171560) with only predicted data given.28
Recrystallization of 4 demonstrated that product 4 is instable and decomposes to pyrazole 5 quite rapidly (Scheme 1). The decomposition reaction took place easily because the nitro group was present at the amide grouping, which activated the proton for electrolytic dissociation. Thus, it can be said that the intrinsic acidity of compound 4 was the promotor of structural acid catalysis to initiate its own denitramidation, thereby fostering a fast hydrolysis reaction.
Most researchers exploit an acid catalyst or semicarbazide hydrochloride in the condensation of acetylacetone with semicarbazide, in which case the principal reaction products are 3,5-dimethylpyrazole-1-carboxamide and 3,5-dimethylpyrazole.The presence of acid in the reaction system (added as a catalyst or generated by the hydrolysis of semicarbazide hydrochloride) accelerates the transformation of hydroxypyrazolines into pyrazoles, and the pyrazole formation rate changes in the row: Me > PhCH, CH > t-Bu.12,13
It is, however, known that the reaction between thiosemicarbazide or furan-2-carbohydrazide and 1,1,1-trifluoro-pentane-2,4-dione furnishes the respective 4,5-dihydro-1H-pyrazoles in a quantitative yield.29
The study into the acidity effect gave us an impetus to ascertain some aspects of the condensation reaction of acetylacetone with other semicarbazide derivatives, particularly with semicarbazide itself, Alk- and Ar-substituted semicarbazides and 4-amino semicarbazide, and more specifically to explore this reaction without using acid catalysts.
The reaction between semicarbazide (6) and acetylacetone could proceed in two directions: (I) through intermediate (7a) at a stoichiometric ratio of 1:1 to furnish 5-hydroxy-3,5-dimethyl-2-pyrazoline (7) which precipitated as a crystal in up to 70% yield during the reaction and (II) at a ratio of 1:2 to deliver 3,5-dimethylpyrazole-1-carboxamide (8) in a 56% yield (Scheme 2). In the case of 1, the ring-closure, that is, the formation of C–N bond, took place due to the intramolecular migration of the proton (H) from the nitrogen atom (N) to (γ) carbon (C) of acetylacetone monohydrazone semicarbazide (7a) to form the CH2 group. This can probably be explained by the shift in pH of the reaction mixture toward the alkaline region from pH 6 to 8, in which case acetylacetone semicarbazide monohydrazone (7a) was formed, followed by further intramolecular transposition and preservation of the hydroxyl group of 7. Semicarbazide is rather a strong base and its twofold excess results in a stronger pH shift of the reaction mixture to an alkaline medium and in the activation of the hydroxyl group of acetylacetone monohydrazone semicarbazide (7a), followed by the condensation, H2O molecule release, and formation of compound 8 (direction II). The reaction took place at room temperature.
Scheme 2. General Synthetic Protocol for 5-Hydroxy-3,5-dimethyl-2-pyrazoline (7) and Acetone Semicarbazone (9).

A temperature rise of the reaction medium had a positive effect on the yield improvement of product 8, but in this case, there was a side reaction associated with the alkaline hydrolysis of acetylacetone to the starting acetone that reacted with residual semicarbazide to deliver acetone semicarbazone 9.
Compound 7 is stable, and even the multiple heating of its aqueous solutions does not lead to its decomposition, and only the 8 h boiling in water decomposes it to pyrazole 5.
The IR spectrum of compound 7 showed vibrational bands for the sum of associated bonds of NH2 and NH groups near 3310–3110 cm–1 and absorption bands near 1675 cm–1 for C=O and C=N bonds, which is on a par with the literature data for 3,5-dimethylpyrazole-1-carboxamide.24 In this case, an intense absorption band typical of stretching vibrations of the OH bond at 3464 and 1121 cm–1 appeared, and a doublet of cyclic methylene vibrations of the CH2 bond was observed near 3010, 1014, and 751 cm–1, while CH3 vibrations remained correspondingly at 2965 and 1464 cm–1.
In the1H NMR spectrum of compound 7, the pyrazole moiety was reliably identified via nucleus–nucleus interactions. The H atoms at the methylene group were chemically nonequivalent, suggesting that an asymmetric site was present nearby. The presence of the OH group was corroborated by the considerable downfield shift of the signal of the quaternary carbon atom. In the 1H NMR spectrum, the hydroxo group gave a signal with an elevated integral intensity, equal to 2 (rather than 1), which is most likely due to the signal coalescence of the OH group and water impurity. The spectrum also showed an NH2 signal, which is well identifiable in the 1H, 15N HMBC spectrum, and a carbon atom signal at 155 ppm correlating with the typical location of C=O group signals of urea derivatives (see the Supporting Information for the 1H, 15N HMBC spectrum of 7). In the13C NMR spectrum, the signal manifested itself as a singlet, indicating that no hydrogen atoms are present in the immediate surrounding (there is no cleavage of the NH2 group on the protons due to these atoms being involved in chemical exchange processes).
The IR and NMR spectra of compound 8 are in full agreement with the literature data.12,13 The structure of compound 9 is corroborated by the absence of one signal from the carbon atom of the double bond in the 13C NMR spectrum. Besides, the signal splitting at 146 ppm in the 13C NMR spectrum looks like a doublet of septets (JCH = 6.7 Hz and JCH = 2.9 Hz), suggesting the presence of six H atoms equally spaced at a distance of a double bond, and the presence of one more H atom a little farther. The nonequivalence of the methyls allowed for the assumption of their attachment to one carbon atom of the double bond (see the Supporting Information for the 13C assignment of compound 9).
The presence of the N–H bond was confirmed by the 1H, 15N HMBC spectrum in which the signals at 6.2 and 8.9 ppm exhibited a coupling constant (see the Supporting Information for the 1H, 15N HMBC spectrum of compound 9).
The linkage between the carbamide moiety and the (CH3)2C=N moiety was corroborated by the 1H, 13C HMBC spectrum in which the H signal at 8.9 ppm had cross-peaks with both the carbon at 146 ppm (C=N) and the carbon at 157.5 ppm (C=O) (see the Supporting Information for the 1H, 13C HMBC spectrum of compound 9).
The mass spectrometry measurement results showed that the resultant compound 9 had a molecular weight of 115 amu (atomic mass units) and an elemental composition of C4H9ON3, which correlates with acetone semicarbazone.
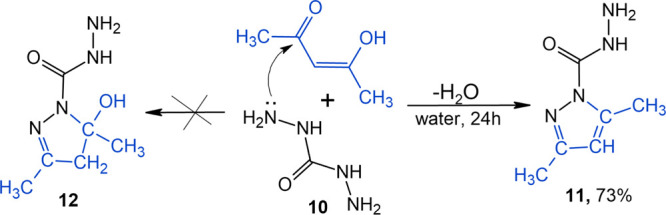
The reaction is known between diaminourea (10) and acetylacetone at an equimolar ratio in an aqueous solution over an acid catalyst, leading to 3,5-dimethylpyrazole-1-carbohydrazide (11) (Scheme 3).30 In the experiment with compound 10, in a manner similar to compound 6 (without an acid catalyst), we expected to derive a hydroxy derivative of 3,5-dimethylpyrazole-1-carbohydrazide (12) similar to Scheme 2 in reaction direction I. However, compound 12 was not isolated, and the major product turned out to be compound 11. The attempts to move the reaction equilibrium toward the formation of hydroxypyrazolines by selecting temperature conditions, solvents, and reagent ratio failed. Probably, the cyclization reaction mechanism was similar to direction II in Scheme 2.
Scheme 3. General Synthetic Protocol for 3,5-Dimethylpyrazole-1-carbohydrazide (11).
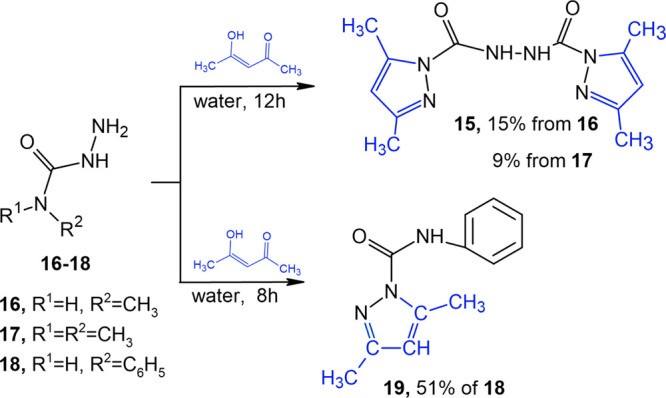
In doing so, a twofold excess of acetylacetone when mixed with diaminourea in aqueous acetonitrile was found to furnish a new bicyclic compound, bis(3,5-dimethylpyrazole-1-carbonyl)hydrazine (15) (Scheme 4), in a maximum yield of 70%. When individual compound 11 and acetylacetone were mixed in a molar ratio of 1:1 in aqueous acetonitrile at room temperature for 7 days, the yield of compound 15 increased to 78%. The reaction temperature rise above 25 °C increased the proportion of pyrazole 5 in the reaction products by breaking the N–CO bond of starting compound 11.
Scheme 4. General Synthetic Protocols for 3,5-Dimethylpyrazole-1-carbohydrazide (11) and bis(3,5-Dimethylpyrazole-1-carbonyl)hydrazine (15).

In order to explore the direction of chemical reactions, the reaction mixture was monitored by NMR spectroscopy throughout the reaction time frame of 7 days (Figure 2). 3,5-Dimethylpyrazole-1-carbohydrazide (11) was found to engage in the reaction with acetylacetone almost immediately to furnish 3,5-dimethylpyrazole-1-carbohydrazide (12). This was evidenced by the nearly instant disappearance of the NH2 signals of starting compound 10 at 4.00 ppm in the NMR spectra, the observed displacement of the broad singlet of NH amide from 8.28 to 8.88 ppm, and the emergence of a resonance signal in 24 h at 7.88 ppm typical of hydrazones.5 In this case, after 3 days, the intensity of that signal reached its maximum and began to decrease, suggesting that intermediate hydrazine 13 was instable in the reaction mixture and the reaction proceeded further. With decreasing intensity of the =CH– singlet of the starting compound at 6.01 ppm, the NMR spectra clearly showed a buildup of a signal at 6.07 ppm in 3 h, relating to =CH– of 1,1-carbonyl-bis(3,5-dimethylpyrazole) (14) in accordance with the literature data.31 Concurrently, a signal at 5.80 ppm referred by us to =CH– of pyrazole 5 began to build up. In this case, the intensity of a signal at 3.62 ppm was steadily increasing, which suggested water release during the reaction. Within a day, the first “trace” signals appear at 6.15 and 5.74 m. d. =CH–1 bis(3,5-dimethylpyrazole-1-carbonyl)hydrazine 15 and pyrazole 5, respectively. These characteristic signals gradually increased over time and finally had a ratio of 2:1 after 168 h (7 days), in which case the signals of the starting and intermediate compounds were not documented, suggesting that the reaction was completed in full (Scheme 4).
Figure 2.

1H assignment of NMR monitoring in CD3CN at 22 °C for 168 h: (11) 3,5-dimethylpyrazole-1-carbohydrazide (11), a precipitate containing 5 and 15.
After reviewing the literature data and detecting the known intermediates and end products of the reaction by NMR spectroscopy, the following reaction mechanism could be hypothesized: the first stage was the reaction between diaminourea with one molecule of acetylacetone to generate 3,5-dimethylpyrazole-1-carbohydrazide (11) and the second stage involved the reaction between the resulting 11 and excess acetylacetone to give presumably 1,1-carbonyl-bis(3,5-dimethylpyrazole) (13) existing in solution. This reaction compound is instable due to the presence of the OH-group mobile hydrogen atom that draws free pair electrons of the NH group. Therefore, at the third stage, the resulting product 13 underwent intramolecular condensation to furnish bicyclic compound 14 and water. Compound 11 residing in solution is a nucleophilic particle with respect to substrate 14. The nucleophilic attack eventually resulted in the breakage of the C–N bond in compound 14 to yield the stable reaction end products 5 and bis(3,5-dimethylpyrazole-1-carbonyl)hydrazine (15) (Scheme 4).
As a result of the experiment, a crystalline precipitate was obtained whose NMR spectra showed three nonequivalent N atoms, with one of the atoms being secondary amide. The carbonyl carbon had a fission in the form of a doublet with a coupling constant of 7 Hz, suggesting an interaction with one H atom via two bonds, which corresponds to the O=C–NH group, so that the 1H, 13C HMBC spectrum showed an interaction between the carbonyl carbon and the hydrogen atom of the NH group. By other chemical shifts, compound 15 was similar to 11.
High-resolution mass spectrometry applied to identify the structure of compound 15 revealed that the spectrum did not change with temperature and related presumably to the same compound. It can be assumed from the analysis results that the molecular ion weighed 276 amu, whereas compounds with 218 amu were not therefore detected. C11H20O6N2 better fits to the measured weight, but the formula C12H16O2N6 is far more possible because a compound with six nitrogen atoms would be most likely nonvolatile; moreover, the weight loss was measured to be 180 m/z, which correlates with the loss of the C5H8N2 diazole moiety.
The structure of compound 15 was reliably validated by XRD. The crystal of 15 contained three independent molecules; the molecular structure of one of them is depicted in Figure 3.
Figure 3.
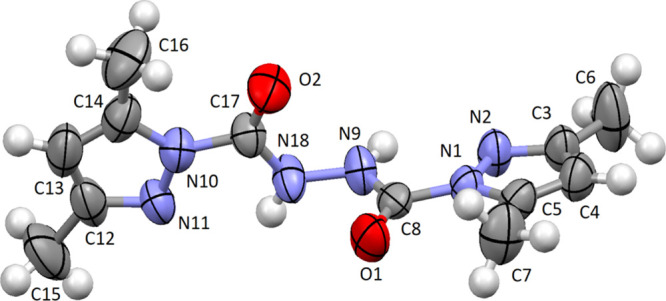
Molecular structure of compound 15. Thermal ellipsoids are shown at 50% probability.
The independent molecules were alike in geometry. All of the pyrazole-1-carbonyl moieties were planar. The conformation of the molecules was distinguished by the C(O)–N(H)–N(H)–C(O) torsion angles of 80.5, 90.2, and 78.0°. A similar conformation was observed in N″-(hydrazinocarbonyl)carbonohydrazide (angle 109.5°).32 However, the formation of complex compound 15 with zinc resulted in a planar structure of ligand 15 (angle 180.0°).33 Note the intramolecular N–H···N hydrogen bonding in the molecules of 15 (H···N 2.19–2.28 Å, N–H···N 105–108°). The intermolecular hydrogen bonds of N–H···O (H···O 2.08, 2.11 Å, N–H···O 152, 152°) and N–H···N (H···N 2.35, 2.41 Å, N–H···N 142, 142°) gave rise to trimeric molecules in the crystal.
Most of the absorption bands typical of pyrazole 5 retained in the IR spectra of compound 15. For instance, the absorption bands of the C=O and C=N bonds appeared around 1750–1702 cm–1. The absorption bands of cyclic methyl vibrations of the CH bond near 3010, 1083, and 762 cm–1also remained, as well as CH3 vibrations correspondingly at 2973–2932 and 1493 cm cm–1. With that, vibrational bands of the NH bonds shifted slightly to the region of 3380–3295, and a doublet at 1593–1574 cm–1 for the NH2 group characteristic vibrations disappeared, but there appeared a singlet at 1580 cm–1 pertaining to the NH group.
The use of Alk- and Ar-substituted semicarbazides corroborated the direct relationship between the structure of the resultant compounds and the basicity of the starting semicarbazide. Although semicarbazides are much stronger bases and nucleophiles than amines because of the additional supply of electrons to the nitrogen atom from adjacent NH2 groups, it can be hypothesized consistently with basicity constant Kb of amines that dimethylsemicarbazide (16) is the strongest base, while methylsemicarbazide (17) has a lower basicity, and phenylsemicarbazide (18) is weaker than semicarbazide. It was found by conducting similar reactions with these compounds (Scheme 5) that phenylsemicarbazide 18 when reacted with acetylacetone in the aqueous medium produces N-anilinocarbonyl-3,5-dimethyl-1-pyrazole carboxamide in 24 h (19). Methylsemicarbazide 17 and dimethylsemicarbazide 16 as stronger bases shifted the reaction pH toward the alkaline medium, thereby catalyzing further reactions whereby bis(3,5-dimethylpyrazole-1-carbonyl)hydrazine (15) could be isolated in 9 and 15% yields, respectively. Unfortunately, N-Alk-substituted-1-pyrazole carboxamides could not be isolated under these conditions. The structure of the derived compounds was validated by NMR spectroscopy and is fully consistent with the literature and our previously obtained results.34−36
Scheme 5. Synthetic Scheme for bis(3,5-Dimethylpyrazole-1-carbonyl)hydrazine (15) and 1-Anilinocarbonyl-3,5-dimethylpyrazole (19).
The promising outlook of the newly synthesized compounds as potentially bioactive agents was tested by computer-aided screening in the PASS software (Prediction of Activity Spectra for Substances) (Table 1) which can predict a good deal of potential biological activities on the basis of a structural formula using a unified description of chemical formula and a universal mathematical algorithm for establishing structure–activity relationship.37 This prediction allowed for conclusions that despite the likeness of the structures under question, they possess a completely different set of pharmacological activities. For instance, new compounds 4 and 15 are highly promising for treating blood strokes, as is compound 11, which is likely due to these compounds structurally bearing a carbazide moiety. The anticonvulsive activity is the most characteristic of compounds 8 and 9 and is likely due to the available carbamide moiety. The hydroxyl inserted into pyrazole 7 favors an antimetastatic activity since this pharmacological action is observed only in this compound. That being said, all the structures under the study share a leucopoiesis-stimulating activity, which is explained by the hydrazone linkage found in these structures.
Table 1. PASS-Predicted Pharmacological Activities.
| pharmacological activity score |
|||||||
|---|---|---|---|---|---|---|---|
| activity | 4 | 5 | 7 | 8 | 9 | 11 | 15 |
| stroke treatment | 0.948 | 0.980 | 0.905 | ||||
| anticonvulsant | 0.359 | 0.734 | 0.644 | 0.570 | 0.543 | ||
| antimitotic | 0.775 | ||||||
| leukopoiesis stimulant | 0.419 | 0.621 | 0.589 | 0.605 | 0.569 | 0.419 | 0.520 |
| antimetastatic | 0.610 | ||||||
Conclusions
In conclusion, monocyclic 3,5-dimethyl-N-nitropyrazole-1-carboxamide (4), monocyclic 5-hydroxy-3,5-dimethyl-2-pyrazoline (7), and bicyclic bis(3,5-dimethylpyrazole-1-carbonyl) hydrazine (15) have been synthesized for the first time through the reaction between acetylacetone and semicarbazide derivatives (4-nitrosemicarbazide, 4-aminosemicarbazide, and semicarbazide) without using acid catalysts. The structures of the said compounds were validated by physicochemical analytical methods, including XRD. The harsher reaction conditions and the alkaline medium were found to decompose acetylacetone to furnish acetone semicarbazone. The computer-aided screening in the PASS software discovered a high predicted biological activity for 3,5-dimethyl-N-nitropyrazole-1-carboxamide (4) and bis(3,5-dimethylpyrazole-1-carbonyl)hydrazine (15) for stroke treatment. Future work is planned to continue similar studies with acylhydrazide and carbazate derivatives, which will more likely undergo similar transformations when reacted with acetylacetone due to the electron-withdrawing group present on amide.
Experimental Section
General Information
The work was done using the equipment provided by the Biysk Regional Center for Shared Use of Scientific Equipment of the SB RAS (IPCET SB RAS, Biysk city). 1H and 13C NMR spectra of the samples were recorded on Bruker AVANCE III 500 and Bruker AV-400 spectrometers operating at 500.03 and 400.13 MHz for 1H and, respectively, at 125.73 and 100.61 MHz for 13C. DMSO-d6 signals were used as the reference standard: δ = 2.50 ppm for residual protons of CHD2 in 1H NMR spectra and δ = 39.51 ppm for residual protons of CD3 in 13C NMR spectra, as well as CDCl3 signals: δ = 7.24 ppm for residual protons in 1H NMR spectra and δ = 77.16 ppm for 13C NMR spectra. 15N NMR spectra were taken relative to formamide as the external standard: δ(15N) = 112.5 ppm. The structural determination and signal assignment in 1H and 13C NMR spectra were done on the basis of 2D heteronuclear 1H, 13C HMBC and 1H, 13C HSQC correlations.
UV absorption spectra were taken on a Varian Cary 50 UV–vis spectrophotometer in water in quartz cells (l = 0.5) at 20 °C. Infrared spectra of the samples were recorded in KBr on a FT-801 Fourier spectrometer (Simex, Russia) at 4000 to 500 cm–1. Elemental analysis was performed on a CHNO FlashEATM 1112 analyzer. The melting point was measured on a Böetius PHMK (VebAnalytik, Dresden) instrument. The decomposition temperature was measured on TGA/SDTA 851e and DSC 822e thermal analyzers (Mettler Toledo, Switzerland) over a temperature range of 25–300 and 25–500 °C under nitrogen at a heating rate of 10 °C/min. The results were digitized and processed in STARe 11.0 thermal analysis software. Mass spectrometry and precise measurements of molecular weights were done on a Thermo Electron Double Focusing System (Thermo Eclectron Corp., USA). The samples contained in metal vials were introduced into the mass spectrometer by direct injection; if necessary, the sample vial can be heated up in the temperature range from 25 to 360 °C. The mass spectrometer was operated in the electron ionization mode at an electron energy of 70 eV (unless otherwise stated). Measurements of accurate ionic masses were performed with respect to perfluorokerosene as the internal mass standard.
Crystallography
The measurements were carried out at the Collective Chemical Service Center of the SB RAS. The XRD of compound 15 was carried out on a Bruker KAPPA APEX II CCD diffractometer with graphite-monochromated Mo Kα (0.71073 Å) radiation at room temperature. The crystals were obtained by the slow evaporation of an ethanol–toluene solution in an open flask under normal conditions. Absorption corrections were applied using SADABS.38 The structure was resolved by the direct method. Positions and temperature factors for the nonhydrogen atoms were refined anisotropically by the full-matrix least-squares method. The hydrogen atoms were refined in the riding model. All computations were done using SHELX-2014 programs.39 Due to the high disorder of solvent molecules, we used the PLATON SQUEEZE tool.40 The atom coordinates and their temperature parameters were deposited with the Cambridge Structural Database.
Crystal Data for 15
C12H16N6O2, Mr = 276.31, monoclinic system, space group C2/c, a = 32.0854 (18), b = 16.6338 (7), c = 18.7396 (11) Å, β = 106.521 (2)°, V = 9588.5 (9) Å3, Z = 24, Dcalc = 1.148 g·cm–3, μ = 0.083 mm–1, ⊖ scan range 2.3 < 25.0°, measured reflections 35,665, independent 8467 (Rint = 0.0545), observed 5329 I ≥ 2σ(I), refined parameters 553, R[I ≥ 2σ(I)] = 0.0701, wR2 = 0.2908, S = 1.09. CCDC 2009938 contains supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk.
Synthetic Methods
The starting 4-nitrosemicarbazide (1) was prepared by the reported procedure.2 Semicarbazide (6) was prepared from semicarbazide hydrochloride by the treatment with an equimolar quantity of KOH in an alcoholic solution at not above 30 °C. Diaminourea (10) was synthesized by the reported procedure.41
3,5-Dimethyl-N-nitropyrazole-1-carboxamide (4) and 3,5-Dimethyl-pyrazole (5)
To a solution of 4-nitrosemicarbazide (1 mmol) in water (100 mL) was added slowly acetylacetone (1 mmol) with vigorous stirring. The whole was held at room temperature for 24 h. The reaction mixture was evaporated to dryness to furnish a light-yellow sediment (4). Yield: 55%. Mp 93–95 °C. FTIR (KBr, cm–1): 3204 (NH), 3115, 3108, 3036, 3000, 2940, 2876, 1666 (C=O) (C=N), 1595, 1492, 1427, 1378, 1353, 1284 (N–NO2), 1224, 1157, 1128, 1030, 1010, 976, 925, 841, and 781.1H NMR (500 MHz, CDCl3, δ, ppm): δ 2.21 (s, 3H, 3–CH3), 2.58 (s, 3H, 5–CH3) 5.90 (s, 1H, 4–CH=), and 12.89 (br s, 1H, NH). 13C NMR (126 MHz, CDCl3, δ, ppm): δ 13.87 (q, 1JCH = 125.8 Hz, 3–CH3), 15.11 (q, 1JCH = 130.8 Hz, 5–CH3), 109.79 (d sept, 1JCH = 174.3, 3JCH = 3.6 Hz, Hz, 4–CH=), 144.91 (quint., 2JCH = 7.2 Hz, 5–C=N), 149.71 (quint., 2JCH = 6.4 Hz, 3–C=N), and 155.62 (s, C=O). 15N NMR (51 MHz, HCONH2, δ, ppm): δ 221.2 (1–N), 250.29 (NH), and 284.15 (2–N). Calcd. for C6H8N4O3, (%): C, 39.13; H, 4.35; and N, 30.43; found, C, 39.47; H, 4.05; and N, 30.78.
Recrystallization of the Resultant Sediment (4) from Diethyl Ether, Water, or Ethanol Gave 3,5-Dimethyl-pyrazole (5)
Mp 108°C. FTIR (KBr, cm–1): 3201 (NH), 3133, 3111, 3040, 2942, 2881, 1665 (C=N), 1596, 1485, 1424, 1307, 1155, 1129, 1010, 856, 780, and 737. 1H NMR (500 MHz, CDCl3, δ, ppm): δ 2.26 (s, 6H, CH3), 5.82 (s, 1H, CH=), and 12.89 (br s, 1H, NH). 13C NMR (126 MHz, CDCl3, δ, ppm): δ 12.03 (CH3), 104.53 (CH=), and 144.42 (C=N). 15N NMR (51 MHz, HCONH2, δ, ppm): δ 234.0.
5-Hydroxy-3,5-dimethyl-2-pyrazoline (7)
To a solution of semicarbazide (1 mmol) in water (40 mL) was added slowly acetylacetone (1 mmol) with vigorous stirring. The whole was held at room temperature for 24 h. The reaction mixture was then evaporated to dryness. Yield: 70%. Mp 130 °C.FTIR (KBr,cm–1): 3464 (OH), 3310 (NH), 3010 (CH2cycle), 2965 (CH3), 1675 (C=O), 1633 (C=N), 1597 (NH2), 1484, 1444, 1382, 1332, 1241, 1221 (C–N), 1194, 1121 (OH), 1054, 1014, 956, 869, and 763. 1H NMR (500 MHz, DMSO, δ, ppm, J/Hz): δ 1.72 [s., 3H, CH3–C(OH)]; 1.91 (s, 3H, CH3–C=N); 2.72 and 2.82 (d, 1H, J = 18.2 Hz, CH2), 3.32 (br s, 2H, OH), and 6.09 (br s, 2H, NH2). 13C NMR (126 MHz, DMSO, δ, ppm): 15.63 (q, CH3–C=N, 1JCH = 128.10 Hz), 26.90 [qd, CH3–C(OH), 1JCH = 127.7 Hz, 3JCH = 4.3 Hz], 52.09 (t quint, CH2, 2JCH = 4.8 Hz), 89.80 [quint, C(OH) (CH3), 2JCH = 6.8 Hz], 150.77 (sext, N=C–CH3, 2JCH = 6.8 Hz), and 155.33 (s, C=O). 15N NMR (51 MHz, HCONH2 δ, ppm, J/Hz): 78.85 (NH2, JNH = 89.8 Hz), 185.61 (N–CO), and 315.64 (C=N). Calcd. for C6H11N3O2, (%): C, 45.86; H, 7.00; and N, 26.75; found, C, 45.81; H, 7.08; and N, 26.63.
3.5-Dimethyl-1-carbamylpyrazole (8) and Acetone Semicarbazone (9)
To a solution of semicarbazide (2 mmol) in water (100 mL) was added slowly acetylacetone (1 mmol) with vigorous stirring. The whole was held at room temperature for 2 h; afterward, the precipitated pyrazole 8 was collected by filtration. Yield: 56%. Mp 112 °C. FTIR (KBr, cm–1): 3419 (NH), 3302, 3241, 2993, 2931, 1701 (C=O) (C=N), 1593, 1573, 1482, 1400, 1330, 1129, 1085, 966, 801, 770, and 707. 1H NMR (500 MHz, DMSO, δ, ppm): δ 2.14 (s, 3H, 3–CH3), 2.44 (5–CH3), 6.03 (s, 1H, 4–CH=), 7.44 (br s, 1H, NH2), and 7.47 (br s, 1H, NH2). 13C NMR (126 MHz, DMSO, δ, ppm, J/Hz): δ 13.17 (q, 1JCH = 127.5 Hz, 3–CH3), 13.60 (q, 1JCH = 130.1 Hz, 5-CH3), 109.52 (d sept, 1JCH = 174.6 Hz, 3JCH = 3.6 Hz, 4–CH=), 142.38 (quint, 2JCH = 6.5 Hz, 5–Cq), 148.90 (quint, 2JCH = 6.5 Hz, 3–Cq), and 151.90 (s, C=O). 15N NMR (51 MHz, HCONH2, δ, ppm): δ 85.84 (NH2), 216.84 (=N–N–CO), and 294.28 (=N–N–CO). Calcd. for C6H9N3O, (%): C, 51.80; H, 6.47; and N, 30.22; found, C, 51.73; H, 6.41; and N, 30.58.
The mother solution was evaporated to dryness and recrystallized from ethanol at 70 °C to give a white sediment (9). Yield: 53%. Mp 85 °C.Mass spectrum: molecular ion peak (m/z) 115. FTIR (KBr, cm–1): 3462 (NH), 3206, 2885 (CH3), 1691 (C=O) (C=N), 1582 (δNH), 1436 (CH3), 1372, 1272, 1124 (C–N), 1046, 987, and 767. 1H NMR (500 MHz, DMSO, δ, ppm): δ 1.78 (s, 3H, CH3), 1.86 (s, 3H, CH3), 6.19 (br s, 2H, NH2), and 8.93 (br s, 1H, NH). 13C NMR (126 MHz, DMSO, δ, ppm, J/Hz): δ 16.81(qq, cis-CH3, 1JCH = 127.3 Hz, 3JCH = 3.2 Hz), 24.85 (qd, trans-CH3, 1JCH = 126.7 Hz, 3JCH = 2.9 Hz), 146.63 (d sept, N=C, 2JCH = 6.7 Hz, 3JCH = 2.9 Hz), and 157.46 (s, C=O). 15N NMR (51 MHz, HCONH2, δ, ppm): δ 78.85 (NH2–CO, JNH = 92.9 Hz), 147.10 (HN–CO, JNH = 88.5 Hz), and 305.35 (C=N). Calcd. for C4H9N3O, (%): C, 41.74; H, 7.83; and N, 36.52; found, C, 41.68; H, 7.51; and N, 36.58.
3,5-Dimethylpyrazole-1-carbohydrazide (11)
To a solution of diaminourea (3 mmol) in water (100 mL) was added slowly acetylacetone (2 mmol) with vigorous stirring. The whole was held at room temperature for 24 h; afterward, precipitated pyrazole 11 was collected by filtration. Yield: 73%. Mp 109 °C. FTIR (KBr, cm–1): 3318, 3209, 2977, 2932, 1704, 1641, 1572, 149 + 5, 1414, 1377, 1348, 1283, 1214, 14185, 1145, 1086, 1030, 923, 891, 788, and 758. 1H NMR (500 MHz, DMSO, δ, ppm): δ 2.14 (s, 3H, CH3–C=N), 2.45 (s, 3H, CH3–C=C), 4.35 (br s, 2H, NH2), 6.05 (s, 1H, CH=), and 9.02 (br s, 1H, NH). 13C NMR (126 MHz, DMSO, δ, ppm, J/Hz): δ 13.22(q, CH3–C=N, 1JCH = 127.4 Hz), 13.30 (q, CH3–C=C, 1JCH = 129.9 Hz), 109.14 (d sept, HC=C, 1JCH = 174.9 Hz, 3JCH = 3.8 Hz), 142.44 (quint, C=N, 2JCH = 6.8 Hz), 149.29 (quint, C=N, 2JCH = 6.2 Hz), and 150.01 (br s, C=O). 15N NMR (51 MHz, HCONH2, δ, ppm): δ 110.00 (NH), 214.24 (N–NH2), and 291.96 (C=N). Calcd. for C6H10N4O, (%): C, 46.75; H, 6.49; and N, 36.36; found, C, 46.79; H, 6.58; and N, 36.52.
Bis(3,5-Dimethylpyrazole-1-carbonyl)hydrazine (15)
(a) To a solution of diaminourea (10) (3 mmol) in aqueous acetonitrile (80 mL) was added slowly acetylacetone (6 mmol) with vigorous stirring. The whole was held at room temperature for 7 days. After acetonitrile was evaporated, compound 15 precipitated in the aqueous layer, collected by filtration, and washed with cold water. Yield: 55%.
When water was used as the reaction medium, the yield improved to 70%. (b) To a solution of 3,5-dimethylpyrazole-1-carbohydrazide (11) (3 mmol) in water (100 mL) was added slowly acetylacetone (3 mmol) with vigorous stirring. The whole was held at room temperature for 7 days. After acetonitrile was evaporated, compound 15 precipitated and was collected by filtration and washed with cold water. Yield: 78%. The use of 80% aqueous acetonitrile as the reaction medium diminished the yield to 21%. (c) To a solution of dimethylsemicarbazide (1 mmol) in water (40 mL) was added slowly acetylacetone (1 mmol). Pyrazole 15 precipitated after a 12 h holding at room temperature. Yield: 9%.
When methylsemicarbazide was used under the same conditions, the yield of pyrazole 15 was 15%.
Mp 145 °C. Mass spectrum: molecular ion peak (m/z) 276 (Figure S7). FTIR (KBr, cm–1): 3380, 3295, 3110, 2974, 2933 (CH), 1751, 1702 (C=O, C=N), 1580 (δNH), 1493, 1413, 1344, 1263, 1083, 1028, 969, 832, and 762. 1H NMR (500 MHz, DMSO, δ, ppm): δ 2.20 (s, 3H, 3-CH3), 2.46 (s, 3H, 5–CH3), 6.15 (s, 1H, CH=), and 10.12 (br s, 1H, NH). 13C NMR (126 MHz, DMSO, δ, ppm, J/Hz): δ 13.24 (q, 1JCH = 127.9 Hz, 3–CH3), 13.37 (q, 1JCH = 129.35 Hz, 5–CH3), 109.83 (d sept, 1JCH = 175.3 Hz, 3JCH = 3.6 Hz, 4–CH=), 143.17 (quint, 2JCH = 7.4 Hz, 3–Cq), 150.13 (quint, 2JCH = 6.4 Hz, 5–Cq), and 150.58 (d, JCH = 7.7 Hz, C=O). 15N NMR (51 MHz, HCONH2, δ, ppm): δ 107.97 (NH–CO), 212.84, and 292.55 (C=N). Calcd. for C4H6N8O6, (%): C, 52.17; H, 5.80; and N, 30.43; found, C, 52.17; H, 5.84; and N, 30.42.
Acknowledgments
The authors acknowledge financial support from the Ministry of Science and Higher Education of the Russian Federation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00518.
The authors declare no competing financial interest.
Supplementary Material
References
- Glukhacheva V. S.; Il’yasov S. G.; Obraztsov A. A.; Gatilov Y. V.; Eltsov I. V. A new synthetic route to heteroanthracenes. Eur. J. Org. Chem. 2018, 2018, 1265–1273. 10.1002/ejoc.201701689. [DOI] [Google Scholar]
- Il’yasov S. G.; Sakovich G. V.; Lobanova A. A. Synthesis, structure, and properties of N,N’-dinitrourea. Propellants, Explos., Pyrotech. 2013, 38, 327–334. [Google Scholar]
- Il’yasov S. G.; Lobanova A. A.; Popov N. I.; Kazantsev I. V.; Varand V. L.; Larionov S. V. Synthesis and properties of Fe(II), Ni(II), Co(II), and Zn(II) chelates with 4-Nitrosemicarbazide. Russ. J. Gen. Chem. 2006, 76, 1719–1723. [Google Scholar]
- Glukhacheva V. S.; Il’yasov S. G.; Sakovich G. V.; Gatilov Y. V. Furfural nitrosemicarbazone salts. Russ. Chem. Bull. 2017, 66, 83–85. 10.1007/s11172-017-1703-1. [DOI] [Google Scholar]
- Glukhacheva V. S.; Il’yasov S. G.; Sakovich G. V.; Tolstikova T. G.; Bryzgalov A. O.; Pleshkova N. V. Synthesis, properties, and application of 4-nitro semicarbazones. Russ. Chem. Bull. 2016, 65, 550–560. 10.1007/s11172-016-1336-9. [DOI] [Google Scholar]
- Il’yasov S. G.; Glukhacheva V. S.; Yermoshina V. A.; Eltsov I. V. Synthesis of nitro-octaaza derivatives of reduced anthracene. Z. Anorg. Allg. Chem. 2019, 645, 792–796. [Google Scholar]
- Obraztsov A.A.; Glukhacheva V. S.; Ilyasov S. G. A new synthetic method for 1,2,4,5,8,9,11,12-octaazacyclotetradeca-5,7,12,14-tetraene-3,10-dione. South-Siberian Scientific Bulletin 2018, 4, 179–183. [Google Scholar]; (in Russian). Article can be accessed via:http://s-sibsb.ru/images/articles/2018/4/S-SibSB_Issue_24.pdf
- Gautam A.; Chandra S. Spectral and biological studies of Ni(II), Pd(II), Pt(IV) and Cu(II) complexes with novel azamacrocyclic Ligand. Asian J. Chem. 2019, 31, 396–402. 10.14233/ajchem.2019.21706. [DOI] [Google Scholar]
- Buttkus H. Reaction of cysteine and methionine with malonaldehyde. J. Am. Oil Chem. Soc. 1969, 46, 88–93. 10.1007/bf02541215. [DOI] [Google Scholar]
- Grinshtein V. Y.; Veveris A. P. A study of monoguanyl hydrazones and some mixed guanyl hydrazones of thiosemi carbazones of β-diketones exhibiting tuberculostatic action. Russ. J. Gen. Chem. 1962, 32, 1056–1062. [Google Scholar]; (in Russian)
- Akhmedzade D. A.; Yasnopolski V. D.; Aslanova A. A.; Yuzbashinskaya P. A.; Salimova M. A. Produkts of the oxidation of p-dimethylcyclohexane by atmospheric oxygen. Azerb. Khim. Zh. 1968, 6, 103–104. [Google Scholar]
- Ried W.; Mühle G. Zurspaltbarkeit von aryl- und acyl-hydrazonenmitacetylaceton. Liebigs Ann 1962, 656, 119–126. 10.1002/jlac.19626560118. [DOI] [Google Scholar]
- Scott F.; O’Donovan G.; Kennedy M.; Reilly J. Notes - Studies in the Pyrazole Series. Part VII. The base-induced scissions of 3,5-dimethyl-1-carbamylpyrazole and -1-thiocarbamylpyrazole. J. Org. Chem. 1957, 22, 820–823. 10.1021/jo01358a601. [DOI] [Google Scholar]
- Coburn M. D.; Ott D. G. An improved synthesis of 3,6diamino 1,2,4,5tetrazine. J. Heterocycl. Chem. 1990, 27, 1941–1945. 10.1002/jhet.5570270717. [DOI] [Google Scholar]
- Kathiravan M. K.; Salake A. B.; Chothe A. S.; Dudhe P. B.; Watode R. P.; Mukta M. S.; Gadhwe S. The biology and chemistry of antifungal agents: a review. Bioorg. Med. Chem. 2012, 20, 5678–5698. 10.1016/j.bmc.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Wright J. B.; Dulin W. E.; Markillie J. H. The antidiabeticactivity of 3,5-dimethylpyrazoles. J. Med. Chem. 1964, 7, 102–105. 10.1021/jm00331a022. [DOI] [PubMed] [Google Scholar]
- do CarmoMalvar D.; Ferreira R. T.; de Castro R. A.; de Castro L. L.; Freitas A. C. C.; Costa E. A.; Florentino I. F.; Mafra J. C. M.; de Souza G. E. P.; Vanderlinde F. A. Antinociceptive, anti-inflammatory and antipyreticeffects of 1,5-diphenyl-1H-pyrazole-3-carbohydrazide, a newheterocyclic pyrazole derivative. Life Sci. 2014, 95, 81–88. 10.1016/j.lfs.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Mondal G.; Jana H.; Acharjya M.; Santra A.; Bera P.; Jana A.; Panja A.; Bera P. Synthesis, in vitro evaluation of antibacterial, antifungal andlarvicidal activities of pyrazole/pyridine based compounds andtheirnanocrystalline MS (M=Cu and Cd) derivatives. Med. Chem. Res. 2017, 26, 3046–3056. [Google Scholar]
- Fesenko A. A.; Shutalev A. D. Different modes of acid-catalyzed cyclization of 4-(γ-oxoalkyl)semicarbazidehydrazones: 7-membered versus 14-membered cyclic semicarbazones formation. Tetrahedron 2015, 71, 9528–9543. [Google Scholar]
- Fesenko A. A.; Shutalev A. D. Base-promoted ring expansion of 3-aminopyrimidine-2-thiones into 1,2,4-triazepine-3-thiones. Tetrahedron 2016, 72, 2560–2573. 10.1016/j.tet.2016.03.082. [DOI] [Google Scholar]
- Bonacorso H. G.; Libero F. M.; Dal Forno G. M.; Pittaluga E. P.; Porte L. M. F.; Martins M. A. P.; Zanatta N. A telescoped protocol for the synthesis of new pyrrolo [3,4-d]pyridazinones by cascade reactions. Tetrahedron Lett. 2015, 56, 5190–5195. 10.1016/j.tetlet.2015.07.035. [DOI] [Google Scholar]
- Calvo L. A.; González-Nogal A. M.; González-Ortega A.; Sañudo M. C. Synthesis of silylatedβ-enaminones and applications to the synthesis of silylheterocycles. Tetrahedron Lett. 2001, 42, 8981–8984. 10.1016/s0040-4039(01)01966-9. [DOI] [Google Scholar]
- Zhang Q.; Hu B.; Zhao Y.; Zhao S.; Wang Y.; Zhang B.; Yan S.; Yu F. Synthesis of N-sulfonylpyrazolesthrough cyclization reactions of sulfonylhydrazines with enaminonespromoted by p-TSA. Eur. J. Org. Chem. 2020, 2020, 1154–1159. 10.1002/ejoc.201901886. [DOI] [Google Scholar]
- Zelenin K. N.; Alekseev V. V.; Kuznetsova O. B.; Saminskaya A. G.; Yakimovich S. I.; Zerova I. V. Carbohydrazones and their ring-chain tautomerism. Russ. J. Org. Chem. 1999, 35, 357–363. [Google Scholar]; (in Russian)
- Alberola A.; Calvo L.; Ortega A. G.; Sádaba M. L.; Sañudo M. C.; Granda S. G.; Rodríguez E. G. Reactions of β-aminoenones with acetylhydrazine, semicarbazide and methoxycarbonylhydrazine. Synthesis of 1-acetyl-, 1-carboxamide- or methyl 1-carboxylated pyrazolederivatives. Heterocycles 2010, 31, 2675–2686. 10.1002/chin.200011113. [DOI] [Google Scholar]
- Souza F. R.; Souza V. T.; Ratzlaff V.; Borges L. P.; Oliveira M. R.; Bonacorso H. G.; Zanatta N.; Martins M. A. P.; Mello C. F. Hypothermic and antipyretic effects of 3-methyl-and 3-phenyl-5-hydroxy-5-trichloromethyl-4, 5-dihydro-1H-pyrazole-1-carboxyamides in mice. Eur. J. Pharmacol. 2002, 451, 141–147. 10.1016/s0014-2999(02)02225-2. [DOI] [PubMed] [Google Scholar]
- Jabłońska-Wawrzycka A.; Rogala P.; Czerwonka G.; Hodorowicz M.; Stadnicka K. J. Zinc(II) complexes with heterocyclic ether, acid and amide. Crystalstructure, spectral, thermal and antibacterial activity studies. J. Mol. Struct. 2016, 1105, 357–369. 10.1016/j.molstruc.2015.10.067. [DOI] [Google Scholar]
- PubChem database of chemical molecules. Electronic resource can be found via https://pubchem.ncbi.nlm.nih.gov/compound/23171560 [date accessed 20.06. 2020].
- Safaei S.; Mohammadpoor-Baltork I.; Khosropour A. R.; Moghadam M.; Tangestaninejad S.; Mirkhani V.; Kia R. Application of a multi-SO3H Bronsted acidic ionic liquid in water: a highlyefficient and reusable catalyst for the regioselective and scaled-up synthesis ofpyrazoles under mild conditions. RSC Adv. 2012, 2, 5610–5616. 10.1039/c2ra20624b. [DOI] [Google Scholar]
- Brown H. W. A study of the regularity of egg-production of Ascaris lumbricoides, Necator americanus and Trichuris trichiura. J. Parasitol. 1927, 14, 110. 10.2307/3271726. [DOI] [Google Scholar]
- Ghorai D.; Mani G. Unsubstitutedquinoidalpyrrole and its reaction with oxygen, charge transfer and palladium(II) complexes via DDQ oxidation. R. Soc. Chem. 2014, 4, 45603–45611. 10.1039/c4ra07808j. [DOI] [Google Scholar]
- Wang Q.-Y.; Wang S.; Feng X.; Wu L.; Zhang S.-H.; Ding N.; Tong W.-C.; Zhou M.-R.; Wang B.; Yang L. A facile method to prepare energetic materials (EMs). RSC Adv. 2017, 7, 48161. 10.1039/c7ra08115d. [DOI] [Google Scholar]
- Zhang W. X.; Ma C. Q.; Wang X. N.; Yu Z. G.; Jing D. H.; Zheng J. M. Template synthesis and structure of dinuclear Zinc complex with three kinds of pyrazoleligands. Chin. J. Struct. Chem. 1996, 15, 404. [Google Scholar]
- Das D.; Kannan S.; Maity D. K.; Drew M. G. B. Steric effects on uranylcomplexation: synthetic, structural, and theoretical studies of carbamoylpyrazolecompounds of the uranyl(VI) ion. Inorg. Chem. 2012, 51, 4869–4876. 10.1021/ic300398a. [DOI] [PubMed] [Google Scholar]
- Böttcher A.; Debaerdemaeker T.; Radziszewski J. G.; Friedrichsen W. Darstellung von 1-Oxo-1H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-3-olaten. Eine Ring-Ketten-Tautomerie in der ReihebicyclischerdipolarerHeterocyclen. Chem. Ber. 1988, 121, 895–908. 10.1002/cber.19881210513. [DOI] [Google Scholar]
- Zelenin K. N.; Solod O. V.; Tomchin A. B. Newdataonreactionbetween 1,4-bifunctionalhydrazinederivativesand 1,3-diketones. Russ. J. Gen. Chem. 1987, 57, 510–522. [Google Scholar]; (in Russian)
- Filimonov D. A.; Lagunin A. A.; Gloriozova T. A.; Rudik A. V.; Druzhilovskii D. S.; Pogodin P. V.; Poroikov V. V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. 10.1007/s10593-014-1496-1. [DOI] [Google Scholar]
- Sheldrick G. M.SADABS, Program for Area Detector Adsorption Correction, Institute for Inorganic Chemistry, University of Gottingen, Germany: 1996.
- Sheldrick G. M. Crystal structure refinement withSHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr C Struct Chem. 2015, 71, 9–18. 10.1107/s2053229614024929. [DOI] [PubMed] [Google Scholar]
- Glukhacheva V. S.; Il’yasov S. G. Synthesis of 1,3-diaminourea from N,N-dinitrourea. PolzunovskiyVestnik 2013, 3, 26–28. [Google Scholar]; (in Russian). Article can be accessed via: http://elib.altstu.ru/journals/Files/pv2013_03/pdf/026gluhachov.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.