

Abstract
Cerebral palsy is a syndrome that encompasses a large group of childhood movement and posture disorders that result from a lesion occurring in the developing brain. The clinical presentation of many metabolic and genetic conditions, particularly in highly consanguineous populations, can mimic cerebral palsy particularly at early age. The aim of this review article is to identify the clinical features that should alert the physician to the possibility of disorders that resemble cerebral palsy, the clinical and neuroimaging red flags, and highlight some metabolic and genetic conditions which may present with spasticity, ataxia and dyskinesia. In the case of metabolic or genetic disorder, making a precise diagnosis is particularly important for the possibility of treatment, accurate prognosis and genetic counseling.
Cerebral palsy (CP) is a group of disorders that affect movement, tone or posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the immature developing brain, most often before birth.1,2 The CP should be better considered as a descriptive syndrome because it is not a specific disease, pathological or etiological entity. The motor component abnormality in CP is often associated with epilepsy, intellectual disability, movement disorders, visual and hearing impairments, and musculoskeletal problems which contribute to the clinical heterogeneity of CP. The presence of these additional features can make the diagnostic process sometimes difficult and challenging. A growing group of metabolic and genetic disorders is being recognized, mainly due to the advancement in the area of metabolic and genetic investigations, to present with exactly the same symptoms and signs as CP.3-12 These CP mimickers are common, particularly in highly consanguineous populations, as in Saudi Arabia, and can be easily missed, mainly in countries lacking national newborn screening programs or advanced metabolic and genetic investigations.13 The possibility of a treatable metabolic or genetic etiology of CP, together with a more accurate diagnosis and genetic counseling, emphasizes the importance of being particularly vigilant in determining the etiology.3 The objective of this review is to provide a practical diagnostic approach to a child with motor delay early in life suggestive of CP, and to summarize the most common metabolic and genetic conditions, focusing on treatable diseases, that may be misdiagnosed as CP.
A brief overview of a CP
Historically, the term CP was used to describe the neurological consequences of an adverse antenatal event (prematurity causing an intraventricular or periventricular hemorrhage) and was later expanded to include other adverse antenatal and perinatal events (cerebral dysgenesis, intracranial infection, and hypoxic-ischemic brain injury).1,2 By definition, the cerebral insult occurred in the developing fetal or infant brain, with infancy spanning from birth to one or even 2 years of age.5 The most common causes of CP are hypoxic-ischemic insult, complications of prematurity, bilirubin toxicity, trauma, and infection.1,2 The prevalence of CP is 2-3 per 1000 live births;1,2 slightly higher in Saudi Arabia, 4-5 per 1000 live births.13 Although the highest risk (40-100 per 1000 live births) is among very preterm and low birth weight babies, most CP patients are born term or near to term.13 Insults are antenatal in approximately 80% of all cases of CP, perinatal in 10%, and occur in infancy in 10% of cases.2 The CP is grouped according to the most predominant pattern of motor disturbance as spastic (diplegia, tetraplegia, hemiplegia), dyskinetic (dystonia, choreoathetosis) or ataxic, although the overall clinical picture is not always pure.1,2 Neuroimaging, mainly MRI, is considered as a standard investigation in all children with CP.14 The most common cerebral abnormalities observed in these children are periventricular white matter injury (19%), diffuse gray matter insult (14%), cerebral malformation (11%), and cerebral vascular accident (11%).14 Interestingly, in around 18% of patients with CP, the MRI findings are nonspecific, or even normal in 15% of cases.14
Metabolic and genetic disorders that mimic CP
Inherited metabolic and genetic diseases are common in highly consanguineous populations. Their clinical presentation may be nonspecific or very slowly progressive, especially in the early age. They can therefore be misdiagnosed as CP.
Prevalence of metabolic and genetic disorders that mimic CP
A growing number of metabolic and genetic disorders may present with symptoms and signs that resemble a CP phenotype.3-12 In a systematic review, Leach et al3 found that at least 54 treatable inborn errors of metabolism mimicking CP. Whole exome sequencing was performed by McMichael et al9 and Mattews et al11 in 183 and 60 CP patients respectively. The CP-causing variant were found in 14% and 65% of their patients respectively.9,11
Red flags in the history, examination and neuroimaging when another diagnosis should be considered
Table 1 & Table 2 summarize the clinical and neuroimaging features that should alert the physician to a possible underlying metabolic or genetic disorder that mimic CP. Thorough history and a careful neurological examination are essential in the evaluation of a patient suspected to have CP.
Table 1.
Red flags in the history and examination that should prompt the consideration of another diagnosis
| History |
|---|
| No risk factors for CP such as: prematurity, low birthweight, multiple births, hypoglycemia, jaundice and kernicterus, intrapartum asphyxia, intracranial hemorrhage, infection, stroke, or head injuries |
| Positive family history of CP |
| Fluctuation in motor symptoms |
| Paroxysmal symptoms in relation to time of day, diet/fasting, or activity |
| Progressive neurological symptoms |
| Regression of milestones |
| Examination |
| Dysmorphic features |
| Isolated motor dysfunction such as isolated ataxia or isolated hypotonia without dystonia or spasticity |
| Peripheral nervous system abnormalities: absent reflexes, sensory signs |
| Eye movement abnormalities (e.g., oculogyria, oculomotor apraxia, or paroxysmal saccadic eye-head movements) |
| Optic atrophy/retinopathy |
CP- cerebral palsy
Table 2.
Red flags in the MRI brain findings that should prompt consideration of another diagnosis.
| Normal neuroimaging |
| Nonspecific abnormalities, such as isolated globus pallidus involvement, which can suggest methylmalonic aciduria |
| Imaging may demonstrate specific lesions that are inconsistent with perinatal brain injury, but characteristic of a particular genetic disorder, such as leukodystrophies, features of glutaric aciduria type 1, or features of Joubert syndrome |
The following elements in the patient’s medical history are not typical for CP and are an important clue to the possibility of CP mimic: absent history of any risk factors leading to brain injury, such as prematurity, perinatal or infantile hypoxic ischemic insult, intracranial bleeding, kernicterus, cerebral vascular accident, infection or head injury, family history of CP, progressive neurological symptoms, regression of milestones, diurnal variation of symptoms or symptoms fluctuation in relation to activity or fasting. Dysmorphic features, isolated motor dysfunction (hypotonia without dystonia or spasticity or isolated ataxia), peripheral nervous system abnormalities (sensory sign or absent reflexes), eye movement abnormalities (paroxysmal saccadic eye-head movements, oculogyria, or oculomotor apraxia), and optic atrophy or retinopathy are atypically seen in CP children.
The following criteria were described by Person et al6 to diagnose a child with a motor features consistent with CP: symptom onset before 2 years of age, nonprogressive clinical course with no milestone regression, and exclusion of disorders that present with predominant cognitive impairment, autism, encephalopathy, or seizure.
Brain MRI is recommended as a first-line investigation in all children with a clinical feature suggestive of CP.6 If the findings are normal or nonspecific, other investigations should be carried out to rule out an underlying metabolic or genetic disease.
Investigations
Most of the metabolic/genetic diseases have overlapping or unspecific phenotypes. The utility of next-generation sequencing in the clinical field has been widely demonstrated in different groups of diseases, mainly in metabolic/genetic diseases.15 Those metabolic and genetic diseases may present with various phenotypes, some of them resembling CP. The first approach should be based on the history, clinical examination and neuroimaging findings. Based on a detailed clinical phenotyping and neuroimaging findings, 2 phenotypes can be proposed:
Scenario 1
The phenotype is highly suspected based on the clinical and neuroimaging findings. In these cases, a targeted biomarker analysis or single gene analysis may facilitate the diagnostic process of a treatable disorders and the initiation of disease specific treatment. For examples, infant with hypotonia, nystagmus, and dystonia, and MRI showing hypomyelination (Figure 1). This picture is highly suggestive of PLP1-related dysmyelinating disorders. Another example, patient with macrocephaly, severe dystonia/ opisthotonos, and MRI showing widening of the Sylvian fissures and basal ganglia abnormalities; this picture is highly suggestive of glutaric aciduria type 1 (Figure 2).
Figure 1.
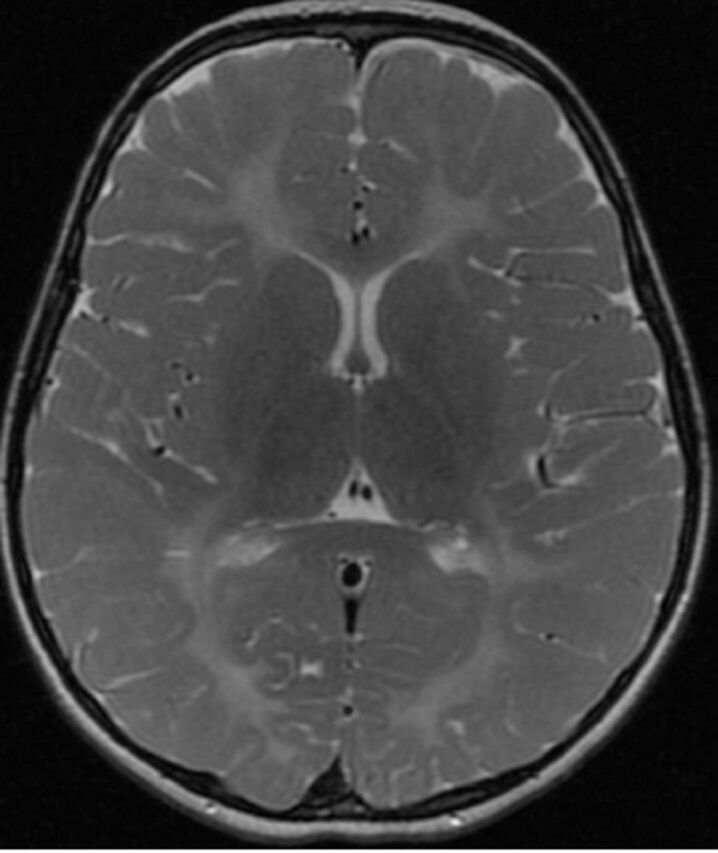
Axial T2-weighted brain magnetic resonance image of a 5-year-old child with Pelizaeus-Merzbacher disease shows bilateral symmetrical hyperintensity of the deep, periventricular and subcortical white matter including the posterior limbs of the internal capsules.
Figure 2.
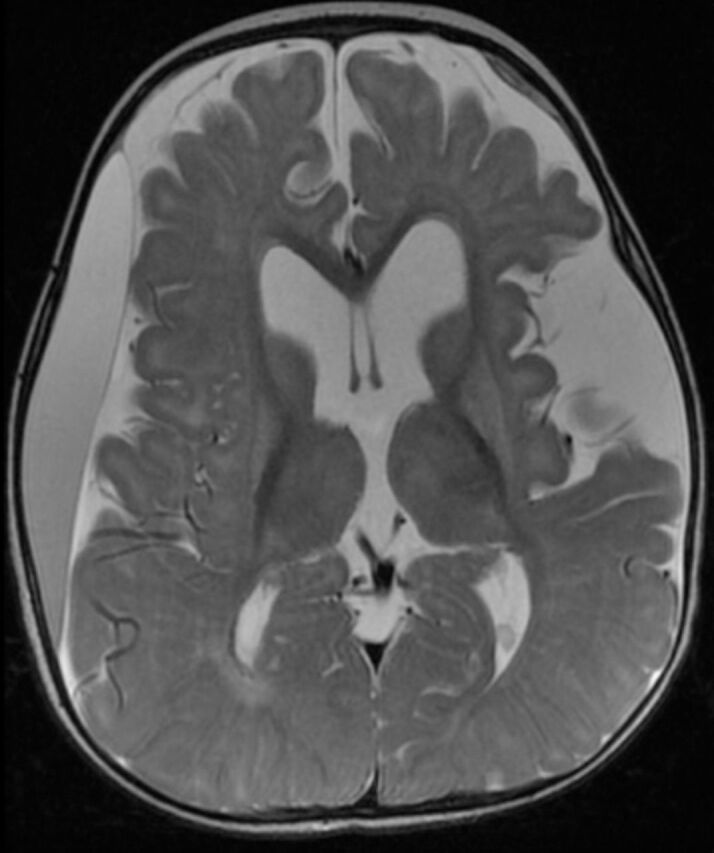
Axial T2-weighted brain magnetic resonance image of a 9-months-old child with glutaric aciduria type1 shows subacute right frontoparietotemporal subdural hematoma with mass effect, diffuse brain atrophy with widening of sylvian fissures bilaterally and secondary ventriculomegaly and increased signal intensity of basal ganglia bilaterally.
Scenario 2
The clinical and neuroimaging findings are non-specific and no specific cause is suspected: the role of next generation sequencing. In recent years, advanced genetic testing techniques have become efficient strategies to diagnose rare disease-causing mutations in children with neurodevelopmental disorders.4 This methodology was recently applied to an atypical CP: Zouvelou et al4 recently analyzed 47 patients with atypical CP and found 23 pathogenic mutations explaining the clinical/MRI phenotype.
Common metabolic and genetic disorders that mimic CP
The metabolic and genetic disorders that may misdiagnosed as CP are outlined in Table 3, classified by the most predominant motor pattern, although in many conditions the clinical features are not always pure.
Table 3.
Common metabolic and genetic disorders that mimic CP according to the prominent motor dysfunction.
| Disorders with prominent spasticity | Disorders with prominent dyskinesia | Disorders with prominent ataxia |
|---|---|---|
| - Hereditary spastic paraglegias - Arginase deficiency - COL4A1-Related spastic CP - Biotinidase deficiency - Aicardi-Goutières syndrome - Sulfite oxidase deficiency/Molybdenum cofactor deficiency22 - Leukodystrophies, such as metachromatic leukodystrophy,23 adrenoleukodystrophy,24 Sjorgen Larsson syndrome25 |
- Dopa-responsive dystonia - Sepiapterin reductase deficiency - Glutaric aciduria type 1 - Glucose transporter deficiency type 1 - Neurodegeneration with brain iron accumulation - Cerebral creatine deficiency syndrome - Lesch Nyhan syndrome - Cerebral folate deficiency - ADCY5-related dyskinesia - PCDH12-related dyskinesia34 - NKX2-1 related ataxic dyskinetic CP35 - TSEN54 Gene-related pontocerebellar hypoplasia type 236 |
- Glucose transporter deficiency type 1 - Ataxia telangiectasia - Pelizaeus-Merzbacher disease - Hereditary ataxias - Joubert syndrome - Mitochondrial cytopathies (mainly 8993 mutation)42 - Pontocerebellar hypoplasia36 - Cockayne syndrome43 - Niemann-Pick disease type C44 - Angelman syndrome12 - Gangliosidosis type 1, juvenile and adult forms45 - Non-ketotic hyperglycinemia3 - Maple syrup urine disease3 - NKX2-1 related ataxic dyskinetic CP35 |
CP- cerebral palsy
Common metabolic/genetic diseases with predominant spasticity
Several neurometabolic disorders can resemble spastic CP, resulting in misdiagnosis Table 3 & Figure 3.16-25
Figure 3.
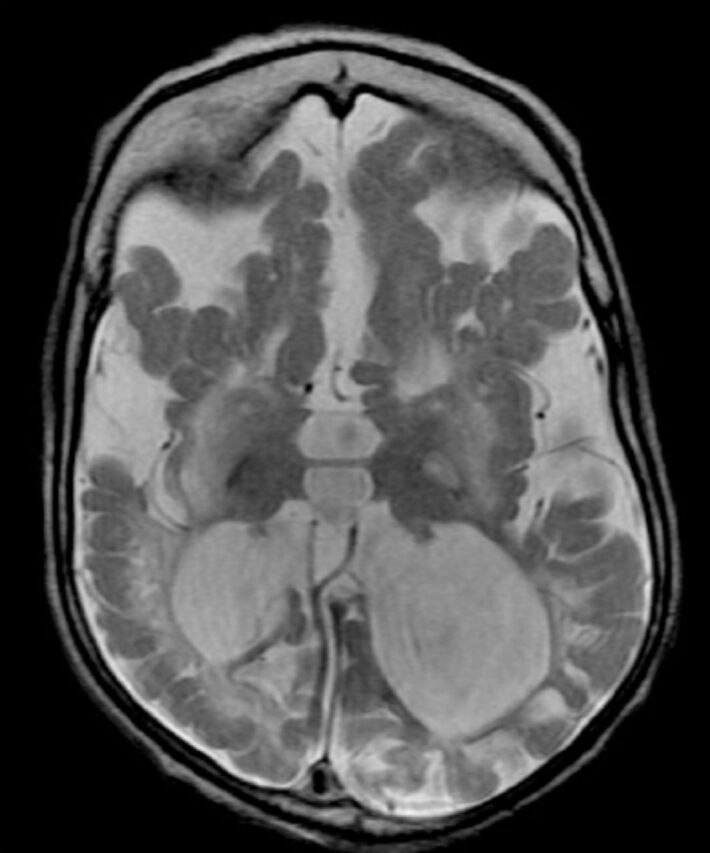
Axial T2-weighted brain magnetic resonance image of a 2-year-old child with molybdenum cofactor deficiency shows extensive brain damage, signifiant loss of white matter with cystic changes, ex vacuo dilatation of ventricles and mushrom-shaped gyri.
Hereditary spastic paraplegias (HSPs) constitute a wide and heterogeneous group of genetically determined neurodegenerative disorders that share a common clinical feature of lower limb spasticity and muscle weakness. There is considerable phenotypic and genetic heterogeneity; there are more than 80 genes, described so far, associated with HSP.16 According to the clinical phenotype, mode of inheritance and the affected gene, the HSP syndromes are classified into: (1) “uncomplicated” HSPs, which exhibit pure progressive lower-extremities spastic weakness, typically occur in the third decade of life, but may start as early as 10 years of age, and are inherited in an autosomal dominant fashion; and (2) “complicated” HSPs, where spastic paraplegia is associated with other neurologic and non-neurologic signs (peripheral neuropathy, cognitive impairment, extrapyramidal manifestations, ataxia, and brain imaging abnormalities); they usually present in early to mid-childhood, and are usually inherited in an autosomal recessive pattern.16 Most HSP patients but not all, have a slowly progressive weakness.6 Some HSPs may be non-progressive, as observed in infantile-onset HSP cases with de novo mutations in SPG3-related spastic paraplegia or DDHD2-related spastic paraplegia.17 Those patients are commonly misdiagnosed as having spastic diplegic/paraplegic CP. Brain and spinal MRI may appear normal or show specific findings suggestive of an HSP syndrome, such as brain atrophy, thin corpus callosum, spinal cord atrophy and a lipid peak, as observed in DDHD2 mutation.17
COL4A1-related spastic CP is a collagen vascular disorder which may cause an antenatal or perinatal cerebrovascular insult, resulting in a neonatal stroke syndrome that manifests as hemiplegia or quadriplegia.18 Screening for the mutation in COL4A1 gene should be undertaken in all neonates and infants with a stroke syndrome in the absence of any antenatal injury, prematurity, or with MRI features that are not characteristic of periventricular leukomalacia or cerebral infarction in the distribution of the anterior or middle cerebral artery.18
Biotinidase deficiency is an autosomal recessive inherited form of multiple carboxylase deficiency. The clinical presentation is usually in infancy with neurological manifestations (hypotonia, seizures, ataxia, and developmental delay), metabolic acidosis, and optic atrophy, in addition to dermatological abnormalities (such as skin rashes and alopecia). Patients with a delayed onset-biotinidase deficiency may develop motor limb weakness and spastic paresis attributed to spinal cord involvement, commonly misdiagnosed as CP.19 The MRI shows signal abnormality in the white matter of the spinal cord. Diagnosis of biotinidase deficiency is confirmed by demonstration of deficient enzyme on a dried blood spot which is included in the newborn screening in many countries, or by analyzing the BTD gene.6 Early treatment with biotin results in resolution of the cutaneous and neurological manifestations, whereas the audiological and ophthalmological manifestations, are more resistant to treatment.19
Arginase deficiency is an autosomal recessive urea cycle disorder. Patients between one and 3 years of age may present with developmental delay, motor and cognitive regression, slowly progressive spastic paresis, acquired microcephaly and failure to thrive.20 Ataxia and seizures may also develop. Arginase deficiency can be confirmed by elevated serum arginine concentrations and through the identification of a mutation on the ARG1 gene. Early diagnosis and early and sustained treatment with a low arginine diet and ‘nitrogen-scavenger’ drugs may ameliorate some of the impairments associated with the disorder.
Aicardi-Goutières syndrome is a genetically inherited autoimmune-mediated encephalopathy. It is a heterogeneous group of disorder characterized by increase cerebrospinal fluid (CSF) white cells and interferon-alpha level, negative serologic screen for prenatal infections, cerebral calcifications and white matter abnormalities.21 Neurological manifestation usually appear in early infancy, with progressive microcephaly, spasticity, dystonia, and severe psychomotor retardation. Non-neurological features include hepatosplenomegaly, elevated hepatic transaminases and thrombocytopenia. Phenotypic variability in Aicardi-Goutières syndrome is well recognized. There are reports of patients that have presented with only mild spasticity and had been diagnosed as CP.21 Genetic heterogeneity is common with mutations identified in the following genes: TREX1, RNASEH2A, RNASEH2B and RNASEH2C.21
Common metabolic/genetic disorders with predominant dyskinesia
Dyskinesia is defined as abnormal involuntary movements, with fluctuating patterns of tone and posture. The dyskinesia can be categorized as dystonia, tremor, chorea, athetosis, ballism, tic, akathisia and orofaciolingual dyskinesia. Table 3 lists some of the disorders mimicking dyskinetic CP.26-36
GCH1-related dopa-responsive dystonia is commonly misdiagnosed as CP.26 It is inherited an autosomal dominant metabolic disorder of the dopamine synthetic pathway. Typically, they present at childhood with gait disorder due to foot dystonia, diurnal fluctuation and improvement with rest or sleep. Patients have a normal intellectual and cognitive function. The confirmation of diagnosis is by molecular testing of the GCH1 gene. Levodopa, usually at low dose, is the mainstay of therapy with complete resolution of symptoms.26
Sepiapterin reductase deficiency is an autosomal recessive disorder caused by mutation in the sepiapterin reductase gene. The triad of oculogyric crises, paroxysmal stiffening and hypotonia is the hallmark of the disease in the early stages.27,28 These features have typical diurnal fluctuations with sleep disturbance. In some patients, the clinical manifestations are nonspecific, and they misdiagnosed as CP with hypotonia or dystonia.28 Neuroimaging is generally normal. Analysis of CSF neurotransmitters and SPR gene confirm the diagnosis. It is a treatable neurometabolic disorder and early treatment with a combination of L-dopa and 5-hydroxytryptophan results in a good neurological outcome.27,28
Glutaric aciduria type 1 is a rare autosomal recessive metabolic disorder caused by alteration in the glutaryl-CoA dehydrogenase gene (GCDH).29 Most untreated affected children present with an encephalopathic crisis, mainly triggered by a febrile illness, in the first 6 years of life. These crises will cause striatal injury and consequent dystonic movement disorder. Other affected individuals may present within the first year of life with delayed milestone, macrocephaly, hypotonia, choreoathetosis or dystonia. The typical MRI finding in glutaric aciduria type 1 is the presence of a widening of the Sylvian fissures and basal ganglia abnormalities (Figure 2). Diagnosis can be made by urine organic acids analysis and genetic study of the GCDH gene which is confirmatory. Early treatment with a diet restriction, riboflavin, carnitine therapy, and aggressive emergency treatment during catabolism improve the neurological outcome.29
Cerebral creatine deficiency syndromes are a group of the creatine synthesis and transport with 3 recognized syndromes: guanidinoacetate methyltransferase (GAMT), arginine glycine amidinotransferase (AGAT), and X-linked creatine transporter deficiency (SLC6A8).30 Patients typically present with an intellectual disability, severe speech delay and abnormal behavior. Additional features include seizures, ataxia dystonia and/or chorea. The brain MRI is usually nonspecific; however, MR spectroscopy shows absent creatine peak, which is the marker of the disease. The diagnosis is based on the analysis of creatine, creatinine and guanidoacetate in the urine and blood, and is confirmed by genetic analysis in the GAMT, AGAT, or SLC6A8 genes.30 Treatment consists of oral supplementation with high-dose creatine for all 3 cerebral creatine deficiency syndromes. Arginine-restricted diet with high-dose ornithine is additionally used in GAMT deficiency. Supplementation with arginine and glycine has been used additionally to treat SLC6A8 deficiency.30
Cerebral folate deficiency typically presents between 4-6 months as an infantile form, mainly in form of behavior changes and refractory epilepsy, whereas after one year of age it presents with a spastic-ataxic syndrome that can mimic CP, progressing to dystonia and schizophrenic syndrome in adolescence.31 The diagnosis is based on the low CSF levels of 5-methyltetrahydrofolate and normal plasma folate levels, and is confirmed by FOLR1 gene analysis. Folic acid therapy results in significant improvement of the clinical symptoms, brain abnormalities and function.31
Lesch-Nyhan syndrome is a rare X-linked recessive disorder of purine metabolism, caused by mutation in the HPRT gene, encoding hypoxanthine-guanine phosphoribosyltransferase. The characteristic clinical features are motor disability, self-injurious behavior and cognitive impairment. The motor symptoms typically start at the age of 3-6 months of life, subsequently, between the ages of 6-24 months, generalized dystonia emerges. Other extrapyramidal and pyramidal signs may occur.32 Children with Lesh-Nyhan syndrome may misdiagnosed initially with dyskinetic CP.32 Neuroimaging is usually normal or shows nonspecific brain atrophy.6 Molecular genetic testing for HPRT1 gene confirms the diagnosis. Current treatments are only symptomatic.
ADCY5-related dyskinesia is a significant genetic cause of early-onset non-progressive hyperkinetic movement disorders. Typically, the onset is around 6 months of age in a patient known to have hypotonia or motor delay.33 The movement disorder, in form of dystonia, chorea or choreoathetosis, is typically episodic and sleep-related. Dyskinesia may exacerbate upon awakening, falling asleep or during intercurrent illnesses for few minutes up to days.6 The exacerbation of dyskinesia in relation to sleep should prompt consideration of an underlying ADCY5 mutation.33 They may have normal cognitive function or mild intellectual disability and normal Brain MRI.6 Patients with ADCY5-related dyskinesia are frequently misdiagnosed as hypotonic or dyskinetic CP.33
Common metabolic/genetic disorders with predominant ataxia and/or mixed motor signs
Up to 10% of patients with CP are diagnosed with the ataxic type.1,2 Interestingly, isolated ataxia is rare feature in a patients with CP associated with birth asphyxia.6 Therefore, further investigation for metabolic/genetic causes should be strongly considered in any patient who presents with predominant ataxia, especially with normal brain MRI.6 Clinically it may be difficult to recognize ataxia in the first year of life and initially they may present with motor developmental delay and truncal hypotonia. Ataxia may also frequently coexist with other involuntary movements. Table 3 lists some disorders mimicking ataxic/hypotonic CP.37-45
Glucose transporter deficiency type 1 (Glut1 deficiency syndrome) is a disorder of the brain energy metabolism caused by mutations in the SLC2A1 gene, which encodes a glucose transporter protein type 1 (Glut1) that is required for glucose to cross the blood-brain barrier.37 Two main phenotypes can be identified. The classic phenotype manifests with an early onset intractable epilepsy, delayed development, acquired microcephaly, and movement disorder (ataxia, spasticity, and dystonia). The non-classic Glut1 phenotype (10-15% of patients) manifests mainly with paroxysmal dyskinesias, including intermittent ataxia, dystonia, exercise-induced dyskinesia, and alternating hemiplegia. The latter is the phenotype that may be misdiagnosed as CP.37 Ataxia and involuntary movements often fluctuate in severity, getting worse with fasting or exercise which is an important clue. Brain MRI is normal or may shows mild cerebral atrophy or subtle white matter abnormalities. The diagnosis is confirmed by a low CSF glucose concentration in the absence of low blood sugar, and analysis of the SLC2A1 gene. Treatment is based on the ketogenic diet or modified Atkins diet which is highly effective in controlling the seizures and improving gait disturbance.37
Ataxia-telangiectasia is a multisystem autosomal recessive inherited disorder, caused by mutations in the ATM gene.38 It presents with progressive ataxia, oculomotor apraxia, and oculocutaneous telangiectasia. Dyskinesia occur frequently, dystonia and choreoathetosis may be more prominent than ataxia at onset:38 thus, this condition can be misdiagnosed as ataxic-dyskinetic CP. Additional features of ataxia telangiectasia include immunodeficiency and risk of malignancy. Brain MRI shows progressive cerebellar atrophy. Blood tests show high levels of alpha-fetoprotein and absent immunoglobulin A. Analysis of the ATM gene confirms the diagnosis.38
Pelizaeus-Merzbacher disease (PMD) is an X-linked inherited disorder of myelination, due to mutations in the PLP1 gene encoding the proteolipid protein. Patients with PLP mutations exhibit phenotypic variability, ranging from the less involved spastic paraplegia type 2 to the more sever classical PMD.39 Classic PMD present during early infancy with head bobbing, nystagmus, psychomotor retardation, tremor, ataxia, and progressive spasticity. Dystonia and athetosis may also occur.6 Regarding neuroimaging, all forms are associated with central hypomyelination (Figure 1).39 PMD can be misdiagnosed as CP in early stages because the spasticity is slowly progressive and the MRI findings may not appreciate abnormalities during the first year of life.39
Hereditary ataxias are a large group of genetic disorders that are commonly characterized phenotypically by gait ataxia, pyramidal tract signs (often spastic diplegia), and incoordination of eye movements. The MRI typically shows cerebellar atrophy. The dominant forms are termed spinocerebellar ataxias, and are usually with an adult onset. The autosomal recessive forms include different diseases, mainly Freiderish ataxia, Refsum disease, ataxia with vitamin E deficiency, cerebrotendinous xanthomatosis, and coenzyme Q10 deficiency. Few of these disorders are amenable to effective treatment.40
Joubert syndrome is genetically heterogeneous group of disorders with an autosomal recessive or X-linked inheritance. Characterized by hypoplasia of the cerebellar vermis and classified in the group of ciliopathies.41 Clinically, ataxia is the predominant neurological sign; however, the initial presentation is typically with a hypotonia, oculomotor apraxia and breathing dysregulation, features that have been misdiagnosed as CP.45 The neurological features may be associated with variable multiorgan involvement mainly the retina, kidneys, liver and skeleton. The hallmark diagnosis of this condition is a pathognomonic midbrain-hind brain malformation on brain MRI “molar tooth sign” (Figure 4).41 Diagnosis may be confirmed by the identification of a mutation in one of the numerous genes associated with the condition.
Figure 4.
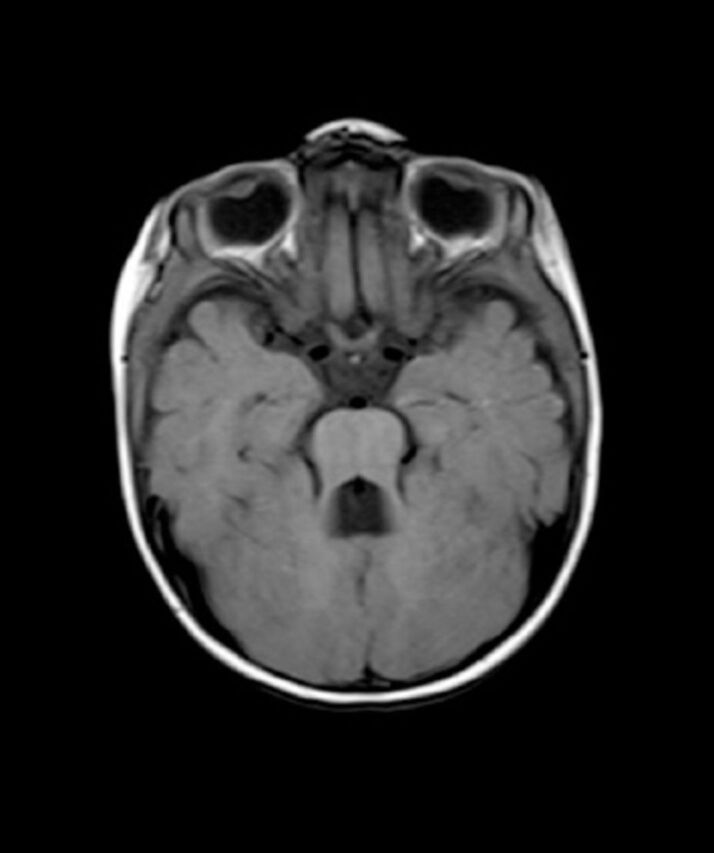
Axial T1-weighted brain magnetic resonance image of a 1-year-old child with Joubert syndrome shows the classical molar tooth sign.
Mitochondrial disorders, one of the most common neurometabolic diseases, are characterized by impaired energy production. They usually manifest as multi-organ disorder, often with neurological dysfunction and are rapidly progressive. Early isolated ataxia, dystonia, or hypotonia with slow course can be observed and confused as CP.42 Mitochondrial disorders affect a wide range of brain structures, including the supra and infratentorial white matter, cortical gray matter, basal ganglia, brainstem and cerebellum (Figure 5). Neuroimaging studies, metabolic and genetic investigations are required for the final diagnosis.
Figure 5.
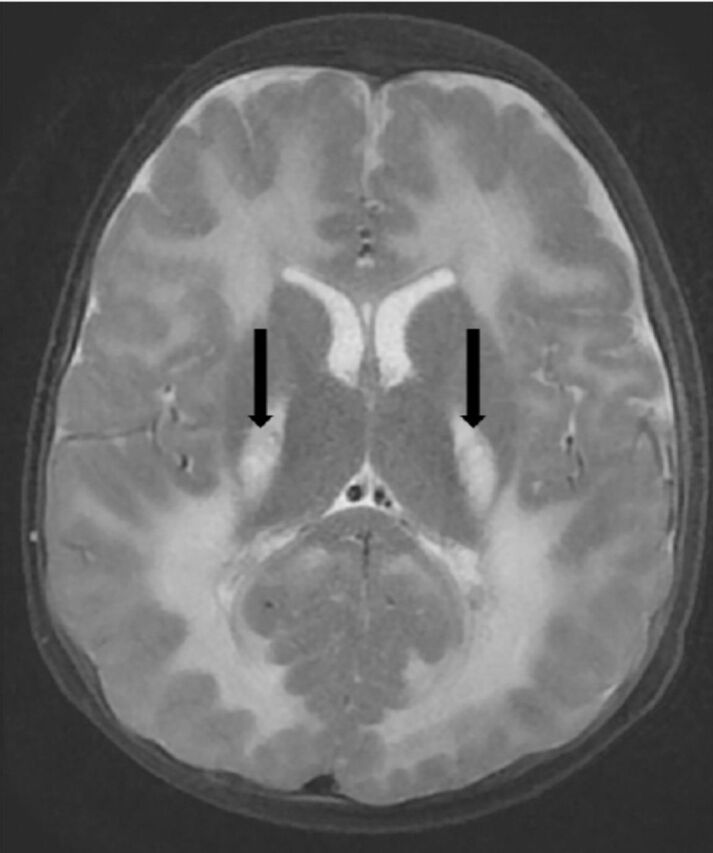
Axial T2-weighted brain magnetic resonance images of a 2-year-old child with mitochondrial leukoencephalopathy, caused by ISCA2 gene mutation shows leukodystrophy with supra and infra tentorial predominantly deep white matter involvement and central areas of cavitation (arrows).
Common metabolic/genetic disorders with predominant bulbar and oromotor dysfunction
Classically, bulbar and oromotor dysfunction, including speech and feeding difficulties, are not a major feature in CP. Bilateral Perisylvian, also called opercular syndrome, includes an oropharyngoglossal dysfunction, moderate to severe dysarthria and bilateral Perislyvian malformations on imaging.46 Children often exhibit a mild spastic diplegia or tetraplegia, as well as variable cognitive impairment, and epilepsy, and may be misdiagnosed as CP. The condition may be congenital or acquired. The most common congenital cause is a dysgenesis of the opercular cortex in the perisylvian region.46
In conclusion, a growing number of metabolic and genetic diseases may present with clinical features that resemble a CP phenotype. Thorough history and a careful neurological examination are essential in the evaluation of a patient suspected to have CP. The increase use of next generation sequencing in neurological conditions will detect more and more CP mimics. Identifying the etiology of CP has practical consequences, such as proper family counseling about the cause of the disease, risk of recurrence, prognosis, and in some diseases provide disease-modifying therapy, and therefore decreasing the total burden from CP.
Acknowledgements
We would like to thank Editage company (www.editage.com) for English language editing.
Footnotes
References
- 1.Graham HK, Rosenbaum P, Paneth N, Dan B, Lin JP, Damiano DL, et al. Cerebral palsy. Nat Rev Dis Primers. 2016;2:15082. doi: 10.1038/nrdp.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colver A, Fairhurst C, Pharoah PO. Cerebral palsy. Lancet. 2014;383:1240–1249. doi: 10.1016/S0140-6736(13)61835-8. [DOI] [PubMed] [Google Scholar]
- 3.Leach EL, Shevell M, Bowden K, Stockler-Ipsiroglu S, van Karnebeek CD. Treatable inborn errors of metabolism presenting as cerebral palsy mimics:systematic literature review. Orphanet J Rare Dis. 2014;9:197. doi: 10.1186/s13023-014-0197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zouvelou V, Yubero D, Apostolakopoulou L, Kokkinou E, Bilanakis M, Dalivigka Z, et al. The genetic etiology in cerebral palsy mimics:The results from a Greek tertiary care center. Eur J Paediatr Neurol. 2019;23:427–437. doi: 10.1016/j.ejpn.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Appleton RE, Gupta R. Cerebral palsy:not always what it seems. Arch Dis Child. 2018 doi: 10.1136/adc.85.5.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson TS, Pons R, Ghaoui R, Sue CM. Genetic Mimics of Cerebral Palsy. Mov Disord. 2019;34:625–636. doi: 10.1002/mds.27655. [DOI] [PubMed] [Google Scholar]
- 7.Segel R, Ben-Pazi H, Zeligson S, Fatal-Valevski A, Aran A, Gross-Tsur V, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84:1660–1668. doi: 10.1212/WNL.0000000000001494. [DOI] [PubMed] [Google Scholar]
- 8.Takezawa Y, Kikuchi A, Haginoya K, Niihori T, Numata-Uematsu Y, Inui T, et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann Clin Transl Neurol. 2018;5:538–551. doi: 10.1002/acn3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. 2015;20:176–182. doi: 10.1038/mp.2014.189. [DOI] [PubMed] [Google Scholar]
- 10.Carecchio M, Mencacci NE. Emerging monogenic complex hyperkinetic disorders. Curr Neurol Neurosci Rep. 2017;17:97. doi: 10.1007/s11910-017-0806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews AM, Blydt-Hansen I, Al-Jabri B, Andersen J, Tarailo-Graovac M, Price M, et al. Atypical cerebral palsy:genomics analysis enables precision medicine. Genet Med. 2018 doi: 10.1038/s41436-018-0376-y. [DOI] [PubMed] [Google Scholar]
- 12.Lee RW, Poretti A, Cohen JS, Levey E, Gwynn H, Johnston MV, et al. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med. 2014;16:821–844. doi: 10.1007/s12017-014-8331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al Salloum AA, El Mouzan MI, Al Omar AA, Al Herbish AS, Qurashi MM. The prevalence of neurological disorders in Saudi children:a community-based study. J Child Neurol. 2011;26:21–24. doi: 10.1177/0883073810371510. [DOI] [PubMed] [Google Scholar]
- 14.Reid SM, Dagia CD, Ditchfield MR, Carlin JB, Reddihough DS. Population-based studies of brain imaging patterns in cerebral palsy. Dev Med Child Neurol. 2014;56:222–232. doi: 10.1111/dmcn.12228. [DOI] [PubMed] [Google Scholar]
- 15.Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–939. doi: 10.1007/s00439-017-1821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Souza PVS, de Rezende Pinto WBV, de Rezende Batistella GN, Bortholin T, Oliveira ASB. Hereditary Spastic Paraplegia:Clinical and Genetic Hallmarks. Cerebellum. 2017;16:525–551. doi: 10.1007/s12311-016-0803-z. [DOI] [PubMed] [Google Scholar]
- 17.Thabet F, Tlili-Graiess K, Tabarki B. Distinct neuroimaging features of DDHD2 gene-related spastic paraplegia, a mimicker of cerebral palsy. Arch Dis Child. 2019 doi: 10.1136/archdischild-2018-316484. [DOI] [PubMed] [Google Scholar]
- 18.Bi D, Wang H, Shang Q, Xu Y, Wang F, Chen M, et al. Association of COL4A1 gene polymorphisms with cerebral palsy in a Chinese Han population. Clin Genet. 2016;90:149–155. doi: 10.1111/cge.12723. [DOI] [PubMed] [Google Scholar]
- 19.Wolf B. Biotinidase deficiency should be considered in individuals exhibiting myelopathy with or without and vision loss. Mol Genet Metab. 2015;116:113–118. doi: 10.1016/j.ymgme.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Jichlinski A, Clarke L, Whitehead MT, Gropman A. “Cerebral Palsy” in a Patient With Arginase Deficiency. Semin Pediatr Neurol. 2018;26:110–114. doi: 10.1016/j.spen.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Al Mutairi F, Alfadhel M, Nashabat M, El-Hattab AW, Ben-Omran T, Hertecant J, et al. Phenotypic and Molecular Spectrum of Aicardi-Goutières Syndrome:A Study of 24 Patients. Pediatr Neurol. 2018;78:35–40. doi: 10.1016/j.pediatrneurol.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Bosley TM, Alorainy IA, Oystreck DT, Hellani AM, Seidahmed MZ, Osman Mel F, et al. Neurologic injury in isolated sulfite oxidase deficiency. Can J Neurol Sci. 2014;41:42–48. doi: 10.1017/s0317167100016243. [DOI] [PubMed] [Google Scholar]
- 23.Van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy:Disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29:261–273. doi: 10.1016/j.beem.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy:inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet. 2015;8:109–121. doi: 10.2147/TACG.S49590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho KH, Shim SH, Kim M. Clinical, biochemical, and genetic aspects of Sjögren-Larsson syndrome. Clin Genet. 2018;93:721–730. doi: 10.1111/cge.13058. [DOI] [PubMed] [Google Scholar]
- 26.Kulshreshtha D, Maurya PK, Singh AK, Thacker AK. Dopa-responsive Dystonia in a Child Misdiagnosed as Cerebral Palsy. J Pediatr Neurosci. 2017;12:172–173. doi: 10.4103/jpn.JPN_123_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman J, Roze E, Abdenur JE, Chang R, Gasperini S, Saletti V, et al. Sepiapterin reductase deficiency:a treatable mimic of cerebral palsy. Ann Neurol. 2012;71:520–530. doi: 10.1002/ana.22685. [DOI] [PubMed] [Google Scholar]
- 28.AlSubhi S, AlShahwan S, AlMuhaizae M, AlZaidan H, Tabarki B. Sepiapterin reductase deficiency:Report of 5 new cases. Eur J Paediatr Neurol. 2017;21:583–586. doi: 10.1016/j.ejpn.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Boy N, Mühlhausen C, Maier EM, Heringer J, Assmann B, Burgard P, et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I:second revision. J Inherit Metab Dis. 2017;40:75–101. doi: 10.1007/s10545-016-9999-9. [DOI] [PubMed] [Google Scholar]
- 30.Fons C, Campistol J. Creatine Defects and Central Nervous System. Semin Pediatr Neurol. 2016;23:285–289. doi: 10.1016/j.spen.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Molero-Luis M, Serrano M, O'Callaghan MM, Sierra C, Pérez-Dueñas B, García-Cazorla A, et al. Clinical, etiological and therapeutic aspects of cerebral folate deficiency. Expert Rev Neurother. 2015;15:793–802. doi: 10.1586/14737175.2015.1055322. [DOI] [PubMed] [Google Scholar]
- 32.Fu R, Chen CJ, Jinnah HA. Genotypic and phenotypic spectrum in attenuated variants of Lesch-Nyhan disease. Mol Genet Metab. 2014;112:280–285. doi: 10.1016/j.ymgme.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang FC, Westenberger A, Dale RC, Smith M, Pall HS, Perez-Dueñas B, et al. Phenotypic insights into ADCY5-associated disease. Mov Disord. 2016;31:1033–1040. doi: 10.1002/mds.26598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki-Muromoto S, Wakusawa K, Miyabayashi T, Sato R, Okubo Y, Endo W, et al. A case of new PCDH12 gene variants presented as dyskinetic cerebral palsy with epilepsy. J Hum Genet. 2018;63:749–753. doi: 10.1038/s10038-018-0432-0. [DOI] [PubMed] [Google Scholar]
- 35.McMichael G, Haan E, Gardner A, Yap TY, Thompson S, Ouvrier R, et al. NKX2-1 mutation in a family diagnosed with ataxic dyskinetic cerebral palsy. Eur J Med Genet. 2013;56:506–509. doi: 10.1016/j.ejmg.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Pacheva IH, Todorov T, Ivanov I, Tartova D, Gaberova K, Todorova A, et al. TSEN54 Gene-Related Pontocerebellar Hypoplasia Type 2 Could Mimic Dyskinetic Cerebral Palsy with Severe Psychomotor Retardation. Front Pediatr. 2018;6:1. doi: 10.3389/fped.2018.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Méneret A, Roze E. GLUT1 Deficiency in a Patient Diagnosed as Cerebral Palsy:Is NGS a Valuable Tool to Be Considered in All Cases of CP to Detect Underlying Genetic Disorders? Mov Disord Clin Pract. 2019;6:277–279. doi: 10.1002/mdc3.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia:a review. Orphanet J Rare Dis. 2016;11:159. doi: 10.1186/s13023-016-0543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen YC, Liang WC, Su YN, Jong YJ. Pelizaeus-Merzbacher disease, easily misdiagnosed as cerebral palsy:a report of a three-generation family. Pediatr Neonatol. 2014;55:150–153. doi: 10.1016/j.pedneo.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan R, Yau WY, O'Connor E, Houlden H. Spinocerebellar ataxia:an update. J Neurol. 2019;266:533–544. doi: 10.1007/s00415-018-9076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar S, Singh D. Joubert Syndrome Mimicking Hypotonic Cerebral Palsy. Indian J Pediatr. 2016;83:1505. doi: 10.1007/s12098-016-2196-x. [DOI] [PubMed] [Google Scholar]
- 42.El-Hattab AW, Scaglia F. Mitochondrial cytopathies. Cell Calcium. 2016;60:199–206. doi: 10.1016/j.ceca.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Vafaee A, Baghdadi T, Norouzzadeh S. Cockayne Syndrome Misdiagnosed as Cerebral Palsy. Iran J Child Neurol. 2018;12:162–168. [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki R, Tanaka A, Matsui T, Gunji T, Tohyama J, Nairita A, et al. Niemann-Pick Disease Type C Presenting as a Developmental Coordination Disorder with Bullying by Peers in a School-Age Child. Case Rep Pediatr 2015. 2015:807591. doi: 10.1155/2015/807591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Regier DS, Proia RL, D'Azzo A, Tifft CJ. The GM1 and GM2 Gangliosidoses:Natural History and Progress toward Therapy. Pediatr Endocrinol Rev. 2016;13:663–673. [PMC free article] [PubMed] [Google Scholar]
- 46.McMillan HJ, Holahan AL, Richer J. Worster-Drought Syndrome Associated With LINS Mutations. Child Neurol Open. 2018;5:2329048X1879108. doi: 10.1177/2329048X18791083. [DOI] [PMC free article] [PubMed] [Google Scholar]