

In this study, Shao et al. investigated the transcriptional regulation of MeCP2, and identified six putative noncoding regulatory elements of Mecp2, two of which are conserved in humans. Their findings provide insight into transcriptional regulation of Mecp2/MECP2 and highlight genomic sites that could serve as diagnostic and therapeutic targets in Rett syndrome (RTT) and MECP2 duplication syndrome (MDS).
Keywords: MeCP2, cis-regulatory elements, noncoding, neurological disorders
Abstract
While changes in MeCP2 dosage cause Rett syndrome (RTT) and MECP2 duplication syndrome (MDS), its transcriptional regulation is poorly understood. Here, we identified six putative noncoding regulatory elements of Mecp2, two of which are conserved in humans. Upon deletion in mice and human iPSC-derived neurons, these elements altered RNA and protein levels in opposite directions and resulted in a subset of RTT- and MDS-like behavioral deficits in mice. Our discovery provides insight into transcriptional regulation of Mecp2/MECP2 and highlights genomic sites that could serve as diagnostic and therapeutic targets in RTT or MDS.
Coding variants in hundreds of genes are known to alter protein levels and lead to intellectual disability (ID) (Schanze et al. 2018). However, only a handful of disease-causing mutations in noncoding cis-regulatory elements (CREs) (Soldner et al. 2016; Oz-Levi et al. 2019) have been identified, and our understanding of how these contribute to ID is limited. MECP2 is an exemplar ID-causing, dosage-sensitive gene, with neurological dysfunction arising from both decreased (RTT) (Amir et al. 1999) and increased (MDS) (Van Esch et al. 2005) levels of MeCP2. This dosage sensitivity underscores the concept that precise control of MeCP2 levels is important for normal brain function (Chao and Zoghbi 2012; Sztainberg et al. 2015). Regulation of MECP2 occurs at the transcriptional, post-transcriptional (Gennarino et al. 2015; Rodrigues et al. 2016), and post-translational levels (Lombardi et al. 2017; Yagasaki et al. 2018), with the latter two being the most well studied. Currently, little is known about the transcriptional regulation of MECP2 beyond its core promoter region (Liu and Francke 2006; Nagarajan et al. 2006; Swanberg et al. 2009).
Results and Discussion
To identify potential CREs of Mecp2 in the brain, we profiled open chromatin in the developing and adult mouse brain using the assay for transposase-accessible chromatin with deep sequencing (ATAC-seq) (Buenrostro et al. 2013). We restricted our search to accessible regulatory elements within the 100-kb genomic region of Mecp2, flanked upstream by the Opn1mw gene and downstream by the Irak1 gene. This genomic region, containing only Mecp2, faithfully drives expression of human MECP2 in a transgenic mouse line, rescuing all neurological defects in Mecp2-null mice (Collins et al. 2004). We identified six putative regulatory elements (Fig. 1A): one at the promoter (Peak-4), three in intron 2 that increase in accessibility during development (Peak-1, Peak-2, and Peak-3), one upstream of Mecp2 (Peak-5) that decreases in accessibility in the adult brain compared with the postnatal day 6 brain, and one peak upstream of Mecp2 (Peak-6) that is consistently accessible throughout development. Importantly, these same peaks are present in previously published ATAC-seq data sets from multiple neuronal cell types in the adult mouse brain (Supplemental Fig. S1; Mo et al. 2015), confirming our findings.
Figure 1.

Identification of putative cis-regulatory elements of Mecp2. (A) ATAC-seq reads mapped to the Mus musculus genome (mm9) are shown from nuclei isolated from embryonic, postnatal, and adult mouse brains (specifically E12.5, P6, and 8 wk of age). Exons are denoted by solid blue rectangles, while introns are between exons. The 3′ UTR is represented by a thinner but longer blue block relative to the coding exons. Note that locus is transcribed in the opposite direction relative to the image shown (i.e., the promoter is at the right, and the 3′ UTR is at the left). Peaks 1, 2, 3, 5, and 6 were pursued for functional analysis of cis-regulatory elements, whereas Peak-4 (which spanned the proximal promoter) was not studied further. (B) RT-qPCR shows Mecp2 mRNA expression in different knockout lines (n = 5). (C) Western blot analysis of mouse brain tissue shows MeCP2 protein expression levels in each different knockout line. Representative Western blot image of MeCP2 protein levels is shown with Gapdh as a loading control, and solid vertical lines indicate areas of the image that have been spliced to remove unneeded lanes (n = 4). All data were analyzed by one-way ANOVA followed by Dunnett post hoc test. Data are presented as mean ± SEM. (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001.
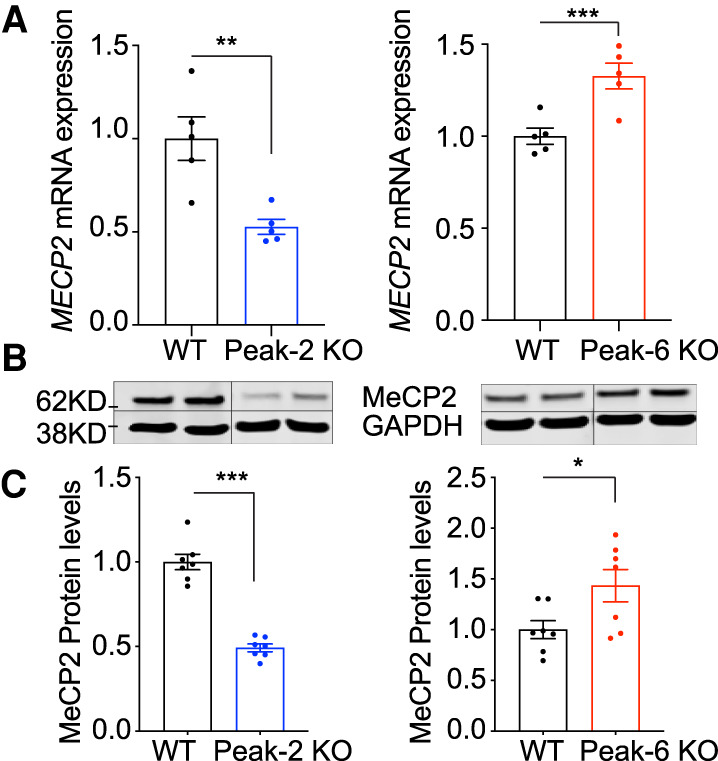
To test whether these putative CREs regulate Mecp2 expression in the mouse brain, we deleted the genomic regions corresponding to each individual peak using CRISPR-Cas9 genome editing (Supplemental Table S1), generating five unique mouse lines. We excluded Peak-4 from our analysis because it corresponds to the Mecp2 promoter, whose deletion would likely ablate Mecp2 expression. We measured Mecp2 mRNA expression in the brains of our knockout (Peak KO) lines and found that Mecp2 expression was decreased by ∼20%–30% in Peak-2KO/y, Peak-3KO/y, and Peak-5KO/y mice, while it was increased by 50% in Peak-6KO/y mice and unchanged in Peak-1KO/y mice (Fig. 1B). Measurement of MeCP2 protein levels by Western blot showed 30% reduction (P < 0.01) in Peak-2KO/y mice and 70% increase (P < 0.0001) in Peak-6KO/y mice, while protein level changes in Peak-1KO/y, Peak-3KO/y, and Peak-5KO/y mice were not significant (Fig. 1C). Interestingly, Peak-2 and Peak-6 are the only two elements with strong sequence conservation at the nucleotide level between mice and humans, suggesting that evolutionary pressure maintained these two gene regulatory elements (Supplemental Fig. S2).
Given that the magnitude of change in MeCP2 levels in both the Peak-2KO/y and Peak-6KO/y mice is less than that of the Mecp2flox/y allele (Samaco et al. 2008) and MECP2 duplication mouse models (Collins et al. 2004), respectively, we wanted to identify and characterize any neurological dysfunction that may arise from more subtle changes in MeCP2 levels. Therefore, we performed a battery of behavioral tests on Peak-2KO/y and Peak-6KO/y male mice, after confirming peak knockout did not result in any gross histological abnormalities as assessed by Cresyl violet staining (Supplemental Fig. S3).
At 10 wk, Peak-2KO/y mice, which have ∼30% reduction in Mecp2 expression, were hyperactive compared with their wild-type littermates (Fig. 2A). At 24 wk, these mice had anxiety-like phenotypes (Fig. 2B) and social deficits (Fig. 2C). At 40 wk, these mice showed social dominance deficits (Fig. 2D). These behavioral deficits are reminiscent of those observed in Mecp2flox/y mice and some RTT (Moretti et al. 2005) and autism mouse models (Spencer et al. 2005; Kazdoba et al. 2016). Unlike Mecp2flox/y mice that express 50% of the normal MeCP2 level (Samaco et al. 2008), we did not observe any sensorimotor gating deficits, motor abnormalities, or learning and memory defects in the Peak-2KO/y mice (Supplemental Fig. S4A–F). Last, we measured the expression of several genes that are dysregulated in Mecp2-null mice (Boxer et al. 2020). Strikingly, expression of these genes in the cortex of Peak-2KO/y mice were altered in the same direction as reported in Mecp2-null animals (Supplemental Fig. S4G,H; Chahrour et al. 2008). These data suggest that a mild reduction in MeCP2 levels mimics a subset of behavioral and molecular changes observed in Mecp2-null mice.
Figure 2.
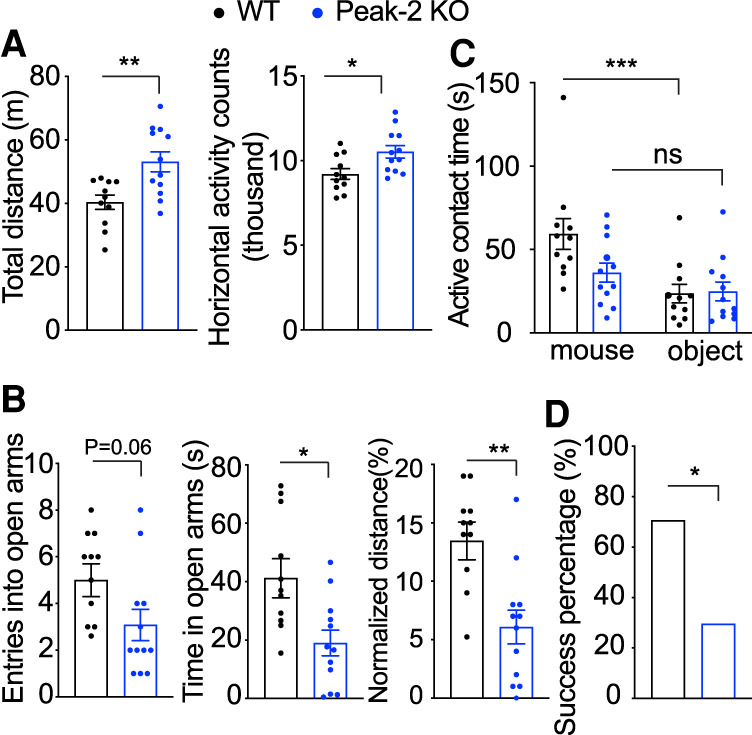
Peak-2KO/y mice show age-related behavioral deficits and abnormal RTT-like gene expression. (A) Peak-2KO/y mice traveled further and displayed more activity counts in the open field assay than their wild-type littermates in open field assay at 10 wk of age (n = 12). (B) Peak-2KO/y mice show anxiety-like phenotypes as measured by decreased entries, less time spent, and decreased distance in the open arms in the elevated plus maze at 24 wk of age (n = 12). (C) Peak-2KO/y mice show social deficits as measured by decreased time investigating a novel mouse in the three-chamber sociability assay at 24 wk of age (n = 12). (D) Peak-2KO/y mice show social dominance deficits as measured by decreased winning percentage in the tube test at 40 wk of age (n = 12). Data are presented as mean ± SEM. All data were analyzed by two-tailed t-test. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (ns) not significant.
In contrast, Peak-6KO/y mice, which display an ∼70% increase in MeCP2, show hypoactivity (Fig. 3A) and anxiety-like phenotypes (Fig. 3B,C) at 10 wk. At 24 wk, these mice showed hippocampal-dependent contextual learning deficits in the contextual fear assay (Fig. 3D). These phenotypes resemble the behavioral profile of MECP2-Tg1 mice (which have a 100% increase of MeCP2 protein). However, unlike the MECP2-Tg1 mice, Peak-6KO/y mice showed no deficits in sensorimotor gating, motor function, or social behavior (Supplemental Fig. S5A–E). When we measured the expression of several genes known to be dysregulated in the MECP2-Tg1 mouse model (Chahrour et al. 2008; Samaco et al. 2012), we found these MeCP2 targets were altered in the same direction as reported in MECP2-Tg1 mice in the cortex of Peak-6KO/y mice (Supplemental Fig. 5F,G). These data suggest that a moderate increase in MeCP2 protein level recapitulates a subset of MDS-like behavioral and molecular phenotypes.
Figure 3.
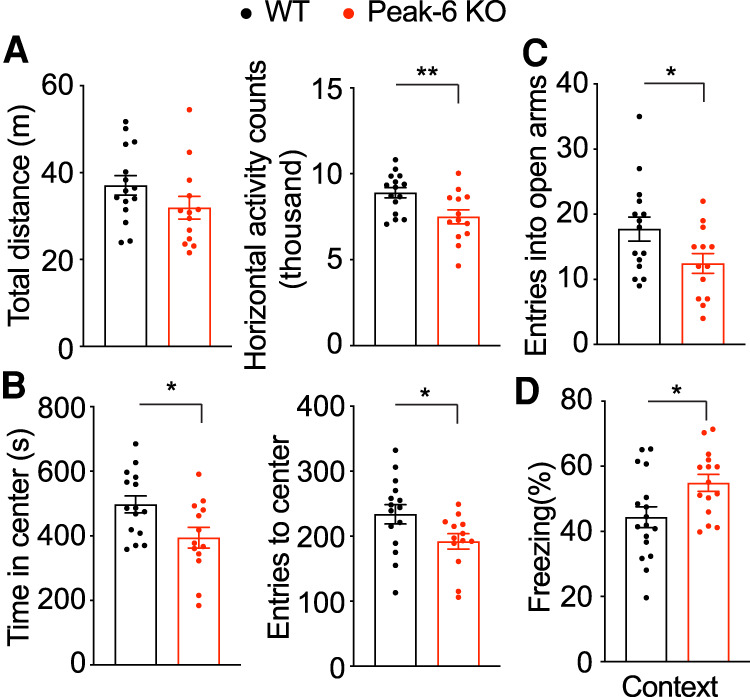
Peak-6KO/y mice show abnormal MDS-like gene expression and behavioral deficits. (A) Peak-6KO/y mice show hypoactivity, with fewer activity counts in open field assay at 9 wk of age (n = 13–15). (B) Peak-6KO/y mice show anxiety-like phenotypes as evidenced by decreased entries and time spent at the center in open field assay at 9 wk of age (n = 13–15). (C) Peak-6KO/y mice show anxiety-like phenotypes as measured by reduced entries to the open arms in the elevated plus maze at 10 wk of age (n = 13–15). (D) Peak-6KO/y mice show learning and memory deficits using a fear conditioning test at 36 wk of age (n = 13–15). Data are presented as mean ± SEM. All data were analyzed by two-tailed t-test. (*) P < 0.05, (**) P < 0.01.
We next subjected the genomic sequences of these peaks to transcription factor motif analysis using HOMER to identify putative factors that bind within these peaks, and this analysis identified a potential CTCF binding site on Peak-6 (Supplemental Fig. S6A; Heinz et al. 2010). To validate this prediction, we conducted CTCF chromatin immunoprecipitation (ChIP) followed by qRT-PCR in the wild-type mouse frontal cortex and found CTCF enrichment on Peak-6 (Supplemental Fig S6B). Disruption of the structural protein CTCF can result in dysregulation of genes near its binding site (Dixon et al. 2012). We measured the expression of genes upstream of Peak-6 and downstream from Mecp2 to see whether their expression is altered. We found the expression of Irak1 (2 kb downstream) and Bgn (480 kb downstream) are increased but not Zfp185, which is 980 kb downstream from Mecp2. Similarly, the expression of Emd (140 kb upstream) and Taz (175 kb upstream) are elevated but not Ikbkg (310 kb upstream), indicating that disruption of this regulatory element can cause dysregulation of some neighboring genes (Supplemental Fig. S6C,D). However, the expression of these genes is not affected in Peak-2 KO mice (Supplemental Fig. S6E). Mouse genetic studies have demonstrated that increasing MeCP2 levels alone is sufficient to cause the neurological phenotypes seen in MDS (Ramocki et al. 2010). These data together with our discovery that the phenotypes seen in Peak-6KO/y mice are a subset of those seen upon doubling MeCP2 support the conclusion that the 70% increase in MeCP2 is the main driver of the disease phenotype in Peak-6KO/y mice.
Given that these two CREs regulate Mecp2 expression and their sequences are highly conserved in humans, we first mined ATAC-seq data generated from human prefrontal cortex to examine whether these peaks are present (Markenscoff-Papadimitriou et al. 2020). Our analyses revealed that both Peak-2 and Peak-6 are present, suggesting both peaks are conserved (Supplemental Fig. S7A). We next tested whether deletion of these conserved CREs also regulates MECP2 expression in cultured human neurons. First, we deleted the two CREs in two male, human induced pluripotent stem cell (iPSC) lines using CRISPR/Cas9 (Supplemental Table S2). Next, we generated Vglut1-positive glutamatergic neurons (iNeurons) from these stem cells using directed differentiation via overexpression of NEUROGENIN2 (NGN2) (Supplemental Fig. S8; Zhang et al. 2013). Consistent with our mouse models, MECP2 mRNA expression is reduced in Peak-2KO/y iNeurons by ∼50%, while it is increased in Peak-6KO/y iNeurons by ∼30% (Fig. 4A). Protein levels of MeCP2 also showed a concomitant change (Fig. 4B,C; Supplemental Fig. S9). These data strongly suggest that Peak-2 and Peak-6 regulate MECP2 expression. Furthermore, motif analysis also identified a potential CTCF binding site on the human sequence of Peak-6 (Supplemental Fig. S7B; Heinz et al. 2010), and we validated that CTCF binds within this region using ChIP-qPCR (Supplemental Fig. S7C).
Figure 4.
Deletion in conserved cis-regulatory elements of MECP2 affect the mRNA and protein levels in human iPSC-derived neurons. (A) MECP2 mRNA expression is reduced in Peak-2 KO and increased in Peak-6 KO iNeurons as measured by RT-qPCR (n = 5). (B) MeCP2 protein expression is reduced in Peak-2 KO and increased in Peak-6 KO iNeurons. Representative Western blot of MeCP2 levels in CRE deletion iNeurons with GAPDH displayed as loading control. Solid vertical line indicates a spliced region of the single gel image to remove unneeded lanes (n = 7). (C) Quantification of MeCP2 protein expression by Western blot normalized to GAPDH loading control (n = 7). All data were analyzed by two-tailed t-test. Data are presented as mean ± SEM. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
To date, several mouse models with varying levels of MeCP2 have been well characterized; notably, Mecp2flox/y mice (50% MeCP2) and transgenic MECP2-Tg1 (200% MeCP2) show progressive behavioral deficits (Collins et al. 2004; Samaco et al. 2008). Our two CRE KO mouse models (Peak-2 and Peak-6) perturb MeCP2 levels in similar directions (albeit milder) than the Mecp2flox/y mice and the MECP2-Tg1 mice, respectively. Our in vivo studies show that even these subtle alterations are sufficient to produce some behavioral phenotypes observed in Mecp2flox/y and MECP2-Tg1 mice (Fig. 5). Our CRE KO lines provide a unique allelic series demonstrating that disease severity directly corresponds with precise MeCP2 levels in the brain. We demonstrate, in agreement with previous work, that normal MeCP2 dosage (100%) is required for normal brain function in mice, and either a 20%–30% decrease or a 50%–70% increase in MeCP2 may lead to some neuropsychiatric phenotypes (altered activity, anxiety, and social and learning deficits), while 50% decrease or 100% increase of MeCP2 leads to severe neurodevelopmental disorders like RTT or MDS, respectively.
Figure 5.

Summary of Peak KO phenotypes and comparison with MeCP2 50% loss (Mecp2flox/y) or MDS mouse model (MECP2-Tg1).
MeCP2 expression has a unique spatial and temporal pattern, where it is the lowest in the liver and highest in the brain (Supplemental Fig. S10A) and increases significantly postnatally (Supplemental Fig. S10B). Accurate postnatal MeCP2 level is critical for normal brain function. Generating ATAC-seq profiles during developmental stages that coincide with the timing of MeCP2 increase identified regions with accessible chromatin structure at the MECP2 gene locus. These accessible regions may contain specific chromatin remodelers or transcription factors that regulate MECP2 expression. Further studies will be needed to identify the putative transcription factor or factors that bind to Peak-2 or Peak-6 to regulate MECP2 expression.
Understanding the phenotypic range that results from varying MeCP2 levels is critical for a number of reasons: first, assessing the type and severity of clinical manifestations of MECP2 dosage-related disorders; second, classifying and treating non-RTT patients with neuropsychiatric disorders due to mild MECP2 mutations or noncoding mutations that affect MeCP2 levels (e.g., in CREs); and third, predicting the clinical benefits of therapeutic interventions based on the degree of modulation of MeCP2 levels. Studies on existing mouse models across a range of MeCP2 levels have shown that phenotypic severity and symptom onset vary in correlation with the deviation from normal (Chao and Zoghbi 2012). Importantly, our CRE KO models provide information about the previously unexplored intermediary range between 50% and 100% (Peak-2 KO) and between 150% and 200% (Peak-6 KO). Our study suggests that reducing the MeCP2 protein level from 200% in MDS patients by even a small degree to 150%–170% could improve many phenotypes, such as motor and sensorimotor deficits. Similarly, in patients with a <50% MeCP2 protein level, boosting MeCP2 level to just 70% could potentially improve some RTT phenotypes. This provides hope that even treatments that slightly correct MeCP2 protein level in MDS or RTT patients could achieve clinically relevant phenotypic improvement.
Noncoding regions account for >98% of the human genome, yet we have limited understanding of their contribution to disease. Here, we demonstrate that disruption of CREs alters MeCP2 levels in cultured human iPSC-derived neurons. Human mutations in these cis-regulatory regions could contribute to atypical RTT patients without mutations in MECP2 coding regions or patients with ID, autism, or neuropsychiatric disorders. Many genes that cause neurodevelopmental disorders and autism (Satterstrom et al. 2020) are dosage sensitive (Han et al. 2013; Rocha et al. 2016; Rice and McLysaght 2017; Raveau et al. 2018; Schnabel et al. 2018). It is possible that unidentified mutations in regulatory elements of these genes may also affect their abundance and lead to disease phenotype. Our research highlights the importance of whole-genome sequencing (WGS) to achieve diagnoses in cases in which exome sequencing fails. Furthermore, the catalog of mutations by WGS on the X chromosome is underrepresented due to the reduced effective population size of sequenced X chromosomes, as males only carry one copy (Telenti et al. 2016). Our study also demonstrates that representation of the noncoding regions on the X-linked gene MECP2 in WGS needs to be reexamined to detect any potential variants in these critical regions.
In conclusion, we identified and functionally characterized two novel, evolutionarily conserved regulatory elements required for normal expression of Mecp2/MECP2. Deletion of either regulatory element in mice caused mild neurological dysfunction, highlighting how small changes in MeCP2 levels in either direction disrupt neurological function. More broadly, this study underscores the potential contribution of mutations in regulatory regions to various neuropsychiatric phenotypes and calls for similar studies of regulatory regions in other dosage sensitive genes involved in autism and ID.
Materials and methods
Animals
Mice were housed in an AAALAS-certified level 3 facility on a 14-h light cycle. All CRE knockout mice were generated in the Genetically Engineered Mouse Core at Baylor College of Medicine, backcrossed with wild-type C57BL6/J mice for five generations. Only male offspring were used for analysis. All procedures to maintain and use these mice were approved by the Institutional Animal Care and Use Committee for Baylor College of Medicine and Affiliates.
Generation of the knockout mice of regulatory elements
All CRE deletion mice were generated via CRISPR/Cas9-mediated gene editing. Briefly, two sgRNAs targeting the 5′ and 3′ (left [L] and right [R]) ends of the putative CREs were designed in Benchling and synthesized by IDT. sgRNAs were in vitro transcribed with the MEGAshortscript T7 transcription kit (Invitrogen). Details about injection are available in the Supplemental Material.
Behavioral assays
All data acquisition and analyses were performed by an individual blinded to the genotype. All behavioral studies were performed during the light period. At least 1 d was given between assays for the mice to recover. All the tests were performed as previously described (Chao et al. 2010) with few modifications. A detailed description is in the Supplemental Material.
ATAC-seq
Nuclear isolation was performed according to Mo et al. (2015) with two biological replicates. Nuclei were collected from fresh brain tissues for ATAC-seq. Detailed descriptions are available in the Supplemental Material.
Statistical analysis
Statistical significance was determined using GraphPad Prism software. The number of animals (n) and the specific statistical tests for each experiment are indicated in the figure legends. Sample size for behavioral studies was determined based on previous experience using mice with the same background.
Data and materials
All data needed to evaluate the conclusions in this study are present here and/or the Supplemental Material. ATAC-seq data from different mouse neuronal cells were obtained from Mo et al. (2015) and GEO GSE63137. ATAC-seq data from human brains were obtained from GSE149268. The accession number for the raw and processed data files reported here is GEO GSE152719. Additional data related to this study are available on request.
Supplementary Material
Acknowledgments
We thank Y. Sun for mouse genotyping and the Genetically Engineered Mouse Core, Human Stem Cell Core, RNA In Situ Hybridization Core, Neurovisualization Core, Neurobehavioral Core, and Human Neuronal Differentiation Core at the Jan and Dan Duncan Neurological Research Institute. This project was funded by the National Institutes of Health (NIH; 5R01NS057819 to H.Y.Z., and 1F32HD100048-01 to S.S.B.), the American Heart Association (19PRE34410104 to M.C.G.), institutional startup funds (to J.D.W.), the Department of Defense (W81XWH-18-1-0350 to J.D.W.), Canadian Institutes of Health Research (PJT-155922 to J.D.W.), the Howard Hughes Medical Institute (to H.Y.Z.), the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (NIH 5P50HD103555), the Henry Engel Fund, and the Ziff family fund.
Author contributions: Y.S. and H.Y.Z. designed the project. Y.S. performed the biochemistry, behavioral, and iNeuron experiments. H.P.T. performed behavioral experiments. S.S.B. performed iNeuron experiments. M.C.G. and J.D.W. performed ATAC-seq experiments and conducted bioinformatic analysis. Y.S. wrote the manuscript and prepared the figures, with contributions from all coauthors according to their area of expertise. H.Y.Z. reviewed all of the data and edited the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.345397.120.
Competing interest statement
The authors declare no competing interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Boxer LD, Renthal W, Greben AW, Whitwam T, Silberfeld A, Stroud H, Li E, Yang MG, Kinde B, Griffith EC, et al. 2020. MeCP2 represses the rate of transcriptional initiation of highly methylated long genes. Mol Cell 77: 294–309.e9. 10.1016/j.molcel.2019.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. 2008. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224–1229. 10.1126/science.1153252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY. 2012. MeCP2: only 100% will do. Nat Neurosci 15: 176–177. 10.1038/nn.3027 [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. 2010. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468: 263–269. 10.1038/nature09582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. 2004. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13: 2679–2689. 10.1093/hmg/ddh282 [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Alcott CE, Chen CA, Chaudhury A, Gillentine MA, Rosenfeld JA, Parikh S, Wheless JW, Roeder ER, Horovitz DD, et al. 2015. NUDT21-spanning CNVs lead to neuropsychiatric disease and altered MeCP2 abundance via alternative polyadenylation. Elife 4: e10782. 10.7554/eLife.10782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Holder JL Jr, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, et al. 2013. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503: 72–77. 10.1038/nature12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazdoba TM, Leach PT, Yang M, Silverman JL, Solomon M, Crawley JN. 2016. Translational mouse models of autism: advancing toward pharmacological therapeutics. Curr Top Behav Neurosci 28: 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Francke U. 2006. Identification of cis-regulatory elements for MECP2 expression. Hum Mol Genet 15: 1769–1782. 10.1093/hmg/ddl099 [DOI] [PubMed] [Google Scholar]
- Lombardi LM, Zaghlula M, Sztainberg Y, Baker SA, Klisch TJ, Tang AA, Huang EJ, Zoghbi HY. 2017. An RNA interference screen identifies druggable regulators of MeCP2 stability. Sci Transl Med 9: eaaf7588. 10.1126/scitranslmed.aaf7588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markenscoff-Papadimitriou E, Whalen S, Przytycki P, Thomas R, Binyameen F, Nowakowski TJ, Kriegstein AR, Sanders SJ, State MW, Pollard KS, et al. 2020. A chromatin accessibility atlas of the developing human telencephalon. Cell 182: 754–769.e18. 10.1016/j.cell.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. 2015. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86: 1369–1384. 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Bouwknecht JA, Teague R, Paylor R, Zoghbi HY. 2005. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum Mol Genet 14: 205–220. 10.1093/hmg/ddi016 [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. 2006. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 1: 172–182. 10.4161/epi.1.4.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz-Levi D, Olender T, Bar-Joseph I, Zhu Y, Marek-Yagel D, Barozzi I, Osterwalder M, Alkelai A, Ruzzo EK, Han Y, et al. 2019. Noncoding deletions reveal a gene that is critical for intestinal function. Nature 571: 107–111. 10.1038/s41586-019-1312-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Tavyev YJ, Peters SU. 2010. The MECP2 duplication syndrome. Am J Med Genet A 152A: 1079–1088. 10.1002/ajmg.a.33184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveau M, Shimohata A, Amano K, Miyamoto H, Yamakawa K. 2018. DYRK1A-haploinsufficiency in mice causes autistic-like features and febrile seizures. Neurobiol Dis 110: 180–191. 10.1016/j.nbd.2017.12.003 [DOI] [PubMed] [Google Scholar]
- Rice AM, McLysaght A. 2017. Dosage-sensitive genes in evolution and disease. BMC Biol 15: 78. 10.1186/s12915-017-0418-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha H, Sampaio M, Rocha R, Fernandes S, Leão M. 2016. MEF2C haploinsufficiency syndrome: report of a new MEF2C mutation and review. Eur J Med Genet 59: 478–482. 10.1016/j.ejmg.2016.05.017 [DOI] [PubMed] [Google Scholar]
- Rodrigues DC, Kim DS, Yang G, Zaslavsky K, Ha KC, Mok RS, Ross PJ, Zhao M, Piekna A, Wei W, et al. 2016. MECP2 is post-transcriptionally regulated during human neurodevelopment by combinatorial action of RNA-binding proteins and miRNAs. Cell Rep 17: 720–734. 10.1016/j.celrep.2016.09.049 [DOI] [PubMed] [Google Scholar]
- Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, Neul JL. 2008. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet 17: 1718–1727. 10.1093/hmg/ddn062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Mandel-Brehm C, McGraw CM, Shaw CA, McGill BE, Zoghbi HY. 2012. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet 44: 206–211. 10.1038/ng.1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. 2020. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180: 568–584.e23. 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanze I, Bunt J, Lim JWC, Schanze D, Dean RJ, Alders M, Blanchet P, Attié-Bitach T, Berland S, Boogert S, et al. 2018. NFIB haploinsufficiency Is associated with intellectual disability and macrocephaly. Am J Hum Genet 103: 752–768. 10.1016/j.ajhg.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel F, Smogavec M, Funke R, Pauli S, Burfeind P, Bartels I. 2018. Down syndrome phenotype in a boy with a mosaic microduplication of chromosome 21q22. Mol Cytogenet 11: 62. 10.1186/s13039-018-0410-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R. 2016. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533: 95–99. 10.1038/nature17939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. 2005. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav 4: 420–430. 10.1111/j.1601-183X.2005.00123.x [DOI] [PubMed] [Google Scholar]
- Swanberg SE, Nagarajan RP, Peddada S, Yasui DH, LaSalle JM. 2009. Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum Mol Genet 18: 525–534. 10.1093/hmg/ddn380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan YW, Liu Z, Rigo F, et al. 2015. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528: 123–126. 10.1038/nature16159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A, Pierce LC, Biggs WH, di Iulio J, Wong EH, Fabani MM, Kirkness EF, Moustafa A, Shah N, Xie C, et al. 2016. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci 113: 11901–11906. 10.1073/pnas.1613365113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gécz J, et al. 2005. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 77: 442–453. 10.1086/444549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagasaki Y, Miyoshi G, Miyata M. 2018. Experience-dependent MeCP2 expression in the excitatory cells of mouse visual thalamus. PLoS One 13: e0198268. 10.1371/journal.pone.0198268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. 2013. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78: 785–798. 10.1016/j.neuron.2013.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.