

Abstract
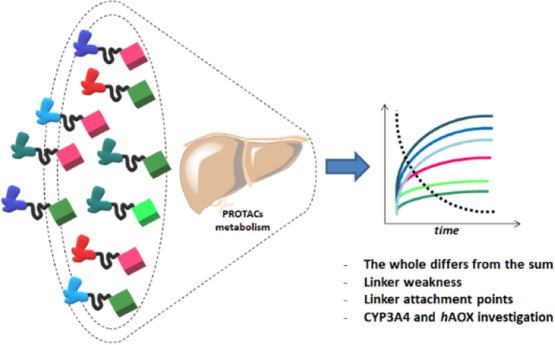
Hetero-bifunctional PROteolysis TArgeting Chimeras (PROTACs) represent a new emerging class of small molecules designed to induce polyubiquitylation and proteasomal-dependent degradation of a target protein. Despite the increasing number of publications about the synthesis, biological evaluation, and mechanism of action of PROTACs, the characterization of the pharmacokinetic properties of this class of compounds is still minimal. Here, we report a study on the metabolism of a series of 40 PROTACs in cryopreserved human hepatocytes at multiple time points. Our results indicated that the metabolism of PROTACs could not be predicted from that of their constituent ligands. Their linkers’ chemical nature and length resulted in playing a major role in the PROTACs’ liability. A subset of compounds was also tested for metabolism by human cytochrome P450 3A4 (CYP3A4) and human aldehyde oxidase (hAOX) for more in-depth data interpretation, and both enzymes resulted in active PROTAC metabolism.
Introduction
Rational drug design represents an essential approach to optimize time and cost in drug discovery and development,1 but it remains a challenging task. Indeed, not only is drug potency a critical feature, but also absorption, distribution, metabolism, and excretion (ADME) properties require optimization by modulating the chemical structure of the candidate. Drugs undergo biotransformations, and thus the optimization of the drug structure per se could be useless when significant metabolic liability, generating novel compounds (metabolites), occurs. In the last two decades, many efforts have been made to decode and predict the metabolic fate of drugs,2 and in silico models,3−6in vitro assays,2 and hybrid approaches (i.e., innovative assays associated with software-assisted data processing)7,8 have been developed to identify the “soft spots” of drugs. Despite signs of progress in the field, all available ADME tools have been calibrated mainly using the chemical space of small molecules, witnessing the outstanding impact that the Lipinski rules9 have had in pharmaceutical research in the past. In the comfortable space of small molecules, the accuracy and sensitivity of the models are usually very good.10−14 Nowadays, the chemical space of the drugs is quickly expanding, ranging from peptides or peptidomimetics15 to Proteolysis Targeting Chimeras (PROTACs)16−26 and their analogues.27−29 PROTACs can be defined as hetero-bifunctional molecules that induce a ligand to bind with the protein of interest (POI), another ligand to recruit an E3 ubiquitin ligase, and a linker to concatenate the two ligands.17 The formation of the ternary complex composed of the POI, the PROTAC, and the E3 ligase allows the E2 ubiquitin-conjugating enzyme to transfer ubiquitin to the surface of the POI, inducing its proteasomal-dependent degradation.30 One of the main advantages of PROTACs is that they can degrade proteins regardless of their function, thus turning into druggable also the “undruggable”, due to their innovative mechanism of action.16 Degradation by PROTACs is a catalytic process, due to the dissociation of the complex after polyubiquitination of the POI, indicating that PROTACs can be recycled for successive rounds of degradation and thus used at reduced doses.31 Therefore, PROTACs represent an innovative class of compounds that overcome traditional limitations, opening a new therapeutic modality and, at the same time, breaking the rules used so far with the potential to revolutionize drug discovery. As extensively reviewed,32−36 hundreds of PROTAC molecules have been developed so far, targeting a wide range of different disease-related protein targets. The entry in phase I clinical trial in 2019 of the first two oral PROTACs (ARV-110 and ARV-471) for the treatment of metastatic castration-resistant cancer and metastatic breast cancer (NCT0888612 and NCT04072952) has focused attention even more on this innovative therapeutic paradigm.37,38 Despite their intriguing capabilities, PROTACs are characterized by a high molecular weight (600–1400 Da),39 making the delivery and bioavailability of PROTACs the most significant hurdles to overcome on the way to the clinic.40 Thus, better understanding and prediction of the ADME properties of PROTACs represent an urgent need for their rational design. To date, the evaluation of the ADME properties of this class of compounds is still minimal, with only a few studies on their experimental physical–chemical properties available41−43 and only one paper about PROTAC metabolism has been published.35 Nevertheless, preliminary studies on small subsets of PROTACs, whose log P was experimentally measured, indicate that traditional in silico tools for property prediction may fail,41 likely due to their peculiar structural features compared with traditional druglike compounds used for generation of predictive models. Therefore, there is an urgent need to collect experimental physicochemical and ADME data on PROTACs to shed light on their peculiar behavior and to be used for modeling purposes. Here, focusing on the human metabolism, a collection of 40 PROTACs (compounds 1–40, Supporting Information Table S1) was studied, assessing their metabolism in cryopreserved human hepatocytes at multiple time points. Their enzymatic biotransformations were also compared with those of the constituent ligands (compounds 41–46, Supporting Information Table S2). Both metabolic rate (half-life value) and soft spot identification were investigated. In addition, a subset of compounds was also tested for metabolism by human CYP3A4 and hAOX for deeper data interpretation, representing the principal isoenzyme involved in liver metabolism (including large substrates)44,45 and one of the emerging enzymes in metabolism studies,46−49 respectively. The complete data set with more experimental details is provided in the Supporting Information.
Results and Discussion
Chemistry
The synthesis of JQ1-based PROTACs 4–6, 8, 9, and 11 was accomplished according to Scheme 1. Briefly, derivative 43 was coupled by amidation reaction with the appropriated E3 ligase ligand properly functionalized with linkers of different lengths in the presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA) at room temperature in dimethylformamide (DMF).
Scheme 1. Syntheses of JQ1-Based PROTACs 4–6, 8, 9, and 11.
Reagents and conditions: (a) HATU, DIPEA, dry DMF, room temperature (rt).
Analogously, as shown in Scheme 2, CX4945-based PROTACs 12–20 were obtained by HATU-mediated amidation reaction between derivative 44 and the appropriated E3 ligase-linker intermediate.
Scheme 2. Syntheses of CX4945-Based PROTACs 12–20.
Reagents and conditions: (a) HATU, DIPEA, dry DMF, rt.
The olaparib-based PROTACs 21–25 were synthesized as depicted in Schemes 3 and 4. For cereblon (CRBN)-addressing PROTAC 21, derivative 45(50) was first reacted with 11-((tert-butoxycarbonyl)amino)undecanoic acid by N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU)-mediated amidation reaction leading to Boc-protected intermediate 60, which after Boc-deprotection reaction gave the key intermediate 61. Then, compound 62 was reacted with fluorothalidomide 62(51) in the presence of DIPEA at 70 °C in DMF (Scheme 3). For the von Hippel-Lindau (VHL)-addressing PROTACs 22 and 23, derivative 45(50) was first reacted with the appropriate dicarboxylic acid monomethyl ester linker by amidation reaction in the presence of HBTU and Et3N at room temperature in dry DMF to furnish intermediates 63–64. The successive basic hydrolysis of methyl esters 63–64 gave intermediates 65–66, which in turn were finally coupled with derivative 42(52) by HATU-mediated amidation reaction to afford PROTACs 22 and 23, respectively (Scheme 3).
Scheme 3. Syntheses of Olaparib-Based PROTACs 21–23.

Reagents and conditions: (a) HBTU, Et3N, dry DMF, rt; (b) 4.0N HCl in dioxane, rt; (c) DIPEA, dry DMF, 70 °C; (d) LiOH monohydrate, tetrahydrofuran (THF):H2O (2:1), rt; (e) HATU, DIPEA, dry DMF, rt.
Scheme 4. Synthesis of Olaparib-Based PROTACs 24 and 25.

Reagents and conditions: (a) 3-bromoprop-1-yne, K2CO3, KI, ACN, reflux; (b) HBTU, Et3N, dry DMF, rt; (c) CuSO4, sodium ascorbate, DMF/tBuOH/H2O (1:1:1), rt.
For the VHL-addressing PROTACs 24 and 25 (Scheme 4), an alkynyl group was introduced by reacting derivative 45(50) with propargyl bromide, generating intermediate 67, or by coupling it with hex-5-ynoic acid, generating intermediate 68. Thus, final PROTACs 24 and 25 were obtained through the copper-assisted click reaction coupling the alkynyl derivatives 67 and 68 with azide-containing VHL derivative 69.
The syntheses of AR ligand-based CRBN-addressing PROTACs 26–32 are shown in Scheme 5. A substitution reaction between derivative 46(53) and tert-butyl 2-bromoacetate in the presence of K2CO3 in acetonitrile at room temperature gave intermediate 70, which after acidic hydrolysis afforded the key intermediate 71. The successive amidation reaction in the presence of HATU and DIPEA at room temperature in DMF between compound 71 and the appropriated thalidomide-linker intermediate furnished PROTACs 26–31. For the synthesis of PROTAC 32, a first Mitsunobu reaction between derivative 46(53) and the hydroxyl poly(ethylene glycol) (PEG)-linker gave the Boc-protected intermediate 73. The Boc-deprotection reaction of 73 furnished intermediate 74, which was then reacted with fluothalidomide 62(51) in the presence of DIPEA at 70 °C in DMF to afford PROTAC 32.
Scheme 5. Syntheses of AR Ligand-Based PROTACs 21–27.

Reagents and conditions: (a) tert-butyl bromoacetate, K2CO3, ACN, rt; (b) 4.0N HCl in dioxane, rt; (c) HATU, DIPEA, dry DMF, rt; (d) PPh3, DIAD, dry THF, 0 °C-rt; (e) DIPEA, dry DMF, 70 °C.
Data Set Selection
With the aim of covering a large chemical diversity, various combinations of ligands for four target proteins, ligands for two E3 ligases, and nineteen linkers were selected to give a final data set of 40 PROTACs (Figure 1). In particular, concerning the selection of ligands for target proteins, the bromodomain and extra-terminal (BET) inhibitor (+)-JQ1,54 the casein kinase 2 (CK2) inhibitor CX4945,55 the FDA-approved poly(ADP-ribose) polymerase (PARP) inhibitor olaparib,50 and an androgen receptor DNA-binding domain binder53 were used. Concerning the ligands for E3 ligases, binders for cereblon (CRBN) and von Hippel-Lindau (VHL) were selected, with these being two of the four most commonly used E3 ligases in PROTAC synthesis, together with cell inhibitor of apoptosis protein (cIAP) and mouse double minute 2 homolog (MDM2).56 Finally, aliphatic, polyethylene glycol (PEG)-based, and cyclic linkers were variably combined, modulating their length and anchor point (Figure 1). The chemical structures of the entire data set are provided in the Supporting Information (Table S1). Among them, five compounds were commercially available (Supporting Information Table S1), entries 1 (dBet1),572 (dBet6),583 (ARV-825),597 (MZ1),60 and 10 (ARV-771),52 15 were kindly provided by Montelino Therapeutics Inc. (Supporting Information Table S1, entries 26–40), while the others were designed and synthesized in house to increase chemical and structural variability.
Figure 1.

Scheme of building blocks characterizing the data set of tested PROTACs.
Optimization of the Metabolic Stability Assay for PROTACs
Metabolic stability of PROTACs has been rarely discussed in the literature, with only one paper published to date.35 In that paper, Zhou et al.35 evaluated the metabolism in mouse liver microsomes (phase I metabolism only) of one PROTAC, known to degrade the BET proteins with thalidomide as the CRBN ligand, after 20 and 40 min incubation times. The major metabolite detected was proposed to be a hydroxylated product, with the site of transformation occurring in the alkyl linker. In the present study, the aim was to test a diverse data set of PROTACs for their metabolic stability in cryopreserved human hepatocytes at multiple time points within a time range of 4 h. Differently from liver microsomes, cryopreserved human hepatocytes contain all phase I and II metabolic enzymes, with all necessary cofactors, and are compatible with longer incubation times.61 In commonly used protocols for metabolic stability assays, enzymatic reactions are quenched at the desired time of incubation by adding an organic solvent (e.g., acetonitrile) to the enzyme-containing solution to induce protein precipitation62−64 and, after centrifugation, the water-containing supernatant is collected and analyzed by liquid chromatography–mass spectrometry (LC–MS). However, such a protocol would not prevent the nonenzymatic degradation of PROTACs in the autosampler during LC–MS analysis.65 Therefore, it is especially critical for high-throughput screenings, in which a large number of samples are collected in the autosampler simultaneously and analyzed in a long sequence of analysis.65 In our studies on PROTAC metabolism, we reasoned that this could be a critical point to be addressed also taking into account that the rapid degradation of thalidomide66 and thalidomide-containing PROTACs in aqueous solution has already been reported elsewhere.43 Therefore, studies on the potential nonenzymatic degradation of substrates in the autosampler during LC–MS analysis were conducted on the commercial PROTAC 1 (dBet1), a potent Bromodomain-containing protein 4 (BRD4) protein degrader that is composed of (+)-JQ1 linked to thalidomide through an aliphatic linker. Compound 1 (dBet1) was incubated in three different solvents: (1) in pure phosphate buffer at pH = 7.4 (PBS); (2) in a mixture of PBS/acetonitrile (1:1 v/v), which is the most common composition of the supernatant injected in the LC–MS instrument in metabolism assays (named here PBS/ACN); and (3) in pure dimethyl sulfoxide (DMSO). A fourth condition entailed the incubation of 1 (dBet1) in PBS/acetonitrile (1:1, v/v) and immediately after the sample was dried under a nitrogen stream (to remove the solvent) and redissolved in DMSO before injection in the LC–MS system. The latter protocol is here named as PBS/ACN-DMSO and was designed to evaluate whether the removal of the first solvent and the resuspension in DMSO could prevent further degradation in the autosampler. Thus, the nonenzymatic stability of 1 (dBet1) in the four solvents and solvent mixtures was analyzed by LC–MS for 12 h at 37 °C, with injections at 0, 3, 6, and 12 h. Figure 2 illustrates the results of the stability of 1 (dBet1). As expected, compound 1 (dBet1) rapidly degrades when stored in the autosampler in pure PBS (Figure 2A). Degradation was also observed in the presence of the PBS/ACN solution, although to a lower extent, and it occurred during the first three hours, becoming constant with time. In DMSO, the solution of 1 (dBet1) became very stable over time. When the PBS/ACN solution was removed by a nitrogen stream and then replaced with an equal volume of DMSO (protocol PBS/ACN-DMSO), the substrate degradation within the first three hours was reduced to about 10%, remaining rather constant with time. The formation of two degradation products was also monitored (Figure 2A), corresponding to the hydrolysis of the phthalimide (degradation product D1) or glutarimide groups (degradation product D2) in the thalidomide moiety (Figure 2A,B). When the PBS/ACN-DMSO protocol was used, the formation of the degradation product was observed in a limited amount, and their concentration was constant over time, indicating that they formed during the PBS/ACN solution removal and not during storage in the autosampler.
Figure 2.

Nonenzymatic stability of 1 (dBet1) in the autosampler during LC–MS analysis acquisitions. (A) Percentage of the remaining 1 and percentage of formation of the degradation products resulted by the hydrolysis of the phthalimide moiety (D1, orange) or of the glutarimide moiety (D2, green) at the four time points (0, 3, 6, 12 h) in the different solutions. (B) Chemical structure of 1 (dBet1), with highlighted phthalimide (orange) and glutarimide (green) rings. (C) Comparison of the metabolic profiles of 1 (dBet1) in cryopreserved human hepatocytes for 4 h using the PBS/ACN (filled circles) or the PBS/ACN-DMSO (empty circles) protocol.
Finally, protocols PBS/ACN and PBS/ACN-DMSO were compared in a real metabolism assay of 1 (dBet1). The study of the metabolic stability of this PROTAC in cryopreserved human hepatocytes was conducted at five time points (0, 30, 60, 120, and 240 min). Monitoring metabolic stability by a kinetic approach allows not only the half-life calculation for the substrate but also reduction in false positives in the characterization of metabolites. Samples for each time point were analyzed by LC–MS/MS, and raw data were analyzed using Mass-MetaSite software7,67 in the WebMetabase platform.68−70Figure 2C shows that the PBS/ACN-DMSO protocol significantly increases the stability of 1 (dBet1) during analysis. Therefore, the final method for the metabolic stability assay used in this study included the PBS/ACN-DMSO protocol (see the Methods section for the whole procedure), to reduce the risk of further degradation of the substrate in the autosampler during analysis.
Metabolic Stability of Constituent Ligands for Tested PROTACs
Before performing an extensive study of the metabolic stability of the 34 PROTACs, the ligands used in PROTACs’ design and synthesis were tested for their metabolism in cryopreserved human hepatocytes, to have a reference on the behavior of the units connected in the final PROTAC structures. The same experimental protocol was used for both PROTACs and ligands (see the methods section). Metabolic stability was first expressed in terms of the half-life (t1/2) of the parent compound, as it represents a commonly used parameter to judge the intrinsic stability of a compound (Supporting Information Table S2).61 Concerning the structure of the tested ligands, an olaparib analogue lacking the carboxycyclopropyl moiety (compound 45, Supporting Information Table S2) was used a reference for the PROTACs (21–25, Supporting Information Table S1) targeting PARP. This carboxycyclopropyl moiety, which represents a solution to improve oral absorption,71 was assumed to only slightly affect the binding with the target72 and was removed to allow the use of the unbound nitrogen of the piperazine ring as the anchor point for the linker. As free compounds, ligands used in this study for PROTAC design (41–46, Supporting Information Table S2) were characterized by good metabolic stability (t1/2 higher than 90 min), with the exception of the AR ligand (compound 46) showing a t1/2 of less than 20 min (Supporting Information Table S2).
Metabolic Stability of PROTACs in Cryopreserved Human Hepatocytes
As for the free ligands, metabolic stability of the whole set of PROTACs in cryopreserved human hepatocytes was studied over a four-hour incubation period (see the Method section). An example of the complete kinetic behavior of the disappearance of the substrate and the appearance of metabolites with time is shown for compound 7 (MZ1) in Figure S1 (Supporting Information). Based on observed kinetic data for the entire data set, the half-life for each PROTAC was calculated, and results are shown in the Supporting Information Table S1.
The analysis of the half-lives led to a number of observations. First, for PROTACs bearing ligands targeting BET, CK2, and PARP (compounds 1–25 in the Supporting Information Table S1), the use of the thalidomide moiety as the E3 ligase binder led to lower t1/2 values compared to PROTACs bearing the VHL ligand (i.e., 5 versus 10, 16 versus 18, 21 versus 22 in the Supporting Information Table S1). PROTAC 1 (dBet1) was the only exception to this trend, possibly due to the very short linker, which could hamper the interaction with metabolism-devoted enzymes. This point will be further discussed in the following paragraphs. The lower half-life values of thalidomide-containing PROTACs suggest that these compounds, in addition to enzymatic transformation, might also undergo partial nonenzymatic degradation during incubation time (the instability of thalidomide was previously shown in Figure 2), but a further analysis of this phenomenon was beyond the scope of this paper. Concerning the PROTAC series containing the AR ligand 46 (compounds 26–40 in the Supporting Information Table S1), they generally showed a higher susceptibility to metabolism in cryopreserved human hepatocytes, with all t1/2 values lower than 100 min independent of the linker or the E3 ligase binder used. This trend, associated with the low metabolic stability of AR ligand 46 as previously discussed (Supporting Information Table S2), suggested that the primary site(s) of metabolism in this series is probably related to metabolic liabilities in the compound 46 moiety rather than in the E3 ligase part of the molecules. Therefore, for a deeper understanding of the t1/2 values, the soft spot analysis was performed.
PROTAC Soft Spot Identification
While metabolic stability data expressed as the half-life of the parent compound represent a valuable parameter to judge the intrinsic stability of a compound,61 the identification of soft spots in a molecule is crucial for the rational design of new and more stable compounds.61 Although it is now always possible to identify the exact site of metabolism by LC–MS/MS analysis, our study allowed us to devise some general indications, which we believe will be useful in the design of new PROTACs. Soft-spot analysis data are provided in the Supporting Information Table S3.
Whole Differs from the Sum of Its Parts
Due to their composed structure, the first natural comparison to make was whether soft spots in a PROTAC could be predicted from the soft spots of the free ligands. As an example, in Table 1, the observed metabolism of compounds 4 and 37, which are composed of totally different building blocks, is compared with that of the corresponding constituent ligands.
Table 1. Half-Life for PROTACs and Single Ligand Components upon Incubation in Cryopreserved Human Hepatocytes.

Concerning PROTAC 4, enzymatic degradation of the thalidomide moiety was not observed, although a low metabolic stability of pomalidomide 41 was detected, due to the opening of the phthalimide and glutarimide rings. Similarly, the JQ1 moiety in PROTAC 4 did not undergo the aliphatic hydroxylation observed for ligand 43, probably due to an increase of the steric hindrance of the JQ1 moiety site of metabolism (however, for other JQ1-based PROTACs, traces of this metabolic route were observed, see the Supporting Information Table S3). Nevertheless, 4 was highly metabolized in cryopreserved human hepatocytes, with the soft spots being identified in the linker and especially in its connection points with the ligands. Concerning PROTAC 37, the liability of the morpholine ring present in the AR ligand 46 was confirmed, as well as that of two out of four points of the VHL ligand structure 42. However, the PEG-like linker also played an important role in the enzymatic degradation of 37, being subjected to O-dealkylation and amide hydrolysis reactions. The preserved liability of the AR ligand 46 moiety is also in agreement with what was discussed in the previous paragraph for the AR degrader series (26–40). In conclusion, translating the well-known Aristotelian concept (“The Whole is Greater than the Sum of its Parts”) to PROTACs’ metabolism, one can derive that “the whole differs from the sum of its parts”. Although this statement may seem trivial, it has a strong impact on medicinal chemists because it confirms that PROTACs represent totally independent chemical entities and that their metabolism cannot be predicted from the one of the ligands used for their design and synthesis. Table 1 also shows that in PROTACs the most labile soft spots are represented by the linker and the chemical connections used to join it to the ligands.
Linker Effect
The linker commonly plays an important role in the biological activities and physicochemical properties of PROTACs. With time, the chemical nature of linkers has been variably modified, changing from initial peptide linkers73 to (un)saturated alkane or PEG-like chains, to variably functionalized linkers.74 In particular, PROTACs containing PEG-based linkers usually display a better solubility profile when compared to those bearing alkyl linkers or even triazole-containing linkers.75 Indeed, introducing PEG moieties (possessing a good safety profile)76 represents a commonly used strategy for improving pharmaceutical properties of small molecules. Recently, piperidine-containing linkers have also proved to be a good option to improve solubility.74 Various bonds have been used to connect a linker to the two ligands, including amide bonds, ether bonds, alkylamines, carbon–carbon bonds, and click-chemistry products.73 Finally, the length of the linker has been extensively changed to adapt the PROTAC to its biological function. In fact, on the one hand, if a linker is too short, the simultaneous binding of the two ligands with their targets will be hampered, and the formation of the ternary complex will not occur. On the other hand, if the linker is too long, the PROTAC will not efficiently move the target and the E3 ligase closer to each other, and thus the target protein will not be ubiquitinated.73,75,77,78 In the previous section, we discussed an example showing a significant metabolic liability of the linkers (Table 1). Therefore, one may ask how the tailoring of the linker may affect the metabolic stability. We already mentioned that the short linker in PROTAC 1 (dBet1) could be a reason for its high metabolic stability. Indeed, the comparison of the t1/2 value of 1 (dBet1) with that for 2 (dBet6) indicated that the extension of the linker from four to eight methylene units reduces the t1/2 value from 135 to 18.2 min, respectively (Figure 3).
Figure 3.

Effect of the length of linear linkers on metabolic stability. The half-life values associated with each compound and expressed in minutes are reported in brackets. Bond cleavages are illustrated as dotted lines, while circles represent atoms subjected to oxidation in a well-defined position by MS/MS fragmentation (blue) or in a defined moiety by MS/MS fragmentation (pink). In the case of pink circles, the displayed position was suggested by MetaSite predictions. Finally, ellipses indicate dehydrogenation reactions, and the same color code for circles was used.
This finding could suggest generalizing that, in the case of linear linkers, a shorter aliphatic linker can be responsible for increased metabolic stability, probably due to steric hindrance of the PROTAC entering into the catalytic site of the metabolic enzymes. The hypothesis was verified comparing two additional PROTACs with a linker composed of four methylene units to long-linker analogues (Figure 3). In particular, the AR-based PROTACs (compounds 33 and 35, Figure 3) confirmed the expected trend, although with a reduced difference in half-life values possibly due to the reduced length difference between the two linkers (from four to six methylene units). However, replacing the AR ligand unit in compounds 33 and 35 with the CX4945 (44) moiety, the increased length of the linear linker did not show the hypothesized effect, with compounds 17 and 18 having equal t1/2 values. Soft spots suggest that, while for the pairs 1/2 and 33/35 the longer linker seems more prone to the N-dealkylation reaction, in the case of 17/18 metabolism is very similar. Thus, this and other possible comparisons of data in Table S1 (Supporting Information) suggest that a very short linker can be commonly associated with improved metabolic stability. However, our data show that, in some cases, PROTACs composed of longer linkers could also be metabolically stable.
Similarly, the nature of the linear linker and its binding moiety seem not to heavily affect the metabolic stability. In Table 2, four PROTACs are displayed, containing the AR ligand and pomalidomide moieties as ligands connected by either an aliphatic or a PEG linker of the same length. In compounds 26 and 27, the hydroxyl thalidomide is linked by an acetamide moiety, while in compounds in 30 and 31, the linker is directly connected through an amine group. Despite this, compounds 27 and 31 share a similar metabolic stability, compound 30 appears only slightly more stable than the two PEG-containing PROTACs, and compound 26 resulted in being very unstable, with a t1/2 of only 8.4 min. Intriguingly, the greater instability of 26 is not related to different soft spots, if compared to 30 (Table 2), suggesting that the different anchor point between the linker and the thalidomide might modify the affinity with the metabolic enzyme(s). As revealed by the comparison of the two PROTACs with an aliphatic linker, compounds 27 and 31, both possessing a PEG-like linker, also share a similar soft spot pattern (Table 2), which is mainly due to O-dealkylation reactions. It is noteworthy that, although in PEG-like PROTACs the number of soft spots is higher due to the multiple O-dealkylation reactions, the overall metabolic rate is not necessarily negatively affected. However, from the medicinal chemistry perspective, a higher number of soft spots make the design of more stable compounds by protection strategies more challenging.
Table 2. Soft Spots for PROTACs 26, 27, 30, and 31.

Finally, six PROTACs with various target and E3 ligase binders and bearing cyclic linkers were synthesized to evaluate their effect on metabolic stability. Among them, compounds 6, 11, and 20 were characterized by the presence of a piperazine moiety in the linker, while compounds 24, 25, and 39 were endowed with a triazole ring. A comparison with similar PROTACs bearing linear linkers (Table S1, Supporting Information) suggested that, with the exception of 39, the presence of cyclic linkers resulted in a higher metabolic stability. Soft spots for compounds 6, 11, 20, 24, 25, and 39 are illustrated in Figure 4. In the BET series (Table S1, entries 1–11 in the Supporting Information), compounds 6 and 11 show higher t1/2 values in the pomalidomide-containing PROTACs and VHL-containing PROTACs, respectively. In addition, the significant increase of stability observed for compound 11 can be explained by the short length of its linker and by the attachment of piperazine to JQ1 through an amide group, which hampers a second N-dealkylation reaction (Figure 4). In the CK2 series (Table S1, entries 12–20 in the Supporting Information), VHL-containing compounds with linear linkers 17–19 were all endowed with short linkers and were characterized by a high metabolic stability. Therefore, compound 20, bearing a short piperazine-containing linker, was synthesized and tested, and again this compound turned out to be the most stable in the series, although the t1/2 value was slightly higher than the one for the linear analogue 17 (218 and 207 min, respectively). In the PARP series (Table S1, entries 21–25 in the Supporting Information), a click-chemistry approach was applied to connect the linker to the target binder through a triazole ring to give compounds 24 and 25. The triazole-containing PROTACs displayed a much greater metabolic stability when compared to their linear analogues (23 and 22, respectively). Indeed, as shown in Table S1, t1/2 values for compounds 24 and 25 were greater than 240 min, with about 84% substrate left. Interestingly, an N-dealkylation reaction occurred at the triazole, similar to that observed for the piperazine-containing linkers (Figure 4). Finally, as anticipated, compound 39 turned out to be the only PROTAC endowed with a cyclic moiety with a lower metabolic stability when compared with its linear analogue 33, considering the triazole ring as a bioisosteric substitution of the amide linkage (Table S1, Supporting Information). Among the detected soft spots (Figure 4), the main site of metabolism was localized at the attachment point of the AR ligand, where the occurrence of an O-dealkylation reaction is likely favored by the presence of two aromatic moieties nearby.
Figure 4.

Effect of the cyclic linkers on metabolic stability. The half-life values associated with each compound and expressed in minutes are reported in brackets. Bond cleavages are illustrated as dotted lines, while circles represent atoms subjected to oxidation in a well-defined position by MS/MS fragmentation (blue) or in a defined moiety by MS/MS fragmentation (pink). In the case of pink circles, the displayed position was suggested by MetaSite predictions. Finally, ellipses indicate dehydrogenation reactions, and the same color code for circles was used.
Effect of the Linker’s Site of Attachment
In the PROTAC design, the site of attachment of the linker to the ligands is typically selected by analyzing the solvent-exposed areas on ligand-protein binding structures.73Figure 5 shows that the selection of the site of attachment might have an impact on the overall metabolic degradation of the PROTAC. Indeed, compound 35 was less stable than 40, although the identified soft spots were comparable.
Figure 5.
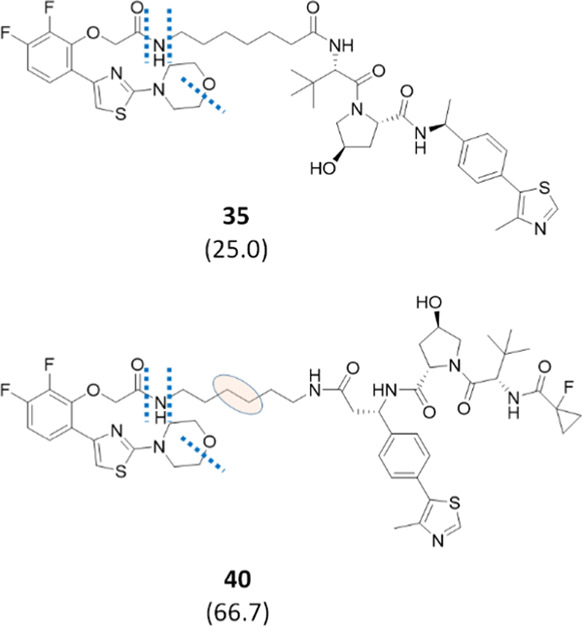
Effect of the linker’s site of attachment on PROTAC stability. Half-life values expressed in minutes are shown in brackets. Bond cleavages are illustrated as dotted lines, while the pink ellipse indicates a dehydrogenation reaction occurring in the linker moiety (the exact position of the soft spot was not elucidated by MS/MS fragmentation, and the displayed position reflects the most probable soft spot according to MetaSite predictions).
PROTAC Degradation by CYP3A4
CYP3A4 represents a major isozyme in the human liver and is known to metabolize a larger variety of xenobiotics.44 An important feature of CYP3A4 is its plasticity, which allows it to accommodate an extensive substrate in its binding site.45 Based on these considerations, we assumed that CYP3A4 could be responsible for most of the phase I metabolism observed in the cryopreserved human hepatocyte data. Therefore, six PROTACs with variable combinations of ligands and linkers were selected, and their metabolism by incubation in the presence of recombinant CYP3A4 for 60 min was studied. Figure 6 shows the soft spots detected, while t1/2 values are provided in brackets. In the experimental conditions used (see the Methods section), all tested PROTACs were significantly metabolized, with half-lives well below 30 min. In addition, the already discussed instability at the level of the linker was proved to be caused by CYP3A4.
Figure 6.

Soft-spot identification of six PROTACs tested for metabolism by CYP3A4. Half-life values expressed in minutes are shown in brackets. Bond cleavages are illustrated as dotted lines. The pink ellipse indicates that a dehydrogenation reaction occurred in the linker. Since the MS/MS fragmentation was not enough to define the exact position of the dehydrogenation’s soft spot, its most probable position was suggested by MetaSite predictions.
PROTAC Degradation by hAOX
In addition to CYP-mediated metabolism, increasing importance has been attributed to human aldehyde oxidase (hAOX), a cytosolic drug-metabolizing enzyme expressed in the human liver.12,79−81 Indeed, strategies designed to reduce CYP-mediated metabolism have resulted in increasing drug reactivity toward AOX.46,47 As a consequence, several compounds have failed due to undetected hAOX-mediated oxidation (e.g., BIBX1382, RO-1, FK3453, carbazeran).48,49 Two reactions are reported to be catalyzed by hAOX: (1) the oxidation of a wide range of azaaromatic scaffolds at the unsubstituted carbon in ortho to the nitrogen (usually the most electron-deficient);82 and (2) the hydrolysis of amides although a few examples have been reported so far.80,81,83 Since the PROTACs commonly contain amide groups and heteroaromatic rings, two PROTACs were selected to be screened for hAOX metabolism. Compounds 33 and 34 were selected as each one contained three amide groups that might be liability spots for hAOX metabolism. In addition, they also contain one 4-aryl or 5-aryl substituted thiazole ring (in the AR ligand moiety (46) and the VHL ligand moiety (42), respectively). Although five-term moieties are commonly considered not prone to be metabolized by hAOX unless it is fused with a phenyl ring to give a benzothiazole,84−86 one exception has been reported by Arora et al.,87 showing that 2H-oxazoles substituted at the C-4 or C-5 position with variably decorated phenyl rings can undergo oxidation by mouse cytosolic AOX to give the corresponding 2-oxazolones. Therefore, we hypothesized that a similar oxidation pattern could occur in the selected PROTACs although this reaction has not been reported for substituted thiazoles to date. Compounds 33 and 34 were therefore incubated in human liver cytosol for 30, 60, and 90 min in the absence and presence of hydralazine, a selective inhibitor of hAOX,88 and the kinetics data are illustrated in Figure S2 (Supporting Information). Two reactions occurred for both compounds, the hydrolysis of an amide and an oxidation, both in the VHL ligand moiety (Figure 7A).
Figure 7.

Metabolism of 33 and 34 in human liver cytosol. (A) Soft-spot identification, with filled circles indicating metabolism by hAOX and empty circles indicating metabolism by other enzymes. Half-life values expressed in minutes are shown in brackets. (B) Pose of 33 in the hAOX cavity according to MetaSite prediction with protein in the surface mode. (C) Pose of 33 in the hAOX cavity according to MetaSite prediction highlighting the main interacting residues and the molybdenum pyranopterin cofactor (MoCo).
While the hydrolysis of the amide was observed also in the presence of hydralazine, indicating that an enzyme other than hAOX is responsible for this cleavage, the hydroxylated product was formed only in the absence of the selective inhibitor (Supporting Information Figures S2 and S3). In addition, the MS/MS fragment ion with m/z 218.0637 revealed that the soft spot for oxidation is located in the 4-methyl-5-phenyl-thiazole moiety that, in its unoxidized form, shows a fragment ion with m/z of 202.0685 (Supporting Information, Figure S2). Based on the findings by Arora et al.,87 it is likely to assume that the metabolites formed for both 33 and 34 are the corresponding 2-thiazolones on the VHL moiety. This finding is noteworthy since VHL is one of the most common E3 ligases exploited for the PROTAC strategy and, therefore, additional larger-scale studies are currently in progress. Finally, MetaSite software was used to generate the most probable binding mode of 33 in the hAOX cavity. Figure 7B illustrates that 33 nicely fits the hAOX cavity and that the pose exposing the 4-methyl-5-phenyl-thiazole toward the molybdenum pyranopterin cofactor (MoCo) is stabilized by several favored interactions, including a H-bond between the hydroxyl group of the pyrrolidine moiety and Glu882 and a π–π stacking between the thiazole ring in the AR ligand moiety and Phe885. These two residues were recently hypothesized to stabilize other hAOX substrates (Figure 7C).80,82
Conclusions
This study represents the first analysis of the metabolic stability of PROTACs applied to a collection of compounds with large chemical variability. Metabolism assays were first performed in cryopreserved human hepatocytes that, containing all hepatic drug-metabolizing enzymes and cofactors at physiological levels, represent the “gold standard” even for the early screening of metabolic stability. Additional studies were conducted to evaluate whether CYP3A4 and hAOX could be involved in the observed metabolic biotransformations. A comprehensive analysis of the data in terms of half-life values and soft spot identification allowed us to highlight general trends in PROTAC metabolism. The linker resulted in being the most liable moiety in a PROTAC molecule. Its instability is mainly localized at the attachment points to ligands, involving N-dealkylation and amide hydrolysis reactions. Such reactions also occurred in CYP3A4 incubation, indicating that this isoform can play an essential role in PROTAC degradation. In the case of PEG-like linkers, a large number of O-dealkylation reactions was observed, indicating that multiple fragmentation points are possible. Nevertheless, the most significant number of soft spots in PEG-like linker-based PROTACs compared to aliphatic-based ones seems not to negatively affect the overall metabolic stability of a compound, with the half-life values being comparable or even better. From the medicinal chemistry perspective, however, the soft spot protection strategies might be more challenging for PEG-like-based PROTACs. The length of the linker also played a role in metabolic stability, with 4-unit linkers being very stable compared to longer ones. Unfortunately, longer linkers are mostly used in PROTAC design for activity optimization, thus limiting the application of this evidence. The use of linkers endowed with cyclic moieties may represent a good strategy to increase metabolic stability; however, we demonstrated that exceptions are possible, as in the case of compound 39. Concerning the E3 ligands, thalidomide-based PROTACs suffered from nonenzymatic degradation in aqueous solutions. Nevertheless, the protocol developed in this study reduces this degradation during storage in the autosampler for LC–MS analysis. This will not prevent the eventually occurring nonenzymatic hydrolysis of the thalidomide moiety during incubation in cryopreserved human hepatocytes, but it will improve reproducibility of the results. When the VHL ligand is used, we discovered that PROTACs could undergo hAOX metabolism at the 5-phenyl-thiazole moiety. This finding not only represents the first evidence of the metabolism on a substituted thiazole by hAOX based on our knowledge but also indicates that further studies are required to verify the affinity of the hydroxylated metabolism with VHL aimed at evaluating the impact on PROTAC efficiency. The metabolic degradation of PROTACs by hAOX on a large scale is currently under investigation. To conclude, we believe that the study herein reported represents a solid base to start considering metabolism in rational PROTAC design.
Experimental Section
Chemistry
General
Unless otherwise noted, starting materials, reagents, and solvents were purchased from commercial suppliers and were used as received without further purification.
Compound 41 was purchased from Fluorochem, compound 42 was prepared according to the reported procedure,52 while compounds 43 and 44 were purchased from Ambeed and Fluorochem, respectively. PROTACs 26–40 were kindly provided by Montelino Therapeutics Inc.
Reactions were routinely monitored by thin-layer chromatography (TLC) performed on a silica gel 60 F254 (layer 0.2 mm) precoated aluminum foil (with a fluorescent indicator UV254) (Sigma-Aldrich). Developed plates were air-dried and visualized under UV light (254/365 nm) or using KMnO4, ninhydrin, or phosphomolybdic acid stain solutions. Flash column chromatography was performed on Merck silica gel 60 (mesh 230–400). 1H NMR and 13C NMR spectra were recorded at room temperature at 400 and 101 MHz, respectively, on a Bruker Avance 400 spectrometer using tetramethylsilane (TMS) or residual solvent peak as the internal standard. Chemical shifts are reported in ppm (δ), and the coupling constants (J) are given in Hertz (Hz). Peak multiplicities are abbreviated as follows: s (singlet), bs (broad singlet), d (doublet), dd (double doublet), t (triplet), dt (double triplet), q (quartet), p (pentet), and m (multiplet). High-resolution mass spectroscopy (HRMS) analyses were carried out on the Agilent Technologies 6540 UHD Accurate Mass Q-TOF LC–MS system. The purity of all final compounds was confirmed to be >95% by ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS). The analyses were carried out according to the method listed below. The mobile phase was a mixture of water (solvent A) and acetonitrile (solvent B), both containing formic acid at 0.1%. Method: Acquity UPLC BEH C18 1.7 μm (C18, 150 mm × 2.1 mm) column at 40 °C using a flow rate of 0.65 mL/min in a 10 min gradient elution. Gradient elution was as follows: 99.5:0.5 (A/B) to 5:95 (A/B) over 8 min, 5:95 (A/B) for 2 min, and then reversion back to 99.5:0.5 (A/B) over 0.1 min. The UV detection is an averaged signal from a wavelength of 190–640 nm, and mass spectra are recorded on a mass spectrometer using positive-mode electrospray ionization. The chemical names were generated using ChemBioDraw 12.0 from CambridgeSoft.
General Procedure A: HATU-Mediated Amidation
Under a nitrogen atmosphere, to a stirred solution of the appropriate carboxylic acid (1.0 equiv), suitable amine (1.0 equiv), and DIPEA (4.0 equiv) in dry DMF, HATU (1.25 equiv) was added and the reaction mixture was stirred at room temperature (1–18 h). The mixture was poured in ice-water, yielding a precipitate collected by filtration. When no precipitate formed, the mixture was extracted with EA (×3) and the reunited organic phases were washed with water (×3) and brine (×3), dried over Na2SO4, and evaporated to dryness. The crude was purified as described below.
2-((S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octyl)acetamide (4)51
General Procedure A (1 h) was followed using 43 (0.021 g, 0.053 mmol) and 47 (0.023 g, 0.053 mmol) to afford the title compound as a yellow solid (0.019 g, 45% yield) after purification by flash column chromatography on SiO2 (DCM/acetone/MeOH, 92:4:4). 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.15 (t, J = 5.3 Hz, 1H), 7.61–7.53 (m, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 7.07 (d, J = 8.7 Hz, 1H), 7.01 (d, J = 7.1 Hz, 1H), 6.52 (t, J = 5.9 Hz, 1H), 5.04 (dd, J = 13.0, 5.3 Hz, 1H), 4.53–4.44 (m, 1H), 3.31–3.01 (m, 7H), 2.93–2.81 (m, 1H), 2.63–2.55 (m, 4H), 2.40 (s, 3H), 2.06–1.97 (m, 1H), 1.61 (s, 3H), 1.59–1.49 (m, 2H), 1.49–1.38 (m, 2H), 1.32 (d, J = 25.3 Hz, 8H). 13C NMR (101 MHz, DMSO-d6) δ 173.26, 170.55, 169.77, 169.41, 167.77, 163.42, 155.60, 150.25, 146.90, 137.19, 136.73, 135.70, 132.73, 132.66, 131.17, 130.58 (2C), 130.25, 130.03, 128.90 (2C), 117.62, 110.83, 109.49, 54.41, 49.01, 42.30, 38.88, 38.16, 31.45, 29.71, 29.22, 29.16, 26.81, 26.76, 22.62, 14.51, 13.14, 11.76. HRMS (ESI) m/z [M + H]+ calcd for C40H43ClN8O5S 783.28384, found 783.28533. UPLC retention time: 5.943 min.
2-((S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)acetamide (5)89
General Procedure A (1 h) was followed using 43 (0.023 g, 0.057 mmol) and 48(90) (0.025 g, 0.057 mmol) to afford the title compound as a yellow solid (0.013 g, 29% yield) after purification by flash column chromatography on SiO2 (DCM/acetone/MeOH, 92:3:5). 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.26 (t, J = 5.2 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.6 Hz, 1H), 7.03 (d, J = 7.0 Hz, 1H), 6.62 (t, J = 5.4 Hz, 1H), 5.06 (dd, J = 12.8, 5.2 Hz, 1H), 4.50 (t, J = 6.9 Hz, 1H), 3.69–3.40 (m, 10H), 3.31–3.21 (m, 5H), 2.92–2.80 (m, 1H), 2.59 (s, 3H), 2.41 (s, 3H), 2.07–1.97 (m, 1H), 1.62 (s, 3H), 1.30–1.22 (m, J = 8.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 173.26, 170.55, 170.12, 169.39, 167.76, 163.46, 155.57, 150.28, 146.86, 137.23, 136.68, 135.67, 132.73, 132.56, 131.15, 130.61 (2C), 130.29, 130.01, 128.91 (2C), 117.90, 111.12, 109.72, 70.18, 70.10, 69.71, 69.37, 54.29, 49.01, 42.17, 39.09, 37.97, 31.44, 22.60, 14.52, 13.14, 11.77. HRMS (ESI) m/z [M + H]+ calcd for C38H39ClN8O7S 787.24237, found 787.24302. UPLC retention time: 5.053 min.
4-((3-(4-(3-Aminopropyl)piperazin-1-yl)propyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione Hydrochloride (49)
Under a nitrogen atmosphere, a solution of 62 (0.246 g, 0.891 mmol), tert-butyl (3-(4-(3-aminopropyl)piperazin-1-yl)propyl)carbamate91 (0.295 g, 0.979 mmol), and DIPEA (0.3 mL, 1.780 mmol) in dry DMSO (2.0 mL) was stirred at 60 °C for 3 h. After cooling to room temperature, the reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/MeOH, 97:3) to give tert-butyl(3-(4-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4yl)amino) propyl)piperazin-1-yl)propyl)carbamate (0.085 g, 56% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.70 (bs, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.08 (d, J = 7.1 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.77 (bs, 1H), 5.35 (bs, 1H), 4.91 (dd, J = 12.0, 5.4 Hz, 1H), 3.43–3.29 (m, 2H), 3.26–3.13 (m, 2H), 2.93–2.36 (m, 14H), 2.19–2.06 (m, 1H), 1.92–1.59 (m, 5H), 1.44 (s, 9H). HRMS (ESI) m/z [M + H]+ calcd for C28H40N6O6 557.30876, found 557.31024. UPLC retention time: 2.993 min.
Then, the solution of the obtained compound (0.078 g, 0.140 mmol) in 4.0N HCl in dioxane (0.78 mL) was stirred at room temperature overnight. The solvent was evaporated to dryness, and the solid was tritured with diethyl ether (DEE) and collected by filtration, yielding 49 as a white solid (0.068 g, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.92 (bs, 2H), 11.11 (s, 1H), 8.04 (bs, 3H), 7.61 (t, J = 7.8 Hz, 1H), 7.19 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 7.0 Hz, 1H), 6.79 (bs, 1H), 5.06 (dd, J = 12.7, 5.3 Hz, 1H), 3.90–3.47 (m, 8H), 3.37–3.02 (m, 8H), 2.97–2.83 (m, 3H), 2.70–2.54 (m, 1H), 2.10–1.92 (m, 4H). HRMS (ESI) m/z [M + H]+ calcd for C23H32N6O4 457.25633, found 457.25670. UPLC retention time: 1.78 min.
2-((S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-N-(3-(4-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)piperazin-1-yl)propyl)acetamide (6)
General Procedure A (overnight) was followed using 49 (0.069 g, 0.140 mmol) and 43 (0.055 g, 0.140 mmol) to afford the title compound as a fluorescent-yellow solid (0.023 g, 20% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 95:5). 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.29 (bs, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.6 Hz, 1H), 7.04 (d, J = 7.1 Hz, 1H), 6.81 (bs, 1H), 5.06 (dd, J = 13.0, 5.2 Hz, 1H), 4.51 (t, J = 7.0 Hz, 1H), 3.43–3.35 (m, 3H), 3.30 (s, 2H), 3.28–3.02 (m, 6H), 2.94–2.84 (m, 2H), 2.65–2.53 (m, 5H), 2.42 (s, 3H), 2.08–2.01 (m, 1H), 1.86–1.67 (m, 3H), 1.63 (s, 3H). HRMS (ESI) m/z [M + H]+ calcd for C42H47ClN10O5S 839.32184, found 839.32312. UPLC retention time: 4.118 min.
(2S,4R)-1-((S)-2-(7-(2-((S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)heptanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (8)
General Procedure A (2 h) was followed using 43 (0.023 g, 0.057 mmol) and 50 (0.035 g, 0.057 mmol) to afford the title compound as a white solid (0.024 g, 43% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 97:3 to 95:5). 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.36 (d, J = 7.7 Hz, 1H), 8.15 (t, J = 5.4 Hz, 1H), 7.79 (d, J = 9.2 Hz, 1H), 7.49 (d, J = 8.6 Hz, 2H), 7.42 (dd, J = 8.1, 5.0 Hz, 4H), 7.37 (d, J = 8.2 Hz, 2H), 5.09 (d, J = 3.5 Hz, 1H), 4.96–4.85 (m, 1H), 4.56–4.47 (m, 2H), 4.43 (t, J = 7.9 Hz, 1H), 4.27 (s, 1H), 3.60 (s, 2H), 3.26–3.02 (m, 4H), 2.59 (s, 3H), 2.45 (s, 3H), 2.41 (s, 3H), 2.30–2.20 (m, 1H), 2.16–2.06 (m, 1H), 2.04–1.95 (m, 1H), 1.84–1.75 (m, 1H), 1.61 (s, 3H), 1.53–1.40 (m, 4H), 1.37 (d, J = 7.0 Hz, 3H), 1.32–1.24 (m, 4H), 0.93 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 172.50, 171.09, 170.09, 169.77, 163.47, 155.59, 150.28, 148.22, 145.12, 137.24, 135.71, 132.72, 131.58, 131.17, 130.57 (2C), 130.29, 130.16, 130.05, 129.28 (2C), 128.93 (2C), 126.84 (2C), 126.74, 69.22, 59.00, 56.79, 56.70, 54.39, 48.16, 38.95, 38.19, 38.12, 35.64, 35.32, 29.60, 28.87, 26.92 (3C), 26.61, 25.85, 22.90, 16.45, 14.51, 13.15, 11.75. HRMS (ESI) m/z [M + H]+ calcd for C49H60ClN9O5S2 954.39201, found 954.39400. UPLC retention time: 5.427 min.
(2S,4R)-1-((S)-2-(tert-Butyl)-14-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-4,13-dioxo-6,9-dioxa-3,12-diazatetradecanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (9)
General Procedure A (2 h) was followed using 43 (0.056 g, 0.14 mmol) and 51 (0.089 g, 0.014 mmol) to afford the title compound as a white solid (0.025 g, 18% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH 95:5). 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.44 (d, J = 7.6 Hz, 1H), 8.27 (t, J = 5.7 Hz, 1H), 7.49 (d, J = 8.6 Hz, 2H), 7.46–7.38 (m, 5H), 7.36 (d, J = 8.3 Hz, 2H), 5.14 (d, J = 3.5 Hz, 1H), 4.96–4.86 (m, 1H), 4.57 (d, J = 9.6 Hz, 1H), 4.54–4.48 (m, 1H), 4.46 (d, J = 8.0 Hz, 1H), 4.29 (s, 1H), 3.98 (s, 2H), 3.60 (dd, J = 17.0, 12.0 Hz, 6H), 3.54–3.46 (m, 2H), 3.31–3.19 (m, 4H), 2.60 (s, 3H), 2.45 (s, 3H), 2.41 (s, 3H), 2.11–2.00 (m, 1H), 1.84–1.75 (m, 1H), 1.63 (s, 3H), 1.35 (d, J = 7.0 Hz, 3H), 0.95 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.90, 170.13, 169.53, 169.03, 163.42, 155.61, 150.27, 148.21, 145.14, 137.23, 135.68, 132.70, 131.57, 131.17, 130.61 (2C), 130.30, 130.15, 130.07, 129.36, 129.27 (2C), 128.93 (2C), 126.79 (2C), 126.74, 70.90, 70.05, 69.88, 69.79, 69.24, 59.02, 57.01, 56.17, 54.28, 48.23, 39.07, 38.20, 37.92, 36.69, 36.23, 26.71 (3C), 22.87, 16.45, 14.51, 13.15, 11.75. HRMS (ESI) m/z [M + H]+ calcd for C48H58ClN9O7S2 972.36619, found 972.36785. UPLC retention time: 5.344 min.
(2S,4R)-1-((S)-3,3-Dimethyl-2-(3-(piperazin-1-yl)propanamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide Hydrochloride (52)
Under a nitrogen atmosphere, a solution of 42 (0.200 g, 0.416 mmol), 3-(1-tert-butoxycarbonylpiperazin-4-yl)propionic acid (0.124 g, 0.458 mmol), HATU (0.209 g, 0.520 mmol), and DIPEA (0.3 mL, 1.664 mmol) in dry DMF (2.0 mL) was stirred at room temperature for 1 h. Then, the reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with water (×2) and brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/acetone/MeOH, 75:20:5) to give tert-butyl-4-(3-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropyl)piperazine-1-carboxylate (0.145 g, 51% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 9.02 (bs, 1H), 8.67 (s, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.56–7.30 (m, 4H), 5.13–5.01 (m, 1H), 4.78 (t, J = 8.0 Hz, 1H), 4.47 (s, 1H), 4.44–4.36 (m, 1H), 4.21 (d, J = 11.2 Hz, 1H), 3.74–3.14 (m, 6H), 2.91–2.30 (m, 11H), 2.14–2.04 (m, 1H), 1.78–1.63 (m, 1H), 1.53–1.41 (m, 12H), 1.07 (s, 9H). HRMS (ESI) m/z [M + H]+ calcd for C35H52N6O6S 685.37473, found 685.37551. UPLC retention time: 3.716 min.
Then, the solution of the obtained compound (0.140 g, 0.204 mmol) in 4.0N HCl in dioxane (1.5 mL) was stirred at room temperature for 2 h. The solvent was evaporated to dryness, and the solid was tritured with DEE and collected by filtration, yielding 52 as a white solid (0.124 g, 98% yield). 1H NMR (400 MHz, MeOD) δ 9.86 (s, 1H), 7.68–7.46 (m, 4H), 5.09–4.99 (m, 1H), 4.66–4.53 (m, 2H), 4.50–4.38 (m, 1H), 3.96 (d, J = 10.9 Hz, 1H), 3.82–3.47 (m, 12H), 2.93 (t, J = 6.5 Hz, 2H), 2.61 (s, 3H), 2.25 (dd, J = 12.8, 7.8 Hz, 1H), 2.01–1.90 (m, 1H), 1.67–1.47 (m, 3H), 1.18–0.95 (m, 9H). HRMS (ESI) m/z [M + H]+ calcd for C30H44N6O4S 585.32230, found 585.32540. UPLC retention time: 2.852 min.
(2S,4R)-1-((S)-2-(3-(4-(2-((S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetyl)piperazin-1-yl)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (11)
General Procedure A (2 h) was followed using 52 (0.062 g, 0.099 mmol) and 43 (0.040 g, 0.099 mmol) to afford the title compound as a white solid (0.020 g, 21% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 93:7). 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.40 (t, J = 8.5 Hz, 2H), 7.52–7.35 (m, 8H), 5.13 (d, J = 3.0 Hz, 1H), 4.99–4.86 (m, 1H), 4.61–4.53 (m, 2H), 4.45 (t, J = 7.9 Hz, 1H), 4.29 (s, 1H), 3.78–3.55 (m, 5H), 3.55–3.46 (m, 2H), 3.46–3.36 (m, 2H), 2.71–2.53 (m, 5H), 2.48–2.39 (m, 8H), 2.38–2.23 (m, 2H), 2.09–2.98 (m, 1H), 1.85–1.74 (m, 1H), 1.63 (s, 3H), 1.38 (d, J = 6.9 Hz, 3H), 0.97 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 171.26, 171.06, 169.99, 168.54, 163.31, 155.71, 150.22, 148.20, 145.16, 137.22, 135.64, 132.65, 131.57, 131.13, 130.61 (2C), 130.33, 130.13, 130.09, 129.33, 129.27 (2C), 128.93(2C), 126.82 (2C), 69.22, 58.98, 56.71, 54.63, 54.37, 52.99, 52.64, 48.20, 45.43, 41.63, 38.22, 35.92 (2C), 35.19, 32.85, 26.92 (3C), 22.92, 16.45, 14.49, 13.16, 11.75. HRMS (ESI) m/z [M + H]+ calcd for C49H59ClN10O5S2 967.38781, found 967.38655. UPLC retention time: 4.492 min
5-((3-Chlorophenyl)amino)-N-(8-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)octyl)benzo[c][2,6]naphthyridine-8-carboxamide (12)
General Procedure A (overnight) was followed using 44 (0.028 g, 0.081 mmol) and 53 (0.040 g, 0.081 mmol) to afford the title compound as a yellow solid (0.027 g, 42% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH 95:5 to 93:7). 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 10.19 (s, 1H), 9.66 (s, 1H), 8.98 (d, J = 5.6 Hz, 1H), 8.84 (d, J = 8.5 Hz, 1H), 8.73 (t, J = 5.3 Hz, 1H), 8.58 (d, J = 5.6 Hz, 1H), 8.27 (s, 1H), 8.22 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.99–7.89 (m, 2H), 7.79 (t, J = 7.9 Hz, 1H), 7.53–7.41 (m, 2H), 7.37 (d, J = 8.5 Hz, 1H), 7.14 (d, J = 7.9 Hz, 1H), 5.11 (dd, J = 12.9, 5.3 Hz, 1H), 4.75 (s, 2H), 3.31–3.26 (m, 2H), 3.21–3.06 (m, 2H), 2.97–2.82 (m, 1H), 2.69–2.52 (m, 2H), 2.11–1.96 (m, 1H), 1.63–1.51 (m, 2H), 1.48–1.38 (m, 2H), 1.38–1.19 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 173.23, 170.34, 167.19, 167.06, 166.11, 165.96, 155.50, 150.54, 148.18, 147.72, 143.78, 142.39, 137.36, 136.16, 133.48, 133.27, 130.58, 127.56, 126.25, 124.19, 123.39, 122.78, 122.68, 121.44, 120.82, 120.70, 119.70, 117.27, 116.83, 116.49, 68.09, 49.27, 38.79 (2C), 31.41, 29.57, 29.45, 29.22, 29.16, 26.97, 26.75, 22.46. HRMS (ESI) m/z [M + H]+ calcd for C42H40ClN7O7 790.27560, found 790.27511. UPLC retention time: 5.247 min.
5-((3-Chlorophenyl)amino)-N-(10-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)decyl)benzo[c][2,6]naphthyridine-8-carboxamide (13)
General Procedure A (overnight) was followed using 44 (0.019 g, 0.053 mmol) and 54 (0.028 g, 0.053 mmol) to afford the title compound as a yellow solid (0.023 g, 51% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH 95:5). 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 10.19 (s, 1H), 9.65 (s, 1H), 8.98 (d, J = 5.6 Hz, 1H), 8.84 (d, J = 8.8 Hz, 1H), 8.73 (t, J = 5.6 Hz, 1H), 8.58 (d, J = 5.7 Hz, 1H), 8.28 (s, 1H), 8.22 (s, 1H), 8.09 (d, J = 8.3 Hz, 1H), 7.96–7.87 (m, 2H), 7.82–7.75 (m, 1H), 7.48 (d, J = 7.3 Hz, 1H), 7.44 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 8.7 Hz, 1H), 7.14 (d, J = 7.5 Hz, 1H), 5.11 (dd, J = 12.8, 5.4 Hz, 1H), 4.75 (s, 2H), 3.32–3.26 (m, 2H), 3.18–3.07 (m, 2H), 2.96–2.83 (m, 1H), 2.67–2.51 (m, 2H), 2.11–1.98 (m, 1H), 1.61–1.52 (m, 2H), 1.46–1.37 (m, 2H), 1.36–1.16 (m, 12H). 13C NMR (101 MHz, DMSO-d6) δ 173.22, 170.33, 167.18, 167.05, 166.11, 165.96, 155.50, 150.53, 148.18, 147.72, 143.77, 142.39, 137.36, 136.17, 133.48, 133.27, 130.57, 127.56, 126.25, 124.19, 123.38, 122.77, 122.67, 121.44, 120.82, 120.69, 119.68, 117.27, 116.82, 116.49, 68.10, 49.27, 38.78 (2C), 31.42, 29.57, 29.44 (3C), 29.26, 29.18, 27.01, 26.77, 22.46. HRMS (ESI) m/z [M + Na]+ calcd for C44H44ClN7O7 840.28830, found 840.28881. UPLC retention time: 5.71 min.
5-((3-Chlorophenyl)amino)-N-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-2-oxo-6,9,12-trioxa-3-azatetradecan-14-yl)benzo[c][2,6]naphthyridine-8-carboxamide (14)
General Procedure A (overnight) was followed using 44 (0.014 g, 0.040 mmol) and 55 (0.022 g, 0.040 mmol) to afford the title compound as a yellow solid (0.014 g, 42% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH 98:2 to 95:5). 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 10.19 (s, 1H), 9.66 (s, 1H), 8.99 (d, J = 5.5 Hz, 1H), 8.89–8.76 (m, 2H), 8.58 (d, J = 5.6 Hz, 1H), 8.28 (s, J = 13.7 Hz, 1H), 8.24 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.03–7.90 (m, 2H), 7.78 (t, J = 7.9 Hz, 1H), 7.51–7.40 (m, 2H), 7.37 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 5.11 (dd, J = 12.8, 5.1 Hz, 1H), 4.76 (s, 2H), 3.64–3.38 (m, 12H), 3.32–3.24 (m, 4H), 2.99–2.80 (m, 1H), 2.69–2.54 (m, 2H), 2.11–1.95 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 173.21, 170.32, 167.30, 167.15, 166.31, 165.85, 155.38, 150.54, 148.17, 147.74, 143.75, 142.36, 137.34, 135.81, 133.45, 133.26, 130.55, 127.52, 126.33, 124.20, 123.35, 122.80, 122.67, 121.53, 120.73, 120.66, 119.67, 117.17, 116.81, 116.45, 70.23, 70.21, 70.08 (3C), 69.31, 69.26, 67.91, 49.25, 38.85, 31.41, 22.45. HRMS (ESI) m/z [M + Na]+ calcd for C42H40ClN7O10 860.24174, found 860.24314. UPLC retention time: 4.355 min.
5-((3-Chlorophenyl)amino)-N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)benzo[c][2,6]naphthyridine-8-carboxamide (15)
General Procedure A (1 h) was followed using 44 (0.020 g, 0.057 mmol) and 48(90) (0.028 g, 0.057 mmol) to afford the title compound as a yellow solid (0.024 g, 57% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 97:3). 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 10.16 (s, 1H), 9.65 (s, 1H), 8.98 (d, J = 5.4 Hz, 1H), 8.81 (d, J = 8.4 Hz, 1H), 8.77 (t, J = 5.6 Hz, 1H), 8.57 (d, J = 5.3 Hz, 1H), 8.27 (s, 1H), 8.23 (s, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.93 (d, J = 8.6 Hz, 1H), 7.45 (q, J = 8.0 Hz, 2H), 7.14 (d, J = 7.4 Hz, 1H), 7.01 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 7.1 Hz, 1H), 6.53 (t, J = 5.8 Hz, 1H), 5.02 (dd, J = 13.1, 5.4 Hz, 1H), 3.70–3.55 (m, 8H), 3.54–3.45 (m, 2H), 3.45–3.36 (m, 2H), 3.17 (d, J = 5.3 Hz, 1H), 2.93–2.80 (m, 1H), 2.62–2.53 (m, 1H), 2.07–1.97 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 173.24, 170.54, 169.32, 167.67, 166.30, 150.53, 148.16, 147.73, 146.73, 143.74, 142.38, 136.49, 135.81, 133.26, 132.43, 130.55, 127.51, 126.30, 124.19, 123.33, 122.76, 122.66, 121.51, 120.67, 119.67, 117.70, 116.81, 110.98, 109.63, 70.18, 70.12, 69.33 (3C), 48.98, 42.15, 31.44, 22.58. HRMS (ESI) m/z [M + H]+ calcd for C38H34ClN7O7 736.22865, found 736.22880. UPLC retention time: 4.820 min.
5-((3-Chlorophenyl)amino)-N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)benzo[c][2,6]naphthyridine-8-carboxamide (16)
General Procedure A (2 h) was followed using 44 (0.011 g, 0.032 mmol) and 56 (0.013 g, 0.032 mmol) to afford the title compound as a yellow solid (0.012 g, 53% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 97:3). 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 10.19 (s, 1H), 9.66 (s, 1H), 8.99 (d, J = 5.6 Hz, 1H), 8.85 (d, J = 8.6 Hz, 1H), 8.74 (t, J = 5.4 Hz, 1H), 8.58 (d, J = 5.8 Hz, 1H), 8.27 (s, 1H), 8.22 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.3 Hz, 1H), 7.60–7.52 (m, 1H), 7.44 (t, J = 8.1 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 7.1 Hz, 1H), 6.54 (t, J = 5.8 Hz, 1H), 5.04 (dd, J = 12.8, 5.4 Hz, 1H), 3.30 (s, 2H), 2.93–2.80 (m, 1H), 2.68–2.53 (m, 2H), 2.05–1.97 (m, 1H), 1.65–1.36 (m, 8H), 1.26–1.20 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.28, 170.57, 169.41, 167.76, 166.12, 150.55, 148.19, 147.73, 146.89, 143.78, 142.39, 136.73, 136.15, 136.10, 133.27, 132.66, 130.59, 127.56, 126.26, 124.20, 123.39, 122.79, 122.69, 121.45, 120.70, 119.70, 117.64, 116.84, 110.81, 109.48, 48.99, 42.27, 31.44, 29.50, 29.13, 26.74, 26.56, 22.61. HRMS (ESI) m/z [M + H]+ calcd for C38H34ClN7O5 704.23882, found 704.23804. UPLC retention time: 5.690 min.
5-((3-Chlorophenyl)amino)-N-(5-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentyl)benzo[c][2,6]naphthyridine-8-carboxamide (17)
General Procedure A (overnight) was followed using 44 (0.036 g, 0.103 mmol) and 57 (0.060 g, 0.103 mmol) to afford the title compound as a yellow solid (0.036 g, 40% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 93:7 to 9:1). 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.66 (s, 1H), 9.01–8.95 (m, 2H), 8.85 (d, J = 8.5 Hz, 1H), 8.76 (t, J = 5.8 Hz, 1H), 8.59 (d, J = 5.9 Hz, 1H), 8.36 (d, J = 8.1 Hz, 1H), 8.26 (s, 1H), 8.23 (s, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.82 (d, J = 9.2 Hz, 1H), 7.48–7.40 (m, 3H), 7.37 (d, J = 8.3 Hz, 2H), 7.15 (d, J = 8.5 Hz, 1H), 5.10 (s, 1H), 4.96–4.85 (m, 1H), 4.52 (d, J = 9.3 Hz, 1H), 4.42 (t, J = 8.0 Hz, 1H), 4.27 (s, 1H), 3.60 (s, 2H), 3.32–3.27 (m, 2H), 2.45 (s, 3H), 2.35–2.26 (m, 1H), 2.23–2.13 (m, 1H), 2.04–1.96 (m, 1H), 1.83–1.74 (m, 1H), 1.61–1.53 (m, 4H), 1.36 (d, J = 7.0 Hz, 3H), 0.93 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 172.43, 171.09, 170.06, 166.13, 150.55, 148.22, 148.19, 148.17, 147.74, 145.12, 143.79, 142.40, 136.11, 133.28, 131.58, 130.59, 130.15, 129.28 (2C), 127.57, 126.84 (2C), 126.28, 124.21, 123.40, 122.79, 122.68, 121.47, 120.69, 119.70, 116.84, 69.24, 59.01, 56.85, 56.73, 48.15, 38.17, 35.67 (2C), 35.18, 29.39, 26.93 (3C), 23.64, 22.91, 16.45. HRMS (ESI) m/z [M + Na]+ calcd for C47H51ClN8O5S 897.32893, found 897.32891. UPLC retention time: 5.092 min.
5-((3-Chlorophenyl)amino)-N-(7-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-7-oxoheptyl)benzo[c][2,6]naphthyridine-8-carboxamide (18)
General Procedure A (overnight) was followed using 44 (0.020 g, 0.057 mmol) and 50 (0.035 g, 0.057 mmol) to afford the title compound as a yellow solid (0.018 g, 35% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 94:6). 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.66 (s, 1H), 9.07–8.94 (m, 2H), 8.85 (d, J = 8.6 Hz, 1H), 8.79–8.69 (m, 1H), 8.59 (d, J = 5.3 Hz, 1H), 8.37 (d, J = 7.8 Hz, 1H), 8.27 (s, 1H), 8.23 (s, 1H), 8.11 (d, J = 7.3 Hz, 1H), 7.94 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 9.2 Hz, 1H), 7.48–7.39 (m, 8.0 Hz, 3H), 7.37 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 7.6 Hz, 1H), 5.08 (bs, 1H), 4.96–4.87 (m, 1H), 4.52 (d, J = 9.2 Hz, 1H), 4.42 (t, J = 7.8 Hz, 1H), 4.27 (s, 1H), 3.60 (s, 2H), 3.33–3.26 (m, 2H), 2.44 (s, 3H), 2.29–2.21 (m, 1H), 2.17–2.09 (m, 1H), 2.03–1.96 (m, 1H), 1.82–1.75 (m, 1H), 1.59–1.43 (m, 4H), 1.39–1.20 (m, 7H), 0.93 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 172.51, 171.09, 170.08, 166.11, 150.54, 148.21, 148.18 (2C), 147.71, 145.13, 143.78, 142.39, 136.16, 133.28, 131.57, 130.60, 130.14, 129.28 (2C), 127.57, 126.84 (2C), 126.27, 124.21, 123.40, 122.79, 122.68, 121.45, 120.69, 119.70, 116.85, 69.21, 59.00, 56.81, 56.71, 48.15, 38.17, 35.64 (2C), 35.34, 29.53, 28.93, 26.92 (3C), 26.80, 25.90, 22.91, 16.45. HRMS (ESI) m/z [M + H]+ calcd for C49H55ClN8O5S 941.33417, found 941.33469. UPLC retention time: 5.413 min.
5-((3-Chlorophenyl)amino)-N-(3-(2-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)propyl)benzo[c][2,6]naphthyridine-8-carboxamide (19)
General Procedure A (2 h) was followed using 44 (0.020 g, 0.070 mmol) and 58 (0.042 g, 0.070 mmol) to afford the title compound as a white solid (0.017 g, 27% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 94:6). 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.66 (s, 1H), 9.05–8.94 (m, 2H), 8.90–8.79 (m, 2H), 8.59 (d, J = 5.6 Hz, 1H), 8.44 (d, J = 7.5 Hz, 1H), 8.27 (s, 1H), 8.24 (s, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.48–7.28 (m, 6H), 7.14 (d, J = 7.6 Hz, 1H), 5.13 (d, J = 3.1 Hz, 1H), 4.94–4.85 (m, 1H), 4.55 (d, J = 9.6 Hz, 1H), 4.45 (t, J = 8.1 Hz, 1H), 4.27 (s, J = 15.7 Hz, 1H), 3.97 (s, 2H), 3.66–3.35 (m, 6H), 2.45 (s, 3H), 2.09–1.97 (m, 1H), 1.94–1.83 (m, 2H), 1.82–1.71 (m, 1H), 1.36 (d, J = 7.0 Hz, 3H), 0.93 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.91, 169.50, 168.95, 166.26, 150.55, 148.21, 148.20, 147.75, 145.21, 143.79, 142.40, 136.02, 133.28, 131.57, 130.59, 130.14, 129.30 (2C), 127.56, 126.78 (2C), 126.72, 126.34, 124.22, 123.39, 122.79, 122.67, 121.51, 120.67, 119.68, 116.84, 69.96, 59.01, 57.02, 56.15, 48.23, 38.20, 37.00, 36.31, 29.81, 26.71 (3C), 22.96, 16.45. HRMS (ESI) m/z [M + H]+ calcd for C47H51ClN8O6S 913.32385, found 913.32523. UPLC retention time: 5.260 min.
(2S,4R)-1-((S)-3,3-Dimethyl-2-(2-(piperazin-1-yl)acetamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide Hydrochloride (59)
Under a nitrogen atmosphere, a solution of 42 (0.200 g, 0.416 mmol), 2-(1-tert-butoxycarbonylpiperazin-4-yl)acetic acid (0.117 g, 0.458 mmol), HATU (0.209 g, 0.520 mmol), and DIPEA (0.3 mL, 1.664 mmol) in dry DMF (2.0 mL) was stirred at room temperature for 1 h. Then, the reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with water (×2) and brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/acetone/MeOH, 63:30:7) to give tert-butyl-4-(2-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethyl)piperazine-1-carboxylate (0.137 g, 46% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 7.89 (bs, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 5.12–5.02 (m, 1H), 4.76 (t, J = 7.9 Hz, 1H), 4.50 (s, 1H), 4.45 (d, J = 8.3 Hz, 1H), 4.15 (d, J = 11.4 Hz, 1H), 3.68–2.98 (m, 8H), 2.62–2.41 (m, 6H), 2.14–2.02 (m, 1H), 1.66–1.58 (m, 1H), 1.50–1.42 (m, 12H), 1.07 (s, 9H). HRMS (ESI) m/z [M + H]+ calcd for C34H50N6O6S 671.35908, found 671.36080. UPLC retention time: 3.804 min
Then, the solution of the obtained compound (0.130 g, 0.194 mmol) in 4.0N HCl in dioxane (1.3 Ml) was stirred at room temperature for 2 h. The solvent was evaporated to dryness, and the solid was tritured with DEE and collected by filtration, yielding 59 as a white solid (0.115 g, 98% yield). 1H NMR (400 MHz, MeOD) δ 10.00–9.85 (m, 1H), 7.57 (d, J = 7.7 Hz, 2H), 7.53 (d, J = 8.1 Hz, 2H), 5.11–5.00 (m, 1H), 4.67 (s, 1H), 4.63–4.55 (m, 1H), 4.50–4.36 (m, 1H), 4.25–3.99 (m, 2H), 3.92 (d, J = 10.9 Hz, 1H), 3.80–3.59 (m, 11H), 2.62 (s, 3H), 2.34–2.19 (m, 1H), 2.01–1.89 (m, 1H), 1.66–1.50 (m, 3H), 1.13–1.03 (m, 9H). HRMS (ESI) m/z [M + H]+ calcd for C29H42N6O4S 571.30665, found 571.30783. UPLC retention time: 3.013 min.
(2S,4R)-1-((S)-2-(2-(4-(5-((3-Chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carbonyl)piperazin-1-yl)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (20)
General Procedure A (2 h) was followed using 59 (0.067 g, 0.110 mmol) and 44 (0.040 g, 0.110 mmol) to afford the title compound as a yellow solid (0.054 g, 55% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 95:5). 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 9.69 (s, 1H), 8.98 (s, 2H), 8.89–8.81 (m, 1H), 8.60 (d, J = 5.6 Hz, 1H), 8.43 (d, J = 7.5 Hz, 1H), 8.31 (s, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.81–7.71 (m, 2H), 7.50 (d, J = 8.2 Hz, 1H), 7.48–7.29 (m, 5H), 7.14 (d, J = 7.8 Hz, 1H), 5.11 (d, J = 2.9 Hz, 1H), 4.96–4.84 (m, 1H), 4.51 (d, J = 9.6 Hz, 1H), 4.44 (t, J = 8.1 Hz, 1H), 4.28 (s, 1H), 3.85–3.36 (m, 8H), 3.16–2.98 (m, 2H), 2.69–2.53 (m, 2H), 2.46 (s, 3H), 2.10–1.96 (m, 1H), 1.83–1.71 (m, 1H), 1.50–1.33 (m, 3H), 0.94 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 170.94, 169.63, 168.98, 168.83, 150.56, 148.22, 148.18, 148.05, 147.58, 145.23, 143.81, 142.35, 137.72, 133.24, 131.58, 130.60, 130.15, 129.30 (2C), 127.60, 126.77 (2C), 125.73, 124.06, 123.12, 123.09, 122.64, 120.55, 120.19, 119.58, 116.81, 69.22, 65.38, 60.87, 58.99, 56.97 (2C), 56.32, 48.22, 38.20, 36.57, 36.17 (2C), 26.77 (3C), 22.97, 16.46. HRMS (ESI) m/z [M + H]+ calcd for C48H52ClN9O5S 902.35789, found 902.35808. UPLC retention time: 4.494 min.
tert-Butyl (11-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-11-oxoundecyl)carbamate (60)
Under a nitrogen atmosphere, a solution of 45(50) (0.267 g, 0.663 mmol), methyl 11-((tert-butoxycarbonyl)amino)undecanoate92 (0.200 g, 0.663 mmol), HBTU (0.314 g, 0.829 mmol), and Et3N (0.185 mL, 0.1.327 mmol) in dry DMF (2.5 mL) was stirred at room temperature for 3 h. The reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with water (×2) and brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/MeOH, 97:3) to give 60 (0.266 g, 62% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 10.66 (s, 1H), 8.47 (d, J = 6.2 Hz, 1H), 7.85–7.67 (m, 3H), 7.37–7.29 (m, 2H), 7.04 (t, J = 8.6 Hz, 1H), 4.54 (bs, 1H), 4.29 (s, 2H), 3.51 (dd, J = 125.0, 74.0 Hz, 8H), 3.09 (t, J = 7.0 Hz, 2H), 2.41–2.23 (m, 2H), 1.44 (s, 12H), 1.27 (s, 13H).
2-(2,6-Dioxopiperidin-3-yl)-4-((11-(4-(2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-11-oxoundecyl)amino)isoindoline-1,3-dione (21)
A solution of 60 (0.260 g, 0.400 mmol) in 4.0N HCl in dioxane was stirred at room temperature for 3 h. The solvent was evaporated to dryness, and the solid was tritured with DEE and collected by filtration, yielding 61 as a white solid (0.218 g, 93% yield).
Under a nitrogen atmosphere, a mixture of 61 (0.117 g, 0.199 mmol), 62(51) (0.055 g, 0.199 mmol), and DIPEA (87 μL, 0.498 mmol) in dry DMF (1.0 mL) was stirred at 70 °C for 2 h. After cooling to room temperature, the reaction mixture was poured in ice-water, yielding a yellow precipitate, which was collected by filtration and purified by flash column chromatography on SiO2 (DCM/acetone, 7:3 to 6:4) followed by preparative TLC purification on SiO2 (DCM/MeOH, 97:3) to give 21 as a fluorescent-yellow solid (0.005 g, 3% yield). 1H NMR (400 MHz, CDCl3) δ 10.30 (s, 1H), 10.12 (s, 1H), 9.02 (s, 1H), 8.63 (s, 1H), 8.46 (s, 1H), 7.87–7.67 (m, 3H), 7.55–7.43 (m, 1H), 7.34–7.30 (m, 1H), 7.09 (d, J = 7.1 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 6.23 (s, 1H), 4.93 (dd, J = 12.0, 5.2 Hz, 1H), 4.28 (s, 2H), 3.86–3.52 (m, 6H), 3.44–3.30 (m, 2H), 3.30–3.21 (m, 2H), 2.94–2.69 (m, 3H), 2.39–2.25 (m, 2H), 2.15–2.07 (m, 1H), 1.71–1.59 (m, 4H), 1.27 (d, J = 11.5 Hz, 12H). HRMS (ESI) m/z [M + H]+ calcd for C44H48FN7O7 806.367752, found 806.36739. UPLC retention time: 5.535 min.
Methyl 12-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-12-oxododecanoate (63)
Under a nitrogen atmosphere, a solution of 45(50) (0.150 g, 0.372 mmol), 12-methoxy-12-oxododecanoic acid (0.091 g, 0.372 mmol), HBTU (0.176 g, 0.465 mmol), and Et3N (0.1 mL, 0.745 mmol) in dry DMF (1.5 mL) was stirred at room temperature for 3 h. The reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with water (×2) and brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/MeOH, 97:3) to give 63 (0.113 g, 51% yield) as a yellow oil, which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 10.68 (bs, 1H), 8.52–8.40 (m, 1H), 7.83–7.67 (m, 3H), 7.40–7.28 (m, 2H), 7.04 (t, J = 8.8 Hz, 1H), 4.29 (s, 2H), 3.89–3.69 (m, 2H), 3.66 (s, 3H), 3.61–3.22 (m, 6H), 2.39–2.26 (m, 4H), 1.67–1.55 (m, 4H), 1.33–1.21 (m, 12H). HRMS m/z [M + Na]+ calcd for C33H41FN4O5 615.29532, found 615.29651. UPLC retention time: 5.408 min.
Methyl 8-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-8-oxooctanoate (64)
Compound 64 was prepared from 45(50) (0.300 g, 0.745 mmol) and 8-methoxy-8-oxooctanoic acid (0.140 g, 0.745 mmol) in a similar manner to that described for compound 63 and obtained as a white solid (0.279 g, 70% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 98:2 to 97:3). 1H NMR (400 MHz, CDCl3) δ 12.09 (d, J = 22.4 Hz, 1H), 8.45 (d, J = 5.3 Hz, 1H), 7.82–7.62 (m, 3H), 7.39–7.25 (m, 2H), 7.08–6.93 (m, 1H), 4.28 (s, 2H), 3.89–3.19 (m, 11H), 2.40–2.20 (m, 4H), 1.67–1.52 (m, 4H), 1.36–1.25 (m, 4H). HRMS m/z [M + Na]+ calcd for C29H33FN4O5 559.23327, found 559.23345. UPLC retention time: 4.304 min.
12-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-12-oxododecanoic Acid (65)
To a stirring solution of 63 (0.097 g, 0.164 mmol) in THF (1.0 mL) at 0 °C was added a solution of LiOH monohydrate (0.069 g, 1.636 mmol) in water (0.5 mL). The reaction mixture was stirred at room temperature for 6 h. The organic solvent was evaporated under reduced pressure, the residue was diluted with water (10 mL), and at 0 °C it was acidified with 2 N HCl (pH = 3) to afford 65 as a white precipitate, collected by filtration and dried (0.084 g, 88% yield). 1H NMR (400 MHz, CDCl3) δ 11.79 (bs, 1H), 8.44 (d, J = 6.8 Hz, 1H), 7.89–7.63 (m, 3H), 7.41–7.28 (m, 2H), 7.03 (t, J = 8.5 Hz, 1H), 4.29 (s, 2H), 3.90–3.18 (m, 8H), 2.46–2.18 (m, 4H), 1.67–1.53 (m, 4H), 1.26 (s, 12H). HRMS m/z [M + H]+ calcd for C32H39FN4O5 579.29773, found 579.29912. UPLC retention time: 4.676 min.
8-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-8-oxooctanoic Acid (66)
Compound 66 was prepared from 64 (0.120 g, 0.224 mmol) in a similar manner to that described for compound 65 and obtained as a white solid (0.076 g, 65% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.61 (s, 1H), 11.99 (bs, 1H), 8.26 (d, J = 7.7 Hz, 1H), 7.96 (d, J = 7.9 Hz, 1H), 7.90 (t, J = 7.5 Hz, 1H), 7.83 (t, J = 7.4 Hz, 1H), 7.48–7.41 (m, 1H), 7.39–7.32 (m, 1H), 7.24 (t, J = 8.7 Hz, 1H), 4.33 (s, 2H), 3.69–3.45 (m, 4H), 3.22–2.86 (m, 4H), 2.37–2.12 (m, 4H), 1.54–1.38 (m, 4H), 1.31–1.18 (m, 4H).
(2S,4R)-1-((S)-2-(12-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-12-oxododecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (22)
General Procedure A (2 h) was followed using 65 (0.050 g, 0.086 mmol) and 42(52) (0.041 g, 0.086 mmol) to afford the title compound as a white solid (0.034 g, 40% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 95:5 to 94:6). 1H NMR (400 MHz, CDCl3) δ 10.75 (d, J = 55.3 Hz, 1H), 8.81 (s, 1H), 8.45 (d, J = 9.1 Hz, 1H), 7.74 (d, J = 29.9 Hz, 3H), 7.60–7.48 (m, 1H), 7.43–7.37 (m, 4H), 7.37–7.28 (m, 2H), 7.08–6.96 (m, 1H), 6.50–6.32 (m, 1H), 5.14–5.04 (m, 1H), 4.75 (t, J = 8.0 Hz, 1H), 4.65–4.57 (m, 1H), 4.52 (s, 1H), 4.27 (s, 2H), 4.16 (d, J = 10.5 Hz, 1H), 3.77 (s, 2H), 3.61 (d, J = 12.1 Hz, 1H), 3.57–3.50 (m, 2H), 3.48–3.19 (m, 4H), 2.59–2.48 (m, 4H), 2.40–2.28 (m, 2H), 2.27–2.19 (m, 2H), 2.13–2.03 (m, 2H), 1.67–1.54 (m, 4H), 1.48 (d, J = 6.5 Hz, 3H), 1.39–1.17 (m, 12H), 1.05 (s, 9H). HRMS m/z [M + Na]+ calcd for C55H69FN8O7S 1027.48862, found 1027.49135. UPLC retention time: 5.230 min.
(2S,4R)-1-((S)-2-(8-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-8-oxooctanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (23)
General Procedure A (2 h) was followed using 66 (0.114 g, 0.217 mmol) and 42(52) (0.105 g, 0.217 mmol) to afford the title compound as a white solid (0.010 g, 5% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 95:5) followed by preparative TLC purification on SiO2 (DCM/MeOH, 9:1). 1H NMR (400 MHz, CDCl3) δ 10.79 (d, J = 64.4 Hz, 1H), 8.72 (s, 1H), 8.46 (d, J = 5.7 Hz, 1H), 7.83–7.66 (m, 3H), 7.56–7.47 (m, 1H), 7.36 (d, J = 17.2 Hz, 5H), 7.05 (t, J = 8.8 Hz, 1H), 6.57–6.42 (m, 1H), 5.14–5.04 (m, 1H), 4.75 (t, J = 7.8 Hz, 1H), 4.62 (d, J = 8.5 Hz, 1H), 4.51 (s, 1H), 4.27 (s, 2H), 4.15 (d, J = 10.9 Hz, 1H), 3.81–3.20 (m, 10H), 2.53 (s, 4H), 2.38–2.21 (m, 4H), 2.13–2.06 (m, 1H), 1.67–1.56 (m, 4H), 1.47 (d, J = 6.7 Hz, 3H), 1.35–1.27 (m, 4H), 1.05 (s, 9H). HRMS m/z [M + H]+ calcd for C51H61FN8O7S 949.44462, found 949.44427. UPLC retention time: 4.612 min.
4-(4-Fluoro-3-(4-(prop-2-yn-1-yl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (67)
To a solution of 45 (0.100 g, 0.248 mmol) in ACN (3.0 mL), K2CO3 (0.086 g, 0.620 mmol), KI (0.021 g, 0.124 mmol), and propargyl bromide solution (80 wt.% in toluene) (0.031 g, 0.258 mmol) were added, and the mixture was refluxed for 4 h. Then, the solvent was evaporated to dryness and the crude residue was diluted with water, yielding a solid, which was collected by filtration, tritured by DEE, and filtered off to afford the title compound as a yellow-orange solid (0.069 g, 69% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.59 (s, 1H), 8.27 (d, J = 7.6 Hz, 1H), 7.98 (d, J = 7.8 Hz, 1H), 7.90 (t, J = 7.4 Hz, 1H), 7.83 (t, J = 7.3 Hz, 1H), 7.46–7.38 (m, 1H), 7.33 (d, J = 5.7 Hz, 1H), 7.22 (t, J = 8.9 Hz, 1H), 4.33 (s, 2H), 3.63 (s, 2H), 3.31 (s, 2H), 3.19 (s, 3H), 2.48 (s, 1H), 2.34 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ 164.24, 159.83, 156.76 (d, J = 244.4 Hz), 145.36, 135.31 (d, J = 3.2 Hz), 133.96, 132.04, 132.00, 129.53, 129.23 (d, J = 3.8 Hz), 128.36, 126.55, 125.95, 124.25 (d, J = 18.5 Hz), 116.35 (d, J = 21.6 Hz), 79.29, 76.54 (2C), 51.43 (d, J = 50.4 Hz), 46.57 (d, J = 51.4 Hz), 41.58, 36.85. HRMS (ESI) m/z [M + H]+ calcd for C23H21FN4O2 405.17268, found 405.17332. UPLC retention time: 2.569 min.
4-(4-Fluoro-3-(4-(hex-5-ynoyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (68)
Under a nitrogen atmosphere, a solution of 45 (0.150 g, 0.372 mmol), 5-hexynoic acid (0.042 g, 0.372 mmol), HBTU (0.175 g, 0.465 mmol), and Et3N (0.1 mL, 0.744 mmol) in dry DMF (2.0 mL) was stirred at room temperature for 2 h. Then, the reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with water (×2) and brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/MeOH, 96:4) to afford the title compound (0.112 g, 66% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 11.70–11.51 (m, 1H), 8.46 (d, J = 6.2 Hz, 1H), 7.83–7.65 (m, 3H), 7.40–7.28 (m, 2H), 7.02 (t, J = 9.0 Hz, 1H), 4.29 (s, 2H), 3.91–3.66 (m, 3H), 3.67–3.50 (m, 3H), 3.45 (s, 1H), 3.39–3.16 (m, 2H), 2.50 (t, J = 7.3 Hz, 2H), 2.33–2.22 (m, 2H), 1.91–1.78 (m, 2H). HRMS (ESI) m/z [M + H]+ calcd for C26H25FN4O3 461.19889, found 461.19983. UPLC retention time: 3.763 min.
(2S,4R)-1-((S)-2-(2-Azidoacetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (69)
Under a nitrogen atmosphere, a solution of 42 (0.150 g, 0.372 mmol), 5-azidopentanoic acid (0.053 g, 0.372 mmol), HOBt (0.011 g, 0.074 mmol), EDC hydrochloride (0.142 g, 0.744 mmol), and NMM (0.40 mL, 3.700 mmol) in dry DMF (2.0 mL) was stirred at room temperature for 5 h. Then, the reaction mixture was poured in ice-water and extracted with EA (×3). The reunited organic phases were washed with water (×2) and brine, dried over Na2SO4, and evaporated to dryness. The crude residue was purified by flash column chromatography on SiO2 (DCM/acetone/MeOH, 75:20:5) to afford the title compound (0.086 g, 44% yield) as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (s, 1H), 8.38 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 9.2 Hz, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 5.10 (d, J = 3.3 Hz, 1H), 4.98–4.86 (m, 1H), 4.53 (d, J = 9.2 Hz, 1H), 4.43 (t, J = 8.0 Hz, 1H), 4.28 (s, 1H), 3.66–3.55 (m, 2H), 3.38–3.34 (m, 1H), 3.32–3.28 (m, 1H), 2.46 (s, 3H), 2.36–2.24 (m, 1H), 2.23–2.10 (m, 1H), 2.07–1.97 (m, 1H), 1.86–1.75 (m, 1H), 1.61–1.44 (m, 4H), 1.38 (d, J = 6.9 Hz, 3H), 0.94 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 172.16, 171.08, 170.02, 148.22, 145.13, 131.58, 130.16, 129.29 (2C), 126.85 (2C), 126.73, 69.22, 59.00, 56.84, 56.73, 50.78, 48.15, 38.19, 35.66, 34.67, 28.29, 26.91, 23.06, 22.91, 16.45. HRMS (ESI) m/z [M + H]+ calcd for C28H39N7O4S 570.28625, found 570.28767. UPLC retention time: 4.475 min.
(2S,4R)-1-((S)-2-(5-(4-((4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (24)
To a solution of 67 (0.041 g, 0.101 mmol) and 69 (0.057 g, 0.101 mmol) in a mixture of DMF/tBuOH/H2O (1:1:1), CuSO4 (0.013 g, 0.050 mmol) and sodium ascorbate (0.059 g, 0.300 mmol) were added, and the reaction mixture was stirred at room temperature for 2 h. Then, the reaction mixture was poured in ice-water, yielding a gray precipitate, which was collected by filtration and purified by flash column chromatography on SiO2 (DCM/MeOH, 95:5 to 90:10) to give 24 (0.025 g, 26% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.59 (s, 1H), 8.99 (s, 1H), 8.37 (d, J = 7.7 Hz, 1H), 8.27 (d, J = 7.7 Hz, 1H), 8.01–7.94 (m, 2H), 7.92–7.78 (m, 3H), 7.48–7.35 (m, 5H), 7.32 (d, J = 6.3 Hz, 1H), 7.21 (t, J = 9.0 Hz, 1H), 5.10 (d, J = 3.2 Hz, 1H), 4.97–4.87 (m, 1H), 4.50 (d, J = 9.3 Hz, 1H), 4.42 (t, J = 8.1 Hz, 1H), 4.36–4.31 (m, 4H), 4.28 (s, 1H), 3.66–3.54 (m, 4H), 3.19–3.09 (m, 2H), 2.48–2.38 (m, 4H), 2.34–2.25 (m, 2H), 2.21–2.11 (m, 1H), 2.05–1.96 (m, 1H), 1.83–1.74 (m, 2H), 1.50–1.40 (m, 2H), 1.38 (d, J = 7.0 Hz, 3H), 0.92 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 172.10, 171.07, 170.01, 164.19, 159.84, 156.76 (d, J = 244.5 Hz), 148.22, 145.37, 145.12, 143.31, 135.27 (d, J = 3.1 Hz), 133.97, 132.04, 131.58, 130.16, 129.52, 129.29 (2C), 129.21, 128.35, 126.84 (2C), 126.70, 126.54, 125.94, 124.27 (d, J = 18.5 Hz), 124.21, 116.34 (d, J = 21.6 Hz), 69.22, 59.01, 56.85, 56.74, 52.80, 52.30 (2C), 49.40, 48.15, 46.96, 41.72, 38.19, 36.86, 35.63 (2C), 34.55, 29.79, 26.88 (3C), 22.90, 22.79, 16.45. HRMS (ESI) m/z [M + H]+ calcd for C51H60FN11O6S 974.45110, found 974.45129. UPLC retention time: 3.76 min.
(2S,4R)-1-((S)-2-(5-(4-(4-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)-4-oxobutyl)-1H-1,2,3-triazol-1-yl)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (25)
Compound 25 was prepared from 68 (0.083 g, 0.180 mmol) and 69 (0.098 g, 0.180 mmol) following the same procedure described for compound 24 and obtained as a crystalline pale-green solid (0.067 g, 36% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 95:5 to 91:9). 1H NMR (400 MHz, DMSO-d6) δ 12.60 (s, 1H), 9.03 (s, 1H), 8.37 (d, J = 7.7 Hz, 1H), 8.27 (d, J = 7.7 Hz, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.93–7.77 (m, 4H), 7.49–7.40 (m, 3H), 7.40–7.32 (m, 3H), 7.24 (t, J = 9.0 Hz, 1H), 5.10 (d, J = 3.3 Hz, 1H), 4.99–4.88 (m, 1H), 4.55–4.46 (m, 1H), 4.42 (t, J = 8.1 Hz, 1H), 4.36–4.24 (m, 5H), 3.68–3.56 (m, 4H), 3.55–3.45 (m, 2H), 3.42–3.36 (m, 2H), 3.22–3.10 (m, 2H), 2.71–2.55 (m, 2H), 2.46 (s, 3H), 2.42–2.24 (m, 3H), 2.22–2.14 (m, 1H), 2.04–1.97 (m, 1H), 1.89–1.71 (m, 5H), 1.52–1.40 (m, 2H), 1.37 (d, J = 6.9 Hz, 3H), 0.93 (s, 9H). HRMS (ESI) m/z [M + H]+ calcd for C54H64FN11O7S 1030.47732, found 1030.47773. UPLC retention time: 4.292 min.
tert-Butyl 2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)acetate (70)
tert-Butyl bromoacetate (0.434 mL, 0.575 g, 2.95 mmol) was added to a stirred suspension of 46(53) (0.800 g, 2.68 mmol) and K2CO3 (0.927 g, 6.71 mmol). The suspension was stirred for 18 h at rt and filtered, and the filtrate was evaporated to dryness. The residue was purified by flash column chromatography on SiO2 (PE/EA, 95:5 to 90:10) to afford 70 (0.800 g, 73% yield) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.92 (ddd, J = 2.3, 6.2, 8.8 Hz, 1H), 7.68 (s, 1H), 7.10–6.87 (m, 1H), 4.67 (d, J = 1.7 Hz, 2H), 4.00–3.79 (m, 4H), 3.68–3.47 (m, 4H), 1.53 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.77, 167.40, 150.32 (dd, J = 11.8, 249.8 Hz), 145.30, 144.64–144.23 (m), 143.98 (dd, J = 14.4, 246.1 Hz), 124.35, 124.04 (dd, J = 3.9, 7.8 Hz), 111.42 (d, J = 17.0 Hz), 107.71, 82.55, 70.00 (d, J = 7.5 Hz), 66.22 (2C), 48.61 (2C), 28.09 (3C).
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)acetic Acid (71)
A solution of 4.0N HCl in dioxane (15 mL) was added to 70 (0.780 g, 1.89 mmol), and the resulting suspension was stirred at room temperature for 16 h. The solvent was evaporated to dryness, and the residue was tritured with DEE and collected by filtration to afford 71 (0.672 g, 91% yield) as a light-yellow solid. 1H NMR (400 MHz, CD3OD) δ 7.48 (ddd, J = 2.3, 5.7, 8.1 Hz, 1H), 7.27 (s, 1H), 7.18 (td, J = 7.5, 9.2 Hz, 1H), 5.01 (d, J = 1.5 Hz, 2H), 4.02–3.86 (m, 4H), 3.86–3.67 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 171.72, 169.44, 152.57 (dd, J = 11.6, 252.2 Hz), 144.55 (d, J = 1.7 Hz), 143.30 (dd, J = 15.0, 248.0 Hz), 136.41 (d, J = 13.3 Hz), 124.82 (dd, J = 3.9, 8.6 Hz), 118.47, 111.66 (d, J = 18.2 Hz), 106.15, 69.24 (d, J = 9.0 Hz), 65.05 (2C), 49.17 (2C).
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)-N-(8-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)octyl)acetamide (26)
General Procedure A (16 h) was followed using 71 (0.032 g, 0.091 mmol) and 53 (0.045 g, 0.091 mmol) to afford the title compound as a yellow solid (0.012 g, 17% yield) following purification by flash column chromatography on SiO2 (DCM/acetone, 75:25). 1H NMR (400 MHz, CDCl3) δ 8.66 (bs, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.66–7.53 (m, 2H), 7.49–7.39 (m, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.08–6.90 (m, 3H), 4.98 (dd, J = 12.0, 5.0 Hz, 1H), 4.65 (s, 2H), 4.60 (s, 2H), 3.94–3.78 (m, 4H), 3.60–3.47 (m, 4H), 3.40–3.25 (m, 4H), 3.02–2.73 (m, 3H), 2.26–2.12 (m, 1H), 1.56–1.47 (m, 2H), 1.45–1.28 (m, 10H). 13C NMR (101 MHz, CDCl3) δ 170.91, 170.82, 168.05, 167.97, 166.64, 166.57, 165.99, 154.51, 150.69 (dd, J = 251.69, 11.5 Hz), 146.51–146.44 (m), 144.19 (dd, J = 47.3, 14.0 Hz), 144.63 (d, J = 10.6 Hz), 137.04, 133.60, 125.25 (d, J = 3.5 Hz), 124.47 (dd, J = 7.6, 4.3 Hz), 119.47, 118.14, 117.38, 112.41 (d, J = 17.1 Hz), 106.11, 72.41 (d, J = 5.1 Hz), 68.01, 66.13 (2C), 49.34, 48.55 (2C), 39.16, 39.10, 31.49, 29.44, 29.23, 29.13, 29.06, 26.70, 26.68, 22.59. HRMS (ESI) m/z [M + H]+ calcd for C38H42F2N6O9S 797.27748, found 797.27834. UPLC retention time: 5.208 min.
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)-N-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethoxy)ethyl)acetamide (27)
General Procedure A (16 h) was followed using 71 (0.025 g, 0.070 mmol) and 72 to afford the title compound as a light-yellow solid (0.009 g, 17% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 97:3). 1H NMR (400 MHz, CDCl3) δ 8.75 (bs, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.68–7.52 (m, 3H), 7.40 (bs, 1H), 7.18 (d, J = 8.3 Hz, 1H), 7.07–6.91 (m, 2H), 4.96 (dd, J = 11.7, 5.1 Hz, 1H), 4.63 (s, 2H), 4.58 (s, 2H), 3.94–3.80 (m, 4H), 3.79–3.63 (m, 8H), 3.60–3.43 (m, 8H), 2.97–2.64 (m, 3H), 2.22–2.10 (m, 1H). HRMS (ESI) m/z [M + H]+ calcd for C36H38F2N6O11S 801.23601, found 801.23745. UPLC retention time: 4.333 min.
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)-N-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-2-oxo-6,9,12-trioxa-3-azatetradecan-14-yl)acetamide (28)
General Procedure A (16 h) was followed using 71 (0.033 g, 0.092 mmol) and 55 to afford the title compound as a light-yellow solid (0.012 g, 16% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 97:3). 1H NMR (400 MHz, CDCl3) δ 8.96 (bs, 1H), 7.75 (t, J = 7.9 Hz, 1H), 7.70–7.59 (m, 2H), 7.55 (d, J = 7.3 Hz, 1H), 7.46 (bs, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.08 (s, 1H), 6.98 (dd, J = 16.7, 9.0 Hz, 1H), 4.92 (dd, J = 12.1, 5.3 Hz, 1H), 4.64 (s, 2H), 4.56 (s, 2H), 3.90–3.82 (m, 4H), 3.71–3.47 (m, 20H), 2.92–2.61 (m, 3H), 2.19–2.07 (m, 1H). HRMS (ESI) m/z [M + H]+ calcd for C38H42F2N6O12S 845.26222, found 845.26303. UPLC retention time: 4.395 min.
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)-N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)acetamide (29)
General Procedure A (16 h) was followed using 71 (0.048 g, 0.122 mmol) and 56 (0.050 g, 0.122 mmol) to afford the title compound as a yellow solid (0.015 g, 18% yield) after purification by flash column chromatography on SiO2 (DCM/acetone/MeOH, 90:10:0 to 89:10:1). 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.63–7.56 (m, 1H), 7.56–7.48 (m, 1H), 7.11 (d, J = 7.0 Hz, 1H), 7.05 (s, 1H), 7.04–6.96 (m, 1H), 6.93 (s, 1H), 6.90 (d, J = 8.5 Hz, 1H), 6.25 (s, 1H), 4.93 (dd, J = 5.3, 11.9 Hz, 1H), 4.60 (s, 2H), 3.89–3.78 (m, 4H), 3.64–3.51 (m, 4H), 3.36 (q, J = 6.8 Hz, 2H), 3.33–3.26 (m, 2H), 2.97–2.68 (m, 3H), 2.18–2.11 (m, 1H), 1.79–1.35 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 170.87, 170.74, 169.50, 168.25, 167.92, 167.59, 150.76 (dd, J = 11.6, 251.4 Hz), 146.96, 146.37–146.09 (m), 144.62 (dd, J = 1.5, 9.1 Hz), 144.15 (dd, J = 14.0, 247.3 Hz), 136.15, 132.48, 125.04, 124.53 (dd, J = 4.0, 7.8 Hz), 116.63, 112.43 (d, J = 17.2 Hz), 111.46, 109.90, 105.96, 72.42 (d, J = 4.9 Hz), 66.11 (2C), 48.87, 48.62 (2C), 42.52, 38.95, 31.41, 29.42, 29.13, 26.62, 26.55, 22.82. HRMS (ESI) m/z [M + H]+ calcd for C34H36F2N6O7S 711.2407, found 711.2412. UPLC retention time: 5.564 min.
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octyl)acetamide (30)
General Procedure A (16 h) was followed using 71 (0.020 g, 0.056 mmol) and 47 (0.024 g, 0.056 mmol) to afford the title compound as a yellow solid (0.016 g, 50% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 99:1 to 96:4) followed by further HPLC purification (Agilent Technologies 1200; column, Eclipse XDB-C18 4.6 mm × 150 mm (5 μm); flow rate, 1.0 mL/min; DAD 190-650 nm; isocratic eluent, ACN/H2O 70:30). 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.64–7.55 (m, 1H), 7.55–7.45 (m, 1H), 7.11 (d, J = 7.1 Hz, 1H), 7.04 (s, 1H), 7.04–6.97 (m, 1H), 6.93 (s, 1H), 6.90 (d, J = 8.6 Hz, 1H), 6.25 (s, 1H), 5.02–4.86 (m, 1H), 4.60 (s, 2H), 3.97–3.76 (m, 4H), 3.64–3.48 (m, 4H), 3.34 (q, J = 6.8 Hz, 2H), 3.28 (q, J = 6.6 Hz, 2H), 2.99–2.67 (m, 3H), 2.21–2.08 (m, 1H), 1.81–1.31 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 170.90, 170.71, 169.50, 168.28, 167.88, 167.61, 150.78 (dd, J = 10.5, 249.5 Hz), 147.01, 146.45–145.86 (m), 144.63 (d, J = 10.5 Hz), 144.14 (dd, J = 14.0, 247.4 Hz), 136.12, 132.49, 124.99, 124.52 (dd, J = 3.8, 7.8 Hz), 116.65, 112.41 (d, J = 17.2 Hz), 111.39, 109.86, 105.96, 72.41 (d, J = 5.1 Hz), 66.11 (2C), 48.86, 48.62 (2C), 42.59, 39.10, 31.42, 29.45, 29.14, 29.12, 29.11, 26.78, 26.72, 22.84. HRMS (ESI) m/z [M + H]+ calcd for C36H40F2N6O7S 739.27200, found 739.27369. UPLC retention time: 6.040 min.
2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octyl)acetamide (31)
General Procedure A (16 h) was followed using 71 (0.018 g, 0.049 mmol) and 48(90) (0.022 g, 0.049 mmol) to afford the title compound as a yellow solid (4.5 mg, 12% yield) after purification by flash column chromatography on SiO2 (DCM/MeOH, 97:3) followed by HPLC purification (Agilent Tecnologies 1200; column, Eclipse XDB-C18 4.6 mm × 150 mm (5 μm); flow rate, 0.8 mL/min; DAD 190-650 nm; isocratic eluent, ACN/H2O 70:30). 1H NMR (400 MHz, CDCl3) δ 8.44 (bs, 1H), 7.64 (t, J = 6.5 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.36 (bs, 1H), 7.09 (d, J = 7.1 Hz, 1H), 7.05–6.95 (m, 2H), 6.83 (d, J = 8.5 Hz, 1H), 6.51 (bs, 1H), 4.98–4.85 (m, 1H), 4.56 (s, 2H), 3.93–3.80 (m, 4H), 3.77–3.64 (m, 8H), 3.62–3.51 (m, 6H), 3.45–3.34 (m, 2H), 2.96–2.66 (m, 3H), 2.20–2.05 (m, 1H). HRMS (ESI) m/z [M + H]+ calcd for C34H36F2N6O9S 743.23053, found 743.23191. UPLC retention time: 4.945 min.
tert-Butyl (2-(2-(2-(2,3-difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)ethoxy)ethoxy)ethyl)carbamate (73)
DIAD (0.087 mL, 0.442 mmol) was slowly added to a stirred ice-cooled solution of 46(53) (0.120 g, 0.402 mmol), tert-butyl (2-(2-(2-hydroxyethoxy)ethoxy)ethyl)carbamate (0.110 g, 0.442 mmol), and PPh3 (0.116 g, 0.442 mmol) in dry THF (5.0 mL). The solution was stirred at 0 °C for 30 min and then at room temperature for 16 h. The reaction mixture was quenched with water and extracted with EA (×3). The reunited organic phases were dried over anhydrous Na2SO4 and concentrated to dryness, affording a crude residue, which was purified by flash column chromatography on SiO2 (DCM/EA, 9:1 to 8:2) to give 73 (0.100 g, 70% yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.89 (ddd, J = 8.8, 6.3, 2.3 Hz, 1H), 7.58 (s, 1H), 7.00–6.88 (m, 1H), 5.10–4.94 (m, 1H), 4.35–4.24 (m, 2H), 3.92–3.79 (m, 6H), 3.72–3.61 (m, 4H), 3.60–3.48 (m, 6H), 3.39–3.25 (m, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.71, 155.99, 150.35 (dd, J = 11.6, 249.5 Hz), 145.52, 144.65 (dd, J = 13.6, 246.2 Hz), 125.04 (d, J = 2.1 Hz), 123.84 (dd, J = 4.0, 7.8 Hz), 111.42 (d, J = 17.0 Hz), 107.47, 79.17, 72.56 (d, J = 6.0 Hz), 70.46, 70.34, 70.30 (2C), 66.22 (2C), 48.60 (2C), 40.35, 28.39 (3C).
2-(2-(2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)ethoxy)ethoxy)ethan-1-amine hydrochloride (74)
A solution of 4.0N HCl in dioxane (2.0 mL) was added to 73 (0.100 g, 0.189 mmol), and the resulting suspension was stirred at room temperature overnight. The solvent was evaporated to dryness, and the residue was tritured with DEE and collected by filtration to afford 74 (0.080 g, 91% yield) as a white solid. 1H NMR (400 MHz, MeOD) δ 7.49–7.40 (m, 1H), 7.28 (s, 1H), 7.04 (dd, J = 16.9, 9.0 Hz, 1H), 4.25 (s, 1H), 3.86–3.50 (m, 18H), 3.02 (s, 2H).
4-((2-(2-(2-(2,3-Difluoro-6-(2-morpholinothiazol-4-yl)phenoxy)ethoxy)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (32)
General Procedure A (6 h) was followed using 74 (0.044 g, 0.095 mmol) and 62(51) (0.024 g, 0.086 mmol), to afford the title compound as a yellow solid (0.06 g, 54% yield) following purification by flash column chromatography on SiO2 (DCM/acetone, 9:1). 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 7.89 (ddd, J = 8.7, 6.3, 2.1 Hz, 1H), 7.58 (s, 1H), 7.52–7.44 (m, 1H), 7.11 (d, J = 7.1 Hz, 1H), 6.99–6.88 (m, 2H), 6.51 (t, J = 5.3 Hz, 1H), 4.90 (dd, J = 12.1, 5.3 Hz, 1H), 4.40–4.24 (m, 2H), 3.98–3.81 (m, 6H), 3.79–3.62 (m, 6H), 3.59–3.38 (m, 6H), 2.96–2.65 (m, 3H), 2.19–2.03 (m, 1H). HRMS (ESI) m/z [M + H]+ calcd for C32H33F2N5O8S 686.20907, found 686.20855. UPLC retention time: 5.749 min.
Metabolic Stability in Cryopreserved Human Hepatocytes
Cryopreserved human hepatocytes (pooled suspension hepatocytes, Gibco) were thawed placing in a 37 °C shaking water bath according to the manufacturer’s specifications and resuspended in Williams E medium (WEM) to have 1 × 106 cells/mL. Samples with the test compound at 1 μM were incubated at 37 °C, and aliquots of 50 μL were collected at 0, 10, 30, 60, 120, and 240 min. The incubations were quenched 1:3 with ice-cold acetonitrile (containing 1 μM labetalol as the internal standard). Samples were then centrifuged at 12000 rpm for 5 min at 4 °C. The supernatant was concentrated by evaporation under a nitrogen stream and reconstituted with DMSO. Blank was prepared similarly but in the absence of the investigated compounds.
Metabolic Stability in CYP3A4 and Human Liver Cytosol
Metabolism of the selected compounds was evaluated upon incubation with human liver cytosol (Gibco) and supersome recombinant CYP isoforms 3A4 expressed in baculovirus-infected insect cells (Gibco). For metabolism in CYP3A4, test compounds (1 μM) were incubated in 0.1 M potassium phosphate buffer (pH 7.4) and allowed to equilibrate at 37 °C. Freshly prepared 25 mM nicotinamide adenine dinucleotide phosphate (NADPH, 1 mM final concentration) was added to the incubation mixture to start the reaction. For metabolism in human liver cytosol, reactions were initiated by adding test compounds (1 μM) to the mix reaction constituted by the human liver cytosol (1 mg/mL) prewarmed in potassium phosphate buffer (pH 7.4). Similarly, in the inhibition experiments of the AOX1, test compounds were incubated in the presence or absence of the selective inhibitor hydralazine (50 μM). In both procedures, aliquots (50 μL) of the incubation mix were removed at 0, 10, 20, 30, and 60 min (CYP3A4) or 0, 30, 60, and 90 min (human liver cytosol) and added to 50 μL of ice-cold acetonitrile quench solution (internal standard, 1 μM labetalol) to stop the reaction. Samples were then centrifuged at 12 000 rpm for 5 min at 4 °C. The supernatant was concentrated by evaporation under a nitrogen stream and reconstituted in DMSO. The blank was prepared similarly but in the absence of the investigated compounds. Samples were then analyzed by LC–MS/MS (see the Analytical procedure).
Stability of Compound 1 (dBet1) in Four Different Solutions during LC–MS Acquisitions
dBet1 (1) (10 mM in DMSO) was diluted 1:1000 in PBS/ACN (50:50, vol/vol) to afford a final concentration of 10 μM; 100 μL was drawn and dried under a gentle nitrogen stream at 40 °C for 30 min. After that evaporation was completed, the dry residue was reconstituted with 100 μL of DMSO. Additional three solutions of dBet1 (1) (10 μM) were prepared in PBS, PBS/ACN (50:50, vol/vol), and DMSO. The four solutions were stored into the autosampler of the LC apparatus at 37 °C for 12 h. Injections (2 μL) were programmed every 3 h. At each time point (0, 3, 6, and 12 h), the samples underwent analyses with a Thermo Q-exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA) as described in the dedicated section.
UHPLC-MS Analysis
A Thermo Q-exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA) was used. The LC system, governed by Chromeleon X-press software, consists of a Binary pump, a thermostated autosampler, and a column compartment, all Dionex Ultimate 3000 series modules (Thermo Fisher Scientific, Waltham, MA). A volume of 2 μL was injected for each sample. Chromatographic separation of analytes was conducted in reverse-phase chromatography. In brief, a Luna Omega 1.6 μm Polar (C18, 2.1 mm × 150 mm) was used, and the mobile phases consisted of water (A) and acetonitrile (B), both containing formic acid at 0.1%. The LC flow was set at 0.400 mL/min in a 12 min gradient elution as follows: 99.5:0.5 (A/B) to 5:95 (A/B) over 10 min, 5:95 (A/B) for 2 min, and then reversion back to 99.5:0.5 (A/B) over 2.5 min. The column was operating at a constant temperature of 40 °C. The LC effluents were introduced into the Q-Exactive mass spectrometer by an H-ESI source that operated in the positive mode with a sheath gas flow rate of 45; an auxiliary gas flow rate of 15; a spray voltage of 3.5 kV; capillary temperature and auxiliary gas heater temperature, respectively, of 320 and 350 °C; and S-lens RF level 50. The Q-Exactive mass spectrometer operates in the data-dependent scan (DDS) mode, with a resolution of 70.000 in full mass and 17.500 in MS/MS, in the scan mass range of 100–1500 at collision energies of 15, 60, and 120 V. The MS/MS data were processed using “MetaSite 5.1.8 Mass 3.3.5” and “WebMetabase release-4.0.4” (MolecularDiscovery, Ltd.).
Acknowledgments
The Università degli Studi di Perugia and MIUR are gratefully acknowledged for financial support to the project AMIS, through the program “Dipartimenti di Eccellenza-2018–2022”. We gratefully acknowledge Molecular Discovery Ltd. for Mass-MetaSite and WebMetabase software and Montelino Therapeutics Inc. for providing several PROTACs tested in this study. We thank Alessandra Di Veroli (University of Perugia) and Simone Moretti (Molecular Horizon, srl) for technical assistance in LC–MS analysis.
Glossary
Abbreviations Used
- ACN
acetonitrile
- ADME
absorption, distribution, metabolism, and excretion
- AR
androgen receptor
- BET
bromodomain and extraterminal
- Boc
tert-butoxycarbonyl
- BRD4
bromodomain-containing protein 4
- cIAP
cell inhibitor of apoptosis protein
- CK2
casein kinase 2
- CRBN
cereblon
- CYP3A4
human cytochrome P450 3A4
- Da
Dalton unified atomic mass unit
- DAD
diode-array detection
- DCM
dichloromethane
- DEE
diethyl ether
- DIAD
diisopropyl azodicarboxylate
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DNA
deoxyribonucleic acid
- EA
ethyl acetate
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide
- ESI
electrospray ionization
- Et3N
triethylamine
- h
hour
- hAOX
human aldehyde oxidase
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- H-ESI
heated electrospray ionization
- HRMS
high-resolution mass spectroscopy
- Hz
Hertz
- LC–MS
liquid chromatography–mass spectrometry
- LC–MS/MS
liquid chromatography with tandem mass spectrometry
- logP
negative logarithm of the partition coefficient
- MDM2
mouse double minute 2 homolog
- MeOH
methanol
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- MoCo
molybdenum pyranopterin cofactor
- MS/MS
tandem mass spectrometry
- NMM
N-methyl morpholine
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PE
petroleum ether
- PEG
poly(ethylene glycol)
- POI
protein of interest
- PPh3
triphenylphosphine
- PROTACs
proteolysis targeting chimeras
- Q-TOF LC–MS
quadrupole time-of-flight liquid chromatography–mass spectrometry
- RF
radio frequency
- rt
room temperature
- t1/2
half life
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- TMS
tetramethylsilane
- UPLC-MS
ultraperformance liquid chromatography-tandem mass spectrometry
- VHL
von Hippel-Lindau
- WEM
Williams E medium
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00793.
PROTACs’ chemical structures and half-life values (t1/2) upon incubation in cryopreserved human hepatocytes (Table S1); structure and half-life values (t1/2) of the constituent ligands upon incubation in cryopreserved human hepatocytes (Table S2); metabolism identification data upon incubation in cryopreserved human hepatocytes (Table S3); metabolic profile for compound 7 (MZ1) (Figure S1); kinetics of the metabolism of compounds 33 and 34 in human liver cytosol and their chromatograms, respectively (Figures S2 and S3) (PDF)
Molecular formula strings (CSV)
Author Contributions
L.G. and J.D. contributed equally to this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Z.; Tang W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B. 2018, 8, 721–732. 10.1016/j.apsb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant-Basak A.; Obach R. S. Emerging models of drug metabolism, transporters, and toxicity. Drug Metab. Dispos. 2018, 46, 1556–1561. 10.1124/dmd.118.084293. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Carosati E.; De Boeck B.; Ethirajulu K.; Mackie C.; Howe T.; Vianello R. MetaSite: understanding metabolism in human cytochromes from the perspective of the chemist. J. Med. Chem. 2005, 48, 6970–6979. 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Baroni M.; Benedetti P.; Goracci L.; Fortuna C. G. Exposition and reactivity optimization to predict sites of metabolism in chemicals. Drug Discovery Today: Technol. 2013, 10, e155–e165. 10.1016/j.ddtec.2012.11.001. [DOI] [PubMed] [Google Scholar]
- T’Jollyn H.; Boussery K.; Mortishire-Smith R. J.; Coe K.; De Boeck B.; Van Bocxlaer J. F.; Mannens G. Evaluation of three state-of-the-art metabolite prediction software packages (Meteor, MetaSite, and StarDrop) through independent and synergistic use. Drug Metab. Dispos. 2011, 39, 2066–2075. 10.1124/dmd.111.039982. [DOI] [PubMed] [Google Scholar]
- Peach M. L.; Zakharov A. V.; Liu R.; Pugliese A.; Tawa G.; Wallqvist A.; Nicklaus M. C. Computational tools and resources for metabolism-related property predictions. 1. Overview of publicly available (free and commercial) databases and software. Future Med. Chem. 2012, 4, 1907–1932. 10.4155/fmc.12.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora I.; Fontaine F.; Serra B.; Plasencia G. High-throughput, computer assisted, specific MetID. A revolution for drug discovery. Drug Discovery Today: Technol. 2013, 10, e199–e205. 10.1016/j.ddtec.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Paiva A. A.; Klakouski C.; Li S.; Johnson B. M.; Shu Y.-Z.; Josephs J.; Zvyaga T.; Zamora I.; Shou W. Z. Development, optimization and implementation of a centralized metabolic soft spot assay. Bioanalysis 2017, 9, 541–552. 10.4155/bio-2016-0299. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Lucas A. J.; Sproston J. L.; Barton P.; Riley R. J. Estimating human ADME properties, pharmacokinetic parameters and likely clinical dose in drug discovery. Expert Opin. Drug Dis. 2019, 14, 1313–1327. 10.1080/17460441.2019.1660642. [DOI] [PubMed] [Google Scholar]
- Alqahtani S. In silicoADME-Tox modeling: progress and prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. 10.1080/17425255.2017.1389897. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Milani N.; Benedetti P.; Lepri S.; Cesarini L.; Baroni M.; Spyrakis F.; Tortorella S.; Mosconi E.; Goracci L. From experiments to a fast easy-to-use computational methodology to predict human aldehyde oxidase selectivity and metabolic reactions. J. Med. Chem. 2018, 61, 360–371. 10.1021/acs.jmedchem.7b01552. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Crivori P.; Carrupt P. A.; Testa B. Molecular fields in quantitative structure–permeation relationships: the VolSurf approach. J. Mol. Struct.: THEOCHEM 2000, 503, 17–30. 10.1016/S0166-1280(99)00360-7. [DOI] [Google Scholar]
- Sjögren E.; Thörn H.; Tannergren C. In silico modeling of gastrointestinal drug absorption: predictive performance of three physiologically based absorption models. Mol. Pharmaceutics 2016, 13, 1763–1778. 10.1021/acs.molpharmaceut.5b00861. [DOI] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discovery Today 2015, 20, 122–128. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Paiva S. L.; Crews C. M. Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H.; Peng Y.; Zhao Q.; Chen Y. Small molecule PROTACs: an emerging technology for targeted therapy in drug discovery. RSC Adv. 2019, 9, 16967–16976. 10.1039/C9RA03423D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R.; Mohl D.; Deshaies R. J. Harnessing the power of proteolysis for targeted protein inactivation. Mol. Cell 2020, 77, 446–460. 10.1016/j.molcel.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Teng M.; Jiang J.; He Z.; Kwiatkowski N. P.; Donovan K. A.; Mills C. E.; Victor C.; Hatcher J. M.; Fischer E. S.; Sorger P. K.; Zhang T.; Gray N. S. Development of CDK2 and CDK5 dual degrader TMX-2172. Angew. Chem., Int. Ed. 2020, 59, 13865–13870. 10.1002/anie.202004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M.; Xiong Y.; Safaee N.; Nowak R. P.; Donovan K. A.; Yuan C. J.; Nabet B.; Gero T. W.; Feru F.; Li L.; Gondi S.; Ombelets L. J.; Quan C.; Jänne P. A.; Kostic M.; Scott D. A.; Westover K. D.; Fischer E. S.; Gray N. S. Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–31.e16. 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Brand M.; Jiang B.; Bauer S.; Donovan K. A.; Liang Y.; Wang E. S.; Nowak R. P.; Yuan J. C.; Zhang T.; Kwiatkowski N.; Müller A. C.; Fischer E. S.; Gray N. S.; Winter G. E. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppi V.; Hughes S. J.; Maniaci C.; Testa A.; Gmaschitz T.; Wieshofer C.; Koegl M.; Riching K. M.; Daniels D. L.; Spallarossa A.; Ciulli A. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa A.; Hughes S. J.; Lucas X.; Wright J. E.; Ciulli A. Structure-based design of a macrocyclic PROTAC. Angew. Chem., Int. Ed. 2020, 59, 1727–1734. 10.1002/anie.201914396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M.; Tomoshige S.; Demizu Y.; Naito M. Selective degradation of target proteins by chimeric small-molecular drugs, PROTACs and SNIPERs. Pharmaceuticals 2020, 13, 74. 10.3390/ph13040074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M.; Ohoka N.; Shibata N.; Tsukumo Y. Targeted protein degradation by chimeric small molecules, PROTACs and SNIPERs. Front. Chem. 2019, 7, 849. 10.3389/fchem.2019.00849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L.; Liu W.; Song Y.; Zhu H.; Duan Y. The present and future of novel protein degradation technology. Curr. Top. Med. Chem. 2019, 19, 1784–1788. 10.2174/1568026619666191011162955. [DOI] [PubMed] [Google Scholar]
- Takahashi D.; Moriyama J.; Nakamura T.; Miki E.; Takahashi E.; Sato A.; Akaike T.; Itto-Nakama K.; Arimoto H. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell 2019, 76, 797–810.e710. 10.1016/j.molcel.2019.09.009. [DOI] [PubMed] [Google Scholar]
- Tovell H.; Testa A.; Maniaci C.; Zhou H.; Prescott A. R.; Macartney T.; Ciulli A.; Alessi D. R. Rapid and reversible knockdown of endogenously tagged endosomal proteins via an optimized haloPROTAC degrader. ACS Chem. Biol. 2019, 14, 882–892. 10.1021/acschembio.8b01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Loh C.; Chen J.; Mainolfi N. Targeted protein degradation mechanisms. Drug Discovery Today: Technol. 2019, 31, 53–60. 10.1016/j.ddtec.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Sun X.; Rao Y. PROTACs as potential therapeutic agents for cancer drug resistance. Biochemistry 2020, 59, 240–249. 10.1021/acs.biochem.9b00848. [DOI] [PubMed] [Google Scholar]
- Konstantinidou M.; Li J.; Zhang B.; Wang Z.; Shaabani S.; Ter Brake F.; Essa K.; Dömling A. PROTACs– a game-changing technology. Expert Opin. Drug Dis. 2019, 14, 1255–1268. 10.1080/17460441.2019.1659242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt G. F.; Scott-Stevens P.; Gaohua L. Targeted protein degradation in vivo with proteolysis targeting chimeras: current status and future considerations. Drug Discovery Today: Technol. 2019, 31, 69–80. 10.1016/j.ddtec.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Liu J.; Ma J.; Liu Y.; Xia J.; Li Y.; Wang Z. P.; Wei W. PROTACs: A novel strategy for cancer therapy. Semin. Cancer Biol. 2020, 10.1016/j.semcancer.2020.02.006. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Hu J.; Xu F.; Chen Z.; Bai L.; Fernandez-Salas E.; Lin M.; Liu L.; Yang C. Y.; Zhao Y.; McEachern D.; Przybranowski S.; Wen B.; Sun D.; Wang S. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2018, 61, 462–481. 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Hughes S. J.; Testa A.; Chen W.; Lamont D. J.; Rocha S.; Alessi D. R.; Romeo R.; Ciulli A. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T.; Snyder L. B.; Willard R. R.; Vitale N.; Pizzano J.; Gordon D. A.; Bookbinder M.; Macaluso J.; Dong H.; Ferraro C.; Wang G.; Wang J.; Crews C. M.; Houston J.; Crew A. P.; Taylor I. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. 10.1200/JCO.2019.37.7_suppl.259. [DOI] [Google Scholar]
- Flanagan J. J.; Qian Y.; Gough S. M.; Andreoli M.; Bookbinder M.; Cadelina G.; Bradley J.; Rousseau E.; Willard R.; Pizzano J.; Crews C. M.; Crew A. P.; Taylor I.; Houston J.. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer Poster Session Abstracts 2019, P5-04-18.
- Maple H. J.; Clayden N.; Baron A.; Stacey C.; Felix R. Developing degraders: principles and perspectives on design and chemical space. MedChemComm 2019, 10, 1755–1764. 10.1039/C9MD00272C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Winkler J. D.; Crews C. M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Steinebach C.; Sosic I.; Lindner S.; Bricelj A.; Kohl F.; Ng Y. L. D.; Monschke M.; Wagner K. G.; Kronke J.; Gutschow M. A MedChem toolbox for cereblon-directed PROTACs. MedChemComm 2019, 10, 1037–1041. 10.1039/C9MD00185A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C.; Kehm H.; Lindner S.; Vu L. P.; Kopff S.; Lopez Marmol A.; Weiler C.; Wagner K. G.; Reichenzeller M.; Kronke J.; Gutschow M. PROTAC-mediated crosstalk between E3 ligases. Chem. Commun. 2019, 55, 1821–1824. 10.1039/C8CC09541H. [DOI] [PubMed] [Google Scholar]
- Steinebach C.; Ng Y. L. D.; Sosic I.; Lee C. S.; Chen S. R.; Lindner S.; Vu L. P.; Bricelj A.; Haschemi R.; Monschke M.; Steinwarz E.; Wagner K. G.; Bendas G.; Luo J.; Gutschow M.; Kronke J. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11, 3474–3486. 10.1039/D0SC00167H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A. Substrates of human hepatic cytochrome P450 3A4. Toxicology 1995, 104, 1–8. 10.1016/0300-483X(95)03155-9. [DOI] [PubMed] [Google Scholar]
- Hsu M.-H.; Johnson E. F. Active-site differences between substrate-free and ritonavir-bound cytochrome P450 (CYP) 3A5 reveal plasticity differences between CYP3A5 and CYP3A4. J. Biol. Chem. 2019, 294, 8015–8022. 10.1074/jbc.RA119.007928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutzler J. M.; Obach R. S.; Dalvie D.; Zientek M. A. Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin. Drug Metab. Toxicol. 2013, 9, 153–168. 10.1517/17425255.2013.738668. [DOI] [PubMed] [Google Scholar]
- Barr J. T.; Choughule K.; Jones J. P.. Enzyme kinetics, inhibition, and regioselectivity of aldehyde oxidase. In Enzyme Kinetics in Drug Metabolism; Methods in Molecular Biology; Humana Press, 2014; Vol. 1113, pp 167–186. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Liu H. H.; Weller P.; Zheng M.; Tao W.; Wang J.; Liao G.; Monshouwer M.; Peltz G. In silico and in vitro pharmacogenetics: aldehyde oxidase rapidly metabolizes a p38 kinase inhibitor. Pharmacogenomics J. 2011, 11, 15–24. 10.1038/tpj.2010.8. [DOI] [PubMed] [Google Scholar]
- Dittrich C.; Greim G.; Borner M.; Weigang-Köhler K.; Huisman H.; Amelsberg A.; Ehret A.; Wanders J.; Hanauske A.; Fumoleau P. Phase I and pharmacokinetic study of BIBX 1382 BS, an epidermal growth factor receptor (EGFR) inhibitor, given in a continuous daily oral administration. Eur. J. Cancer 2002, 38, 1072–1080. 10.1016/S0959-8049(02)00020-5. [DOI] [PubMed] [Google Scholar]
- Menear K. A.; Adcock C.; Boulter R.; Cockcroft X.-l.; Copsey L.; Cranston A.; Dillon K. J.; Drzewiecki J.; Garman S.; Gomez S.; Javaid H.; Kerrigan F.; Knights C.; Lau A.; Loh V. M.; Matthews I. T. W.; Moore S.; O’Connor M. J.; Smith G. C. M.; Martin N. M. B. 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable Inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- Bradner J.; Buckley D.; Winter G.. Methods to induce targeted protein degradation through bifunctional molecules. PCT. Int. Appl. WO2016105518, Jun 30, 2016.
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Ban F.; Dalal K.; Leblanc E.; Frewin K.; Ma D.; Adomat H.; Rennie P. S.; Cherkasov A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 2014, 57, 6458–6467. 10.1021/jm500802j. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre F.; Chua P. C.; O’Brien S. E.; Siddiqui-Jain A.; Bourbon P.; Haddach M.; Michaux J.; Nagasawa J.; Schwaebe M. K.; Stefan E.; Vialettes A.; Whitten J. P.; Chen T. K.; Darjania L.; Stansfield R.; Anderes K.; Bliesath J.; Drygin D.; Ho C.; Omori M.; Proffitt C.; Streiner N.; Trent K.; Rice W. G.; Ryckman D. M. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. 10.1021/jm101251q. [DOI] [PubMed] [Google Scholar]
- Chopra R.; Sadok A.; Collins I. A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discovery Today: Technol. 2019, 31, 5–13. 10.1016/j.ddtec.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Mayer A.; Buckley D. L.; Erb M. A.; Roderick J. E.; Vittori S.; Reyes J. M.; di Iulio J.; Souza A.; Ott C. J.; Roberts J. M.; Zeid R.; Scott T. G.; Paulk J.; Lachance K.; Olson C. M.; Dastjerdi S.; Bauer S.; Lin C. Y.; Gray N. S.; Kelliher M. A.; Churchman L. S.; Bradner J. E. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 2017, 67, 5–18.e19. 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranczewski P.; Stanczak A.; Sundberg K.; Svensson R.; Wallin A.; Jansson J.; Garberg P.; Postlind H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [PubMed] [Google Scholar]
- Jin X.; Pybus B. S.; Marcsisin S. R.; Logan T.; Luong T. L.; Sousa J.; Matlock N.; Collazo V.; Asher C.; Carroll D.; Olmeda R.; Walker L. A.; Kozar M. P.; Melendez V. An LC–MS based study of the metabolic profile of primaquine, an 8-aminoquinoline antiparasitic drug, with an in vitro primary human hepatocyte culture model. Eur. J. Drug Metab. Pharmacokinet. 2014, 39, 139–146. 10.1007/s13318-013-0139-8. [DOI] [PubMed] [Google Scholar]
- Jia L.; Liu X. The conduct of drug metabolism studies considered good practice (II): In vitro experiments. Curr. Drug Metab. 2007, 8, 822–829. 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strano-Rossi S.; Anzillotti L.; Dragoni S.; Pellegrino R. M.; Goracci L.; Pascali V. L.; Cruciani G. Metabolism of JWH-015, JWH-098, JWH-251, and JWH-307 in silico and in vitro: a pilot study for the detection of unknown synthetic cannabinoids metabolites. Anal. Bioanal. Chem. 2014, 406, 3621–3636. 10.1007/s00216-014-7793-9. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Chen H.; Petusky S. L. Development and application of an automated solution stability assay for drug discovery. J. Biomol. Screening 2006, 11, 40–47. 10.1177/1087057105281363. [DOI] [PubMed] [Google Scholar]
- Lepper E.; Smith N.; Cox M.; Scripture C.; Figg W. Thalidomide metabolism and hydrolysis: mechanisms and implications. Curr. Drug Metab. 2006, 7, 677–685. 10.2174/138920006778017777. [DOI] [PubMed] [Google Scholar]
- Bonn B.; Leandersson C.; Fontaine F.; Zamora I. Enhanced metabolite identification with MS(E) and a semi-automated software for structural elucidation. Rapid Commun. Mass Spectrom. 2010, 24, 3127–3138. 10.1002/rcm.4753. [DOI] [PubMed] [Google Scholar]
- Cece-Esencan E. N.; Fontaine F.; Plasencia G.; Teppner M.; Brink A.; Pahler A.; Zamora I. Software-aided cytochrome P450 reaction phenotyping and kinetic analysis in early drug discovery. Rapid Commun. Mass Spectrom. 2016, 30, 301–310. 10.1002/rcm.7429. [DOI] [PubMed] [Google Scholar]
- Radchenko T.; Kochansky C. J.; Cancilla M.; Wrona M. D.; Mortishire-Smith R. J.; Kirk J.; Murray G.; Fontaine F.; Zamora I. Metabolite identification using an ion mobility-enhanced data-independent acquisition strategy and automated data processing. Rapid Commun. Mass Spectrom. 2020, 34, e8792 10.1002/rcm.8792. [DOI] [PubMed] [Google Scholar]
- Radchenko T.; Fontaine F.; Morettoni L.; Zamora I. WebMetabase: cleavage sites analysis tool for natural and unnatural substrates from diverse data source. Bioinformatics 2019, 35, 650–655. 10.1093/bioinformatics/bty667. [DOI] [PubMed] [Google Scholar]
- Talele T. T. The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- Thorsell A.-G.; Ekblad T.; Karlberg T.; Löw M.; Pinto A. F.; Trésaugues L.; Moche M.; Cohen M. S.; Schüler H. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J. Med. Chem. 2017, 60, 1262–1271. 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Jiang X.; Feng F.; Liu W.; Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm. Sin. B. 2020, 10, 207–238. 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Wang C.; Qin C.; Xiang W.; Fernandez-Salas E.; Yang C.-Y.; Wang M.; Zhao L.; Xu T.; Chinnaswamy K.; Delproposto J.; Stuckey J.; Wang S. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- Wurz R. P.; Dellamaggiore K.; Dou H.; Javier N.; Lo M. C.; McCarter J. D.; Mohl D.; Sastri C.; Lipford J. R.; Cee V. J. A “click chemistry platform” for the rapid synthesis of bispecific molecules for inducing protein degradation. J. Med. Chem. 2018, 61, 453–461. 10.1021/acs.jmedchem.6b01781. [DOI] [PubMed] [Google Scholar]
- Turecek P. L.; Bossard M. J.; Schoetens F.; Ivens I. A. PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs. J. Pharm. Sci. 2016, 105, 460–475. 10.1016/j.xphs.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N.; Nagai K.; Morita Y.; Ujikawa O.; Ohoka N.; Hattori T.; Koyama R.; Sano O.; Imaeda Y.; Nara H.; Cho N.; Naito M. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J. Med. Chem. 2018, 61, 543–575. 10.1021/acs.jmedchem.7b00168. [DOI] [PubMed] [Google Scholar]
- Terao M.; Romão M. J.; Leimkühler S.; Bolis M.; Fratelli M.; Coelho C.; Santos-Silva T.; Garattini E. Structure and function of mammalian aldehyde oxidases. Arch. Toxicol. 2016, 90, 753–780. 10.1007/s00204-016-1683-1. [DOI] [PubMed] [Google Scholar]
- Lepri S.; Ceccarelli M.; Milani N.; Tortorella S.; Cucco A.; Valeri A.; Goracci L.; Brink A.; Cruciani G. Structure–metabolism relationships in human-AOX: Chemical insights from a large database of aza-aromatic and amide compounds. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E3178–E3187. 10.1073/pnas.1618881114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao M.; Garattini E.; Romao M. J.; Leimkuhler S. Evolution, expression, and substrate specificities of aldehyde oxidase enzymes in eukaryotes. J. Biol. Chem. 2020, 295, 5377–5389. 10.1074/jbc.REV119.007741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T.-D. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Sodhi J. K.; Wong S.; Kirkpatrick D. S.; Liu L.; Khojasteh S. C.; Hop C. E. C. A.; Barr J. T.; Jones J. P.; Halladay J. S. A novel reaction mediated by human aldehyde oxidase: Amide hydrolysis of GDC-0834. Drug Metab. Dispos. 2015, 43, 908–915. 10.1124/dmd.114.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gristwood W.; Wilson K. Kinetics of some benzothiazoles, benzoxazoles, and quinolines as substrates and inhibitors of rabbit liver aldehyde oxidase. Xenobiotica 1988, 18, 949–954. 10.3109/00498258809167518. [DOI] [PubMed] [Google Scholar]
- Dalvie D. K.; Kalgutkar A. S.; Khojasteh-Bakht S. C.; Obach R. S.; O’Donnell J. P. Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol. 2002, 15, 269–299. 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- Manevski N.; King L.; Pitt W. R.; Lecomte F.; Toselli F. Metabolism by aldehyde oxidase: drug design and complementary approaches to challenges in drug discovery. J. Med. Chem. 2019, 62, 10955–10994. 10.1021/acs.jmedchem.9b00875. [DOI] [PubMed] [Google Scholar]
- Arora V. K.; Philip T.; Huang S.; Shu Y.-Z. A novel ring oxidation of 4- or 5-substituted 2H-oxazole to corresponding 2-oxazolone catalyzed by cytosolic aldehyde oxidase. Drug Metab. Dispos. 2012, 40, 1668–1676. 10.1124/dmd.112.044545. [DOI] [PubMed] [Google Scholar]
- Strelevitz T. J.; Orozco C. C.; Obach R. S. Hydralazine as a selective probe inactivator of aldehyde oxidase in human hepatocytes: estimation of the contribution of aldehyde oxidase to metabolic clearance. Drug Metab. Dispos. 2012, 40, 1441–1448. 10.1124/dmd.112.045195. [DOI] [PubMed] [Google Scholar]
- Crews C. M.; Burslem G.; Cromm P. M.; Jaime-Figueroa S.; Toure M.. Preparation of imide-based compounds as modulators of proteolysis and methods of use. PCT. Int. Appl. WO2019148055, 2019.
- Mainolfi N.; Ji N.; Kluge A. F.; Weiss M. M.; Zhang Y.. Preparation of peptide conjugates as IRAK4 protein kinase degraders. US Patent, US20190192668, Jun 30, 2019.
- Ryckebusch A.; Deprez-Poulain R.; Debreu-Fontaine M.-A.; Vandaele R.; Mouray E.; Grellier P.; Sergheraert C. Synthesis and antimalarial evaluation of new 1,4-bis(3-aminopropyl)piperazine derivatives. Bioorg. Med. Chem. Lett. 2003, 13, 3783–3787. 10.1016/j.bmcl.2003.07.008. [DOI] [PubMed] [Google Scholar]
- Gavande N.; Kim H.-L.; Doddareddy M. R.; Johnston G. A. R.; Chebib M.; Hanrahan J. R. Design, synthesis, and pharmacological evaluation of fluorescent and biotinylated antagonists of ρ1GABAc receptors. ACS Med. Chem. Lett. 2013, 4, 402–407. 10.1021/ml300476v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.