

Abstract
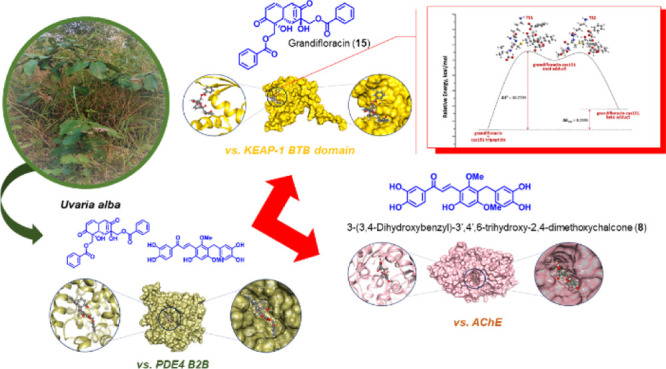
Inhibition of the major cyclic adenosine monophosphate-metabolizing enzyme PDE4 has shown potential for the discovery of drugs for cancer, inflammation, and neurodegenerative disorders such as Alzheimer’s disease. As a springboard to explore new anti-cancer and anti-Alzheimer’s chemical prototypes from rare Annonaceae species, the present study evaluated anti-PDE4B along with antiproliferative and anti-cholinesterase activities of the extracts of the Philippine endemic species Uvaria alba using in vitro assays and framed the resulting biological significance through computational binding and reactivity-based experiments. Thus, the PDE4 B2B-inhibiting dichloromethane sub-extract (UaD) of U. alba elicited antiproliferative activity against chronic myelogenous leukemia (K-562) and cytostatic effects against human cervical cancer (HeLa). The extract also profoundly inhibited acetylcholinesterase (AChE), an enzyme involved in the progression of neurodegenerative diseases. Chemical profiling analysis of the bioactive extract identified 18 putative secondary metabolites. Molecular docking and molecular dynamics simulations showed strong free energy binding mechanisms and dynamic stability at 50-ns simulations in the catalytic domains of PDE4 B2B, ubiquitin-specific peptidase 14, and Kelch-like ECH-associated protein 1 (KEAP-1 Kelch domain) for the benzylated dihydroflavone dichamanetin (16), and of an AChE and KEAP-1 BTB domain for 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) and grandifloracin (15), respectively. Density functional theory calculations to demonstrate Michael addition reaction of the most electrophilic metabolite and kinetically stable grandifloracin (15) with Cys151 of the KEAP-1 BTB domain illustrated favorable formation of a β-addition adduct. The top-ranked compounds also conferred favorable in silico pharmacokinetic properties.
1. Introduction
The cell cycle is tightly regulated by cyclic nucleotide signaling ascribed as a composite component of tumor pathogenesis. Impairments in the synthesis of cyclic adenosine monophosphate (cAMP) or cyclic guanosine monophosphate by regulating phosphodiesterase enzymes (PDEs) have been linked to cancer pathologies.1,2 Thus, the cellular regulation of cAMP signaling has been an attractive target in designing small molecules for a wide array of diseases, including cancer and Alzheimer’s.3,4 Among the 11 PDE enzyme families, PDE4 specifically catalyzes the hydrolysis of cAMP.1,2,5 Tumor cells originating from the central nervous system, lung, blood, and breast have an upregulated PDE4 level that primarily drives cAMP hydrolysis.5 Thus, PDE4 is an attractive target for cancer chemotherapy. PDE4 inhibitors suppress tumor growth and induce apoptosis in cancer cells.6 The ubiquitin-proteasome pathway is another potential drug target for carcinogenesis involved in synchronized proteolysis of cyclins and cyclin-dependent kinase inhibitors critical for cell cycle progression.7,8 Protein ubiquitination is primarily regulated by deubiquitinases (DUBs). Thus, inhibiting DUBs, such as ubiquitin-specific peptidase 14 (USP14), may induce apoptosis in cancer cells while conferring low cytotoxicity to normal cells.8 The cytoprotective mechanism of the body against oxidative stress involving the Kelch-like ECH-associated protein 1 (KEAP-1)–nuclear factor erythroid 2 (Nrf2)–antioxidant response element pathway is another related interesting avenue for cancer therapy.9 Oxidative stress is the underlying contributor to virtually every chronic disease. Thus, disrupting the KEAP-1–Nrf2–ARE (antioxidant response element) pathway and consequently enhancing Nrf2 activity are promising strategies for preventing chronic diseases involving oxidative stress and inflammation.10 Natural products, particularly those with unsaturated electrophilic moieties, have been identified to target USP14 and KEAP-1.9,11
Meanwhile, there have been studies indicating that selective inhibition of PDE4 may potentially treat neurodegenerative disorders, including Alzheimer’s disease (AD).12 Similarly, inhibition of acetylcholinesterase enzyme (AChE) represents a therapeutic strategy for managing AD. AChE catalyzes the hydrolysis of acetylcholine that is vital in memory formation and cognitive functions.13 Several pre-clinical studies indicate that PDE4 and AChE inhibitors mitigate deficits in long-term memory caused by oxidative stress, aging, and overexpression of amyloid-β plaques and neurofibrillary tangles in brain tissues. Thus, PDE4 and AChE inhibitors hold promise in the treatment of neurodegenerative and inflammatory diseases.14
The genus Uvaria Linn. (family Annonaceae) is a prolific source of pharmacologically active natural products exhibiting biological activities such anti-cancer, anti-inflammatory, antitubercular, antioxidant, and antimicrobial activities.15−22 Despite numerous studies on several Uvaria species, limited scientific efforts have been described for the endemic Philippine medicinal plant, Uvaria alba. Thus, our present investigation focused on the in vitro evaluation of the anti-phosphodiesterase, antiproliferative, and anti-cholinesterase activities of the dichloromethane (DCM) sub-extract of U. alba along with computational-driven drug discovery experiments to identify specific inhibitors that could target USP14, KEAP-1 domains [Tramtrack and Bric-à-Brac (BTB) and Kelch], PDE4 B2B, and AChE enzymes. A density functional theory (DFT) calculation was also performed to model a possible Michael addition reaction of electrophilic metabolites with KEAP1 and illustrate irreversible, covalent inhibitions.
2. Results and Discussion
2.1. Anti-phosphodiesterase, Antiproliferative, and Anti-cholinesterase Profiles of U. alba Extracts
The phosphodiesterase inhibitory activities of the U. alba extracts were assessed against human recombinant cAMP-specific phosphodiesterase (PDE4 B2). The U. alba crude extract was shown to inhibit PDE4 B2 in comparison with the standard drug rolipram (IC50 = 0.22 μg/mL). Among the sub-extracts, the DCM extract exhibited inhibition against PDE4 B2 with an IC50 of 16.8 μg/mL (Table 1). The role of phosphodiesterases (PDEs) in carcinogenesis has been established in several studies. PDE4 B2 participates in signal transduction by regulating cellular concentrations of cyclic nucleotides2 and is upregulated in colorectal cancer with oncogenic KRAS, thereby inhibiting apoptosis and disrupting cell polarity.23 In addition, PDE4 inhibitors may also induce apoptosis in chronic lymphocytic leukemia by activating PP2A-induced dephosphorylation of proapoptotic Bcl-2.24
Table 1. Antiproliferative, Anti-phosphodiesterase (PDE4 B2) and Anti-AChE Activities of U. alba.
| antiproliferative
activity, GI50f (μg/mL) |
cytotoxic, CC50g (μg/mL) | ||||
|---|---|---|---|---|---|
| test sample | anti-PDE4 B2 IC50e (μg/mL) | HUVECh | K-562i | HeLaj | anti-AChE IC50e (μg/mL) |
| Ua crudea | >20 | >50 | >50 | cytostatic | 0.25 + 0.03 |
| UaBb | >20 | >50 | >50 | >50 | 0.24 + 0.01 |
| UaPc | >20 | 21.3 + 0.7 | 16.7 + 1.6 | cytostatic | 0.25 + 0.02 |
| UaDd | 16.8 + 3.89 | 24.1 + 1.6 | 12.3 + 1.8 | cytostatic | 0.22 + 0.01 |
| doxorubicin | 0.1 + 0.5 | 1.0 + 0.6 | 2.0 ± 0.8 | ||
| rolipram | 0.22 + 0.10 | ||||
| galantamine | 0.14 + 0.01 | ||||
Crude DCM–methanol (1:1) extract.
Petroleum ether sub-extract.
Petroleum ether sub-extract.
DCM sub-extract.
Inhibitory concentration—50.
Growth inhibition—50.
Cytotoxic concentration—50
Human umbilical vein endothelial cells.
Chronic myelogenous leukemia cells.
Cervical cancer cells.
To further evaluate the role of U. alba constituents in carcinogenesis, the extracts were assessed for anti-proliferative and cytotoxic activities using the CellTiter-Blue assay (Table 1, Figure 1). The DCM (UaD) and petroleum ether (UaP) sub-extracts were shown to be antiproliferative against chronic human myelogenous leukemia cells (K-562). Interestingly, the crude extract Ua as well as the sub-extracts UaD and UaP elicited cytostatic effects against HeLa. An extract or plant component is cytostatic if it inhibits cell proliferation over a large range of concentrations compared to a reference drug.
Figure 1.

Cytostatic curve of the crude Ua extract, DCM (UaD), and petroleum ether (UaP) sub-extracts against HeLa.
The downregulation of PDE4 B2 has also been implicated with neuronal plasticity and pro-cognitive development; thus, the PDE4 B2 inhibitory property UaD sub-extract may also confer neuroprotective effects. This claim is further strengthened when UaD inhibited AChE—an enzyme responsible for terminating nerve impulse transmission at the cholinergic synapses by rapid hydrolysis of acetylcholine. A strong AChE inhibition (IC50 = 0.22 μg/mL) was observed in comparison with the positive drug standard galantamine (IC50 = 0.14 μg/mL). Similar to PDE4 B2, inhibition of AChE is also recognized as a strategy for treating Alzheimer’s and Parkinson’s diseases.13 Thus, UaD may exhibit neuroprotective effects by modulating the activity of both PDE4 B2 and AChE.
2.2. HPLC-MS-Quantitative Time-of-Flight Profiling of the UaD Sub-Extract
The UaD sub-extract was chemically profiled to putatively identify secondary metabolites present using LC-HR-ESIMS-quantitative time-of-flight (QToF) analysis (Figures S1 and S2). Eighteen compounds, namely, 7-benzoyloxy-2H-1-benzopyran-2-one (1), uvaridapoxide A (2), microcarpin A (3), microcarpin B (4), grandiuvarone (5), uvaridacane A (6), valderramenol A (7), 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8), albanol A (9), tepanone (10), valderramenol B (11), 5-O-methylchamanetin (12), cyathosthemine (13), bractelactone (14), grandifloracin (15), dichamanetin (16), albanol B (17), and cyathoviridine (18) were identified (Figure 2, Table S1). These compounds have been previously identified from Uvaria and other related genera of Annonaceae and are known to confer cytotoxic and/or anti-inflammatory properties.
Figure 2.
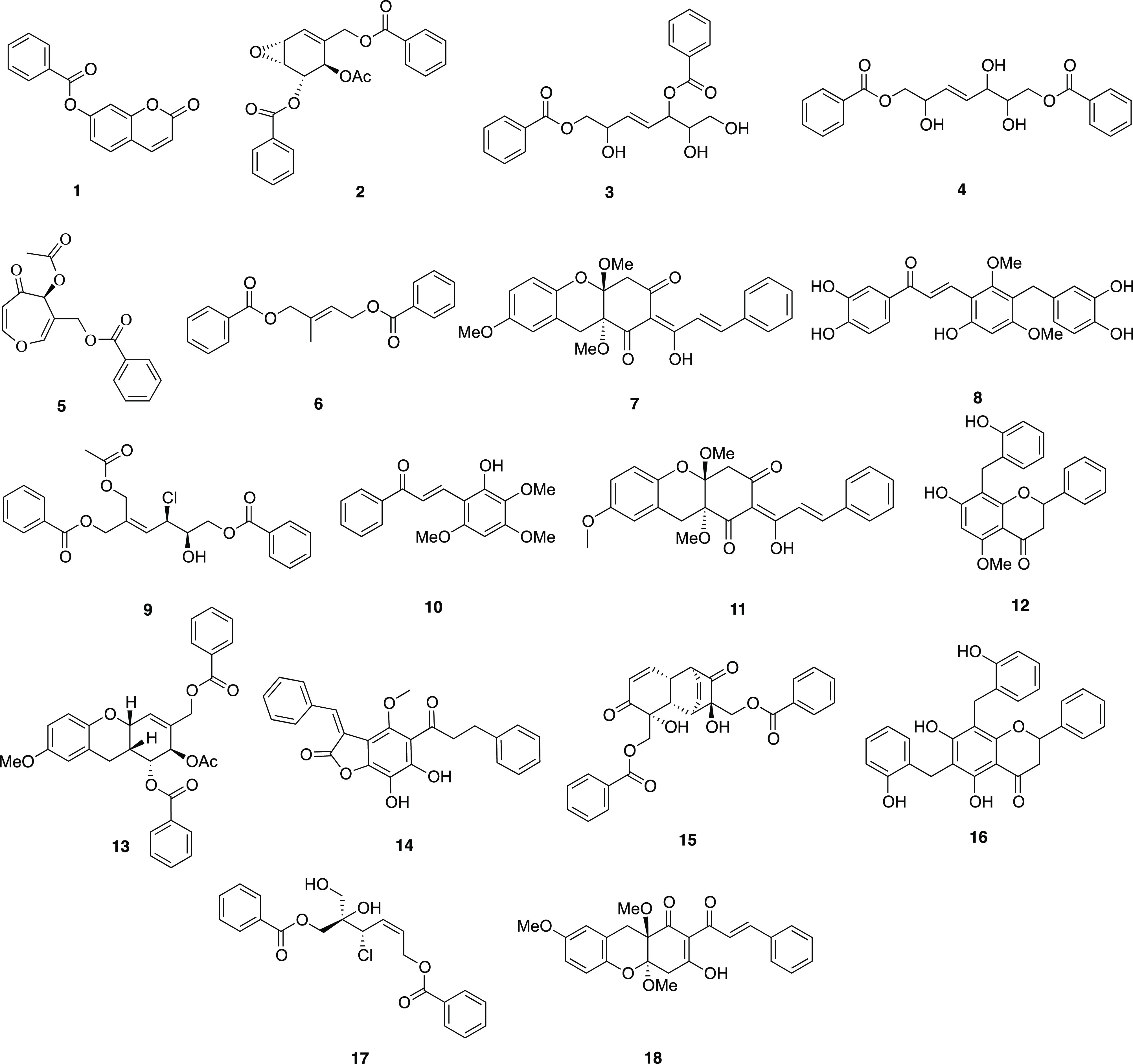
Secondary metabolites 1–18 detected in the DCM sub-extract of U. alba.
2.3. Molecular Docking to PDE4B2
The structure of PDEs comprises three domains, with the catalytic domain being the most conserved among the PDE isoenzymes. PDE4 B2B, in particular, comprises an N-terminal regulatory domain (residues 1–151), a catalytic domain (residues 152–489), and a C-terminal domain (residues 490–568). To assess the binding characteristics of 1–18, a molecular docking approach was performed against PDE4 B2B, directed to the binding domain of the enzyme. Dichamanetin (16) and grandifloracin (15) displayed the highest binding affinity with a binding energy (BE) of −10.2 kcal/mol each (Table 2). Compound 16’s attachment to the binding domain of the enzyme is mostly due to pi–pi interplay occurring in four regions of the molecule: the chromanone core against Met347; a phenyl substituent against Phe414 and Phe446; 8-hydroxybenzyl against Tyr233 and Tyr410; and 6-hydroxyphenyl with Leu303. A hydrogen bonding was also observed between Asp392 and a hydroxyl substituent of the chromanone core. Grandifloracin (15) was strongly bound to the active site of the enzyme through intramolecular conventional hydrogen bonds with (a) a hydroxyl substituent against His234 and (b) the electronegative oxygens of two ester groups against Tyr233, His278, and Asn283. Another notable compound that conferred strong binding affinity against PDE4 B2B is bractelactone (14) with a BE of −10.1 kcal/mol (Table 2, Figure 3). Bractelactone’s (14) attachment to putative binding was markedly strengthened by hydrogen bonding between two hydroxyl substituents of the benzofuranone core against His238, His274, Tyr233, and Asp392. For compound 14, a pi–anion and a pi–sulfur intermolecular attraction was observed between Glu304 against the compound’s xanthone core and Cys432 against a benzoyl substituent. Of the three top-scoring ligands, bractelactone (14) was further subjected to molecular dynamics (MD) simulations because it showed a better ADMETox profile.
Table 2. BE Values of 1–18 against PDE4 B2B, USP14, KEAP-1, and AChE.

Figure 3.

Docked poses of (a) bractelactone (14) against PDE4 B2 (PDB ID:1RO6); (b) dichamanetin (16) against USP14 (PDB ID: 6IIM); (c) dichamanetin (16) against the KEAP-1 Kelch domain (PDB ID: 47LB); (d) grandifloracin (15) against the KEAP-1 BTB domain (PDB ID: 5DAD); and (e) 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) against AChE (PDB ID: 4EY6).
2.4. Molecular Docking to Cancer Targets USP14 and KEAP-1 (Kelch and BTB Domains)
USP14, a member of the deubiquitinating enzymes, is an important potential drug target due to its involvement in cancer development and neurodegenerative diseases. Regulation of the ubiquitin-proteasome system is an essential control mechanism and potential inhibitor of DUBs, including USP14 and thus, may balance and prevent disruption of this regulatory system. The benzylated hydroxyflavones, dichamanetin (16) and 5-O-methylchamanetin (12), exhibited favorable binding to the putative binding domains of USP14 with BEs of −9.8 and −9.4 kcal/mol, respectively (Table 2, Figure 3). The chromanone core of 16 is affixed to the binding pocket through pi–anion interaction against Asp199 and stacked pi–amide bonding with Gln198. The hydroxybenzyl moiety is also bound through pi–alkyl and stacked pi–pi attraction with Tyr476, Lys342, and Arg330. There are two prominent stacked pi–pi interactions responsible for compound 12’s attachment to the binding site: (a) Phe331 with the phenyl ring B of the hydroflavone moiety and (b) Tyr476 and the benzyl group.
Molecular docking was also performed for KEAP-1, a member of the BTB-Kelch family of proteins known to interact with Nrf2.25,26 Activation with Nrf2 leads to a coordinated antioxidant and anti-inflammatory responses of the enzyme. The structure of KEAP-1 is divided into four discrete domains: the N-terminal broad complex, Tramtrack and Bric-à-Brac (BTB) domain, a BACK domain, and a C-terminal Kelch domain.27 The Kelch domain of the protein, showcasing a six-bladed propeller-like structure, is required for substrate capture and can be the structural motifs of Nrf2.28 We investigated the binding properties of the secondary metabolites to the Nrf2-binding region of the C-terminal Kelch domain of KEAP-1. Dichamanetin (16) and cyathostemmine (13) conferred strong binding affinities to the said binding region of the protein with BEs of −11.1 and −10.6 kcal/mol, respectively (Table 2, Figure 3). Dichamanetin’s (16) attachment to the Kelch domain’s active site is primarily through H-bonding with the ketone moiety of the chromanone core against Arg415 and the hydroxyl of two benzyl substituents against Asn414 and Ile416. A distinct pi–sigma interaction between the chromanone core and Ala556 was also observed along with stacked pi–pi attraction between the phenyl substituent and Tyr572. With cyathostemmine (13), the complex was stabilized through polar pi affinities with the tyrosines of the Nrf2-binding region of the domain, specifically between (a) the xanthene core and Tyr 334 and (b) the two benzoyl groups against Tyr525 and Tyr572. In addition, 13 is further strengthened through H-bonding of two ester groups against Arg415 and Ser602.
The central BTB domain is another important region of KEAP-1, particularly for its role in providing sensing mechanisms to oxidative stress. This domain houses the nucleophilic residue, Cys151, which was found to be one of the most reactive residues in the regulation of cellular stress.29,30
This residue is a vital component for the post-translational modification of KEAP-1 particularly upon exposure to oxidative stress.31 Thus, the docking behavior of the secondary metabolites was tested against the BTB domain of the enzyme, particularly directed in and around Cys151. Interestingly, grandifloracin (15) exhibited the highest affinity in the binding region (BE = −7.3 kcal/mol) (Table 2, Figure 3). Compound 15 interacted with Cys151 through its tricyclododecenone core and one of the benzoyl substituents via pi–alkyl interplay. Several notable interactions include H-bonding and pi–cation with Lys131, pi–sigma with His154, pi–pi stacking with His129, and alkyl interactions with Ala88, Pro89, and Pro130. Two other compounds showed significant binding to the BTB active region, namely, valderramenol A (7) and 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8), both with a BE of −7.1 kcal/mol. With compound 7, Cys151 interacted via pi–alkyl with the compound’s core and caused a steric bump with a hydroxy substituent. Several stabilizing intermolecular forces were also observed such as pi–cation with Lys131, pi stacking with His129, H-bonding with His154, and pi–alkyl interactions with Ala88 and Arg135. Meanwhile, compound 8 is hooked to the Cys151 residue through pi–sulfur and pi–alkyl interactions with the B ring of the chalcone core and the phenol substituent, respectively. H-bonding with His154, pi–pi stacking with Tyr85, and pi–alkyl interaction with Arg135 were observed between the compound and the active surface of the BTB domain. All three top-scoring metabolites interacting with the KEAP-1 BTB domain active surface, centered around the nucleophilic Cys151, were enone-bearing Michael acceptors. Thus, we speculate that a possible Michael addition may occur, aside from conventional intermolecular forces of attraction, between the electrophilic metabolites and the nucleophilic Cys151 residue.
2.5. Frontier Molecular Orbital Energy Calculation of Electrophilic U. alba Metabolites
The nucleophilic Cys151 residue of the BTB domain of KEAP-1 is considered as the main cysteine sensor for the enone-bearing class of electrophilic inducers which upregulate cytoprotective responses and inhibit pro-inflammatory pathways of the enzyme. This nucleophilic residue is structurally positioned at the surface and not on a deep pocket, making it available for covalent bond formation with incoming electrophiles, aside from the usual intermolecular interactions predicted by molecular docking. Frontier orbitals HOMO and LUMO were used to calculate for the electrophilic potentials of the Michael acceptor enone-bearing secondary metabolites of U. alba. Parr and co-workers introduced the concept of the global electrophilicity index based on a compound’s chemical potential and hardness, which are functions of the energetics of the molecular orbitals.32 The HOMO–LUMO energies, hardness, chemical potential, and electrophilicity index are presented in Table S7 and Figure 4. The parameters were obtained by optimizing the target nucleophiles using B3LYP calculation via DFT. In the present study, 7-benzoyloxy-2H-1-benzopyran-2-one (1), grandifloracin (15), and valderramenol A (7) showed the highest electrophilicity indices among the metabolites. Of the three, however, grandifloracin (15) conferred the highest HOMO–LUMO band gap, which suggests the kinetic stability of the compound.33
Figure 4.

Frontier molecular orbitals of the grandifloracin (LUMO) and active nucleophilic residue of KEAP-1 (HOMO) calculated at the B3LYP/STO-3G level of theory.
Based on molecular docking analysis, which is grounded on intermolecular forces, against the BTB domain of KEAP-1, grandifloracin (15) and valderammenol A (7) were the top binding ligands. Thus, we further investigated the potentiality of a Michael addition reaction between these electrophilic metabolites and the nucleophilic Cys151 residue of the enzyme.
2.6. Reaction Thermochemistry on the Michael Addition of Grandifloracin (15) to Cys151 of the KEAP-1 BTB Domain
α,β-Unsaturated carbonyl compounds have received interest as drug candidates because of their ability to interact with heteroatomic nucleophiles of enzymes, such as the thiol group of cysteine residues, potentially through Michael addition.34 We explored the possibility of a covalent interaction between the electrophilic U. alba metabolites with known anti-proliferative activity and the reactive cysteine residues of KEAP-1. Among the main considerations for exploiting the Michael acceptor reactivity is the presence of a highly reactive, easy-to-access thiol group, particularly the Cys151 found on the surface of the BTB domain of KEAP-1.
Grandilfloracin (15) was used as a Michael acceptor to model the Michael addition to Cys151 of KEAP-1, represented as a truncated tripeptide (Lys150-Cys151-Val152, 19). The ground-state geometries of 15 and 19 were carried out using DFT at the 6-31G**(d,p) level of theory in an aqueous medium (polarizable continuum model, PCM, the solvent is water) to mimic biological conditions. Ground-state geometries are characterized though frequency calculation as those obtaining real frequencies only. The proposed mechanism involved the formation of an enol intermediate (20) through concerted addition of a thiol group to the β-carbon of 15 and a proton transfer to carbonyl oxygen, followed by a keto–enol tautomerization to yield the final Cys151-grandifloracin keto adduct (21) (Scheme 1).
Scheme 1. Proposed Michael Addition of Grandifloracin (15) to the Cys151 Tripeptide Segment (19) of the BTB Domain of KEAP-1.
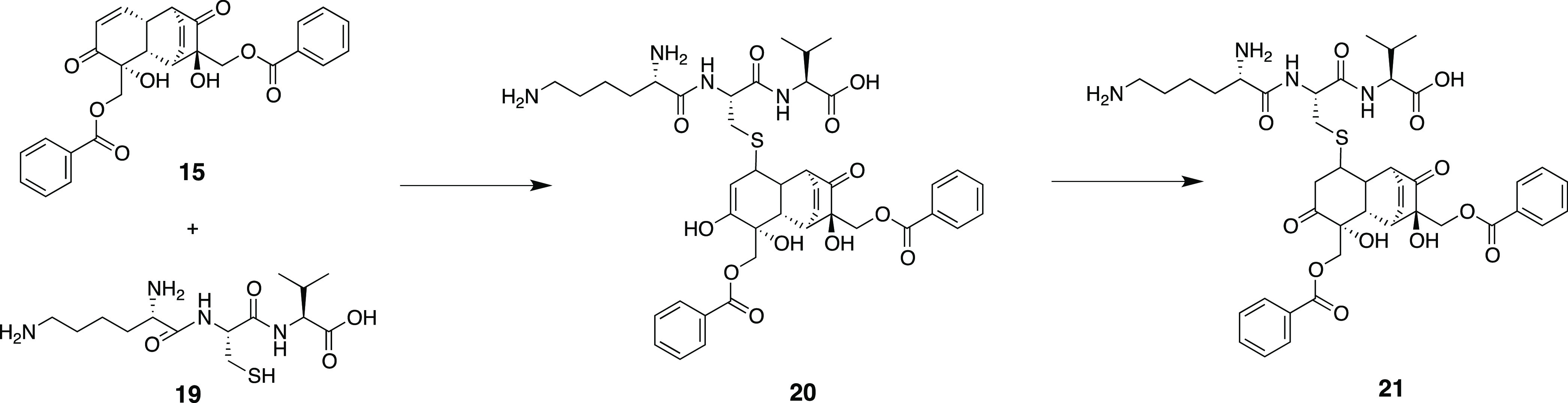
Transition state (TS) geometries were predicted using the QST3 method guided by the optimized geometries of two minima: the reactants and the intermediate/product. The TS was confirmed as having one, and only one, imaginary frequency (a negative vibrational eigenvalue) corresponding to the vibration along the reaction coordinate connecting the two minima. The geometry of the first TS was searched by positioning the thiol group (S32) of Cys151 close to the β-carbon (C31) of grandifloracin with an initial distance rS···C = 2.50 Å (Figure S4). The energy of activation of the reaction is defined as the energy difference, ΔE⧧, of the first TS grandifloracin-Cys151 adduct E⧧ and the ground-state Eo of the reactants. This TS proceeds toward the formation of the intermediate. The formation of intermediate is rate-determining (ΔE⧧ = 30.77 kcal/mol). Unlike most conjugate addition reactions, this step is endothermic probably due to steric interactions of the bulky grandifloracin structure 19 with tripeptide 20, which means that steric accessibility of the reactant could be a major driving force for the feasibility of the reaction. A similar finding was also observed in the Michael addition of deoxyguanosine to catechol estrogen-3,4-quinone35 and the aza-Michael addition of Lys16 (of β-amyloid Aβ42 fibrils) to oxidized (+)-taxifolin36 where in the formation of the enol adduct is endothermic. The final step of the mechanism is keto–enol tautomerization, with ΔEketo–enol⧧ = −20.02 kcal/mol. As expected, the keto form of the adduct (product) is more thermodynamically stable than the enol adduct (intermediate). The reaction path and energetics of the Michael addition reaction between grandifloracin and the Cys151-containing tripeptide of the BTB domain of KEAP-1 are shown in Figure 5 and Table S8.
Figure 5.

DFT/6-31G**(d,p) reaction profile of the Michael addition of grandifloracin (15) and Cys151 tripeptide (19) (KEAP-1 BTB domain).
2.7. Molecular Docking to AChE
The secondary metabolites from the UaD sub-extract of U. alba were also tested for in silico binding toward AChE, of which, 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) had a BE of −9.8 kcal/mol (Table 2, Figure 3). The chalcone core of 8 is bound to the binding site via hydrogen bonding formed between Ser293 and Arg296 and the two hydroxyl substituents, pi interactions with Tyr124 and Trp286 and a pi–lone pair attraction with Tyr337. Bractelactone (14) also exhibited significant binding affinity with AChE (BE = −9.8 kcal/mol). Post-docking analysis revealed that compound 14 is completely nestled onto the AChE’s active site gorge with which the enzyme–ligand complex is stabilized mostly by pi–pi stacking interplay with aromatic components of the structure against Trp86, Tyr124, Trp286, and His447.
2.8. MD Simulation and Binding Free Energy Calculations of Top-Ranked Ligands in Complex with Target Enzymes
The dynamics of the five ligand bound systems—bractelactone (14)-PDE4 B2B, 3-(3,4-dihydroxybenzyl)-3′,4′,6- trihydroxy-2,4-dimethoxychalcone (8)-AChE, grandifloracin (15)-KEAP-1 (BTB domain), dichamanetin (16)-KEAP-1 (Kelch domain), and dichamanetin (16)-USP14—were found to be stable during the course of the MD simulation at 50 ns (Figure 6). In the case of bractelactone (14)-PDE4 B2B, a minute fluctuation between 20 and 30 ns was noted. The system, however, remained stable for the rest of the time with an average root mean square deviation (rmsd) of 2.5 Å. In addition, the stability of 3-(3,4-dihydroxy benzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) bound to AChE was also measured. The system was noted to be stable with an average rmsd of 1.5 Å. For this complex, an initial incremental increase in the rmsd of the ligand bound system was observed up to 10 ns, while for the rest of the simulation time, the system attained stability. In the case of dichamanetin (16)-KEAP-1 (Kelch domain), the system was found to be stable with a little convergence at 42 ns. Meanwhile, grandifloracin (15) in complex with the BTB domain of KEAP-1 was noted to be stable throughout the simulation time with little convergence at 30 ns. Lastly, dichamanetin (16) in complex with USP 14 showed convergence at 10–15 ns, while for the rest of the simulation time, the system attained stability.
Figure 6.

rmsd (Å) of top-scoring protein–ligand complexes as determined during 50 ns of MD simulation. The y-axis shows the rmsd value in Å while the x-axis shows the time in nanoseconds.
The residual flexibility of each system was also determined (Figure S8). As shown, the average root mean square fluctuation (RMSF) of bractelactone (14) bound to PDE4 B2B was reported to be between 2.0 and 2.5 Å. On the other hand, the average RMSF of 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) bound to AChE was observed to be 1–2.5 Å. A significant peak was also noted between 195 and 200 residues in the said complex. For the dichamanetin (16)-KEAP-1 (Kelch domain) complex, a higher fluctuation between 52 and 60 residues was noted, whereas grandifloracin (15) in complex with the BTB domain of KEAP-1 showed a much higher fluctuation. The dichamanetin (16)–USP14 complex possessed an even higher fluctuation in the regions of 40–50, 120–130, 180–200, 220–230, and 255–260.
The bractelactone (14)-PDE4 B2B exhibited a degree of compactness up to 30 ns with an average Rg value of 20.8 Å. Meanwhile, 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) bound to AChE had an average Rg of 21.0 Å. Subsequently, the binding of grandifloracin (15) and dichamanetin (16) to the different binding domains of KEAP-1 showed stable but different Rg patterns. The average Rg value for the dichamanetin (16)–KEAP-1 (Kelch) complex was reported to be 14 Å, while for grandifloracin (15) the Rg value was observed to be 18 Å. The higher Rg value for grandifloracin might be due to the higher fluctuation pattern observed in the RMSF of the grandifloracin (15)-KEAP-1 (BTB domain) system. In the case of the dichamanetin (16)–USP14 complex, the average Rg value was observed to be 21.0 Å. Overall, the ligand-bound systems possessed structural compactness favoring the tight binding of these ligands to their respective receptors (Figure S9).
The trajectories from MD simulations were subjected to total binding free energy calculations. The BE for bractelactone (14)-PDE4 B2B was reported to be −66.64 kcal/mol. Furthermore, a relatively higher energy binding was detected for 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) bound to the AChE receptor. The BE for this complex was at −77.46 kcal/mol, suggesting the strong inhibitory property of this compound to the AChE receptor. Meanwhile, for KEAP-1, two of its binding domains were targeted. Dichamanetin (16) bound to the Kelch domain was found to have a BE of −55.76 kcal/mol, whereas grandifloracin (15) targeting the BTB domain was reported to have a total BE of −48.41 kcal/mol. On the other hand, dichamanetin (16) bound to USP14 possessed a very strong BE. The total BE for this system was observed to be −83.98 kcal/mol. The other energy terms such as vdW, electrostatic forces, polar solvation, and solvent-accessible surface area for each system that contributed to the total BE are given in Table S15.
2.8.1. Hydrogen Bonding Analysis
In order to determine the half-life of each interaction with the key residues, we used the hydrogen bonding occupancy approach. A 50-ns trajectory was used for each system to monitor the hydrogen bonding during the course of simulation. It can be seen that most of the ligands formed significant hydrogen bonds for a longer time. In case of bractelactone_PDE4B2B, Tyr233, His238, His274, Asn283, and Met347 were actively involved in hydrogen bonding. It can be seen that Tyr233 was involved in 89% of the MD trajectories. In dimethoxychalcone_AChE residues Asp74, Tyr124, Glu202, Ser293, and Arg296 were involved among which the life time of Ser293 was reported to be 92%. Furthermore, residues Ser363, Asn414, Arg415, and Tyr436 in dichamanetin_KEAP1-Kelch were actively involved in hydrogen bonding. The grandifloracin_KEAP1-BTB complex was reported to be involved in interactions with Ala88, His129, Pro130, Lys131, and Arg135. Only three residues Arg415, Ile416, and His426 were reported in the dichamanetin_USP14 complex. All the results are calculated in percentage trajectories (0.89 mean 89% of the trajectories) and given in Table S17. The intra-molecule H-bonds are given in Figure 7.
Figure 7.

Hydrogen bonding occupancy of the key residues during the course of simulation. Each residue bonding is given in percentage trajectories.
2.9. Drug-likeness and ADMET Prediction of Secondary Metabolites from UaD
The overall pharmacokinetic behavior of 1–18 was determined through in silico absorption, distribution, metabolism, and excretion (ADME) screening. Based on Lipinski’s rule of five, the druggability of the compounds was predicted using the following descriptors: molecular weight, lipophilicity (reported as octanol–water partition coefficient), and the number of hydrogen bond donors and acceptors. Interestingly, all of the 18 secondary metabolites—including the top-ranked compounds such as 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8), cyathostemmine (13), bractelactone (14), grandifloracin (15) and dichamanetin (16)—exhibited good bioavailability and drug-likeness by fulfilling Lipinski’s criteria (Table S18). Furthermore, the BOILED-Egg (brain or intestinal estimated permeation predictive model), an intuitive graphical plot of the functions of lipophilicity and apparent polarity, was used to predict passive intestinal absorption and brain permeation of the compounds (Figure 8). Compounds located in the yellow region (yolk) have a high probability of blood–brain barrier (BBB) penetration while those in the white region have the propensity for passive absorption through the gastrointestinal (GI) tract. Dichamanetin (16), which demonstrated strong binding affinity to the target enzymes PDE4 B2B, USP14, and KEAP-1 (Kelch domain), was predicted to have good GI absorption and poor BBB crossing capacities. Similarly, grandifloracin (15), which showed favorable binding to KEAP-1 (BTB domain), shared the same pattern of bioavailability as dichamanetin (16). Despite having good binding affinity to AChE, compound 8 was predicted to be poorly absorbed in the GI tract, hence explaining its location outside the white portion of the BOILED-Egg model. A prediction of the toxicities, such as mutagenicity, tumorigenicity, irritant effect, and reproductive toxicity, of the 18 secondary metabolites from U. alba was also performed using OSIRIS Property Explorer (Table S19). Among the top-ranked compounds, 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) did not demonstrate any form of toxicity. Grandifloracin (15), however, demonstrated an irritant effect, whereas dichamanetin (16) was predicted to confer tumorigenicity. Both compounds 15 and 16 were projected to induce reproductive toxicity. Thus, iteration of their structures is highly encouraged to improve their toxicity profile.
Figure 8.

Prediction of GI tract and brain permeation of the top-ranked U. alba secondary metabolites—3-3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychal cone (8), cyathostemmine (13), bractelactone (14), grandifloracin (15), and dichamanetin (16)—by brain or the intestinal estimated permeation predictive model (BOILED-Egg) method.
3. Conclusions
The DCM sub-extract of U. alba (UaD) showed anti-phosphodiesterase and consequently antiproliferative and anti-cholinesterase properties in vitro. Eighteen putative secondary compounds were detected and in silico target fishing identified dichamenetin (16) with strong binding affinities to PDE4 B2B, USP14, and the KEAP-1 Kelch domain. On the other hand, 3-(3,4-dihydroxybenzyl)-3′,4′,6-trihydroxy-2,4-dimethoxychalcone (8) and grandifloracin (15) demonstrated favorable binding to AChE and the KEAP-1 BTB domain, respectively. We also demonstrated the potential of the most electrophilic grandifloracin (15) to initiate a Michael addition reaction with the active nucleophilic Cys151-containing tripeptide in the KEAP-1 BTB domain. MD simulation revealed the complexes formed between the top-ranked ligands and their protein targets to be thermodynamically stable. With favorable pharmacokinetic properties of top-ranked ligands, these results warrant further investigations on the secondary metabolites of U. alba against their respective target proteins for the discovery of new drug leads against cancer and AD.
4. Materials and Methods
4.1. Collection and Identification of Plant Material
The leaves of U. alba were collected in the lowlands of Palauig, Zambales, Luzon, Philippines (15°43′ N, 119°91′ E) in October 2013. These were authenticated with voucher specimens (USTH 1631) deposited at the University of Santo Tomas Herbarium and at the Philippine National Herbarium, Manila, Philippines.
4.2. Crude Extraction and Fractionation
The ground air-dried leaves (2.1 kg) were extracted with technical-grade (1:1) DCM–MeOH and concentrated in vacuo at 45 °C. 349 g of crude DCM–MeOH extract (Ua) was yielded. Ua crude extract was suspended in distilled water and partitioned according to increasing polarity, yielding three sub-extracts, namely, petroleum ether (UaP), DCM (UaD), and n-butanol (UaB) sub-extracts. The sub-extracts were concentrated in vacuo and tested for biological activities.
4.3. Phosphodiesterase (PDE4 B2) Inhibition Assay
The effect of U. alba DCM sub-extract on human recombinant cAMP-specific phosphodiesterase (PDE4 B2) was studied with the diluted test sample in 20 μL of 50 mM Tris-HCl buffer (pH 7.5) containing 8.3 mM MgCl2, 1.7 mM EGTA, and 5 U recombinant human cAMP-specific PDE4 B2 (Biocat 60042-BPS, Heidelberg, Germany). The reaction was started with 20 μL of 24 μM cAMP (Sigma A9501, Taufkirchen, Germany) dissolved in 50 mM Tris-HCl buffer (pH 7.5) containing 8.3 mM MgCl2 and 1.7 mM EDTA. After an incubation period of 30 min at 30 °C, the reaction was stopped and the AMP concentration was quantified with the PDELight HTS cAMP Phosphodiesterase Assay Kit (Lonza, LT07-600, Koeln, Germany) following the manufacturer’s instructions. The luminescence was measured using the microtiter plate reader Infinite M200 (TECAN) with 0.1 s integration time. Rolipram was used as the reference PDE inhibitor drug. IC50 value is defined as the concentration of the plant material required to inhibit PDE4 B2 activity by 50%.37
4.4. Antiproliferative and Cytotoxicity Assay
The antiproliferative and cytotoxic properties of the plant samples were assessed using CellTiter-Blue assay as previously described.17,21,38 The antiproliferation assay was carried out by testing the test substance dissolved in DMSO against chronic myelogenous leukemia (K-562, DSM ACC 10) and human umbilical vein endothelial cells (HUVEC, CRL-1730). Meanwhile, cytotoxicity assessment was carried out on the subconfluent monolayers of human cervical cancer cells (HeLa, DSM ACC 57). Optical densities were measured at 660 nm in SUNRISE microplate reader (TECAN, Crailsheim, Germany). The GI50 and CC50 values were defined as being where the dose response curve intersected the 50% line compared to untreated control. These values were determined using the software Magellan (TECAN). Doxorubicin was used as the reference drug. The selectivity index was computed for those samples with antiproliferative property using the formula: GI50 HUVEC/GI50 K-562.17,38
4.5. AChE Inhibition Assay
The AChE inhibitory property was evaluated using slightly modified Ellman’s assay.39 Electric eel AChE (type-VI-S, EC 3.1.1.7, Sigma) was used as the enzyme source, while acetylthiocholine iodide (ATCI, Sigma, St. Louis, MO, USA) was used as the substrate of the reaction. 5,5-Dithiobis-(2-nitrobenzoic acid) (Sigma, St. Louis, MO, USA) was used for the measurement of the cholinesterase activity. Test samples were reconstituted with 50% DMSO. About 140 μL of 0.1 mM sodium phosphate buffer (pH 8.0), 20 μL of 3 mM DTNB, and 20 μL of 0.36 U/mL of AChE were added in the reaction, followed by incubation for 15 min at 37 °C and addition of 10 μL of 15 mM ATCI. The microplates were then read at 412 nm using a Glomax microplate reader (Promega, Madison, Wisconsin). Galantamine was used as the reference drug. The IC50 value is defined as the concentration of the plant material required to inhibit AChE activity by 50%.
4.6. LC—High-Resolution MS Analysis
The U. alba DCM sub-extract (UaD), being the most biologically active, was chemically profiled for the identification of its putative secondary metabolites using LC-HR-ESIMS-QToF performed on a Shimadzu LC-20 AD apparatus equipped with an autosampler (SIL-20A, Shimadzu), diode array detector (SPD-M20AV, Shimadzu), and coupled with a microToF II (Bruker Daltonics) ESI-QToF mass spectrometer. HPLC column Chromolith Performance RP-18e (2.0 × 100 mm i.d.) was used for the analysis. The eluents were acetonitrile and water with 0.1% acetic acid. After injecting 5 μL of the UaD sub-extract, flow elution was set at 0.2 mL/min. The effluents were monitored at 350 nm. The mass spectra were recorded in the mass range m/z 50 to 2000. The Bruker DataAnalysis 4.3 software (Bruker, Germany) was used for data acquisition and analysis. Individual components were identified by comparison of their m/z values in the total ion count profile with those compounds described in literature or by matching their MS/MS spectra with those reported in a public repository of mass spectral data called MassBank.40
4.7. Molecular Docking
The binding affinities of the ligands from UaD to the docking sites of PDE4 B2B (PDB ID: 1RO6), AChE (PDB ID:4EY6), USP14 (PDB ID:6IIM), the KEAP1-Kelch domain (PDB ID: 4L7B), and the KEAP1-BTB domain (PDB ID:5DAD) were calculated (Figure S10). Molecular docking experiments were performed on the UCSF Chimera platform.41 The three-dimensional structures of the protein were added to the docking platform as PDB formats. Each protein crystal structure was processed by removing existing co-crystallized ligands and water molecules. Meanwhile, the ligands were added to the docking platform, rendered from SMILES notation or added as a SYBYL mol2 file. Minimization and dock-prepping of ligand and protein structures were done by adding the missing hydrogen atoms and appropriate charges to the structures employing the Gasteiger charge method computed using Amber’s Antechamber module.42 The docking procedure was done using a flexible ligand into a flexible active site protocol, where the ligand was allowed to be flexible and torsion within a grid box encompassing the ligand-binding cavity of each enzyme. A grid box was set around the bound co-crystallized ligand of the enzyme. The coordinates of the grid box, with the size 25 × 25 × 25 points, is summarized in Table S20. With all docking parameters maintained at default values, (number of binding modes = 10 at maximum exhaustiveness search), molecular docking simulation was performed following the Broyden–Fletcher–Goldfarb–Shanoo algorithm of AutoDock Vina (version 1.1.2).43 After each run, AutoDock Vina provides a set of docking poses for each ligand with calculated binding affinities in which the docking pose with the best affinity was chosen to represent the set. This was then subjected to post-dock analysis. Visualization and analysis of the enzyme–ligand complex conformation were carried out using Biovia Discovery Studios (version 4.1) and LigPlot+ (version 2.2). Validation of the docking protocol was done by extracting the bound co-crystallized ligand and re-docking it to the set grid. rmsd between the co-crystal and the re-docked pose was obtained. The rmsd’s in all the protein complexes were found to be less than 2 Å (ranging from 0.157 to 1.511 Å, Table S21), which indicates that the computed ligand–protein conformation is good.44
4.8. Reaction Thermochemistry and Frontier Molecular Orbital Energy Calculation
All calculations were performed within the Gaussian16W suite of programs.45 Initial conformational searches of all species were performed by scanning all freely rotating dihedral angles at the Hartree–Fock level of theory and the 6-31G** basis set to locate their approximate global energy minimum structures prior to full geometry optimization. Frontier molecular orbital energies (HOMO, EHOMO and LUMO, ELUMO) of the electrophilic U. alba metabolites, the Michael acceptors, were calculated for their empirical global electrophilicity index (ω)32,46
where μ is the chemical potential or the first derivative of total energy, E, with respect to the electron number, N, and η is the hardness or the second derivative of total energy with respect to the electron number.
Geometry optimization and energy calculation of the stable species of the proposed Michael addition mechanism were performed via DFT B3LYP/6-31G**(d,p).47 Approximate locations of TSs were determined by performing relaxed potential energy surface scans as well as refinement of the TS structure using TS Berney and QST3 optimization at the same level of theory. The TS was confirmed by intrinsic reaction coordinate calculations and were distinguished as having a single imaginary vibrational frequency. All potential energy surface scans, geometry optimizations, and single-point calculations were performed at 298.15 K, 1.0 atm pressure, and in a PCM solvent for water.
4.9. MD Simulation and Binding Free Energy Calculation
The MD behavior of the top scoring complexes was simulated using Amber 18.30 Drug topologies were generated using antechamber.48 With TIP3P water box and Na+ ions, the systems were solvated and neutralized. Gentle energy minimization in two steps was performed to relax all the systems followed by heating. The steepest descent algorithm (42) and the conjugate gradient algorithm was used for 6000 and 3000 cycles, respectively (43). The particle mesh Ewald algorithm for short- and long-range interactions was also used with a cutoff distance of 10.0 Å. The system was heated up to 300 K with ntlism 10,000, ntx was set as 1 while ntb was used as 0. Finally, a total of 50 ns simulations for each system were performed.49 The detailed information on the parameters used in this study for MD simulation are given in supplementary file Table S16. CPPTRAJ and PTRAJ were used to evaluate the trajectories for stability (rmsd) and residual flexibility (RMSF).50
Trajectories from MD simulations were subjected to binding free energy calculations using the MMPBSA.py script.51 This method is widely implemented by different studies to calculate the BE of a ligand–protein, protein–protein, and protein–nucleic acid complexes.52−54 The total BE (ΔG) was then calculated using the equation
To further understand each energy term such as electrostatic energy, van der Waals forces, polar and nonpolar interactions, which may contribute to the total energy (G), the equation below was used
Furthermore, hydrogen bonding analysis of each protein–ligand complex was performed to reveal the half-life of each interaction during MD simulation.
4.10. ADMET Profiling
Computational prediction of the ADME properties of secondary metabolites from UaD was carried out using SwissADME software (Molecular Modeling Group, Swiss Institute of Bioinformatics, 2019). Pharmacokinetic profiles were evaluated according to Lipinski’s “rule-of-five”, which analyzes the biochemical features of a drug that may influence its absorption and permeation across cell membranes. For a compound to exhibit drug likeness, at least three of the following Lipinski’s criteria must be fulfilled: molecular weight < 500 Da, calculated lipophilicity (log P) < 5, number of hydrogen-bond acceptors <10, and number of hydrogen-bond donors <5. Furthermore, OSIRIS property explorer program (Thomas Sander, Idorsia Pharmaceuticals Ltd., 2017) was employed for in silico toxicity prediction, which takes into account the potential mutagenicity, tumorigenicity, irritant effects, and reproductive toxicity of the UaD secondary compounds.55,56
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00137.
LC chemical profile UaD; putative compounds 1–18 identified from UaD; and brief description of in silico analysis data (PDF)
Author Contributions
M.T.Q., K.I.N., and A.L. contributed equally. M.T.Q. performed the conceptualization, investigation, formal analysis, writing, and editing; K.I.N. participated in the conceptualization, investigation, formal analysis, writing, and editing; A.G.L. performed the conceptualization, and investigation; R.A.F. carried out the conceptualization, investigation, formal analysis, and writing; D.Y.P., K.R.P., J.C.A., H.-M.D., and A.W.-S. participated in the investigation; D.T. conducted the investigation and editing; A.K. conducted the investigation, formal analysis, and writing; D.-Q.W. participated in the investigation, formal analysis, and writing; and A.P.G.M. performed the supervision, formal analysis, editing, and reviewing.
This work was supported in part by the International Foundation for Science (grant no. F/5376-1).
The authors declare no competing financial interest.
Supplementary Material
References
- Yamanaka Y.; Mammoto T.; Kirita T.; Mukai M.; Mashimo T.; Sugimura M.; Kishi Y.; Nakamura H. Epinephrine inhibits invasion of oral squamous carcinoma cells by modulating intracellular CAMP. Cancer Lett. 2002, 176, 143–148. 10.1016/S0304-3835(01)00764-9. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Murray F.; Zahno A.; Kanter J. R.; Chou D.; Suda R.; Fenlon M.; Rassenti L.; Cottam H.; Kipps T. J.; et al. Cyclic nucleotide phosphodiesterase profiling reveals increased expression of phosphodiesterase 7B in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19532–19537. 10.1073/pnas.0806152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho-Chung Y. S. CAMP signaling in cancer genesis and treatment. Cancer Treat. Res. 2003, 115, 123–143. 10.1007/0-306-48158-8_5. [DOI] [PubMed] [Google Scholar]
- Carniglia L.; Ramírez D.; Durand D.; Saba J.; Turati J.; Caruso C.; Scimonelli T. N.; Lasaga M. Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediators Inflammation 2017, 2017, 1–23. 10.1155/2017/5048616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori K.; Kotera J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- Pullamsetti S. S.; Banat G. A.; Schmall A.; Szibor M.; Pomagruk D.; Hänze J.; Kolosionek E.; Wilhelm J.; Braun T.; Grimminger F.; et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene 2013, 32, 1121–1134. 10.1038/onc.2012.136. [DOI] [PubMed] [Google Scholar]
- Chen D.; Ping Dou Q. The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr. Protein Pept. Sci. 2010, 11, 459–470. 10.2174/138920310791824057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraju K.; Mofers A.; Pellegrini P.; Salomonsson J.; Ahlner A.; Morad V.; Hillert E.-K.; Espinosa B.; Arnér E. S. J.; Jensen L.; et al. Cytotoxic unsaturated electrophilic compounds commonly target the ubiquitin proteasome system. Sci. Rep. 2019, 9, 1–17. 10.1038/s41598-019-46168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadimajd S.; Khazaei M. Oxidative stress and cancer: The role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. 10.2174/1568009617666171002144228. [DOI] [PubMed] [Google Scholar]
- Copple I. M. The Keap1-Nrf2 cell defense pathway - A promising therapeutic target?. Adv. Pharmacol. 2012, 6, 43–79. 10.1016/B978-0-12-398339-8.00002-1. [DOI] [PubMed] [Google Scholar]
- Bello M.; Morales-González J. A. Molecular recognition between potential natural inhibitors of the Keap1-Nrf2 complex. Int. J. Biol. Macromol. 2017, 105, 981–992. 10.1016/j.ijbiomac.2017.07.117. [DOI] [PubMed] [Google Scholar]
- Heckman P. R. A.; Wouters C.; Prickaerts J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: A translational overview. Curr. Pharm. Des. 2014, 21, 317–331. 10.2174/1381612820666140826114601. [DOI] [PubMed] [Google Scholar]
- Roseiro L. B.; Rauter A. P.; Serralheiro M. L. M. Polyphenols as acetylcholinesterase inhibitors: Structural specificity and impact on human disease. Nutr. Aging 2012, 1, 99–111. 10.3233/NUA-2012-0006. [DOI] [Google Scholar]
- Dey A.; Bhattacharya R.; Mukherjee A.; Pandey D. K. Natural products against Alzheimer’s disease: Pharmaco-therapeutics and biotechnological interventions. Biotechnol. Adv. 2017, 35, 178–216. 10.1016/j.biotechadv.2016.12.005. [DOI] [PubMed] [Google Scholar]
- Tudla F. A.; Aguinaldo A. M.; Krohn K.; Hussain H.; Macabeo A. P. G. Highly oxygenated cyclohexene metabolites from Uvaria rufa. Biochem. Syst. Ecol. 2007, 35, 45–47. 10.1016/j.bse.2006.08.006. [DOI] [Google Scholar]
- Notarte K. I.; Devanadera M. K. P.; Mayor A. B. R.; Cada M. C. A.; Pecundo M. H.; Macabeo A. P. G. Toxicity, antibacterial, and antioxidant activities of fungal endophytes Colletotrichum and Nigrospora spp. isolated from Uvaria grandiflora. Philipp. J. Sci. 2019, 148, 503–510. [Google Scholar]
- Macabeo A. P.; Lopez A. D. A.; Schmidt S.; Heilmann J.; Dahse H. M.; Alejandro G. J. D.; Franzblau S. G. Antitubercular and cytotoxic constituents from Goniothalamus gitingensis. Rec. Nat. Prod. 2013, 8, 41–45. [Google Scholar]
- Macabeo A. P. G.; Letada A. G.; Budde S.; Faderl C.; Dahse H.-M.; Franzblau S. G.; Alejandro G. J. D.; Pierens G. K.; Garson M. J. Antitubercular and cytotoxic chlorinated seco-cyclohexenes from Uvaria alba. J. Nat. Prod. 2017, 80, 3319–3323. 10.1021/acs.jnatprod.7b00679. [DOI] [PubMed] [Google Scholar]
- Macabeo A. P. G.; Flores A. I. G.; Fernandez R. A. T.; Budde S.; Faderl C.; Dahse H.-M.; Franzblau S. G. Antitubercular and cytotoxic polyoxygenated cyclohexane derivatives from Uvaria grandiflora. Nat. Prod. Res. 2020, 1–4. 10.1080/14786419.2020.1741579. [DOI] [PubMed] [Google Scholar]
- Macabeo A. P. G.; Tudla F. A.; Alejandro G. J. D.; Kouam S. F.; Hussain H.; Krohn K. Benzoylated derivatives from Uvaria rufa. Biochem. Syst. Ecol. 2010, 38, 857–860. 10.1016/j.bse.2010.08.007. [DOI] [Google Scholar]
- Macabeo A. P. G.; Martinez F. P. A.; Kurtán T.; Tóth L.; Mándi A.; Schmidt S.; Heilmann J.; Alejandro G. J. D.; Knorn M.; Dahse H.-M.; et al. Tetrahydroxanthene-1,3(2 H)-dione derivatives from Uvaria valderramensis. J. Nat. Prod. 2014, 77, 2711–2715. 10.1021/np500538c. [DOI] [PubMed] [Google Scholar]
- Macabeo A. P. G.; Pilapil L. A. E.; Garcia K. Y. M.; Quimque M. T. J.; Phukhamsakda C.; Cruz A. J. C.; Hyde K. D.; Stadler M. Alpha-glucosidase- and lipase-inhibitory phenalenones from a new species of Pseudolophiostoma originating from Thailand. Molecules 2020, 25, 965. 10.3390/molecules25040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda T.; Ota T.; Fujimoto T.; Doi K.; Tanaka Y.; Yoshida Y.; Ogawa M.; Matsuzaki H.; Hamabashiri M.; Tyson D. R.; et al. Inhibition of phosphodiesterase-4 (PDE4) activity triggers luminal apoptosis and akt dephosphorylation in a 3-d colonic-crypt model. Mol. Cancer 2012, 11, 46–58. 10.1186/1476-4598-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon E.-Y.; Lerner A. PDE4 inhibitors activate a mitochondrial apoptotic pathway in chronic lymphocytic leukemia cells that is regulated by Protein Phosphatase 2A. Blood 2003, 101, 4122–4130. 10.1182/blood-2002-10-3208. [DOI] [PubMed] [Google Scholar]
- Adams J.; Kelso R.; Cooley L. The Kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 2000, 10, 17–24. 10.1016/S0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- Canning P.; Sorrell F. J.; Bullock A. N. Structural basis of Keap1 interactions with Nrf2. Free Radicals Biol. Med. 2015, 88, 101–107. 10.1016/j.freeradbiomed.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova A. T.; Kostov R. V.; Canning P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. 10.1016/j.abb.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Zhang D.; Hannink M.; Beamer L. J. Crystal Structure of the Kelch domain of human Keap1. J. Biol. Chem. 2004, 279, 54750–54758. 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- Dayalan Naidu S.; Muramatsu A.; Saito R.; Asami S.; Honda T.; Hosoya T.; Itoh K.; Yamamoto M.; Suzuki T.; Dinkova-Kostova A. T. C151 in Keap1 is the main cysteine sensor for the cyanoenone class of Nrf2 activators, irrespective of molecular size or shape. Sci. Rep. 2018, 8, 1–12. 10.1038/s41598-018-26269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachakonda G.; Xiong Y.; Sekhar K. R.; Stamer S. L.; Liebler D. C.; Freeman M. L. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- Zhang D. D.; Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. 10.1128/mcb.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr R. G.; Szentpály L. V.; Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- Uzzaman M.; Chowdhury M. K.; Belal Hossen M. Thermochemical, molecular docking and ADMET studies of aspirin metabolites. Front. Drug, Chem. Clin. Res. 2019, 2, 1–5. 10.15761/fdccr.1000130. [DOI] [Google Scholar]
- Schwöbel J. A. H.; Madden J. C.; Cronin M. T. D. Examination of Michael addition reactivity towards glutathione by transition-state calculations. SAR QSAR Environ. Res. 2010, 21, 693–710. 10.1080/1062936X.2010.528943. [DOI] [PubMed] [Google Scholar]
- Stack D. E.; Li G.; Hill A.; Hoffman N. Mechanistic insights into the Michael addition of deoxyguanosine to catechol estrogen-3,4-quinones. Chem. Res. Toxicol. 2008, 21, 1415–1425. 10.1021/tx800071u. [DOI] [PubMed] [Google Scholar]
- Ginex T.; Trius M.; Luque F. J. Computational study of the Aza-Michael addition of the flavonoid (+)-taxifolin in the inhibition of β-amyloid fibril aggregation. Chem.—Eur. J. 2018, 24, 5813–5824. 10.1002/chem.201706072. [DOI] [PubMed] [Google Scholar]
- Kim B.-Y.; Willbold S.; Kulik A.; Helaly S. E.; Zinecker H.; Wiese J.; Imhoff J. F.; Goodfellow M.; Süssmuth R. D.; Fiedler H.-P. Elaiomycins B and C, novel alkylhydrazides produced by Streptomyces sp. BK 190. J. Antibiot. 2011, 64, 595–597. 10.1038/ja.2011.53. [DOI] [PubMed] [Google Scholar]
- Krauth F.; Dahse H.-M.; Rüttinger H.-H.; Frohberg P. Synthesis and characterization of novel 1,2,4-triazine derivatives with antiproliferative activity. Bioorg. Med. Chem. 2010, 18, 1816–1821. 10.1016/j.bmc.2010.01.053. [DOI] [PubMed] [Google Scholar]
- Macabeo A. P. G.; Vidar W. S.; Chen X.; Decker M.; Heilmann J.; Wan B.; Franzblau S. G.; Galvez E. V.; Aguinaldo M. A. M.; Cordell G. A. Mycobacterium tuberculosis and cholinesterase inhibitors from Voacanga globosa. Eur. J. Med. Chem. 2011, 46, 3118–3123. 10.1016/j.ejmech.2011.04.025. [DOI] [PubMed] [Google Scholar]
- Horai H.; Arita M.; Kanaya S.; Nihei Y.; Ikeda T.; Suwa K.; Ojima Y.; Tanaka K.; Tanaka S.; Aoshima K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya R.; Chacko S.; Bose P.; Lapenna A.; Pattanayak S. P. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci. Rep. 2019, 9, 1–13. 10.1038/s41598-019-52162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; et al. Gaussian 16, (Revision A.03): Wallingford, CT, 2016.
- Chattaraj P. K.; Duley S.; Domingo L. R. Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index. Org. Biomol. Chem. 2012, 10, 2855–2861. 10.1039/c2ob06943a. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Antechamber, an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Toukmaji A.; Sagui C.; Board J.; Darden T. Efficient particle-mesh ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000, 113, 10913–10927. 10.1063/1.1324708. [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Sun H.; Li Y.; Tian S.; Xu L.; Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. 10.1039/c4cp01388c. [DOI] [PubMed] [Google Scholar]
- Khan A.; Saleem S.; Idrees M.; Ali S. S.; Junaid M.; Chandra Kaushik A.; Wei D.-Q. Allosteric ligands for the pharmacologically important Flavivirus target (NS5) from ZINC database based on pharmacophoric points, free energy calculations and dynamics correlation. J. Mol. Graph. Model. 2018, 82, 37–47. 10.1016/j.jmgm.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Khan A.; Chandra Kaushik A.; Ali S. S.; Ahmad N.; Wei D.-Q. Deep-learning-based target screening and similarity search for the predicted inhibitors of the pathways in Parkinson’s disease. RSC Adv. 2019, 9, 10326–10339. 10.1039/c9ra01007f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Khan A.; Chandra Kaushik A.; Junaid M.; Zhang X.; Wei D.-Q. The systematic modeling studies and free energy calculations of the phenazine compounds as anti-tuberculosis agents. J. Biomol. Struct. Dyn. 2019, 37, 4051–4069. 10.1080/07391102.2018.1537896. [DOI] [PubMed] [Google Scholar]
- Quimque M. T. J.; Notarte K. I. R.; Fernandez R. A. T.; Mendoza M. A. O.; Liman R. A. D.; Lim J. A. K.; Pilapil L. A. E.; Ong J. K. H.; Pastrana A. M.; Khan A.; Wei D.-Q.; Macabeo A. P. G. Virtual screening-driven drug discovery of SARS-CoV-2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2020, 1–18. 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez R. A. T.; Quimque M. T. J.; Notarte K. I. R.; Manzano J. A.; Pilapil D. Y. H.; de Leon V. N.; San Jose J. J. P.; Villalobos O.; Macabeo A. P.. Myxobacterial depsipeptide chondramides interrupt SARS-CoV-2 entry by targeting its broad, cell tropic spike protein: Antagonistic prospects for anti-COVID-19 drug discovery. 2021, ChemRxiv:10.26434/chemrxiv.14043641, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.