

Abstract
Perfluorooctanoic acid (PFOA) was used as a fluoropolymer manufacturing aid at a fluoropolymer production facility in Parkersburg, WV from 1951 to 2013. The manufacturer introduced a replacement surfactant hexafluoropropylene oxide dimer acid (HFPO–DA) that has been in use at this site since 2013. Historical releases of PFOA and related epidemiological work in this area has been primarily focused on communities downstream. To provide an update on the ongoing impacts from this plant, 94 surface water samples and 13 soil samples were collected mainly upstream and downwind of this facility. PFOA was detected in every surface water sample with concentrations exceeding 1000 ng/L at 13 sample sites within an 8 km radius of the plant. HFPO–DA was also found to be widespread with the highest levels (>100 ng/L) found in surface water up to 6.4 km north of the plant. One sample site, 28 km north of the plant, had PFOA at 143 ng/L and HFPO–DA at 42 ng/L. Sites adjacent to landfills containing fluorochemical waste had PFOA concentrations ranging up to >1000 ng/L. These data indicate that downwind atmospheric transport of both compounds has occurred and that the boundaries of the impact zone have yet to be fully delineated.
Graphical Abstract
Introduction
PFAS (per- and polyfluorinated alkyl substances) are anthropogenic organic compounds with most or all carbon–hydrogen bonds replaced by carbon–fluorine bonds. The nature of the carbon–fluorine bond makes these compounds highly stable and unreactive and has led to their use in a wide array of commercial applications.(1) Due to their exceptional resistance to degradation and chemical reaction, PFAS were long assumed to be biologically inert and safe for human exposure.(2) However, animal studies have linked long-chain PFAS—a group that includes perfluorooctanoic acid (PFOA, or C8)—to endocrine disruption, liver toxicity, dislipidemia, immunotoxicity, reproductive effects, and tumorigenesis.(3−5) Epidemiological studies of human populations have found associations between chronic exposure to long-chain PFAS and adverse outcomes similar to those observed in animal studies.(6) In 2016, the U.S. EPA published a drinking water health advisory of 70 ng/L for the sum of PFOA and perfluorooctanesulfonic acid (PFOS) or for either chemical independently.(7) In response to these concerns, many manufacturers have replaced long-chain PFAS with short-chain and polyether PFAS. Many of these “replacement” PFAS are believed to be less bioaccumulative than the long-chain PFAS, but little is known about their potential for human toxicity.(8−10)
At the fluoropolymer production facility outside Parkersburg, WV, which is the focus of this study, PFOA was used as a surfactant in the manufacture of Teflon from 1951 to 2013 (62 years). PFOA was phased out in favor of a replacement PFAS, hexafluoropropylene oxide dimer acid (HFPO–DA, and its ammonium salt, known by the tradename “GenX”, CAS 13252–13-6, 62037–80-3).(11) In 2005, the C8 Science Panel was formed to study the effects of PFOA exposure on the human population near the facility. The Science Panel conducted a series of epidemiological investigations with a base cohort of 69 030 area residents with a population geometric mean blood serum PFOA level of 32.9 ng/mL.(12,13) Participation in these studies was limited to those living within the boundaries of six public water districts—Belpre, Little Hocking, Lubeck, Tuppers Plains, Mason County, and Pomeroy—known to be contaminated by PFOA. These districts are largely downstream from the facility, and the primary sources of contamination were thought to be direct deposition of airborne emissions onto wellfields and groundwater contamination via recharge from the Ohio River.(14) Due to the influential nature of the C8 Science Panel studies, much of the ongoing interest has focused on this area to the southwest of the facility. Less attention has been given to potential impacts near industrial dumps, landfills, or other regions downwind of the facility. The prevailing winds in this area are to the northeast, and PFOA air emissions were estimated to average 5900 kg/year from 1951 to 2003, which exceeds the estimates of average discharge into the Ohio River over the same span by approximately 450 kg/year.(2)
Under three Safe Drinking Water Act (SDWA) Orders and an amended Order, USEPA Regions 3 and 5 have been working with the facility and its contractors since 2001 to identify water supplies (public and private) which may be at risk of PFOA contamination. Ten municipal drinking water systems serving over 74 000 customers have had treatment system upgrades installed and surveillance monitoring put in place to maintain delivered PFOA concentrations of less than 70 ng/L. The facility and its contractors have also installed residential activated carbon treatment systems or have provided alternative drinking water sources to about 270 individual residences in this region. Work continues to identify and remediate public and private drinking water systems that may be using source water with PFOA at 70 ng/L or higher.(15)
The purpose of this paper is 2-fold. The first goal was to more fully characterize historical and ongoing PFAS contamination in surface water and soil that has primarily occurred via atmospheric deposition around this facility. The second goal was to look for evidence of the replacement surfactant, HFPO–DA, as to date very little information on the environmental occurrence of this compound has been made available.
Methods and Materials
Standards and Reagents.
The origins of standards used are listed in the Supplemental Information (SI).
Sample Collection.
A total of 102 water samples (94 surface and 8 drinking water) and 13 soil samples were collected without regard to weather in the area surrounding the fluoropolymer facility on June 6–9, July 12–13, and December 1–3 of 2016 and March 25, 2018. The results from each trip informed the plans for each subsequent trip. The first sampling trip focused on surface water samples along a 188 km stretch of the Ohio River, from 130 km downstream to 58 km upstream of the facility, and tributaries that pass near known PFAS containing landfills (Figure 1).(16) To explore the impacts of air emissions, during the second trip water was collected from lakes, rivers, and creeks to the north and northeast of the facility as far as 16 km downwind. The third trip included both water and soil samples and expanded the collection radius to more than 48 km downwind to the north and northeast of the facility. The fourth trip solely focused on soil samples collected to the north of the facility.
Figure 1.

Sampling locations for the first sampling trip, June 6–9, 2016. Samples taken from the Ohio River are denoted with an open triangle; samples from tributaries are marked with an open circle.
For each sampling trip that included water collection, precleaned 1 L high-density polyethylene (HDPE) bottles,(17) along with a trip blank and two trip spikes were shipped from the USEPA laboratory in Research Triangle Park, NC to The Ohio State University in Columbus, OH for use in the field. Water samples from the main channel of a body of water were collected using a Kemmerer stainless steel sampler or by manually submerging a sample bottle 15–30 cm below the surface of the water. For the third sampling trip only, 5 mL of 35% nitric acid was added to each sample immediately after collection as a safeguard against microbial growth, owing to concerns that the large number of samples taken on this trip could lead to longer storage times prior to analysis. All samples were stored at room temperature and returned to the EPA laboratory for analysis within 10 days of collection.
Composite soil samples were collected on December 1–3, 2016 and March 25, 2018 from public lands or easements following the methods of Strynar et al.(18) Approximately 500 g of soil was collected at each location and placed in a cooler to transport for analysis. The 2016 samples were stored at 4 °C until they were processed with the 2018 sample set.
Quality Assurance.
On each water sampling trip, a trip blank, low spike, and high spike were taken into the field. The trip blank consisted of 1 L of deionized (DI) water. Each of the trip spikes consisted of 1 L of DI water spiked with a known quantity of target analytes—perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluorobutanesulfonic acid (PFBS), perfluorohexanesulfonic acid (PFHxS), and perfluorooctanesulfonic acid (PFOS)—with concentrations of 25 ng/L of each analyte in the low spike and 100 ng/L in the high spike. HFPO–DA was only included in the trip spikes for the third sampling trip due to limited availability of a standard. Trip blanks and spikes were analyzed in the same manner and at the same time as samples.
Water Sample Preparation and Extraction.
Water samples were prepared in accordance with the methods of Nakayama et al.(17) Briefly, for each sample, 1 L was transferred from the sample bottle into a HDPE graduated cylinder. The bottle was rinsed with 10 mL of methanol to release any analytes bound to the container. The combined sample and rinsate was filtered using GF/F glass microfiber filter paper (Whatman Inc., Piscataway, NJ), and 25 μL of 1 ng/μL isotopically labeled internal standards (IS) was added. For the samples of the first and second trips, a labeled standard containing PFBA, PFOA, PFNA, PFDA, PFHxS, and PFOS was used. For the samples collected during the third trip, labeled PFOA, HFPO–DA, and PFOS were used. Five hundred milliliter aliquots of each sample were concentrated onto Oasis WAX (weak anion-exchange) cartridges (Waters Corp., Milford, MA) using a positive displacement pump (Sep-Pak Concentrator, Waters Corp.). Cartridges were eluted with 4 mL of basic methanol (0.3% NH4OH), and the eluate was concentrated to 1 mL under dry nitrogen on a TurboVap LV evaporator (Caliper Life Sciences, Hopkinton, MA). A 100 μL aliquot of the concentrated eluate was combined with 300 μL of 2.5 mM ammonium acetate buffer and added to an LC vial for analysis.
Soil Sample Preparation and Extraction.
Soil samples were prepared and extracted in accordance with Strynar et al.(18) except soil was sieved through 2 mm stainless steel mesh for approximately 1 min with manual agitation. To report results in terms of ng/g dry weight, 3–5 g of subsample of each soil sample was dried overnight in a drying oven at 105 °C; the difference of the subsample mass before and after drying was used to calculate the percent dry solids of each sample. Two of the 13 soil samples were selected for replicate analysis.
Approximately 1 g of each sample was placed in a 15 mL polypropylene Falcon tube with 5 mLof methanol containing 10 ng each of isotopically labeled PFHxA, PFOA, and HFPO–DA as internal standards. The samples were then sonicated in a water bath for 30 min and centrifuged for 5 min at 16 800g. Using a vacuum manifold, Supelco Supelclean ENVI-Carb 3 mL cartridges were preconditioned with 6 mL of methanol and the entire 5 mL aliquot of supernatant was passed through the cartridge and collected in a fresh 15 mL Falcon tube. This extract was then concentrated to 0.5 mL with a Zymark TurboVap LV evaporator. A 100 μL aliquot was combined with ammonium acetate buffer in an LC vial for analysis. Procedural blanks (blank extractions including all reagents and disposable materials) and solvent blanks were included in this analysis to evaluate process control.
Mass Spectrometry.
Analysis for the targeted PFAS was performed with a Waters Acquity ultraperformance liquid chromatograph in tandem with a Waters Quattro Premier XE triple-quadrupole mass spectrometer (UPLC-MS/MS; Waters Corp.) following the methods outlined in Nakayama et al.(17) and Strynar et al.(18)
Quantitation.
Matrix-matched calibration curves were prepared by adding varying amounts of targeted PFAS to 1 L of DI water along with 25 μL of 1 ng/μL IS. For the first water sampling trip, a five-point curve with 10, 25, 50, 75, and 100 ng/L of each targeted PFAS was used to provide a quantifiable range of 10–100 ng/L. For the second trip, due to a lack of availability of IS, quantitation was based on peak area counts alone. The third trip used an eight-point calibration curve for a quantifiable range of 10–1000 ng/L. Soil analysis used a seven-point external solvent-based calibration curve of targeted PFAS and IS for a quantifiable range of 1–100 ng/g dry weight. For all samples, the detection limit was defined as a notable deflection in baseline signal (3× signal-to-noise) while the limit of quantitation (LOQ) was determined to be the concentration of the first calibration point that back-predicted within ±30% of its nominal value.
Results
Quality Assurance.
For water samples, the field blanks (n = 3) showed no measurable levels of targeted PFAS. The percent recovery for the targeted PFAS in trip spikes (low spike, 25 ng/L, n = 3; high spike, 100 ng/L, n = 3) is included in Table S1. Recovery for PFOA ranged from 67% to 129% and from 78% to 115% for HFPO–DA, while other targeted PFAS generally ranged between 70% and 130%. Larger variability in the results from other compounds is partially attributed to the low concentrations of analytes in the trip spikes relative to the higher calibration curves (10–500 and 10–1000 ng/L) needed for the second and third sampling trips.
Comparison of duplicate samples (n = 8) is given in Table S2. Duplicate sampling from the first and third sampling trips and soil samples provided reasonable validation of the methods. For the second sampling trip, the results for three samples, including two duplicates, were discarded due to fouling of the UPLC-MS/MS autosampler injector indicated by a diminished IS response. The remaining duplicate sample pair for this sampling trip showed higher variability in both PFOA and HFPO–DA concentrations, most likely due to the lack of IS in this sample set. For soils, procedural blanks and solvent blanks were routinely below limits of quantitation.
Surface Water.
Trip #1: Ohio River.
Given the historically high levels of PFOA released into the Ohio River directly from the fluoropolymer facility, the first portion of this field survey focused primarily on samples collected from the mainstem of the Ohio River (Figure 1). Resulting Ohio River samples (Table S3) had consistently low concentrations of all PFAS, with the highest sample showing 15.0 ng/L PFOA. Five samples collected upstream of the facility contained no measurable PFOA or HFPO–DA. Downstream of the facility, PFOA was not measured above the LOQ (10 ng/L) until 93–130 km downstream where samples collected near Pomeroy, Larkin, and Crab Creek were found in the range 12–15 ng/L.
In contrast, all of the samples collected from streams downstream of closed and capped landfills used solely by the fluoropolymer facility(19) had PFOA ranging from 51 to 1250 ng/L. Brinker Run, which drains from the Letart Landfill, had PFOA at 103 ng/L; Lee Creek, which receives drainage from the Dry Run Landfill, had an average PFOA concentration of 58.1 ng/L; Pine Run, which is also in the Dry Run basin, had a concentration of 189 ng/L; and Vaughts Run, which drains the Local Landfill, had PFOA at 1250 ng/L. Only Vaughts Run had quantifiable levels of HFPO–DA at 71.2 ng/L.
Other samples collected from watersheds not known to contain PFAS landfills also showed elevated levels of the target compounds. The Little Kanawha River slightly upstream and southeast of Parkersburg in West Virginia had PFOA at 22.6 ng/L. In Ohio both the East and the West Forks of the Little Hocking River (460 km2 drainage area)(20) had elevated PFOA concentrations of 463 and 209 ng/L, respectively, near their confluence. HFPO–DA was also present in the East Fork at 59.4 ng/L.
Trip #2: Little Hocking watershed.
The second round of sampling explored the potential of airborne deposition affecting surface waters based on samples collected as far as 16 km to the northeast of the plant (Figure S1). All surface water samples, from both stationary and moving waters, were found to have PFOA with concentrations ranging from 319 to 2570 ng/L. Most samples (85%) had HFPO–DA detections with concentrations ranging as high as 227 ng/L (Table S4). The highest levels of PFOA and HFPO–DA were found in the direction of prevailing winds, directly across the Ohio River to the north and upstream to the northeast of the plant on the East Fork of the Little Hocking River where PFOA ranged between 902 and 2500 ng/L and HFPO–DA between 37 and 227 ng/L. The most distant sample point along this vector, Browns Run, at 16 km from the plant, had PFOA at 781 ng/L and HFPO–DA at 30.4 ng/L.
A small cluster of sites in Vienna, WV, and north across the Ohio River (Browns Run, Briscoe Run, and Pond Run) had PFHxS (64.5–79.0 ng/L) and PFOS (22.1–71.9 ng/L) in addition to the regionally ubiquitous PFOA (496–781 ng/L, Table S4). The presence of the two sulfonates (particularly PFHxS) in this specific area is consistent with contamination linked to past use of aqueous film-forming foam (AFFF) fire-fighting materials.(21,22) However, we are unaware of any fire-fighting activities in this area using AFFF.
Trip #3: Little Hocking and beyond.
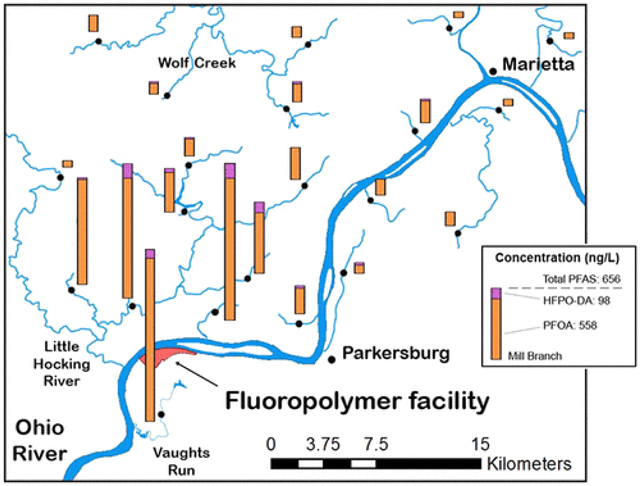
In an additional attempt to find the outer edge of contamination in the direction of the prevailing winds, we expanded the radius for the third sampling trip (Figure 2, Table S5). The highest concentrations were found nearest to the plant and downwind, directly across the Ohio River (as was also the case for Trip 2), with PFOA at 1310 ng/L and HFPO–DA at 137 ng/L. The most distant sample, Wingett Run 48 km northeast in the Little Muskingum River watershed in Ohio, had no PFOA or HFPO–DA above the LOQ. The most distant points where PFOA was found were Fifteen Mile Creek (13.7 ng/L) 41 km northeast in the Little Muskingum River watershed and Cat Creek (33.7 ng/L), 34 km distant along a slightly more north northeastern vector in the Muskingum River watershed (north of Lowell, OH). The most distant point where HFPO–DA was found was the South Branch of Wolf Creek, 24 km due north of the facility and just south of Beverly, OH, where PFOA was 143 ng/L and HFPO–DA was measured at 42.1 ng/L. Those sampling sites visited during trip 2 containing measurable levels PFHxS and PFOS also had measurable levels during trip 3.
Figure 2.

Sampling locations and PFAS concentrations in surface waters from the third sampling trip, December 1–3, 2016: (a) PFOA and (b) HFPO–DA.
In general, PFAS concentrations from this phase of the study were lower than the second round of sampling, as noted in a comparison of sample sites that were visited during both trips. One possible explanation for the lower concentrations in this round of testing was higher amounts of precipitation before this sampling event and correspondingly higher levels of flow in the tributaries at this time. There are no gauging stations for the Little Hocking River, but USGS streamflow data for the Muskingum River at Beverly, 32 km north of the fluoropolymer facility, showed flow during the later trip (mean 84 700 L/s) was nearly double that of the earlier trip (mean 43,300 L/s).(23) Thus, the samples from this later trip were likely diluted. A second possibility is the temporal separation between the two sampling dates (July and December). As Koc, the organic carbon partition coefficient, is dependent on temperature, the lower ambient temperatures of the December sampling trip may have slowed the horizontal migration of PFOA into surface water.(24,25) Lack of the mass-labeled internal standards for quantitation in the second sampling campaign could also have contributed to potential systematic bias between these two sample sets.
Drinking Water.
Eight samples of drinking water, including one duplicate, were taken from various sources over the course of the three sampling trips (Table S6). All PFAS concentrations were below the LOQ except the two private well samples taken in Devola and Lowell, which showed 28.0 and 23.3 ng/L PFOA, respectively. Each location is more than 27 km northeast of the fluorochemical facility.
Soil.
PFOA was quantifiable in all soil samples collected in 2016 and 2018 (Tables 1 and S7) except for duplicate samples collected on Veto Road (across the Ohio River from Parkersburg) in 2016 which both showed PFOA slightly below the LOQ of 1 ng/g. The most distant sample site, Archers Fork 2 (2016) at 48 km northeast of the facility, showed PFOA at 4.96 ng/g with no indication of HFPO–DA. A soil sample collected 45 km due north of the facility in Reinersville, OH (2018), also had PFOA at 7.35 ng/g but no detectable HFPO–DA. The highest levels of PFOA and HFPO–DA were found in 2016 samples from Drag Strip Road (26.9 and 3.20 ng/g, respectively) and Veto Lake (18.4 and 8.14 ng/g, respectively) located 4 and 8.5 km, respectively, to the northeast of the facility. Additionally, a soil sample collected adjacent to the Little Hocking Water Association (LHWA) well field in 2018 had PFOA at 6.68 ng/g and HFPO–DAat 3.09 ng/g. This is in sharp contrast to samples collected on the LHWA site in 2004 (but not analyzed until 2018), which showed much higher levels of PFOA (range 59.1–66.2 ng/g) and no detectable HFPO–DA.
Table 1.
Measured PFOA and HFPO–DA Concentrations in Soil (ng/g dry weight)a
| year | description | distance (km) | PFOA (ng/g) | HFPO–DA (ng/g) |
|---|---|---|---|---|
| 2004 | Little Hocking A | 57.3 | c | |
| Little Hocking B | 66.2 | c | ||
| Little Hocking C | 59.1 | c | ||
| 2016 | Drag Strip Road | 4.0 | 26.9 | 3.20 |
| Veto Lake | 8.0 | 18.4 | 8.14 | |
| Veto Road | 13.0 | b | c | |
| Veto Road (dup.) | 13.0 | b | c | |
| Strouds Run | 15.3 | 5.70 | b | |
| Lookout Park | 24.0 | 2.52 | c | |
| Archers Fork 1 | 35.3 | 6.77 | c | |
| Archers Fork 2 | 48.1 | 4.96 | c | |
| 2018 | LHWA | 1.3 | 6.68 | 3.09 |
| Veto Lake | 8.0 | 9.30 | 1.00 | |
| Veto Lake (dup.) | 8.0 | 10.3 | 1.20 | |
| Watertown | 24.3 | 2.86 | c | |
| Beverly | 32.1 | 11.0 | c | |
| L. Olive Green Creek | 39.9 | 4.88 | c | |
| Reinersville | 45.4 | 7.35 | c |
Distance is measured from fluoropolymer production facility.
Detected but below the LOQ of 1 ng/g.
Not detected.
Discussion
Atmospheric deposition.
Water samples from the mainstem of the Ohio River collected in 2016 showed low or background levels of PFAS contamination above and below the outfall of the facility. The samples from Vaughts Run and the Little Hocking River (Figure 1) gave the first indication in this study that atmospheric transport of HFPO–DA was an important contributor to surface water contamination. Vaughts Run is likely at least partially influenced by materials contained in the Local Landfill, but HFPO–DA was measured at 71.2 ng/L in this stream. Considering that HFPO–DA was first used almost three decades after the landfill closed,(19) it is unlikely to have originated from wastes buried in the landfill but instead is more likely to be originating from atmospheric deposition from the nearby plant. Moreover, the East Fork of the Little Hocking River contained elevated levels of PFAS (including HFPO–DA and other PFAS), but it is not downstream from the facility and not influenced by any known PFAS-containing landfills. Both sampling locations (Vaughts Run and the East Fork of the Little Hocking) are within 3 km of the facility. Figure S1 indicates prevailing winds at the facility are to the northeast, toward the Little Hocking watershed and in the opposite direction of the flow of the Ohio River at that point. These observations in close proximity to the site support previous findings focusing on PFOA within this region.(2,13,26,27) Those studies demonstrated that atmospheric deposition was a primary source of PFOA contamination within this relatively confined area downwind of the facility.
On the basis of this preliminary evidence of atmospheric impacts from the first sampling trip, particularly in the Little Hocking River, the second and third trips explored the range of potential impacts by collecting surface water samples at sites along the prevailing wind directions. The resulting samples support the atmospheric deposition hypothesis in two ways. First, PFAS occurred in separate bodies of water (lakes and streams) in different watersheds. While the East and West Fork of the Little Hocking River and their tributaries were the most heavily impacted, Browns Run and Wolf Creek in the adjacent Muskingum River watershed also showed relatively high levels of both PFOA and HFPO–DA. Additionally, by aggregating the data from the second and third sampling trips, we found a significant exponential decrease in concentration with distance from the facility in the general direction of the prevailing wind for both PFOA (Figure 3a, p < 0.001) and HFPO–DA (Figure 3b, p < 0.001). This exponential decay is consistent with Chen et al.,(28) who demonstrated a significant decline in air, soil, and surface water concentrations downwind from two fluoropolymer manufacturing facilities in Fuxin, China.
Figure 3.

Surface water PFAS concentrations from the second and third trips with distance from the facility. Linear fit for log10(concentration) vs distance was generated using the Microsoft Excel Analysis ToolPak. Surface water samples from the second (red squares) and third trips (blue diamonds) are aggregated in each graph: (a) PFOA and (b) HFPO–DA.
On the basis of our observations, we found the impact zone to be at least 41 km for surface water and 48 km for soil downwind from this facility. To further evaluate the plausibility of atmospheric transport with subsequent deposition, we conducted our own complementary atmospheric transport modeling using CALPUFF View (Lakes Environmental; Waterloo, Ontario, Canada).(29) In this absence of modern data, we used the stack parameters (including stack height, stack diameter, exit velocity, temperature, and emission rate of PFOA) from Paustenbach et al.,(2) in these simulations, along with meteorological data (from the Weather Research and Forecasting model, provided by Lakes Environmental) for a typical year. The key output from CALPUFF View in our analysis is the spatial distribution of total (wet + dry) atmospheric deposition rates. For the historical emissions of PFOA, our estimated deposition rates ranged from roughly 10–7 to 10–4 μg/m2/s. These deposition rates are consistent with those in Paustenbach et al.,(2) specifically Zones 1–15 in their Table 9 (from 3 × 10–6 to 6 × 10–4 μg/m2/s) and slightly greater than Fang et al.(30) (3 × 10–8 μg/m2/s). We note that the latter work focused solely on dry deposition over the Bohai and Yellow Seas, and the specific proximity to sources is unknown.
Because we lacked empirical observations of deposition rates, we evaluated our model using the soil samples collected during both 2016 and 2018 (Figure S2 in SI). There is generally a log–linear relationship between soil PFOA concentration and the predicted PFOA deposition rate with good correlation (R2 = 0.8). The soil sample from the LHWA is suspected as an outlier (soil concentration was 6.68 ng/g and the predicted total deposition rate was 7 × 10–4 μg/m2/s). If we include this value in our regression, the p value for the slope is 0.14, while the p value is <0.001 without this observation. Therefore, our comparison to the soil samples corroborates the predicted spatial patterns.
Next, we sought to gain insight on HFPO–DA atmospheric transport and deposition. Brandsma et al.(31) detected HFPO–DA in plant material 3 km downwind from a fluoropolymer manufacturing plant in The Netherlands, indicating that HFPO–DA undergoes near-field deposition. A limitation in our model is the uncertainty related to HFPO–DA. In particular, specific model inputs for HFPO–DA are uncertain. For example, emission rates were unknown for our HFPO–DA simulations, so we assumed the stack emissions of HFPO–DA were the same as the historical PFOA emissions. The emission rate is simply a scalar value that will affect the deposition rate; therefore, while the magnitude of the deposition rate may vary, the underlying spatial distribution will remain unchanged with a different emission rate. Moreover, there is considerable uncertainty in parameters affecting HFPO–DA’s wet and dry deposition (e.g., Henry’s law coefficient for predicting scavenging into wet precipitation). Sensitivity analyses using these parameters demonstrate that the overall spatial pattern is consistent when values of these are varied (e.g., see Figures 5–12 in Moreno).(29)
Therefore, both our field observations and our computer simulations indicate that atmospheric transport and subsequent deposition of stack emissions from this fluoropolymer production facility along the Ohio/West Virginia border result in both PFOA and HFPO–DA contamination regionally. Our work does expand the boundaries to >40 km downwind when considering the extent of drinking water source contamination within southeast Ohio; prior efforts have largely focused on the area within 20 km from the source. Furthermore, the extent to which current atmospheric emissions of HFPO–DA may be impacting the region is uncertain; we are exploring this using atmospheric air samples in ongoing work.
Legacy PFOA contamination.
The continued occurrence of PFOA contamination in surface water samples as far as 41 km away to the northeast, 10 years after air emissions of PFOA were reduced from 13 600 kg/year in 2000 to 140 kg/year in 2006(19) and 3 years after air emissions of PFOA were ceased entirely at the facility(2,11) suggests a wide-ranging long-term presence in the communities of this region. Given the relatively sparse data set collected during this effort, from areas primarily to the north and northeast of the plant, it is evident that the range of the PFOA-impacted land mass in this region was not completely delineated by this study.
The appearance of HFPO–DA.
To our knowledge, this is the first published study to reveal the presence of the replacement surfactant, HFPO–DA, in surface waters and soil surrounding this facility and the first published indication that HFPO–DA has been released as an airborne contaminant in this area. Although the concentrations of HFPO–DA were lower than those of PFOA, HFPO–DA had only been emitted for 3–5 years, while PFOA was likely emitted for 62 years. The history of high levels of PFOA air emissions, with an estimated peak of 15 000 kg/year in 1999,(2) suggests HFPO–DA will also continue to accumulate in water and soils via atmospheric deposition.
HFPO–DA did not appear in any of the finished public water samples examined. This result is consistent with a recent facility report to USEPA which evaluated the occurrence of HFPO–DA in raw and treated water from 10 private residences (5 in Ohio and 5 in West Virginia) and 4 public water supplies near their facility.(32) HFPO–DA was found in 100% of the raw water from private residences in Ohio (n = 5, range 16–52 ng/L) and in the source water for two of the four public water systems tested (range < 10–81 ng/L). Finished water from all sites reported HFPO–DA below the limit of detection (<10 ng/L), with each location using a previously deployed carbon-based filtration system originally installed to remove PFOA. While encouraging, recent studies have shown shorter chain replacement PFAS like HFPO–DA may be more difficult to remove than PFOA in water treatment processes.(33,34) Thus, breakthrough for HFPO–DA may occur before PFOA, arguing for more frequent monitoring of HFPO–DA concentrations at public water systems that have detected HFPO–DA in source wells.
While this study provides some indication of how far airborne releases of HFPO–DA may travel, a better understanding of the physicochemical properties of HFPO–DA, especially the organic carbon partition coefficient Koc, would help with an evaluation of its persistence and mobility in soils. By means of comparison, PFOA’s value of Koc is widely variable in published laboratory studies with an average log Koc of 2.8.(35) A 2011 study by Shin et al.(16) modeling PFOA transport in the region surrounding the facility determined an optimized log Koc of 0.4, well below the range of values in other publications.(36) Even using this comparatively low value, which would indicate a higher rate of mobility than previously reported, Shin et al.(16) estimated it would take PFOA 11 years to migrate through the vadose zone before it would reach the source wells of the Little Hocking Water Association. HFPO–DA was detected in Little Hocking source wells in 2018,(32) approximately 5 years after its introduction at the fluoropolymer production facility, suggesting that it may have a faster rate of migration than PFOA, although how much faster remains uncertain. If HFPO–DA takes years to migrate into source well and our research supports a pattern of widespread atmospheric deposition of HFPO–DA similar to that of PFOA, this suggests the eventual arrival of HFPO–DA into groundwater throughout the region with potential for human and ecological exposures.
Landfill Influence.
Samples were taken from tributaries draining regions of three known clay-lined PFAS landfills used by the facility: Local Landfill (1964-mid 1980s), Dry Run Landfill (1988–2006), and Letart Landfill (1968–1995).(19,37) All samples (Figure 1, Table S3) showed elevated levels of PFOA, even though all of these PFAS landfills have been closed and capped to minimize infiltration of precipitation into the waste mass. Vaughts Run, which drains land surrounding the Local Landfill, is likely partially influenced by atmospheric deposition because of its proximity to the facility. Other landfill-passing tributaries are more distant from the facility and not in the direction of prevailing winds, suggesting that landfill leachate is a possible source of PFOA contamination at these sites. Our results support the need for more research near these known PFAS landfills to clarify the reasons for these elevated PFAS levels. USEPA evaluated these three waste sites (Letart, Dry Run, and Local landfills) and areas surrounding them in 2001 and 2002, and PFOA contamination was determined to be localized (within a 1.6 km radius). All impacted drinking water supplies have since been addressed.(15)
These findings also have implications for other landfills known to have accepted PFAS waste from the facility. In 2009 the facility operator submitted information to USEPA Region 3 identifying 11 sites in four states that accepted PFAS waste, beyond those mentioned in this study.(37) Considering that the closed-and-capped landfills investigated in this study appear to be a continuing source of PFAS contamination, further research is needed to investigate these other sites for their potential impact on local water quality and/or human health. USEPA Region 3 investigated the 4 sites named in this report in West Virginia and Virginia and determined that no drinking water supplies were impacted by PFOA contamination.(15)
Soil.
As shown in Table 1, in both the 2016 and the 2018 soil samples, PFOA concentrations above the LOQ of 1 ng/g dry weight were found in soils at all sampling sites save one and HFPO–DA above the LOQ at five distinct sites. Detected PFOA concentrations ranged from 5 to 27 ng/g in 2016–18 samples. Although the small number of soil samples (2016, n = 7; 2018, n = 6) precludes strong conclusions concerning the state of soil in the area, these results are consistent with a hypothesis of widespread atmospheric deposition of PFOA and a similarly emerging pattern for HFPO–DA.
Limited analysis of temporal PFOA soil concentrations was conducted. Comparison between three samples taken from the Little Hocking well field in 2004 (range 57.3–66.2 ng/g) and a sample taken nearby in 2018 (6.68 ng/g) shows an approximately 10-fold lower level of PFOA in the 2018 soil, implying that PFOA soil contamination is decreasing over time. Davis et al.(26) measured PFOA at 110 and 170 ng/g in two soil samples from the Little Hocking well field in August 2002. These results are also consistent with a general decrease in PFOA concentration over time at this location.
While the concentration may be decreasing with time, these soils continue to contain much higher levels of PFOA than concentrations measured globally. Rankin et al.(38) measured a geometric mean concentration of 1.8 ng/g of PFOA in 33 North American soils, including samples taken from known PFAS hot spots near emission sources. The higher soil concentrations in our study indicate that these soils remain highly contaminated in PFAS. Due to the small sample size, more research is needed to reach meaningful conclusions related to the rate of decrease of PFAS in this region.
Implications for the Region.
USEPA Regions 3 and 5 have overseen work by the facility under authority from a SDWA Order to identify contaminated water sources and remediate affected drinking water systems (public and private) since 2006. This work has proceeded systematically, with the most recent series of tests showing PFOA contamination in groundwater at levels above the EPA’s health advisory (70 ng/L) as far north as the Muskingum River near the towns of Lowell and Beverly, approximately 30 km to the north.(15) It is notable that samples from this current effort show PFOA contamination extending well beyond the limits of this SDWA Order testing with elevated soil PFOA found in Reinersville (7.35 ng/g, 45.4 km to the north) and Archers Fork 2, (4.96 ng/g, 48.1 km to the northeast). The extent to which this soil contamination influences groundwater in these areas remains to be determined. This all leads to questions as to whether all potentially exposed individuals have been tested for elevated levels of serum PFAS and whether all potentially exposed households and public water utilities have had their drinking water evaluated. The USEPA will continue to have the fluorochemical facility test further from the plant until data indicate the outer boundary of the contamination in groundwater from air release has been determined. These findings may also have implications for agriculture, as consumption of crops grown in contaminated soil has been found to correlate with higher serum PFOA levels.(39)
Supplementary Material
Acknowledgements.
The Ohio State University (OSU) authors were primarily responsible for planning, data collection, and data analysis for this study. The OSU received no direct funding from USEPA for this project and declare no potential conflicts of interest. USEPA provided technical assistance and in-kind support, including access to sampling equipment and analytical instrumentation. This article was reviewed in accordance with the policy of the Office of Research and Development, U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents necessarily reflect the view and policies of the Agency nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- 1.Lindstrom AB; Strynar MJ; Libelo EL Polyfluorinated Compounds: Past, Present, and Future. Environ. Sci. Technol. 2011, 45 (19), 7954–7961, DOI: 10.1021/es2011622 [DOI] [PubMed] [Google Scholar]
- 2.Paustenbach DJ; Panko JM; Scott PK; Unice KM A Methodology for Estimating Human Exposure to Perfluorooctanoic Acid (PFOA): A Retrospective Exposure Assessment of a Community (1951–2003). J. Toxicol. Environ. Health, Part A 2006, 70 (1), 28–57, DOI: 10.1080/15287390600748815 [DOI] [PubMed] [Google Scholar]
- 3.Post GB; Cohn PD; Cooper KR Perfluorooctanoic Acid (PFOA), an Emerging Drinking Water Contaminant: A Critical Review of Recent Literature. Environ. Res. 2012, 116, 93–117, DOI: 10.1016/j.envres.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 4.Lau C; Anitole K; Hodes C; Lai D; Pfahles-Hutchens A; Seed J. Perfluoroalkyl Acids: A Review of Monitoring and Toxicological Findings. Toxicol. Sci. 2007, 99 (2), 366–394, DOI: 10.1093/toxsci/kfm128 [DOI] [PubMed] [Google Scholar]
- 5.Grandjean P; Andersen EW; Budtz-Jørgensen E; Nielsen F; Mølbak K; Weihe P; Heilmann C. Serum Vaccine Antibody Concentrations in Children Exposed to Perfluorinated Compounds. JAMA 2012, 307 (4), 391–397, DOI: 10.1001/jama.2011.2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum A; Balan SA; Scheringer M; Trier X; Goldenman G; Cousins IT; Diamond M; Fletcher T; Higgins C; Lindeman AE; Peaslee G; de Voogt P; Wang Z; Weber R. The Madrid Statement on Poly- and Perfluoroalkyl Substances (PFASs). Environ. Health Perspect. 2015, 123 (5), A107, DOI: 10.1289/ehp.1509934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drinking Water Health Advisories for PFOA and PFOS; U.S. EPA, 2016. [DOI] [PubMed] [Google Scholar]
- 8.Grandjean P; Clapp R. Perfluorinated Alkyl Substances: Emerging Insights Into Health Risks. New Solut. 2015, 25 (2), 147–163, DOI: 10.1177/1048291115590506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheringer M; Trier X; Cousins IT; de Voogt P; Fletcher T; Wang Z; Webster TF Helsingør Statement on Poly-and Perfluorinated Alkyl Substances (PFASs). Chemosphere 2014, 114, 337–339, DOI: 10.1016/j.chemosphere.2014.05.044 [DOI] [PubMed] [Google Scholar]
- 10.Wang Z; DeWitt JC; Higgins CP; Cousins IT A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs)?. Environ. Sci. Technol. 2017, 51 (5), 2508–2518, DOI: 10.1021/acs.est.6b04806 [DOI] [PubMed] [Google Scholar]
- 11.DuPont. Form 10-K; U.S. Securities and Exchange Commission, 2014. [Google Scholar]
- 12.Frisbee SJ; Brooks AP Jr, Maher A; Flensborg P; Arnold S; Fletcher T; Steenland K; Shankar A; Knox SS; Pollard C; Halverson JA; Vieira VM; Jin C; Leyden KM; Ducatman AM The C8 Health Project: Design, Methods, and Participants. Environ. Health Perspect. 2009, 117 (12), 1873–1882, DOI: 10.1289/ehp.0800379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin H-M; Ryan PB; Vieira VM; Bartell SM Modeling the Air-Soil Transport Pathway of Perfluorooctanoic Acid in the Mid-Ohio Valley Using Linked Air Dispersion and Vadose Zone Models. Atmos. Environ. 2012, 51, 67–74, DOI: 10.1016/j.atmosenv.2012.01.049 [DOI] [Google Scholar]
- 14.Shin H-M; Vieira VM; Ryan PB; Steenland K; Bartell SM Retrospective Exposure Estimation and Predicted versus Observed Serum Perfluorooctanoic Acid Concentrations for Participants in the C8 Health Project. Environ. Health Perspect. 2011, 119 (12), 1760–1765, DOI: 10.1289/ehp.1103729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogers R. USEPA Region 3, Personal Communication. [Google Scholar]
- 16.Shin H-M; Vieira VM; Ryan PB; Detwiler R; Sanders B; Steenland K; Bartell SM Environmental Fate and Transport Modeling for Perfluorooctanoic Acid Emitted from the Washington Works Facility in West Virginia. Environ. Sci. Technol. 2011, 45 (4), 1435–1442, DOI: 10.1021/es102769t [DOI] [PubMed] [Google Scholar]
- 17.Nakayama SF; Strynar MJ; Reiner JL; Delinsky AD; Lindstrom AB Determination of Perfluorinated Compounds in the Upper Mississippi River Basin. Environ. Sci. Technol. 2010, 44 (11), 4103–4109, DOI: 10.1021/es100382z [DOI] [PubMed] [Google Scholar]
- 18.Strynar MJ; Lindstrom AB; Nakayama SF; Egeghy PP; Helfant LJ Pilot Scale Application of a Method for the Analysis of Perfluorinated Compounds in Surface Soils. Chemosphere 2012, 86, 252–257, DOI: 10.1016/j.chemosphere.2011.09.036 [DOI] [PubMed] [Google Scholar]
- 19.Small MJ Final Report of the Peer Consultation Panel Conducting the Review for the Scientific Peer Consultation Process for a Site Environmental Assessment Program as Part of the DuPont-EPA Memorandum of Understanding and Phase II Workplan (EPA-HQ-OPPT-2004–0113-0485); Carnegie Mellon University: Pittsburgh, PA, 2009. [Google Scholar]
- 20.Gazetteer of Ohio Streams, 2nd ed.; Ohio Department of Natural Resources, 2001. [Google Scholar]
- 21.Hu XC; Andrews DQ; Lindstrom AB; Bruton TA; Schaider LA; Grandjean P; Lohmann R; Carignan CC; Blum A; Balan SA; Higgins CP; Sunderland EM Detection of Poly- and Perfluoroalkyl Substances (PFASs) in U.S. Drinking Water Linked to Industrial Sites, Military Fire Training Areas, and Wastewater Treatment Plants. Environ. Sci. Technol. Lett. 2016, 3 (10), 344–350, DOI: 10.1021/acs.estlett.6b00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houtz EF; Higgins CP; Field JA; Sedlak DL Persistence of Perfluoroalkyl Acid Precursors in AFFF-Impacted Groundwater and Soil. Environ. Sci. Technol. 2013, 47 (15), 8187–8195, DOI: 10.1021/es4018877 [DOI] [PubMed] [Google Scholar]
- 23.USGS Current Conditions for USGS 03150500 Muskingum River at Beverly OH; U.S. Geological Survey (USGS), https://waterdata.usgs.gov/oh/nwis/uv?site_no=03150500 (accessed Jul 1, 2018). [Google Scholar]
- 24.Schwarzenbach RP; Gschwend PM; Imboden DM Environmental Organic Chemistry, 2nd ed.; John Wiley & Sons: New York, 2003. [Google Scholar]
- 25.Higgins CP; Luthy RG Modeling Sorption of Anionic Surfactants onto Sediment Materials: An a Priori Approach for Perfluoroalkyl Surfactants and Linear Alkylbenzene Sulfonates. Environ. Sci. Technol. 2007, 41 (9), 3254–3261, DOI: 10.1021/es062449j [DOI] [PubMed] [Google Scholar]
- 26.Davis KL; Aucoin MD; Larsen BS; Kaiser MA; Hartten AS Transport of Ammonium Perfluorooctanoate in Environmental Media near a Fluoropolymer Manufacturing Facility. Chemosphere 2007, 67 (10), 2011–2019, DOI: 10.1016/j.chemosphere.2006.11.049 [DOI] [PubMed] [Google Scholar]
- 27.Barton CA; Zarzecki CJ; Russell MH A Site-Specific Screening Comparison of Modeled and Monitored Air Dispersion and Deposition for Perfluorooctanoate. J. Air Waste Manage. Assoc. 2010, 60 (4), 402–411, DOI: 10.3155/1047-3289.60.4.402 [DOI] [PubMed] [Google Scholar]
- 28.Chen H; Yao Y; Zhao Z; Wang Y; Wang Q; Ren C; Wang B; Sun H; Alder AC; Kannan K. Multimedia Distribution and Transfer of Per- and Polyfluoroalkyl Substances (PFASs) Surrounding Two Fluorochemical Manufacturing Facilities in Fuxin, China. Environ. Sci. Technol. 2018, 52 (15), 8263–8271, DOI: 10.1021/acs.est.8b00544 [DOI] [PubMed] [Google Scholar]
- 29.Moreno AVP Modeling Atmospheric Transport of Perfluorinated Alkyl Substances from Chemours Facilities Using CALPUFF View. Masters Thesis, 2019. http://rave.ohiolink.edu/etdc/view?acc_num=osu1555004834088189. [Google Scholar]
- 30.Fang X; Wang Q; Zhao Z; Tang J; Tian C; Yao Y; Yu J; Sun H. Distribution and dry deposition of alternative and legacy perfluoroalkyl and polyfluoroalkyl substances in the air above the Bohai and Yellow Seas, China. Atmos. Environ. 2018, 192, 128–135, DOI: 10.1016/j.atmosenv.2018.08.052 [DOI] [Google Scholar]
- 31.Brandsma SH; Koekkoek JC; van Velzen MJM; de Boer J. A substitute GenX detected in the environment near a fluoropolymer manufacturing plant in the Netherlands. Chemosphere 2019, 220, 493–500, DOI: 10.1016/j.chemosphere.2018.12.135 [DOI] [PubMed] [Google Scholar]
- 32.Chemours Company Well Sampling Results; U.S. EPA, https://www.epa.gov/pfas/chemours-company-well-sampling-results. [Google Scholar]
- 33.Sun M; Arevalo E; Strynar M; Lindstrom A; Richardson M; Kearns B; Pickett A; Smith C; Knappe DRU Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environ. Sci. Technol. Lett. 2016, 3 (12), 415–419, DOI: 10.1021/acs.estlett.6b00398 [DOI] [Google Scholar]
- 34.Gebbink WA; van Asseldonk L; van Leeuwen SPJ Presence of Emerging Per- and Polyfluoroalkyl Substances (PFASs) in River and Drinking Water near a Fluorochemical Production Plant in the Netherlands. Environ. Sci. Technol. 2017, 51, 11057, DOI: 10.1021/acs.est.7b02488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zareitalabad P; Siemens J; Hamer M; Amelung W. Perfluorooctanoic Acid (PFOA) and Perfluorooctanesulfonic Acid (PFOS) in Surface Waters, Sediments, Soils and Wastewater – A Review on Concentrations and Distribution Coefficients. Chemosphere 2013, 91 (6), 725–732, DOI: 10.1016/j.chemosphere.2013.02.024 [DOI] [PubMed] [Google Scholar]
- 36.Prevedouros K; Cousins IT; Buck RC; Korzeniowski SH Sources, Fate and Transport of Perfluorocarboxylates. Environ. Sci. Technol. 2006, 40 (1), 32–44, DOI: 10.1021/es0512475 [DOI] [PubMed] [Google Scholar]
- 37.Information Request; C-8 Waste Disposal Sites, Washington Works Facility (EPA-HQ-OPPT-2003–0012-1426); DuPont: Washington, WV, 2009. [Google Scholar]
- 38.Rankin K; Mabury SA; Jenkins TM; Washington JW A North American and Global Survey of Perfluoroalkyl Substances in Surface Soils: Distribution Patterns and Mode of Occurrence. Chemosphere 2016, 161, 333–341, DOI: 10.1016/j.chemosphere.2016.06.109 [DOI] [PubMed] [Google Scholar]
- 39.Emmett EA; Shofer FS; Zhang H; Freeman D; Desai C; Shaw LM Community Exposure to Perfluorooctanoate: Relationships Between Serum Concentrations and Exposure Sources. J. Occup. Environ. Med. 2006, 48 (8), 759–770, DOI: 10.1097/01.jom.0000232486.07658.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.