

Abstract
Macrophages comprise a majority of the resident immune cells in adipose tissue (AT) and regulate both tissue homeostasis in the lean state and metabolic dysregulation in obesity. Since the AT environment rapidly changes based upon systemic energy status, AT macrophages (ATMs) must rapidly adapt phenotypically and metabolically. There is a distinct dichotomy in the polarization and bioenergetics of in vitro models, with M2 macrophages utilizing oxidative phosphorylation (OX PHOS) and M1 macrophages utilizing glycolysis. Early studies suggested differential polarization of ATMs, with M2-like macrophages predominant in lean AT and M1-like macrophages in obese AT. However, recent studies show that the phenotypic plasticity of ATMs is far more complicated, which is also reflected in their bioenergetics. Multiple ATM populations exist near the middle of the M2 to M1 continuum and appear to utilize both glycolysis and OX PHOS in obesity. The significance of the dual fuel bioenergetics is unclear and may be related to an intermediate polarization, their buffering capacity, or the result of a mixed population of distinct polarized ATMs. Recent evidence also suggests that lean ATMs serve as a substrate buffer or reservoir to protect the AT from lipid, catecholamine, or iron overload. Furthermore, recent models of weight loss and weight cycling reveal additional roles for ATMs in systemic metabolism. Evaluating ATM phenotype and intracellular metabolism together may more accurately illuminate the consequences of ATM accumulation in obese AT, lending further insight into obesity related comorbidities in humans.
Keywords: immunometabolism, bioenergetics, polarization, phenotype, plasticity
Introduction
Obesity afflicts millions of people worldwide. According to the World Health Organization, the prevalence of obesity has almost tripled since 1975 with approximately 650 million people diagnosed with obesity in 2016. The prevalence of obesity is projected to increase to 1.12 billion people in 2030 when accounting for secular trends (1). Obesity carries a significant risk for comorbidities including, but not limited to, cardiometabolic diseases such as atherosclerosis, hypertension, myocardial infarction, stroke, insulin resistance (IR), Type 2 Diabetes Mellitus (T2DM), dyslipidemia, and non-alcoholic fatty liver disease. Obesity also imparts a profound economic burden due to healthcare costs. Weight loss has been associated with improvements in the aforementioned obesity-associated metabolic complications (2, 3). However, for many, these weight loss efforts are not maintained, resulting in a cycle of weight gain and loss termed weight cycling. In several recent studies, weight cycling was shown to increase the risk of cardiometabolic diseases even beyond obesity itself, reviewed in [(4)]. The inflammatory nature of obesity is thought to contribute to many of its comorbidities and much recent work has focused on immune changes in weight loss and weight cycling. Specifically, obesity is associated with chronic adipose tissue (AT) inflammation.
Over 15 years have passed since the first reports of increased macrophage infiltration into AT in obese compared to lean mice and humans (5, 6). Since that time, almost every known immune cell type has been found in AT. Cumulatively, we now know that in addition to macrophages, T cells (CD4, CD8, Tregs), B cells, natural killer T cells, dendritic cells, neutrophils, eosinophils, and mast cells also reside in AT. Overall, during the transition from the lean to the obese state, the AT milieu becomes more inflammatory. Changes in these immune populations have been thoroughly discussed in many outstanding reviews (7–9) and will not be detailed at length here. Taken together, however, the knowledge that cells of the innate and adaptive immune system co-exist in AT is important and raises many questions regarding the immunogenicity of obesity. Furthermore, most of the immune cell populations change in their numbers and inflammatory status within AT during weight gain, weight loss, and weight cycling. In obesity, the inflammatory response in AT contributes to – if not drives – IR and dysregulated adipocyte lipolysis, which promote ectopic lipid storage and T2DM. The intracellular fuel utilization of immune cells, as well as, the interaction of immune cells within metabolic tissues are termed intrinsic- and extrinsic immunometabolism, respectively. Both are important for understanding mechanisms by which immune cells contribute to immune-mediated diseases. We recently reviewed the interactions between intrinsic and extrinsic immunometabolism (10); however, there is still much to uncover regarding the phenotype, function, and metabolism of adipose tissue macrophages (ATMs) in obesity. While extrinsic immunometabolism has been studied by many groups, including ours, much less is known about intrinsic immunometabolism of ATMs. Targeting the associated metabolic pathways may help improve our understanding of and treatments for obesity, CVD, and T2DM. In the current review article, recent advances in our understanding of ATM phenotypes and immunometabolism are emphasized.
Adipose Tissue Macrophage Polarization and Populations
In pivotal reports published in 2003, Xu et al. and Weisberg et al. (5, 6) showed that macrophages reside in AT, increase in obesity in proportion to body weight, and coincide with systemic IR in both mice and humans (6). Subsequently, many studies in mice and humans have characterized mechanisms by which immune cell populations contribute to inflammation and dysregulation of metabolism in AT. Macrophages are the predominant cell type to accumulate in AT in obesity, and their role in inflammation has been strongly linked to both obesity and IR (11). Early work focused on the chemokine-dependent recruitment of macrophages to AT. In mouse models null for specific chemokines, the results were mixed and have been reviewed in detail by others (12, 13) and us (14, 15). Additional work has shown that resident yolk sac derived ATMs can self-renew and proliferate (16), while hematopoietic derived ATMs can infiltrate and proliferate in situ (17, 18) and can even egress from the tissue (19, 20). Other features of ATMs have been discovered over the years and we will focus the current article on those that may directly affect, or be impacted by, intrinsic immunometabolism.
Polarization
The first report of ATM polarization in obese versus lean mice was published by Lumeng et al. (11). They showed that ATMs from lean mice expressed higher levels of genes such as Ym1, Arg1, and Il10, traditionally attributed to M2-like or “alternatively activated” macrophages. Conversely, ATMs from obese mice expressed higher levels of genes such as TNFa, Nos2 and Ccr2, which are associated with M1-like or “classically activated” macrophages. Two of the key enzymes that distinguish M1-like from M2-like macrophages are iNOS and arginase, respectively. In other contexts, iNOS utilizes arginine for the generation of nitric oxide (NO) and reactive oxygen species (ROS) for bacteriostatic purposes; however, in obese AT, NO contributes to mitochondrial dysfunction and ROS production by macrophages contributes to AT IR and inflammation in obese mice (21, 22). In M2 macrophages, the arginine used by arginase is ultimately converted for use in collagen biosynthesis during tissue remodeling. Interestingly, obese ATM have been reported to express Nos2, not Arg1, despite their role in extracellular matrix remodeling with obesity (11, 23). Others’ early reports, dependent on real-time (RT) PCR for gene expression and flow cytometry revealed a more nuanced and mixed phenotype (24, 25). For example, Shoelson and colleagues found that ATMs from HFD-fed mice had increased expression of Tnfα, Il1β, Cxcl10 and Il6, but downregulation of Nos2 and Il12α – all considered to be markers of M1-like cells (24). Conversely, traditional M2 markers Il10 and Tgfβ were upregulated in the obese AT. Obin and colleagues reported that all ATM subsets they evaluated displayed an enhanced M2 profile in mice fed HFD for 12 compared to 8 weeks (25). Taken together, these data suggest a mixed phenotype of macrophages in obese AT.
As technologies have advanced, so has our ability to discover additional nuances in ATM phenotypes. Becker and colleagues (26) used proteomics of membrane proteins to identify the ATMs from obese mice as being unique from other classically activated macrophages in vivo, and from in vitro polarized M1-like or M2-like macrophages. They named these macrophages “MMe”, for metabolically activated, and showed that exposure of BMDMs to a metabolic cocktail (palmitic acid, glucose, and insulin) creates an in vitro phenotype that most closely mimics ATMs from HFD mice. MMe’s were defined by expression of lipid-related genes, Plin2 and Abca1, as well as typical M1-inflammatory cytokines like Tnfα, Il1β, and Il6. In a follow up paper this group found that MMe macrophages promote not only inflammation, but dead adipocyte clearance through lysosomal exocytosis driven by NADPH oxidase 2 (NOX2) (27). A recent paper by Stienstra’s group supported this mixed phenotype with obese ATMs expressing elevated inflammatory genes like Cd11c, TNF, NLRP3, and IL-6 as well as anti-inflammatory genes like Il1ra, Il10, Akt1, Stat6, Socs1, and Tgfb1 (28). Interestingly, the inflammatory genes Ifny, Nos2, Myd88, and Il1β were downregulated in obese compared with lean ATMs. The differences between ATMs and in vitro polarized macrophages has been further supported by Zhou and colleagues who studied visceral ATMs from chow and HFD-fed mice and directly compared their transcriptomes (29). They developed a high resolution data analysis platform, named “MacSpectrum”, which accounts for a polarization index and an activation-induced differentiation index. Their results demonstrated that ATMs in lean and obese mice cluster separately from in vitro polarized M2 and M1 BMDMs. In addition, they revealed that ATMs and circulating monocytes have a unique signature that correlates with BMI and homeostasis model assessment of insulin resistance in humans. Thus, the field at large is accepting that simple M1/M2 nomenclature does not accurately describe ATMs, although the simplified categorization can make phenotypic and metabolic descriptions easier to describe and categorize.
Moreover, additional data is surfacing that human ATM polarization is also more variable than the M1 or M2 paradigm. Human ATMs have phenotypic plasticity or a mixed activation state with the differential expression of both M1 and M2 markers [reviewed in (30)]. In lean humans, a presumably anti-inflammatory state, markers for both M1 and M2 were identified comprising approximately 60% of interstitial macrophages with a shift in obese humans to more of an M2 phenotype in fibrotic areas (31). Moreover, an M2 marker in mice, CD206, is found on ATMs of both lean and obese humans. Higher expression of CD206 correlated with another M2 marker CD163 and M1 markers CD11c, CD80, CD86 and HLA-DR (32). The CD11c+CD206+ ATMs described by Wentworth et al. are another example of macrophages harboring both markers, existing on the continuum of M1/M2 (33). It is interesting to note differential expression of other markers on human CD11c+ and CD206+ ATMs, such as the CD45 leukocyte marker, CD1c lipid antigen presenting cell marker, and CD86 general antigen presenting cell marker that likely assist with handling excess lipid (33).
Macrophage lipid production and utilization is also linked to their polarization. For example, M1 macrophages synthesize lipids and produce inflammatory lipid mediators like eicosanoids while M2 macrophages tend to take up and oxidize lipids (34). This relationship appears in the context of obesity as well. Chawla and colleagues demonstrated that the transcription factor peroxisome proliferator activated receptor (PPAR) gamma is required for alternative activation of ATMs, and that mice deficient in macrophage PPARγ have accelerated development of obesity and IR (35). In similar studies by Lee and colleagues, macrophages lacking PPARδ were not able to increase lipid handling and the mice developed IR and hepatic steatosis (36). Further evidence of the importance of lipid handling in obese ATMs was provided by Becker and colleagues in their description of MMe ATMs (26). This phenotype was characterized as having an upregulation of lipid metabolism typically observed in M2-like macrophages including the cholesterol export protein, ABC-A1, and the lipid droplet protein, perilipin 2. In particular, palmitic acid recreated the obesogenic phenotype, indicating that the lipid-rich milieu of AT shapes the identity of the macrophages. In addition, PPARγ and p62 were shown to promote a lipid-metabolism phenotype in MMe macrophages (26). Thus, M2-like ATMs in lean AT require lipid-related transcriptional regulation for their protective function, while at the same time, the lipid-rich environment of obese AT drives a pro-inflammatory MMe phenotype, indicating the importance of lipid balance for ATM phenotype and function.
Newly identified populations
Single cell data has propelled the field forward by verifying, further characterizing, and identifying novel ATMs and other immune cell populations (Table 1). Using flow cytometry coupled with single cell RNA sequencing (scRNA-seq) and followed by imaging, Lazar and colleagues identified CD11b+Ly6c−CD9− ATMs to be interstitially spaced, while CD11b+Ly6c−CD9+ ATMs were within crown-like structures and increased with obesity – this was true in both mouse and human AT (37). The CD9+ ATMs expressed genes related to lipid metabolism (including Lpl, Plin2, Cd63), lysosomal pathways (Acp5, Ctss, Lamp2, Lipa), pro-inflammatory mediators (Il1α, Il18, Tnfα, Ccl2), and intracellular vesicle formation. In contrast, the CD9− ATMs had tissue-homeostatic gene programs, such as vascular development and organization. This group also performed Assay for Transposase-Accessible Chromatin (ATAC-seq) to assess chromatin regulation and demonstrated Ly6c+ and CD9+ ATMs displayed distinct profiles: CD9+ cells were enriched for binding at transcription sites for inflammatory mediators while Ly6C+ cells were enriched for tissue regulatory gene expression pathways, data that confirmed their scRNA-seq results. CD9+ ATMs were also found in human visceral AT, and as in mice, had higher lipid content and localized to CLSs.
Table 1.
Populations of adipose tissue macrophages identified by single cell RNA sequencing
| Citation | Mouse or Human | Populations | Function/ Role | Obesity Phenotype | Associated genes/ Other info |
|---|---|---|---|---|---|
| 37 | Both | Cd11b+Ly6c+ | monocyte derived cells | Increased | |
| CD11b+Ly6C−F480+ CD64+ CD9− | interstitially spaced, vascular development/organization | No difference | |||
| CD11b+Ly6C−F480+ CD64+ CD9+ | surrounding CLS, lipid metabolism | Increased | -Lpl, Plin2, CD63, ACp5, Ctss, Lamp2, Lipa, IL1ra, IL18, Tnf, Ccl2 | ||
| 39 | Mouse | Lyve lo MHCII hi | nerve associated macs | Unknown | |
| Lyve hi MHC II lo | vascular associated macs | Unknown | |||
| 38 | Mouse | “Mon1” | Decreased | -Retnla, Fn1 | |
| “Mon2” | Decreased | -Plac8, Clec4e | |||
| “Mac1” | Decreased | -Retnla, Lyve1, CD209f, Cd163 -associated with perivascular macrophages from Chakarov 2019 [ref 39] | |||
| “Mac 2” | surrounding CLS | Increased | -Nche1, Cd9 | ||
| “Mac 3” | surrounding CLS, lipid metabolism/ phagocytosis (LAMs) | Increased | -Nche1, Cd9, Spp1, Trem2, Lipa, Lpl, Ctsb, Ctsl, Fabp5, Fabp5, Lgals1/3, Cd36 -associated with CD9+ from Hill 2018 [ref 37] -KO Trem2 increased weight, body fat, glucose intolerance, insulin, cholesterol, LDL |
This past year, Amit and colleagues published another comprehensive scRNA-seq analysis of CD45+ immune cells from lean and obese mice (38). This group performed a time course analysis of AT from mice placed on HFD for 6, 12, or 18 weeks; each time point was compared to age-matched chow-fed controls. They detected the most extensive reorganization of immune cells in mice on diet for 12 weeks. As expected, the largest changes were an expansion of ATMs and a reduction in regulatory T cells and type 2 innate lymphoid cells. Importantly, ATMs from leptin deficient obese mice, fed a chow diet, also showed these changes. They identified 2 monocyte and 3 macrophage clusters increased in obese mice. One cluster had a perivascular phenotype similar to a recent report (39), and another had elevated CD9 as was shown by Lazar’s group (37). A CD9+CD63+ subset they called Lipid Associated Macrophages (LAM) was characterized by an expression pattern reflecting lipid metabolism and phagocytosis. LAMs were also found in AT from obese but not lean humans. This scRNA-seq technique allowed them to identify the gene Trem2 to be uniquely identified in this LAM population. They showed Trem2 was required to generate LAMs during obesity and was essential for AT remodeling, as knockout mice displayed adipocyte hypertrophy, accelerated AT accumulation, and dramatically impaired systemic glucose tolerance.
Relationship between polarization and bioenergetics
In vitro polarized macrophage energetics
The concept that “fuel feeds function”, was first proposed by Craig Thompson (40) for T cell responses, and we now know that intracellular bioenergetic pathways are intricately linked to differentiation and effector function for all immune cells. Indeed, in vitro, M1-like and M2-like macrophages display very different metabolic profiles, whereby M1-like macrophages utilize glycolysis for rapid ATP generation while M2-like macrophages use oxidative phosphorylation (OX PHOS) (41). These differences were first shown by Boscá and colleagues who utilized tracer-based metabolomics to show that classically activated macrophages are glycolytic (42). We also know that macrophages, like other immune cells, utilize many metabolic pathways and fuel sources for purposes beyond ATP generation, summarized nicely by Viola et al. (43). In addition to ATP generation from glycolysis, M1-like macrophages rely on the oxidative pentose phosphate pathway (PPP) for both ATP and NADPH generation, contributing to ROS generation as well as glutathione reduction. Additionally, breaks in the TCA cycle allow for accumulation of itaconate and succinate, thereby facilitating microbial killing and HIF1α stabilization, respectively. Citrate/ acetyl-coA facilitate the acetylation of histones and proteins that regulate M1 function. Finally, lipid signaling mediators are produced by fatty acid synthesis, and additional ROS is generated by the inhibition of the electron transport chain (ETC). In M2 macrophages, the TCA cycle is intact and respiration is coupled efficiently in the ETC, supporting enhanced OX PHOS and diminished ROS production. Additionally, glutamine derived α-ketoglutarate drives epigenetic reprogramming on the promoters of M2-specific marker genes (44), favors prolyl hydroxylase activity that inhibits HIF1α expression, and provides a substrate for UDP-GlcNAc synthesis, a process essential for the glycosylation of many M2 associated proteins (45).
Interestingly, while M1 macrophages require glucose utilization for cytokine production, ROS formation, and bacterial killing (46), M2 macrophages appear fairly flexible in their fuel utilization. Several studies have shown that M2 cells breakdown glucose and that glycolytic blockade with 2-deoxyglucose (2-DG) may inhibit M2 polarization and function (47, 48); however, it appears that M2 differentiation does not require glycolysis, as long as OX PHOS remains intact (46). Moreover, fatty acid oxidation has long been considered the primary source of energy for M2 cells. In 2014, Pearce and colleagues showed that CD36-mediated uptake of fatty acids and their subsequent lipolysis in lysosomes via lysosomal acid lipase was required for the elevated OX PHOS in IL-4 differentiated M2 macrophages (49). Further, pharmacological inhibition of carnitine palmitoyltransferase (CPT), the transporter for fatty acids into the mitochondria, impairs M2 polarization (50, 51). However, subsequent studies challenge the importance of CPT utilization, as high doses of etomoxir, a CPT inhibitor, suppress M2 polarization by reducing coenzyme A, and genetic ablation of CPT2 does not prevent M2 differentiation by IL-4 (52). Interestingly, the same study showed that direct OX PHOS blockade did not impair IL-4-associated genes or cell surface markers, suggesting our understanding of M2 metabolic requirements is incomplete.
Metabolic profiles of ATMs
While utilization of glycolysis versus OX PHOS in in vitro polarized or transfected cells appears relatively straight forward, much less is known about in vivo ATM metabolism. Obese ATMs appear to have a unique metabolic phenotype, partially M1 and M2 like, similar to their polarization as previously discussed. Leitinger’s group was one of the first to assess the bioenergetic profile of ATMs from lean and obese mice (53). Linking their work to oxidized phospholipids and even iron handling (via heme oxygenase), they show that Cd11b+ “Mox” macrophages (54) predominate in lean AT and have low glycolytic capacity and low OX PHOS. In contrast, Cd11b+CD11c+CD206+ mixed-phenotype ATMs from obese mice are characterized by elevated glycolysis and OX PHOS. Their studies uniquely show that treatment of macrophages with certain truncated oxidized phospholipids results in suppressed bioenergetics, phenocopying ATMs from lean mice, while treatment with full-length oxidized phospholipids results in enhanced bioenergetics, phenocopying ATMs from obese mice. More recent work by Stienstra and colleagues utilized transcriptome and metabolic flux analysis of ATMs from lean and obese mice, as well as ex vivo co-culture of BMDMs with AT explants to determine the metabolic signature of macrophages from various conditions (28, 54). Similar to Leitinger’s work, they showed F480+ ATMs from obese mice or from co-culture with obese AT explants had increases in both glycolysis and OX PHOS. In fact, expression of genes in these fuel utilization pathways were sufficient for them to distinguish between macrophages from lean and obese mice. Interestingly, these changes were only noted in macrophages from AT, and not those from the peritoneal cavity. We have included example data from our laboratory of both the oxidative and glycolytic profiles of in vitro polarized M1 and M2 BMDMs and of lean and obese ATMs (Figure 1). These closely recapitulate what has been shown by others (28, 49, 53), and draw attention to the idea that obese ATMs are not specifically M1- or M2-like, similar to what was suggested by gene expression and flow cytometry results discussed above.
Figure 1. Obese adipose tissue macrophages (ATMs) display a hypermetabolic phenotype, different from M1 or M2 polarized bone marrow derived macrophages (BMDMs).
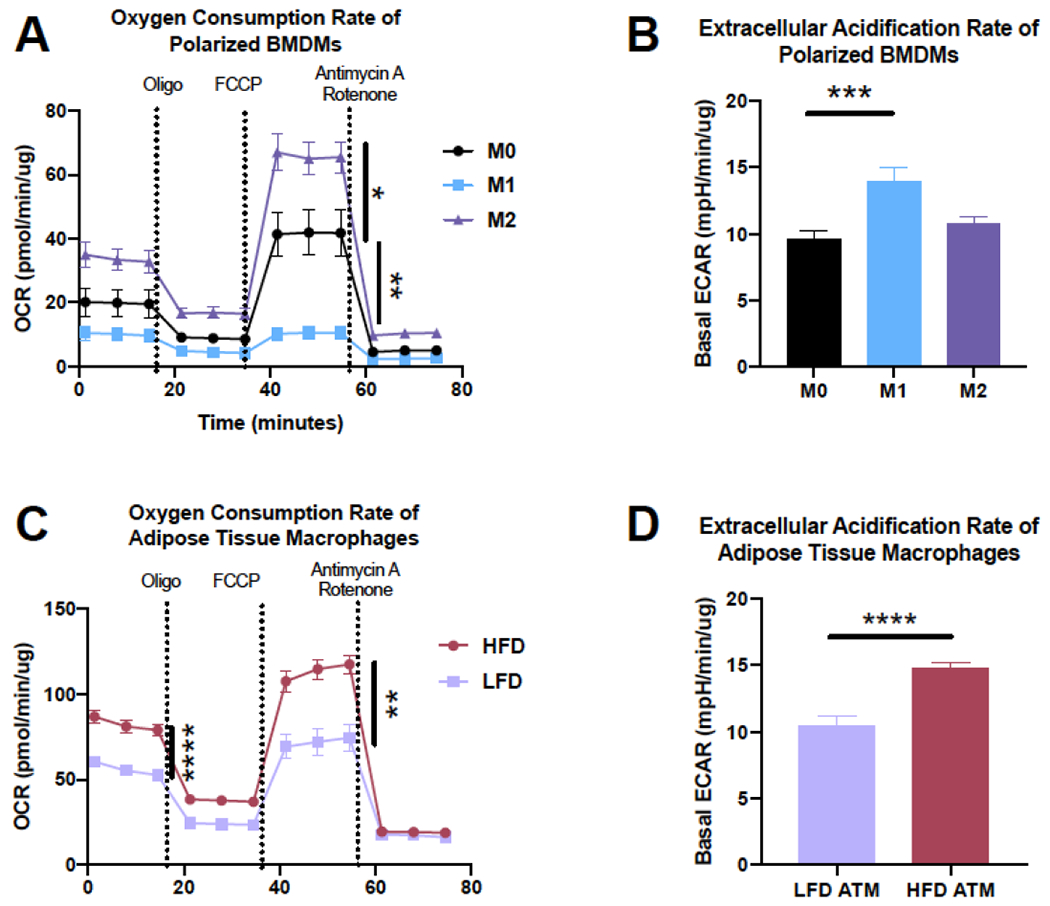
A-B) BMDMs from C57BL/6J mice were polarized for 24 hr with 10 ng/mL LPS/IFNy (M1) or IL-4/IL1-3 (M2). A) A mitochondrial stress test was completed on a Seahorse metabolic flux analyser for basal and maximal oxygen consumption rate (OCR) and B) basal extracellular acidication rate (ECAR). C-D) ATMs were isolated from C57BL/6J mice on a low fat diet (LFD) or high fat diet (HFD) for 9 wks and C) OCR and D) ECAR were assessed.
*p<0.05, **p<0.01, ***p<0.001, ***p<0.0001 by two-way ANOVA (A and C), one way ANOVA (B), or t-test (C and D) and post-hoc testing where appropriate.
Interestingly, the study by Stienstra’s group also suggests that cytokine production in lean ATMs is supported by fatty acid, glucose, and glutamine utilization (more similar to M2 cells), while cytokine production in obese ATMs are supported by glycolysis (more similar to M1 cells (28). Microarray analysis also highlighted genes associated with many metabolic pathways, including carbon metabolism, amino acid metabolism, OX PHOS, glycolysis, amino/nucleotide sugar metabolism, PPP, fructose/mannose metabolism, mTOR signaling, glycerolipid metabolism, and sphingolipid metabolism; however, little is known about the requirements of these pathways for metabolic and inflammatory function. Only two studies modulate metabolic proteins in ATMs. Makowski’s group found a similar phenotype to Stienstra’s group, with glycolytic macrophages expressing high levels of the glucose transporter, Glut 1; however, paradoxically, Glut1 myeloid knockout mice were not protected against diet induced obesity (55). Moreover, while obese ATMs have increased expression of the fatty acid transporter Fatp1, Fatp1 deficient mice have worsened weight gain and glucose tolerance following HFD-feeding (56). Fatp1 deficiency increases glycolysis, Nos2, and the prostaglandin 15-HETE in BMDMs, and IL-6, IL-1β, NLRP3 activation, and oxidative stress following HFD-feeding in vivo; however, Fatp1 deficiency also increased OX PHOS, despite suppressing fatty acid uptake and oxidation in BMDM. Together, these results suggest that obese ATM metabolism is not unequivocal and ATMs may be metabolically flexible; however, the contribution of each metabolic pathway to ATM function in lean and obese settings is not known. Lipid metabolism has also been assessed in ATMs by Ferrante and colleagues, due to the lipid rich environment of AT (57, 58). HFD-feeding results in lysosome biogenesis and lipid catabolism in ATMs induced by factors secreted by AT, but is not dependent on autophagy. Further understanding of mechanistic links between obese ATM metabolism and function may suggest potential targets to alleviate macrophage-associated AT inflammation and dysfunction.
No studies have examined human ATM bioenergetics, although a few studies link macrophages with adipocyte metabolism. One study showed an increase in ATP-linked respiration but not glycolysis in a human adipocyte cell line treated with LPS/IFN-gamma activated macrophage-conditioned media. Conversely, there was a decrease in ATP-linked respiration in the context of IL-10 and TGFβ treatment (59). As these cytokines are present in AT during obesity and can be released by ATMs, this suggests a role for macrophage function influencing adipocyte function. Moreover, gene expression analysis of human subcutaneous AT from subjects with varying BMIs show a negative correlation between the macrophage markers CD163 and CD40 and the mitochondrial genes UQCRC2 (encoding complex III core protein of the electron transport chain) and NDUFB8 (encoding complex I subunit of the electron transport chain) primarily found in adipocytes, which also supports the ideas that macrophages may influence adipocyte metabolism. DNA microarray analysis in AT from human obese subjects undergoing calorie restriction showed decreased gene expression of adipocyte and mitochondrial metabolism, namely OX PHOS; however, it is unclear if these genes change in macrophages, adipocytes, or both (60). Concomitantly there was an increase in macrophage markers during calorie restriction indicating a possible activation of ATMs by FFA released during increased lipolysis in the local environment, but this did not translate to systemic inflammation, as there was a decrease in serum C reactive protein (60). No work has directly examined bioenergetics in obese human macrophages, which may be especially important to our overall understanding of ATM metabolism in obesity. Moreover, newer work seeks to understand not only ATMs in obesity, but the role and function of ATMs in lean healthy AT as well as following weight loss, calorie restriction, bariatric surgery, and weight cycling.
Role of Resident Immune Cells in AT Homeostasis
For many years, investigators in the field of extrinsic immunometabolism have focused on mechanisms by which the inflammatory milieu of obese AT promotes local IR resulting in increased basal lipolysis, ectopic lipid storage, and concomitant impairment in systemic insulin action. However, more recent work is exploring the concept that resident immune cells in lean AT play important homeostatic roles. These roles could include anti-inflammatory cytokine secretion, antigen presentation, and extracellular matrix maintenance. Newer areas of investigation have highlighted the role of ATMs in buffering concentrations of locally required substrates such as catecholamines, lipid, and iron. Whether the bioenergetics of these special ATMs influences their buffering may be important for these functions as well.
Catecholamine Buffering
Sympathetic innervation of white AT is important for lipolysis, beiging, and thermogenesis, and this occurs via norepinephrine (NE) signaling. While one early study reported that ATMs themselves can produce NE (61), several other groups have refuted this (62–64). Now, the prevailing thought is that a special population of ATMs can recycle NE delivered from sympathetic nerves. These specialized ATMs have variously been called sympathetic neuron-associated macrophages [SAMs; (63)] and nerve-associated macrophages [NAMs; (64)]. These cells do not express a key enzyme required for NE synthesis, but rather, have a unique gene expression profile characterized by increases in neural- and adrenergic-related genes (63). Thus, they can import and degrade NE, thereby controlling local sympathetic tone. Interestingly, depletion of the NE importer in SAMs resulted in increased serum NE, thermogenesis, lipolysis, and weight loss during food restriction (63). In contrast to iron-recycling ATMs discussed below, SAMs have a pro-inflammatory phenotype that is exaggerated in obesity (63). The metabolic phenotype of these cells has not been assessed, however given the intricate link between metabolism and function, it may be of interest to understand how catecholamine buffering influences the metabolic phenotype of these cells and vice versa.
Lipid Buffering
As mentioned above, obese ATMs express transcription factors important for lipid metabolism and have a distinct fatty acid-mediated metabolic phenotype. However, whether the ATMs actually contribute to lipid metabolism in AT in the lean or obese state, or whether their phenotype was merely a consequence of their lipid-rich environment was not known. Ferrante and colleagues answered this question in a series of papers starting in 2013. First, they showed that obesity induces a program of lysosomal biogenesis in ATMs (57). Like Becker, they defined a lipid catabolism phenotype, rather than a classically activated M1-like phenotype. Importantly, when this lysosome-dependent lipid metabolism was inhibited, adipocyte lipolysis was also depressed. In Ferrante’s subsequent work, his group demonstrated that adipocytes can release exosome-sized, lipid-filled vesicle they refer to as “AdExos” (65). These AdExos are actually the vehicle for lipid transfer from adipocytes to ATMs and they also can convert BMDMs into the MMe phenotype. Using electron microscopy, they revealed invaginated lipid structures budding off adipocytes and many vesicular structures that looked like exosomes (65). We also detect similar structures in transmission electron microscopy work from our group (Figure 2). These exosomes are released at more than twice the rate from obese leptin deficient AT compared to lean, a rate that is suggested to turnover the entire lipid content of adipocytes from obese mice every 42 days compared to the turnover from lean mice every 104 days (65)! In other related work, Maxfield and colleagues have shown that the ATMs can actually exocytose lysosomal enzymes in extracellular synapses that join ATMs with dead adipocytes, in a process called exophagy (66). Thus, this work cumulatively reveals ATMs as a buffer and recycler of lipids in AT, however, we have little understanding of the contribution of lipid uptake and metabolism to the production of ATP, metabolic intermediates, and inflammatory lipid mediators.
Figure 2: Electron microscopy of closly interacting adipocytes and macrophages: invaginated lipid structures budding off adipocytes and many vesicular structures resembling exosomes.

Adipose tissue was collected from lean chow fed C57BL/6J mice, cut into 1 mm pieces, and immediately fixed in 2.5% glutaraldehyde, before being prepared by Vanderbilt Imaging Core for TEM imaging. Images were captured on a Philips/FEI T-12 transmission electron microscope.
Iron Buffering
Macrophages also play an important role in iron homeostasis by sequestering iron from pathogens, recycling iron for use to generate new red blood cells, and preventing toxicity as unbound iron can act as an oxidant [reviewed in (67)]. Consistent with the plasticity of macrophages and their ability to adapt to the tissue microenvironment, we have recently published data suggesting that a special population of ATMs might recycle iron (68, 69). We identified phenotypic M2-like macrophages in the AT with increased intracellular iron and iron handling genes including iron uptake (CD163, transferrin receptor), metabolism (heme oxygenase), storage (ferritin light and heavy chains), and export (ferroportin and ceruloplasmin) from the stromal vascular fraction of lean mice fed normal chow (68). These ferromagnetic ATMs were isolated via magnetic column separation and are termed MFehi with the nonferromagnetic population termed MFelo. In obesity, we observed the expected increase in total ATMs from obese mice fed HFD, with a majority representing MFelo macrophages that were of the M1-phenotype. Moreover, there was also a phenotypic shift in the MFehi cells from the M2-like phenotype identified in lean mice to a more pro-inflammatory phenotype in obese mice. These inflammatory MFehi cells also had a decrease in iron content as well as the aforementioned iron handling genes. Concomitantly, atomic absorption spectrometry showed an increase in iron content in adipocytes of mice fed HFD. These data suggest that in addition to the role of ATMs in protecting adipocytes from lipotoxicity and promoting tissue repair, they also play a role in protecting adipocytes from iron overload. However, during obesity, inflammatory ATM polarization impairs homeostatic iron handling in MFehi cells and promotes adipocyte iron overload. Consistent with this hypothesis, we have shown that MFehi cells from lean AT increase their iron content and upregulate iron handling genes following a high iron diet or intraperitoneal injection of iron, protecting the adipocytes from iron overload (69). The phenomenon of iron handling macrophages is not unique to AT as other studies have shown increased iron uptake by M2 macrophages in human atherosclerotic plaque hemorrhages and in the context of iron overload (70). In addition, emerging evidence suggests that macrophage iron handling plays a critical role in other setting such as muscle, brain, bone marrow and pancreas [reviewed in (article in press)].
Since iron can affect the polarization of macrophages and macrophage polarization dictates fuel metabolism, it seems logical that iron affects immunometabolism. One study has shown that iron depletion of human macrophages results in increased glycolysis, enhanced extracellular acidification rate, and citrate accumulation, all of which characterize metabolic flux deviation from the TCA cycle, likely due to inhibition of iron-dependent mitochondrial aconitase. Conversely, there was a decrease in genes involved in mitochondrial oxidation (71). This is logical given that one of functions of the mitochondria is the formation of Fe-S clusters that acts as a co-factor in many cellular pathways, namely in energy production. More specifically, the Fe-S clusters are necessary for the efficient function of Complexes I-III of the electron transport chain (72). However, a recent study of hemoglobin-haptoglobin treated monocytes shows that glucose transporters, fatty aldehyde dehydrogenase, and glucose-6-phosphate-1 dehydrogenase are also within the top 50 upregulated proteins (73). More work will need to be done to determine how these genes and their associated pathways influence cellular iron uptake and cycling.
Weight Loss
Weight loss is the most logical and effective way to ameliorate the metabolic effects of obesity. Weight loss has been associated with an improvement in obesity-associated metabolic derangements such as T2DM and cardiovascular disease [reviewed in (2, 3)]. Weight loss can be accomplished by calorie restriction such as switching from a HFD to a LFD, exercise, medications, or bariatric surgery. Despite the systemic benefits, emerging studies in mice and humans suggest the immune activation of obesity is not readily resolved, which has implications for weight cycling as discussed below.
The first mouse study investigating ATMs after weight loss was published by Ferrante and colleagues in 2010 (74). They discovered an interesting dichotomy, whereby early phases of weight loss via caloric restriction – marked by increased lipolysis – resulted in an increase in ATMs, and only after extended weight loss did the ATM numbers decrease. Mechanistically, these investigators demonstrated that the increase in fatty acid flux due to lipolysis resulted in increased adipocyte chemokines and thereby the initial increase in ATMs. One of the conclusions of this work was that ATMs can act as buffers for fatty acids lipolyzed from adipocytes – an idea corroborated by their later work discussed above. In a later study, Lumeng and colleagues showed that IR was not completely ameliorated even though mice had reached a lean weight after 8 weeks of weight loss by switching to a chow diet (75). With regards to immune cell populations, ATMs and T cells persisted even after weight loss and the ATMs retained an inflammatory phenotype (75). In this report, even 24 weeks after switching the diet, the weight loss animals had increased inflammatory ATMs compared to weight-matched controls that had never been obese. CD40 and CD80, co-stimulatory molecules were also maintained in an elevated state in the weight loss mice. Their data suggest that T cell activation is also maintained after weight loss. Finally, they showed that ATM populations were maintained in weight loss via proliferation. Bruning and colleagues found a similarly sustained inflammatory ATM phenotype in mice that lost weight for 14 weeks with a combination of switching to a diet with intermediate fat content and pair-feeding (76). Similar to what Lumeng found, Bruning’s group showed that some inflammatory cytokines were even higher in the weight loss mice than in obese mice maintained on HFD for the entire study. Together, these studies demonstrate persistent macrophage-dominated inflammation in AT even after prolonged weight loss, an observation not found for the liver, in which inflammation and fat content were normalized after weight loss.
Fisher and colleagues used both weight gain and weight loss models to assess visceral AT immune cell populations (77). They detected 15 different leukocyte populations, all of which were changed in obesity. However, macrophages were the most abundant (7 different clusters) and were also the most diverse in their gene expression profiles. Similar to what was shown by Jaitin et al. (38), in obesity, the predominant macrophage subpopulation in obese AT was characterized by a lipid binding and metabolism phenotype. Interestingly after only 2 weeks of caloric restriction, this population was lost, and a new population — characterized by phagocytosis — was gained, indicating not only that ATMs don’t revert to a lean phenotype after caloric restriction, they take on a completely different phenotype. With regards to metabolic processes, they showed that many of the macrophage clusters were enriched for genes related to OX PHOS and also lipid metabolism.
In a model of 24-weeks of weight loss via voluntary exercise wheel running, Vieira-Potter and Padilla demonstrated a decrease in ATMs and inflammatory cytokines at the end of the study (78). Interestingly, this decrease was not found in mice that were allowed to exercise only intermittently, despite having the same body weight and adiposity as the regularly exercised animals. More work should examine how different mechanisms of exercise influence ATM inflammation, and more broadly, how other mechanisms of weight loss may influence immunometabolism.
There have also been some studies investigating the consequence of weight loss on ATMs in humans. Most of these studies have analyzed AT from patients undergoing bariatric surgery as this intervention has been shown to result in an average decrease of total body weight by 30 to 35% after 1 year (79, 80). There is increasing evidence that the total number of macrophages in the AT decreases after bariatric surgery with a decrease in pro-inflammatory ATMs (81–83). Specifically, Aron-Wisnewsky et al. showed an increase in classical M2 markers CD163 and CD206 from subcutaneous ATMs after 15% weight loss induced 3 months after bariatric surgery (84). Further studies showed a significant association between Il10 mRNA in visceral AT and the magnitude of weight loss at 1 to 3 months after bariatric surgery when compared to baseline and that a higher expression of Il1 and leptin mRNA, Th2 cells, and total macrophages could be a predictor of greater improvement of BMI within months after bariatric surgery (81). In addition to ATMs, neutrophils were also found to decrease in both visceral and subcutaneous tissue after weight loss due to bariatric surgery; however, no changes in T cell populations were observed (82). Together, these results suggest that while weight loss may improve physiological dysfunction, the effects on AT immune cells are not all restorative and should be further classified.
Another way to model the negative energy balance associated with weight loss is via activating lipolysis. Dr. Granneman and colleagues performed scRNA-seq on over 33,000 cells from the stromal vascular fraction of epidydimal and inguinal AT in lean mice with or without β-adrenergic stimulation (85). The purpose of this study was to evaluate the recruitment of brown/beige adipocytes during lipolytic stimulation, and they indeed found that they could classify a proliferating population of cells that would differentiate into brown/beige adipocytes. In addition to lipolysis, adrenergic stimulation also causes adipocyte death, and thereby resident ATMs take on a role in efferocytosis, the phagocytosis and clearance of dead cells. Lipolysis is considered one of the mechanisms by which weight loss, as in the studies above, changes the phenotype of macrophages and other immune cells in the AT. Granneman’s group performed scRNA-seq also on the Lin+ immune cells from eWAT and iWAT of control and stimulated mice. Lipolysis increased the number of proliferating macrophage/denritic cells, while reducing the number of NKT cells. Interestingly, their data showed that some M2-like markers, Chil3, Clec10a, and Arg1, segregated on different cell populations, indicating more heterogeneity that was previously appreciated. Finally, a macrophage population identified as proliferating was also enriched for genes involved in lipid metabolism: Cd36, Fabp5, Lpl, and Lipa – possibly the population acting as efferocytes. In addition, this population was associated with expression of genes involved in extracellular matrix remodeling, migration and adhesion. There have been no studies to determine how intrinsic immunometabolism changes with weight loss, which may elucidate interesting pathways tied to the function of AT immune populations.
Weight Cycling
Weight cycling, also termed “weight fluctuation” or “weight variability” is not a new phenomenon in human weight patterns. A recent meta-analysis sought to evaluate the effect of weight cycling on obesity-related metabolic risk factors and T2DM from studies conducted between 1994 and 2015. Mackie et al. concluded that 11 out of 19 publications showed a positive correlation between weight cycling and increased body fat and deposition of AT centrally but did not find a correlation with increased risk of T2DM (86). Many other studies have seen a correlation between weight cycled and worsened cardiometabolic disease, atherosclerosis, and even mortality (87–90), suggesting that it is still an important area of study.
Our lab and others have consistently shown that weight cycled obese mice have impaired glucose tolerance, increased AT IR and increased pro-inflammatory T cell infiltration compared to non-weight cycled obese mice. Our mouse model is of extreme weight cycling (91): 9 weeks of HFD (weight gain), followed by 9 weeks of LFD (weight loss), followed by an additional 9 weeks of HFD (weight regain). Control mice were on HFD for the final 18 weeks of the study. Both the weight cycling and the obese models were on HFD for a total of 18 weeks and their body and fat mass were equivalent for the last 4–5 weeks of the study. Despite similar body mass, the weight cycled mice had dramatically reduced glucose clearance during intraperitoneal glucose tolerance tests and also displayed severely impaired insulin signaling in the AT. In our initial report, we demonstrated increased CD8+ T cells and memory T cells, a finding we can reproduce and are pursuing. Aguilas’s group has also shown that alternating 8 wk high fat diet bouts increase glucose intolerance, blood lipids, and TNFα (92) and Gao’s group has published that weight cycled mice regain weight faster and also have an increase in CD4+ T cells (93). Moreover, transcriptome analyses suggested that weight regain increases macrophages, T cells, CLS, and dead/dying adipocytes (94). An unanswered question is whether the intrinsic immunometabolism of T cells and/or ATMs is altered during weight cycling and further, whether these potential changes contribute to the accelerated metabolic disease noted in these models.
Conclusions and Emerging Questions
One thing that is clear from the studies detailed in this review is that resident macrophages in lean AT do not have transcriptional profiles of in vitro M2-polarized macrophages and recruited or proliferated macrophages in obese AT do not have transcriptional profiles of in vitro M1-polarized macrophages. Nor do they recapitulate the metabolic profiles of in vitro polarized cells. As ATMs in obesity have both increased glycolysis and OX PHOS, this represents a novel finding in the field of immunometabolism as compared to previously published data regarding fuel utilization in M1 favoring glycolysis and M2 favoring OX PHOS. Typically, cells upregulate one pathway over the other in order to facilitate the production of both ATP and other mediators. This dichotomy may be due to: a) analyzing different ATM subpopulations cumulatively; with some subpopulations having increased glycolysis (possibly associated with CLS) whereas other populations have increased OX PHOS (possibly represent interstitial ATMs) within the same tissue or b) versus the same cell utilizing both glycolysis and OX PHOS (Figure 3). The latter hypothesis may seem less likely; however, the dual metabolic phenotype appears to parallel the studies that suggest ATMs secrete both inflammatory and anti-inflammatory mediators and have both M1- and M2- related gene and protein expression. Moreover, there is currently evidence that Th17 cells (at least those polarized in vitro) as well as atherogenic monocytes can increase both pathways simultaneously (95–99). Interestingly, isolating ATM by CD11b (53), F480+ (28), or adherence (Figure 1) all produce the same hyperinflammatory phenotype; however, these are likely still mixed populations. Future work with ATMs sorted by marker such as CD9 will help to determine if the different functional phenotypes have similar or different metabolic pathway utilization. Furthermore, cell sorting or deeper analysis of single cell data may help to determine if the cell pathways are upregulated similarly in the monocyte- macrophage differentiation following recruitment or if the hypermetabolic phenotype is due to change in function. Assays like metabolomics, metabolic flux analysis, or tracer studies can also be used to determine the fuel sources pathways and intermediates that contribute to ATM function.
Figure 3: Obese ATM have a unique hypermetabolic phenotype.
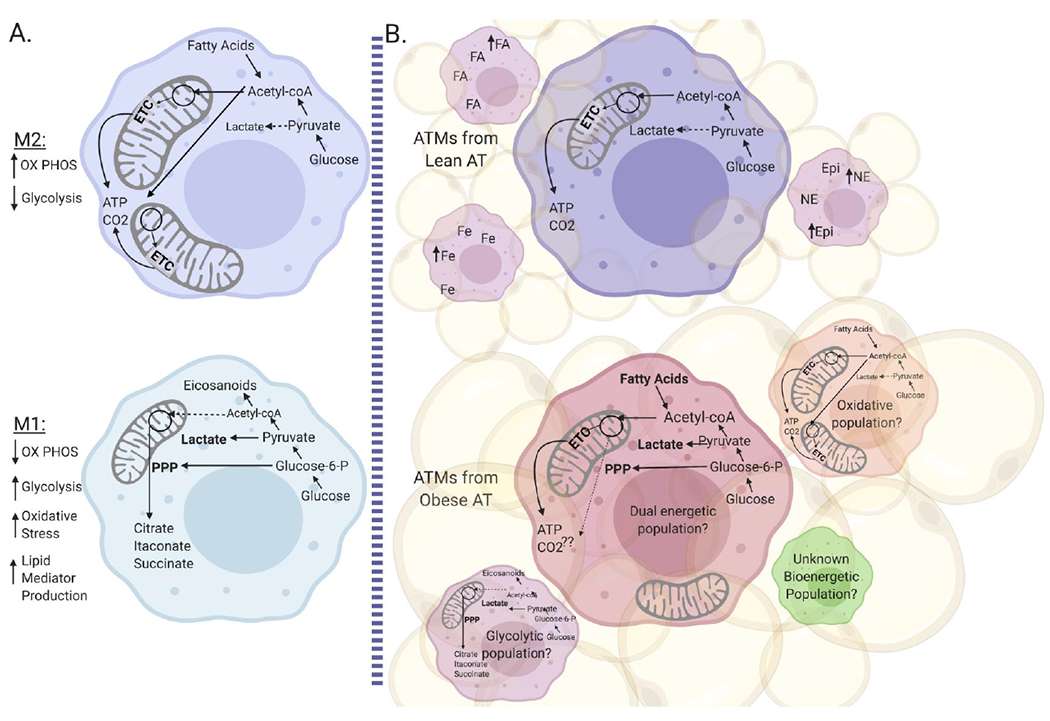
A) Metabolic pathways are well-defined in BMDM polarized to M1 and M2 macrophages in vitro. B) Lean ATMs have low metabolic utilization, and may have multiple populations that handle catecholamines, iron, and lipids. In contrast, obese ATMs are hypermetabolic. Recent single cell RNA-sequencing data suggests that obese ATM also have multiple mixed populations. Future work should define 1) if the hypermetabolic phenotype is a result of pooled populations or if it is present in single populations and 2) how obese ATMs use glycolysis and OX PHOS for energy production, intermediate mediators, and both phagocytic and inflammatory functions.
Furthermore, a deep dive into recent literature suggests that at least one of the primary roles of macrophages in AT is to store and/or recycle a variety of substances from other cells in the AT: lipids from adipocytes, catecholamines from neurons, and iron from the local environment. Another thing that is quite clear is that lipid metabolism is a major defining feature of macrophages from AT as compared to macrophages from other tissues or those that are in vitro polarized. From the oxidized phospholipids in Leitinger’s work, to the palmitate-driven MMe phenotype of Becker’s work, to the lipid recycling ATM phenotype of Ferrante’s work and the scRNAseq studies discussed here, lipids clearly define the phenotype, function, and bioenergetics of ATMs. Not surprising given the tissue within which they reside. Thus, future studies should examine not only the role of obese inflammatory ATMs, but tissue resident homeostatic functions as well. And, as function is linked closely to bioenergetic utilization, understanding the contribution of different pathways to buffering/recycling may expand our knowledge of macrophage functions.
Finally, new studies examining weight loss and weight cycling, human AT immune populations, and new technologies like single cell sequencing are dramatically progressing the field of knowledge regarding extrinsic immunometabolism. Further studies regarding intrinsic immunometabolism should help provide a mechanistic understanding of how AT immune tissue function is changed with weigh; however, there is much to still uncover. Technologies like single cell sequencing, CITE-sequencing, and CyTOF may help to uncover how many immune cell populations change along with body weight. Technologies like CODEX and MALDI-TOF Mass Spectrometry may help to determine how these populations change spatially within the AT, and technologies like ATAC-sequencing may help to define if these changes are driven by epigenetic regulation. Additionally, future studies should continue to examine how AT immune cells contribute to weight loss and regain with different diets, with exercise, and following both pharmacological and surgical weight loss interventions, both in mice and in humans. Finally, there has not been clear work studying the influence of age or sex on the outcomes reviewed, which may also influence not only the results, but may have implications for how we evaluate and treat obesity-associated diseases.
Acknowledgements
AH Hasty is supported by a Research Program Grant from the NIDDK (R01DK121520), a Merit Award from the Veterans Affairs (5I01BX002195) and an Innovative Project Award from the American Heart Association (19IPLO134760376). M Bhanot is partially supported by an American Heart Association Strategically Focused Research Network award (17SFRN33520017). HL Caslin is supported by the Molecular Endocrinology Training Grant (DK07563). TEM imaging was performed through the use of the VU Cell Imaging Shared Resource (supported by NIH grants DK20593, DK58404, and DK59637). We would like to acknowledge ©BioRender for the figure creation software.
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
References Cited
- 1.Kelly T, Yang W, Chen CS, Reynolds K, and He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond). 2008;32(9):1431–7. [DOI] [PubMed] [Google Scholar]
- 2.Vidal J Updated review on the benefits of weight loss. Int J Obes Relat Metab Disord. 2002;26 Suppl 4:S25–8. [DOI] [PubMed] [Google Scholar]
- 3.Goodpaster BH, Kelley DE, Wing RR, Meier A, and Thaete FL. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes. 1999;48(4):839–47. [DOI] [PubMed] [Google Scholar]
- 4.Rhee EJ. Weight Cycling and Its Cardiometabolic Impact. J Obes Metab Syndr. 2017;26(4):237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation. 2003;112(12):1796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanneganti TD, and Dixit VD. Immunological complications of obesity. Nat Immunol. 2012;13(8):707–12. [DOI] [PubMed] [Google Scholar]
- 8.Dalmas E Role of innate immune cells in metabolism: from physiology to type 2 diabetes. Semin Immunopathol. 2019;41(4):531–45. [DOI] [PubMed] [Google Scholar]
- 9.Ivanov S, Merlin J, Lee MKS, Murphy AJ, and Guinamard RR. Biology and function of adipose tissue macrophages, dendritic cells and B cells. Atherosclerosis. 2018;271:102–10. [DOI] [PubMed] [Google Scholar]
- 10.Caslin HL, and Hasty AH. Extrinsic and Intrinsic Immunometabolism Converge: Perspectives on Future Research and Therapeutic Development for Obesity. Curr Obes Rep. 2019;8(3):210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lumeng CN, Bodzin JL, and Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao L, Herlea-Pana O, Heuser-Baker J, Chen Y, and Barlic-Dicen J. Roles of the chemokine system in development of obesity, insulin resistance, and cardiovascular disease. J Immunol Res. 2014;2014:181450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai Y, and Sun Q. Macrophage recruitment in obese adipose tissue. Obes Rev. 2015;16(2):127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surmi BK, and Hasty AH. The role of chemokines in recruitment of immune cells to the artery wall and adipose tissue. Vascul Pharmacol. 2010;52(1-2):27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill AA, Reid Bolus W, and Hasty AH. A decade of progress in adipose tissue macrophage biology. Immunol Rev. 2014;262(1):134–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassnain Waqas SF, Noble A, Hoang AC, et al. Adipose tissue macrophages develop from bone marrow-independent progenitors in Xenopus laevis and mouse. J Leukoc Biol. 2017;102(3):845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amano SU, Cohen JL, Vangala P, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014;19(1):162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haase J, Weyer U, Immig K, et al. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia. 2014;57(3):562–71. [DOI] [PubMed] [Google Scholar]
- 19.Appari M, Channon KM, and McNeill E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid Redox Signal. 2018;29(3):297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramkhelawon B, Hennessy EJ, Menager M, et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20(4):377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang JE, Ko MS, Yun JY, et al. Nitric Oxide Produced by Macrophages Inhibits Adipocyte Differentiation and Promotes Profibrogenic Responses in Preadipocytes to Induce Adipose Tissue Fibrosis. Diabetes. 2016;65(9):2516–28.27246913 [Google Scholar]
- 22.Han CY. Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue. Diabetes Metab J. 2016;40(4):272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Santibanez G, Singer K, Cho KW, et al. Obesity-induced remodeling of the adipose tissue elastin network is independent of the metalloelastase MMP-12. Adipocyte. 2015;4(4):264–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrero L, Shapiro H, Nayer A, Lee J, and Shoelson SE. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc Natl Acad Sci U S A. 2010;107(1):240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaul ME, Bennett G, Strissel KJ, Greenberg AS, and Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet--induced obesity in mice. Diabetes. 2010;59(5):1171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kratz M, Coats BR, Hisert KB, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20(4):614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, et al. Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep. 2017;20(13):3149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boutens L, Hooiveld GJ, Dhingra S, et al. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61(4):942–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C, Menoret A, Farragher C, et al. Single cell transcriptomics based-MacSpectrum reveals novel macrophage activation signatures in diseases. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Catrysse L, and van Loo G. Adipose tissue macrophages and their polarization in health and obesity. Cell Immunol. 2018;330:114–9. [DOI] [PubMed] [Google Scholar]
- 31.Spencer M, Yao-Borengasser A, Unal R, et al. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab. 2010;299(6):E1016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blaszczak AM, Jalilvand A, Liu J, et al. Human Visceral Adipose Tissue Macrophages Are Not Adequately Defined by Standard Methods of Characterization. J Diabetes Res. 2019;2019:8124563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wentworth JM, Naselli G, Brown WA, et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59(7):1648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remmerie A, and Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7(6):485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill DA, Lim HW, Kim YH, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A. 2018;115(22):E5096–E105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaitin DA, Adlung L, Thaiss CA, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell. 2019;178(3):686–98 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakarov S, Lim HY, Tan L, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363(6432). [DOI] [PubMed] [Google Scholar]
- 40.Fox CJ, Hammerman PS, and Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5(11):844–52. [DOI] [PubMed] [Google Scholar]
- 41.O’Neill LA, Kishton RJ, and Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Prados JC, Traves PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–14. [DOI] [PubMed] [Google Scholar]
- 43.Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, and Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol. 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu PS, Wang H, Li X, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–94. [DOI] [PubMed] [Google Scholar]
- 45.Jha AK, Huang SC, Sergushichev A, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–30. [DOI] [PubMed] [Google Scholar]
- 46.Mills EL, and O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol. 2016;46(1):13–21. [DOI] [PubMed] [Google Scholar]
- 47.Tan Z, Xie N, Cui H, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. 2015;194(12):6082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang SC, Smith AM, Everts B, et al. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity. 2016;45(4):817–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kerner J, and Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486(1):1–17. [DOI] [PubMed] [Google Scholar]
- 51.Malandrino MI, Fucho R, Weber M, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab. 2015;308(9):E756–69. [DOI] [PubMed] [Google Scholar]
- 52.Divakaruni AS, Hsieh WY, Minarrieta L, et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018;28(3):490–503 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serbulea V, Upchurch CM, Schappe MS, et al. Macrophage phenotype and bioenergetics are controlled by oxidized phospholipids identified in lean and obese adipose tissue. Proc Natl Acad Sci U S A. 2018;115(27):E6254–E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kadl A, Meher AK, Sharma PR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107(6):737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freemerman AJ, Zhao L, Pingili AK, et al. Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype Are Fueled by GLUT1. J Immunol. 2019;202(4):1265–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson AR, Qin Y, Cozzo AJ, et al. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol Metab. 2016;5(7):506–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu X, Grijalva A, Skowronski A, et al. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18(6):816–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grijalva A, Xu X, and Ferrante AW Jr. Autophagy Is Dispensable for Macrophage-Mediated Lipid Homeostasis in Adipose Tissue. Diabetes. 2016;65(4):967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keuper M, Sachs S, Walheim E, et al. Activated macrophages control human adipocyte mitochondrial bioenergetics via secreted factors. Mol Metab. 2017;6(10):1226–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capel F, Klimcakova E, Viguerie N, et al. Macrophages and adipocytes in human obesity: adipose tissue gene expression and insulin sensitivity during calorie restriction and weight stabilization. Diabetes. 2009;58(7):1558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen KD, Qiu Y, Cui X, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480(7375):104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fischer K, Ruiz HH, Jhun K, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med. 2017;23(5):623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pirzgalska RM, Seixas E, Seidman JS, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. 2017;23(11):1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Camell CD, Sander J, Spadaro O, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. 2017;550(7674):119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flaherty SE 3rd, Grijalva A, Xu X, et al. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363(6430):989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haka AS, Barbosa-Lorenzi VC, Lee HJ, et al. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J Lipid Res. 2016;57(6):980–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soares MP, and Hamza I. Macrophages and Iron Metabolism. Immunity. 2016;44(3):492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orr JS, Kennedy A, Anderson-Baucum EK, et al. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes. 2014;63(2):421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hubler MJ, Erikson KM, Kennedy AJ, and Hasty AH. MFe(hi) adipose tissue macrophages compensate for tissue iron perturbations in mice. Am J Physiol Cell Physiol. 2018;315(3):C319–C29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bories G, Colin S, Vanhoutte J, et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ Res. 2013;113(11):1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pereira M, Chen TD, Buang N, et al. Acute Iron Deprivation Reprograms Human Macrophage Metabolism and Reduces Inflammation In Vivo. Cell Rep. 2019;28(2):498–511 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cronin SJF, Woolf CJ, Weiss G, and Penninger JM. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front Mol Biosci. 2019;6:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaempfer T, Duerst E, Gehrig P, et al. Extracellular hemoglobin polarizes the macrophage proteome toward Hb-clearance, enhanced antioxidant capacity and suppressed HLA class 2 expression. J Proteome Res. 2011;10(5):2397–408. [DOI] [PubMed] [Google Scholar]
- 74.Kosteli A, Sugaru E, Haemmerle G, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120(10):3466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zamarron BF, Mergian TA, Cho KW, et al. Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes. 2017;66(2):392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmitz J, Evers N, Awazawa M, et al. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol Metab. 2016;5(5):328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weinstock A, Brown EJ, Garabedian ML, et al. Single-Cell RNA Sequencing of Visceral Adipose Tissue Leukocytes Reveals that Caloric Restriction Following Obesity Promotes the Accumulation of a Distinct Macrophage Population with Features of Phagocytic Cells. Immunometabolism. 2019;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Porter JW, Rowles JL 3rd, Fletcher JA, et al. Anti-inflammatory effects of exercise training in adipose tissue do not require FGF21. J Endocrinol. 2017;235(2):97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mahawar KK, Graham Y, Carr WR, et al. Revisional Roux-en-Y Gastric Bypass and Sleeve Gastrectomy: a Systematic Review of Comparative Outcomes with Respective Primary Procedures. Obes Surg. 2015;25(7):1271–80. [DOI] [PubMed] [Google Scholar]
- 80.Sjostrom CD, Lissner L, Wedel H, and Sjostrom L. Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: the SOS Intervention Study. Obes Res. 1999;7(5):477–84. [DOI] [PubMed] [Google Scholar]
- 81.Lasselin J, Magne E, Beau C, et al. Adipose inflammation in obesity: relationship with circulating levels of inflammatory markers and association with surgery-induced weight loss. J Clin Endocrinol Metab. 2014;99(1):E53–61. [DOI] [PubMed] [Google Scholar]
- 82.Garcia-Rubio J, Leon J, Redruello-Romero A, et al. Cytometric analysis of adipose tissue reveals increments of adipocyte progenitor cells after weight loss induced by bariatric surgery. Sci Rep. 2018;8(1):15203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Labrecque J, Laforest S, Michaud A, Biertho L, and Tchernof A. Impact of Bariatric Surgery on White Adipose Tissue Inflammation. Can J Diabetes. 2017;41(4):407–17. [DOI] [PubMed] [Google Scholar]
- 84.Aron-Wisnewsky J, Tordjman J, Poitou C, et al. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. 2009;94(11):4619–23. [DOI] [PubMed] [Google Scholar]
- 85.Burl RB, Ramseyer VD, Rondini EA, et al. Deconstructing Adipogenesis Induced by beta3-Adrenergic Receptor Activation with Single-Cell Expression Profiling. Cell Metab. 2018;28(2):300–9 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mackie GM, Samocha-Bonet D, and Tam CS. Does weight cycling promote obesity and metabolic risk factors? Obes Res Clin Pract. 2017;11(2):131–9. [DOI] [PubMed] [Google Scholar]
- 87.Bangalore S, Fayyad R, Laskey R, et al. Body-Weight Fluctuations and Outcomes in Coronary Disease. N Engl J Med. 2017;376(14):1332–40. [DOI] [PubMed] [Google Scholar]
- 88.Byun SS, Bello NA, Liao M, Makarem N, and Aggarwal B. Associations of weight cycling with cardiovascular health using American Heart Association’s Life’s Simple 7 in a diverse sample of women. Prev Med Rep. 2019;16:100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park KY, Hwang HS, Cho KH, et al. Body Weight Fluctuation as a Risk Factor for Type 2 Diabetes: Results from a Nationwide Cohort Study. J Clin Med. 2019;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delahanty LM, Pan Q, Jablonski KA, et al. Effects of weight loss, weight cycling, and weight loss maintenance on diabetes incidence and change in cardiometabolic traits in the Diabetes Prevention Program. Diabetes Care. 2014;37(10):2738–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anderson EK, Gutierrez DA, Kennedy A, and Hasty AH. Weight cycling increases T-cell accumulation in adipose tissue and impairs systemic glucose tolerance. Diabetes. 2013;62(9):3180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barbosa-da-Silva S, Fraulob-Aquino JC, Lopes JR, Mandarim-de-Lacerda CA, and Aguila MB. Weight cycling enhances adipose tissue inflammatory responses in male mice. PLoS One. 2012;7(7):e39837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zou J, Lai B, Zheng M, et al. CD4+ T cells memorize obesity and promote weight regain. Cell Mol Immunol. 2018;15(6):630–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kyung DS, Sung HR, Kim YJ, et al. Global transcriptome analysis identifies weight regain-induced activation of adaptive immune responses in white adipose tissue of mice. Int J Obes (Lond). 2018;42(4):755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koelwyn GJ, Corr EM, Erbay E, and Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19(6):526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Binger KJ, Corte-Real BF, and Kleinewietfeld M. Immunometabolic Regulation of Interleukin-17-Producing T Helper Cells: Uncoupling New Targets for Autoimmunity. Front Immunol. 2017;8:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kumar V T cells and their immunometabolism: A novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur J Cell Biol. 2018;97(6):379–92. [DOI] [PubMed] [Google Scholar]
- 98.Liu RT, Zhang M, Yang CL, et al. Enhanced glycolysis contributes to the pathogenesis of experimental autoimmune neuritis. J Neuroinflammation. 2018;15(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Franchi L, Monteleone I, Hao LY, et al. Inhibiting Oxidative Phosphorylation In Vivo Restrains Th17 Effector Responses and Ameliorates Murine Colitis. J Immunol. 2017;198(7):2735–46. [DOI] [PMC free article] [PubMed] [Google Scholar]