

In this study, Driskill et al. studied whether the TAZ-CAMTA1 gene fusion is a driver of epithelioid hemangioendothelioma (EHE), a poorly understood and devastating vascular cancer. They show that TAZ-CAMTA1 expression in endothelial cells is sufficient to drive the formation of vascular tumors with the distinctive features of EHE, and inhibition of TAZ-CAMTA1 results in the regression of these vascular tumors, and their findings provide the first genetic model of a TAZ fusion oncoprotein driving its associated human cancer, pinpointing TAZ-CAMTA1 as the key driver and a valid therapeutic target of EHE.
Keywords: TAZ, YAP, CAMTA1, epithelioid hemangioendothelioma, Hippo pathway, gene fusion, vascular anomalies, vascular malformations, cancer, endothelial cells
Abstract
Epithelioid hemangioendothelioma (EHE) is a poorly understood and devastating vascular cancer. Sequencing of EHE has revealed a unique gene fusion between the Hippo pathway nuclear effector TAZ (WWTR1) and the brain-enriched transcription factor CAMTA1 in ∼90% of cases. However, it remains unclear whether the TAZ-CAMTA1 gene fusion is a driver of EHE, and potential targeted therapies are unknown. Here, we show that TAZ-CAMTA1 expression in endothelial cells is sufficient to drive the formation of vascular tumors with the distinctive features of EHE, and inhibition of TAZ-CAMTA1 results in the regression of these vascular tumors. We further show that activated TAZ resembles TAZ-CAMTA1 in driving the formation of EHE-like vascular tumors, suggesting that constitutive activation of TAZ underlies the pathological features of EHE. We show that TAZ-CAMTA1 initiates an angiogenic and regenerative-like transcriptional program in endothelial cells, and disruption of the TAZ-CAMTA1-TEAD interaction or ectopic expression of a dominant negative TEAD in vivo inhibits TAZ-CAMTA1-mediated transformation. Our study provides the first genetic model of a TAZ fusion oncoprotein driving its associated human cancer, pinpointing TAZ-CAMTA1 as the key driver and a valid therapeutic target of EHE.
Epithelioid hemangioendothelioma (EHE) is a malignant cancer that appears to be of endothelial origin and affects patients of any age (Corrin et al. 1979; Weldon-Linne et al. 1981; Weiss and Enzinger 1982; Sardaro et al. 2014). This tumor can be found in multiple organs, including the liver (21%), lungs (12%), and bone (14%), although many other primary sites have been identified, and tumors grow within or around blood vessels (Weiss and Enzinger 1982; Sardaro et al. 2014). EHE is usually clinically asymptomatic, and patients are often diagnosed with metastatic disease (Groeschl et al. 2014; Sardaro et al. 2014). Because of the rarity of this tumor, a lack of large clinical studies, and the absence of animal models, the only definitive treatment for EHE is surgical resection, though recurrence after surgery has been reported (Groeschl et al. 2014; Rude et al. 2014; Sardaro et al. 2014). Therefore, new treatments are urgently needed to combat EHE.
Sequencing analyses of EHE have revealed a disease-defining group of chromosomal abnormalities resulting in gene fusions (Errani et al. 2011; Tanas et al. 2011; Antonescu et al. 2013; Suurmeijer et al. 2020). The most common gene fusion (in 90% of cases) occurs between the genes WWTR1 (TAZ) and CAMTA1 on chromosomes 1 and 3, respectively (Errani et al. 2011; Tanas et al. 2011). The TAZ-CAMTA1 fusion notably consists of the N terminus of TAZ, containing its TEAD-binding domain, WW domain, and three Hippo-responsive phosphorylation sites, and the C terminus of CAMTA1, containing its transcriptional activation domain and a nuclear localization signal (Tanas et al. 2016). Other sequencing analyses revealed that TAZ-CAMTA1-negative cases of EHE harbor YAP1-TFE3 (in ∼10% of cases) (Antonescu et al. 2013) or less commonly TAZ-MAML2 or TAZ-ACTL6A gene fusions (Suurmeijer et al. 2020). No other recurrent genetic abnormalities have been noted (Seligson et al. 2019; Rosenbaum et al. 2020). Since their discovery, the expression of the products of these gene fusions has been validated as specific markers for the clinical diagnosis of EHE (Shibuya et al. 2015; Doyle et al. 2016).
Recent studies have identified a variety of YAP and TAZ fusions with different protein partners in additional tumor types, including supratentorial ependymoma (Pajtler et al. 2015, 2019), poroma and porocarcinoma (Sekine et al. 2019), and other vascular tumors including epithelioid hemangioma (Antonescu et al. 2014), pseudomyogenic hemangioendothelioma (Panagopoulos et al. 2019), and angiomyxoma (Lee et al. 2019). While some of these YAP/TAZ fusions have been shown to be oncogenic in vitro or in ectopic tissues (Tanas et al. 2016; Pajtler et al. 2019; Sekine et al. 2019; Szulzewsky et al. 2020), none of them, including TAZ-CAMTA1, has been shown to drive the development of the respective tumor in which the gene fusion was initially identified. Thus, whether a given YAP/TAZ fusion can cause the corresponding tumor remains unknown. In the absence of such models, it remains unclear whether these fusions are viable therapeutic targets.
Another open question concerns the molecular mechanism by which the YAP/TAZ gene fusions may confer gain-of-function activities. The oncoproteins TAZ and its paralog YAP, both of which are involved in gene fusions found in EHE, act as the key nuclear effectors of the Hippo pathway, an evolutionarily conserved kinase cascade that controls overall organ size, tissue homeostasis, and regeneration in a variety of contexts (Johnson and Halder 2014; Koo and Guan 2018; Zheng and Pan 2019). TAZ and YAP act as transcriptional coactivators that bind to their cognate transcription factors, the TEF/TEAD family of transcription factors, to promote cellular proliferation and inhibit cell death. Under homeostatic conditions, the LATS1/2 kinases phosphorylate and ultimately lead to 14-3-3-mediated cytoplasmic sequestration and β-TrCP-mediated degradation of YAP/TAZ. Upstream of LATS1/2 are the kinases MST1/2 and a series of protein regulators that tie regulation of the kinase cascade to such signals as mechanical forces, GPCR signaling, and metabolic status. Whereas YAP and TAZ have been intensely studied, CAMTA1 is a transcription factor that is highly expressed in the brain, and its function remains largely unknown (Tanas et al. 2011; Long et al. 2014). Previous molecular characterization of TAZ-CAMTA1 suggested that the strong nuclear localization signal of CAMTA1 prevents the fusion protein from being regulated by the Hippo pathway and therefore results in the constitutive nuclear localization and activation of TAZ-CAMTA1 (Tanas et al. 2016).
In this study, we used a combination of transgenic mice and a mammalian endothelial cell line to show that expression of TAZ-CAMTA1 is sufficient to cause vascular tumors with the features of EHE. Furthermore, we demonstrate that continued expression of the fusion protein is required for tumor growth. The expression of activated TAZ also leads to EHE-like vascular tumors, and the TAZ-CAMTA1 fusion protein requires its interaction with TEAD to initiate tumorigenesis. We demonstrate that TAZ-CAMTA1 activates an angiogenic and regenerative gene program in endothelial cells. Last, we show that TAZ-CAMTA1 is still susceptible to inhibition by Hippo pathway hyperactivation, and treatment with known YAP/TAZ activity-modulating drugs, like statins, can inhibit TAZ-CAMTA1-mediated endothelial transformation.
Results
TAZ-CAMTA1 expression in endothelial cells drives the formation of EHE-like vascular tumors in the lungs of mice
To investigate whether TAZ-CAMTA1 is a driver of EHE, we tested whether conditional overexpression of TAZ-CAMTA1 in mouse endothelial cells could lead to the formation of vascular tumors. We therefore generated transgenic mice that expressed a FLAG epitope-tagged human TAZ-CAMTA1 cDNA (Fig. 1A; Tanas et al. 2016) under the control of a minimal CMV promoter and a tetracycline (Tet) response element (TRE). To express TAZ-CAMTA1 throughout the mouse vasculature, we used an established, well-characterized transgenic driver line, Cdh5-tTA, in which the tetracycline-controlled transactivator (tTA) is under control of the Cdh5 promoter and is expressed specifically in mouse endothelial cells (Sun et al. 2005). We crossed the two single-transgenic lines to generate double-transgenic TRE-TAZ-CAMTA1;Cdh5-tTA (TAZ-CAMTA1iEC) offspring in which expression of TAZ-CAMTA1 is repressed upon feeding the mice doxycycline in their drinking water (Tet-off system) (Fig. 1B). After crossing the two single-transgenic lines, pregnant mothers were maintained on normal water to initiate TAZ-CAMTA1 expression (Supplemental Fig. S1A). However, no viable double-transgenic offspring were obtained, suggesting that TAZ-CAMTA1 expression in endothelial cells during development generates an embryonic-lethal phenotype (Supplemental Fig. S1B).
Figure 1.
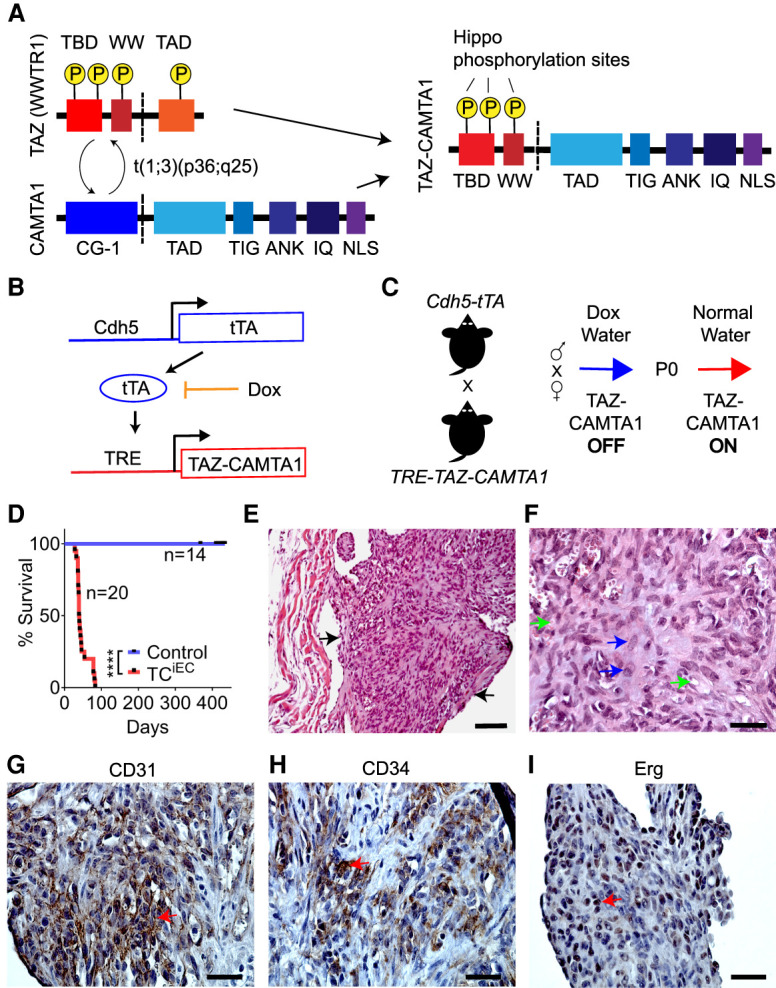
TAZ-CAMTA1 expression in endothelial cells drives the formation of EHE-like vascular tumors in the lungs of mice. (A) Structure of TAZ, CAMTA1, and the resulting TAZ-CAMTA1 fusion protein formed from the t(1;3)(p36;q25) chromosomal translocation. TAZ-CAMTA1 contains the N terminus of TAZ with its TEAD-binding domain (TBD), WW domain, and three LATS1/2 phosphorylation sites, and the C terminus of CAMTA1 with its transcription activation domain (TAD), TIG domain, ankyrin repeats (ANK), IQ motifs, and a nuclear localization signal (NLS). TAZ-CAMTA1 has lost the TAD of TAZ, one of TAZ's LATS1/2 phosphorylation sites, and the CG-1 domain of CAMTA1. (B) Schematics of the Cdh5-tTA and TRE-TAZ-CAMTA1 alleles for endothelial-specific expression of TAZ-CAMTA1. (C) Schematic for induction of TAZ-CAMTA1 expression after birth. (D) Survival curve for Ctrl (single transgenic Cdh5-tTA or TRE-TAZ-CAMTA1 mice, n = 14, median survival = undefined) or TAZ-CAMTA1iEC mice (Cdh5-tTA;TRE-TAZ-CAMTA1, n = 20, median survival = 39.5 d). (****) P < 0.0001, Mantel-Cox test. (E–I) Representative images showing H&E (E,F), CD31 (G), CD34 (H), and Erg (I) immunohistochemistry of a tumor found within a vessel of the lungs of 7-wk-old TAZ-CAMTA1iEC mice. Black arrows in E point to the tumor, and blue arrows in F show plump spindled and epithelioid cells, while green arrows show cytoplasmic vacuoles, some with red blood cells. Red arrows in G– I show positive staining for the respective markers. Scale bars: E, 100 µm; F––I, 35 µm.
To overcome this embryonic lethality, we crossed the single-transgenic parents and maintained pregnant mothers on 1.5 mg/mL doxycycline water, which resulted in double-transgenic offspring at the proper Mendelian ratio. At P0, we removed doxycycline water from the mating cages and followed the offspring for the development of any phenotypes (Fig. 1C). We observed that TAZ-CAMTA1iEC mice all showed lethality before day 83, with a median survival of 39.5 d (Fig. 1D). Before death, many TAZ-CAMTA1iEC mice developed lethargy, quick breathing, and edema (Supplemental Fig. S1C). To understand the cause of these phenotypes, we systematically isolated and analyzed the hearts, lungs, livers, skin, adipose tissue, bones, ovaries, and intestines of the double-transgenic mice at P40 or when the mice began to develop symptoms. Some mice were observed to exhibit pleural effusions upon dissection. We consistently observed the presence of widespread microscopic tumors within the blood vessels of the lungs, particularly those with at least one smooth muscle layer (Fig. 1E). Tumor cells showed an epithelioid or spindled morphology and exhibited cytoplasmic vacuoles, sometimes with red blood cells, features that are characteristic of EHE of the lung (Fig. 1F; Rosenbaum et al. 2020). These tumors stained positive for CD31, CD34, and Erg and were negative for pan-cytokeratin (Pan-CK) (Fig. 1G–I; Supplemental Fig. S1F–I), confirming their endothelial origin. Thus, expression of TAZ-CAMTA1 in endothelial cells is sufficient to lead to the formation of EHE-like tumors.
TAZ-CAMTA1 is required for the maintenance of vascular tumors
To investigate whether the gene fusion is required for the maintenance of these EHE-like vascular tumors, we subjected the TAZ-CAMTA1iEC mice described above that had TAZ-CAMTA1 expression from P0 to P40, a time point at which tumors have been established, to either normal water to continue TAZ-CAMTA1 expression or to doxycycline water to repress TAZ-CAMTA1 expression (Fig. 2A). All mice maintained on normal water had died within 35 d (P75), while mice given doxycycline water showed no lethality (Fig. 2B). Consistent with these results, histological examination of P40 TAZ-CAMTA1iEC mice revealed widespread CD31+ tumors within the vessels of the lung, whereas such tumors were absent in the lungs of TAZ-CAMTA1iEC mice that had received doxycycline for 2 wk (Fig. 2C–H). Quantitatively, P40 TAZ-CAMTA1iEC mice exhibited approximately two tumors per 40× field of view (Fig. 2I), with approximately two independent tumors detected in ∼51% of medium-sized vessels (Fig. 2J–K), while we observed no tumors within the vessels of TAZ-CAMTA1iEC mice treated for 2 wk with doxycycline (P54 TAZ-CAMTA1iEC). We also examined Ki67 staining within the endothelial cells of P40 TAZ-CAMTA1iEC mice and 2 wk after turning off TAZ-CAMTA1 expression. While we observed a significant increase in Ki67 staining in the endothelial cells of P40 TAZ-CAMTA1iEC compared with control mice, we observed no difference in staining within the blood vessels of TAZ-CAMTA1iEC mice after 2 wk of doxycycline treatment as compared with treated nontransgenic control mice (Fig. 2L–N). Thus, TAZ-CAMTA1 expression is required for continued growth of these vascular tumors, and inhibition of expression leads to tumor regression.
Figure 2.

TAZ-CAMTA1 is required for tumor progression and maintenance. (A) Schematic for removal of TAZ-CAMTA1 transgene expression after tumor formation. (B) Survival curve for mice that were maintained on doxycycline water (median survival = undefined, n = 6) or normal water (median survival = 16 d, n = 5) after P40. (**) P < 0.01, Mantel-Cox test. (C–H) H&E (C–E) and CD31 immunostaining (F–H) of Ctrl (C,F), TAZ-CAMTA1iEC (D,G), and TAZ-CAMTA1iEC mice given doxycycline for 2 wk (E,H). All P40 TAZ-CAMTA1iEC mice (n = 5) exhibited tumors, while no P40 Ctrl mice (n = 5) or P54 TAZ-CAMTA1iEC mice (receiving 2 wk of doxycycline after P40, n = 3) had any lung tumors. Black arrows point to examples of tumors within blood vessels. Scale bar, 50 µm. (I) Quantification of tumors observed per 40× field of view in P40 Ctrl (0 ± 0, n = 5), P40 TAZ-CAMTA1iEC (2.5 ± 0.7, n = 5, [**] P = 0.01), P54 Ctrl (0 ± 0, n = 3), or P54 TAZ-CAMTA1iEC mice (0 ± 0, n = 3, P > 0.05). (J) Quantification of noncapillary vessels affected in P40 Ctrl (0% ± 0, n = 5), P40 TAZ-CAMTA1iEC (51.3% ± 8.9, n = 5, [***] P < 0.001), P54 Ctrl (0% ± 0, n = 3), or P54 TAZ-CAMTA1iEC mice (0% ± 0, n = 3, P > 0.05). (K) Quantification of independent tumors observed per vessel in P40 Ctr (0 ± 0, n = 5), P40 TAZ-CAMTA1iEC (2.1 ± 0.3, n = 5, [****] P < 0.0001), P54 Ctrl (0 ± 0, n = 3), or P54 TAZ-CAMTA1iEC mice (0 ± 0, n = 3, P > 0.05). (L–N) Endomucin (Emcn) and Ki67 immunofluorescence of P40 and P54 TAZ-CAMTA1iEC vessels. P40 TAZ-CAMTA1iEC (14.4 ± 2.6, n = 3) vessels had significantly more Ki67 positivity than Ctrl (1.8 ± 0.2, n = 3, [**] P < 0.01), while P54 TAZ-CAMTA1iEC (1.4 ± 1.4, n = 3) vessels had no difference in Ki67 positivity compared with control (0.8 ± 0.8, n = 3, P > 0.05). White arrows point to Ki67+Emcn+ endothelial cells. Scale bars, 25 µm. All data are presented as mean ± S.E.M, and P-values were calculated with unpaired t-tests.
TAZ-CAMTA1 is highly tumorigenic in endothelial cells
To complement our analysis of transgenic mice, we examined whether the expression of TAZ-CAMTA1 could transform cultured endothelial cells. We expressed the fusion protein in a doxycycline-inducible manner (Tet-on system) in a well-characterized, SV40-immortalized mouse endothelial cell line known as MS1 (Arbiser et al. 1997). Indeed, TAZ-CAMTA1 expression enabled MS1 cells to grow in soft agar (Fig. 3A–C). We next tested whether TAZ-CAMTA1 expression would allow these cells to form progressive vascular tumors in vivo. Thus, we injected MS1 cells subcutaneously into the flanks of nude mice and gave the mice water with sucrose as a control or water with sucrose and doxycycline to induce the expression of TAZ-CAMTA1. TAZ-CAMTA1-expressing MS1 cells formed vascular tumors that grew over time (Fig. 3D–F), whereas control cells formed benign hemangiomas that did not progress as previously described (Arbiser et al. 1997). While control cells formed well-circumscribed masses with a maximum diameter of ∼5 mm, TAZ-CAMTA1-expressing MS1 cells formed poorly circumscribed nodules that showed areas of ulceration and necrosis and reached up to ∼1 cm in diameter by about 3 mo of growth. Tumors formed from TAZ-CAMTA1-expressing MS1 were largely composed of epithelioid and spindled CD31+ cells growing in nests and cords that grew as a solid mass, while control MS1 cells formed blood-filled hemangiomas (Fig. 3G–L). Unlike the parental MS1 cells, we also observed metastasis of TAZ-CAMTA1-expressing MS1 cells to the lung at 3 mo, forming large, poorly defined nodules that showed areas of central necrosis (Fig. 3M–O). Therefore, TAZ-CAMTA1 drives anchorage-independent growth in endothelial cells and confers these cells the ability to both grow and metastasize.
Figure 3.

TAZ-CAMTA1 is highly tumorigenic in endothelial cells. (A–C) Ctrl (−Dox) (3.3 colonies ± 0.7, n = 3) or TAZ-CAMTA1-expressing (+Dox) (338 colonies ± 27.1, n = 3) MS1 cells grown on soft agar. TAZ-CAMTA1 expression significantly increased the colony forming ability of MS1 cells on soft agar. (***) P < 0.001. (D,E) Representative images of tumors established in nude mice from Ctrl (−Dox) or TAZ-CAMTA1-expressing (+Dox) MS1 cells. Scale bar, 1 cm. Red arrows point to tumors. (F) Growth curve of tumors formed from Ctrl (13.4 ± 8.9, n = 5) or TAZ-CAMTA1-expressing (213.8 ± 76.7, n = 5) MS1 cells. (*) P < 0.05. Values represent final time point, day 100. All values are mean ± SEM, and P-value was calculated with an unpaired t-test. (G–L) Representative H&E (G,H), CD31 (I,J), and FLAG (K,L) immunohistochemistry of tumors derived from Ctrl (−Dox) (G,I,K) or TAZ-CAMTA1-expressing (+Dox) (H,J,L) MS1 cells. Red arrows point to positive immunostaining. Scale bars,100 µm. (M–O) Representative H&E (M), CD31 (N), and FLAG (O) immunohistochemistry of a lung metastasis identified in a nude mouse injected subcutaneously with MS1 cells expressing TAZ-CAMTA1. Red arrows point to positive immunostaining. Scale bars,100µm.
TAZ-CAMTA1 drives a proliferative endothelial cell population with a YAP/TAZ target gene signature
To understand how TAZ-CAMTA1 transforms endothelial cells, we performed droplet-based single-cell RNA sequencing (scRNA-seq) by the 10× genomics platform on lung endothelial cells dissociated from 5-wk-old TRE-H2B-GFP;TAZ-CAMTA1iEC mice that had been enriched by flow-activated cell sorting (FACS) (Fig. 4A). Using t-SNE analysis, we identified 10 transcriptionally distinct cell populations (Fig. 4B). Nine of the populations expressed endothelial cell markers, such as CD31, CD34, Cdh5, and Cldn5 (Supplemental Fig. S2A), while one population expressed markers consistent with immune cells but not endothelial cells (Supplemental Fig. S2B). Based on differential marker gene analysis and a previous publication (Goveia et al. 2020), we were able to identify cell populations consistent with arterial endothelial cells, venular endothelial cells, lymphatic endothelial cells, and capillary cells (Fig. 4C; Supplemental Fig. S2C,D; Supplemental Table S4; Supplemental Data File 1). We also identified an interferon-activated endothelial cell population, an immature endothelial population, and a population with both of these features, all of which have been suggested to be characteristic of lung carcinoma-associated endothelial cells Altogether, this suggests that we isolated normal and untransformed endothelial cells as well as reactive endothelial cells that may be associated with the transformed endothelial cell population.
Figure 4.

TAZ-CAMTA1 drives a proliferative endothelial cell population with a YAP/TAZ target gene signature. (A) Schematic for isolation of lung ECs for scRNA-seq. (B) t-SNE plot of clusters of cells observed from scRNA-seq. (C) t-SNE plots showing expression of various marker genes. Red arrows point to cluster showing positive expression. (D) Volcano plot of 806 differentially expressed genes in TAZ-CAMTA1-induced proliferative subpopulation. Wilcoxon rank sum test, adjusted P-value < 0.05. Genes in red have a log(FC) > 1 and genes in blue have a log(FC) < 1. Some pertinent genes and their corresponding points are marked. (E) t-SNE plots of markers from proliferative subpopulation. (F) Immunostaining validation of differentially expressed genes identified from scRNA-seq analysis. White arrows point to positive staining of Pdgfa, Cryab, or S100a6 in P40 TAZ-CAMTA1iEC but not Ctrl lung vessels. Scale bars, 25 µm. (G) GSEA plot showing normalized enrichment score (NES) of YAP-conserved signature in the proliferative subpopulation, NES = 1.8. P < 0.01, FDR = 0.1. (H) t-SNE plots of canonical YAP/TAZ target genes. Red arrows point to the proliferative cluster showing high expression of these genes.
To identify the tumor population that had been observed on histology, we performed an analysis of cell cycle genes that might be predictive of a proliferative endothelial cell population, since only the tumor endothelial cells had been observed to show widespread staining for Ki67. We found that genes indicative of proliferation all clustered in a particular and distinct population, which we named the “proliferative population,” and to a lesser extent in its adjacent population, the “preproliferative population” (Fig. 4C; Supplemental Fig. S3A,B). Based on this identification, we performed differential gene expression testing and identified 806 differentially expressed genes in this proliferative population (Fig. 4D). Highly and uniquely expressed genes within this population included Pdgfa, Cryab, and S100A6, and we confirmed that Pdgfa and Cryab were also overexpressed in MS1 cells expressing TAZ-CAMTA1 (Fig. 4E; Supplemental Fig. S3C,D). Based on this analysis, we performed immunofluorescence staining of lungs from P40 TAZ-CAMTA1iEC mice and confirmed that Pdgfa, Cryab, and S100a6 were all specifically expressed in emergent vascular tumors and not in control or nontumor tissue (Fig. 4F).
We then performed gene set enrichment analysis (GSEA) (Subramanian et al. 2005) on the differentially expressed genes identified in this proliferative endothelial cell population. Prominently, we observed a significant increase in YAP target gene signatures previously defined in two independent studies in nonendothelial cells (Fig. 4G; Supplemental Fig. S3E; Cordenonsi et al. 2011; Tremblay et al. 2014). Indeed, we confirmed that many canonical YAP/TAZ target genes were specifically increased in this endothelial cell population (Fig. 4H). Besides the YAP/TAZ gene signature, the proliferative endothelial cell population also showed gene signatures of MTOR activation, KRAS activation, P53 down-regulation, and late embryonic stem cell identity (Supplemental Fig. S3F).
TAZS4A phenocopies TAZ-CAMTA1
The activation of YAP/TAZ target genes in TAZ-CAMTA1-expressing endothelial cells led us to investigate whether TAZ can also affect the function of endothelial cells. Thus, we overexpressed either empty vector (Ctrl), TAZ, or TAZS4A, a gain-of-function variant in which the four Hippo-regulated phosphorylation sites of TAZ are mutated, in the MS1 endothelial cell line (Supplemental Fig. S4A). Similar to TAZ-CAMTA1, TAZS4A, but not TAZ or empty vector, enabled MS1 cells to grow in soft agar (Fig. 5A–D). Consistent with this result, both TAZS4A and TAZ-CAMTA1 activated the expression of canonical YAP/TAZ target genes Amotl2 and Cyr61 (Supplemental Fig. S4B,C).
Figure 5.
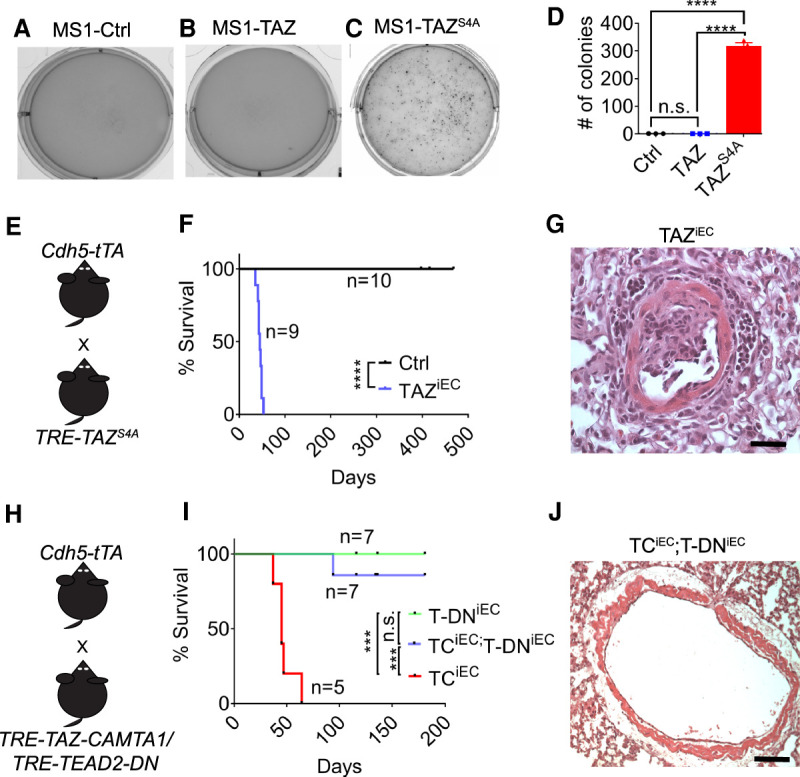
TAZS4A phenocopies TAZ-CAMTA1, and TAZ-CAMTA1 requires the TEAD family of transcription factors to drive tumorigenesis. (A–D) Representative images of Ctrl (1.0 ± 0.1, n = 3), TAZ (0.7 ± 0.1, n = 3), or TAZS4A-expressing (318.6 ± 11.3, n = 3) MS1 cells growing in soft agar. Only TAZS4A is sufficient to promote anchorage-independent growth. (****) P < 0.0001, one-way ANOVA with post-hoc Tukey test. (E) Schematic for generation of endothelial-specificTAZS4A (TAZiEC) transgenic mice. (F) Survival curve for Ctrl (median survival = undefined, n = 10) and TAZiEC (median survival = 45 days, n = 9) mice. (****) P < 0.0001, Mantel-Cox test. (G) Representative H&E of tumor located in noncapillary vessel of a P35 TAZiEC mouse. Scale bar, 35 µm. (H) Schematic for generation of TRE-TEAD2DN;Cdh5-tTA (T-DNiEC) and TRE-TEAD2DN;TRE-TAZ-CAMTA1;Cdh5-tTA (TCiEC;T-DNiEC) transgenic mice. (I) Survival curve of T-DNiEC mice (median survival = undefined, n = 7), TAZ-CAMTA1iEC mice (median survival = 45 d, n = 5), and TEAD2DNiEC, TAZ-CAMTA1iEC mice (median survival = undefined, n = 7). (***) P < 0.001, Mantel-Cox test. (J) Representative H&E of a vessel in a TCiEC;T-DNiEC mouse showing the absence of any vascular tumors. Scale bar, 100 µm. None of the TCiEC;T-DNiEC mice examined (n = 3) showed lung tumors at P40.
Next, we sought to test whether TAZS4A expression in vivo could produce similar EHE-like vascular tumors. We therefore adopted a similar strategy to express TAZS4A in the endothelial cells as we used for TAZ-CAMTA1, by generating TRE-TAZS4A;Cdh5-tTA (TAZiEC) mice (Fig. 5E). We maintained pregnant mothers on doxycycline water to silence the transgene and replaced the doxycycline-containing water with normal water at birth to turn on the transgene, as we have done for testing the oncogenic activity of TAZ-CAMTA1. Under such regimen, the TAZiEC mice all died by P53 with a median survival of 45 d (Fig. 5F). Similar to the TAZ-CAMTA1iEC mice, the TAZiEC mice also formed vascular tumors within the noncapillary vessels of the lung, showing that TAZS4A phenocopies TAZ-CAMTA1 in driving EHE-like vascular tumors (Fig. 5G).
TAZ-CAMTA1 requires the TEAD family of transcription factors to drive tumorigenesis
Given that TAZ-CAMTA1 and TAZS4A can drive similar phenotypes, we tested whether TAZ-CAMTA1, like TAZ, requires the TEAD family of DNA-binding transcription factors. First, we generated a transgenic model expressing TAZ-CAMTA1 in the Drosophila compound eye, a well-established model to dissect the growth regulatory function of the Hippo signaling pathway. Unlike wild-type TAZ, expression of TAZ-CAMTA1 drove massive eye overgrowth that resembles the phenotype caused by TAZS89A, a gain-of-function TAZ mutant that lacks the most critical Hippo-responsive phosphorylation site. Furthermore, knockdown of Sd, the Drosophila ortholog of TEAD, or overexpression of a TAZ-CAMTA1S51A mutant that has lost TAZ's ability to bind to TEAD (Tanas et al. 2016), inhibited that overgrowth (Supplemental Fig. S4D–K). To study this TEAD dependence in mammals, we introduced the previously reported TRE-TEAD2DN line that expresses a dominant-negative form of TEAD2 (Liu-Chittenden et al. 2012) into the TAZ-CAMTA1iEC line (TRE-TAZ-CAMTA1;TRE-TEAD2DN;Cdh5-tTA, TCiEC;T-DNiEC) (Fig. 5H). While all TAZ-CAMTA1iEC mice that had expressed TAZ-CAMTA1 starting at P0 had died by P64, six out of seven of the TAZ-CAMTA1iEC mice that coexpressed TEAD2DN survived through the time course of the experiment, showing that expression of the TEAD2DN abrogates TAZ-CAMTA1-induced lethality (Fig. 5I). Moreover, unlike the TAZ-CAMTA1iEC mice (Fig. 5J), we observed no tumors in P40 TAZ-CAMTA1iEC mice that coexpressed TEAD2DN (n = 3), demonstrating that TEAD2DN overexpression rescues TAZ-CAMTA1-induced tumor formation. Importantly, expression of TEAD2DN alone (TRE-TEAD2DN;Cdh5-tTA mice [T-DNiEC]) did not lead to physical or behavioral abnormalities, suggesting that inhibition of TEAD function in endothelial cells under homeostatic conditions has limited toxicity.
TAZ-CAMTA1 and TAZS4A drive a gene program associated with angiogenesis and regeneration
To take a broader approach to understand the similarities between TAZ and TAZ-CAMTA1 and to understand how their overexpression can drive endothelial cell transformation, we performed mRNA sequencing on MS1 cells expressing either the empty vector, TAZS4A, or TAZ-CAMTA1. After differential expression testing comparing either TAZS4A or TAZ-CAMTA1 with control, we compared the identity of the differentially expressed genes. This analysis unveiled significant overlap in the transcriptional programs of TAZS4A and TAZ-CAMTA1, revealing 965 similarly affected genes accounting for a Jaccard index of 0.24 and a P-value (hypergeometric test) of 1.3 × 10−235 (Fig. 6A; Supplemental Data File 2).
Figure 6.

TAZ-CAMTA1 and TAZS4A drive a gene program associated with angiogenesis and regeneration. (A) Number of differentially expressed genes (DEG; FDR < 0.01) in MS1 cells expressing TAZS4A or TAZ-CAMTA1 as compared with control MS1 cells. Overlapped region indicates genes expressed in both populations. Hypergeometric test P-value and Jaccard index are indicated. (BC) Volcano plot of DEGs of TAZS4A or TAZ-CAMTA1. Red (overexpressed) and blue (underexpressed) are genes with −log10(p) > 25. (D,E) Normalized enrichment scores (NES) for selected oncogenic pathways in MS1 cells expressing TAZS4A or TAZ-CAMTA1 as determined by GSEA. Red indicates positive enrichment and blue indicates negative enrichment. (F) qPCR for Vegfa expression in control MS1 cells (1.0 ± 0.0, n = 3) and MS1 cells expressing TAZS4A (3.1 ± 0.1, n = 3) or TAZ-CAMTA1 (4.9 ± 0.1, n = 3). (***) P < 0.001. one-way ANOVA with post-hoc Tukey test. All data are mean ± SEM. (G) Enrichment of an angiogenesis gene signature in TAZ-CAMTA1-expressing MS1 cells, NES = 2.6. (**) P < 0.01, FDR < 0.01. (H) Heat map showing expression of selected regenerative endothelial cell markers in TAZS4A- or TAZ-CAMTA1-expressing MS1 cells.
Between TAZS4A and TAZ-CAMTA1, we observed overlap in the most differentially expressed genes; for example, genes such as Steap4, Anxa6, Serpina3g, Mmp2, and Pdgfa were highly elevated in MS1 cells expressing either TAZS4A or TAZ-CAMTA1 (Fig. 6B,C). We thus wanted to determine whether TAZS4A and TAZ-CAMTA1 regulate the same downstream transcriptional programs. We performed a gene set enrichment analysis (Subramanian et al. 2005) and saw that both TAZS4A and TAZ-CAMTA1 up-regulated such gene programs as those related to KRAS activation, embryonic stem cell identity, and VEGFA activation (Fig. 6D,E).
The VEGFA signature induced by TAZS4A or TAZ-CAMTA1 in MS1 cells is interesting since VEGFA overexpression has been previously shown to transform MS1 cells (Arbiser et al. 2000). We thus tested whether TAZ-CAMTA1 and TAZS4A regulate VEGFA expression. Indeed, we confirmed by qPCR that Vegfa was significantly up-regulated in TAZS4A- or TAZ-CAMTA1-expressing MS1 cells (Fig. 6F). Consistent with endothelial cell activation by Vegfa, we also observed significant enrichment of an angiogenesis gene signature in TAZ-CAMTA1-expressing MS1 cells (Fig. 6G). Furthermore, recent work has sought to understand whether there is an endothelial stem cell or a population of endothelial cells that can regenerate upon injury (McDonald et al. 2018; Wakabayashi et al. 2018). We observed significant up-regulation of genes in TAZS4A- or TAZ-CAMTA1-expressing MS1 cells that correspond to regenerative populations of endothelial cells (Fig. 6H). We detected up-regulation of Bst1, also known as CD157, a marker of endothelial stem cells (Wakabayashi et al. 2018), and up-regulation of Atf3, a required gene for endothelial cell regeneration after injury (McDonald et al. 2018). Taken together, TAZ-CAMTA1 drives gene programs in endothelial cells associated with angiogenesis and regeneration.
TAZ-CAMTA1 is resistant to proteasomal degradation
Hippo-regulated phosphorylation inhibits TAZ activity by inducing cytoplasmic sequestration (through a 14-3-3 binding site) and proteasomal degradation (through a β-TrCP-mediated phosphodegron site) (Lei et al. 2008; Liu et al. 2010). Previous molecular characterization of TAZ-CAMTA1 and other YAP/TAZ fusions suggested that the strong nuclear localization signal of CAMTA1 (and other fusion partners) prevents the fusion proteins from being regulated by the Hippo pathway and therefore results in the constitutive nuclear localization and activation of TAZ-CAMTA1 (Tanas et al. 2016; Pajtler et al. 2019; Szulzewsky et al. 2020). Indeed, we found that TAZ-CAMTA1 showed more nuclear localization compared with TAZ (Supplemental Fig. S5A–D). However, given our previous finding that nuclear localization of endogenous YAP by mutating its single 14-3-3-binding site (S112 of mouse YAP, equivalent to S127 of human YAP) does not cause tumorigenesis in vivo due to a compensatory mechanism that increases YAP degradation (Chen et al. 2015), nuclear localization alone is unlikely to explain the oncogenic activity of YAP/TAZ fusions. Thus, we speculated that additional mechanisms likely contribute to the gain-of-function activity of YAP/TAZ fusions. We noted that many of these fusions, including TAZ-CAMTA1, have lost the C-terminal phosphodegron site (Supplemental Fig. S5E; Szulzewsky et al. 2020), suggesting that loss of proteasomal degradation may contribute to their gain-of-function activation. Indeed, when the phosphodegron is mutated in TAZ, TAZ becomes more stable and exhibits gain-of-function activities (Liu et al. 2010).
To test whether TAZ-CAMTA1 is more resistant to proteasomal degradation since it has lost the C-terminal phosphodegron that is normally present on TAZ, we expressed TAZ-CAMTA1 or TAZ in MCF10A cells and treated the cells with cycloheximide, a translation inhibitor. While TAZ degraded over the time course of the experiment, TAZ-CAMTA1 remained stable (Supplemental Fig. S5F). We also treated these cells with bortezomib, a proteasomal degradation inhibitor, and while we saw TAZ's protein level increase, TAZ-CAMTA1's protein level showed little change (Supplemental Fig. S5G). Thus, both enhanced nuclear localization and loss of proteasomal degradation likely contribute to the oncogenic activity of TAZ-CAMTA1.
TAZ-CAMTA1 is still susceptible to regulation by Hippo signaling
As an unbiased way of identifying regulators and effectors of TAZ-CAMTA1 that may be therapeutically targeted, we sought to identify its protein interaction partners. We therefore expressed FLAG-tagged TAZ-CAMTA1 or empty vector in 293T cells, enriched the proteins by coimmunoprecipitation with a FLAG antibody, and subjected the immunoprecipitates to label-free mass spectrometry (Supplemental Data File 3). After eliminating proteins that were also identified by the control empty vector, we performed gene ontology to identify the regulatory processes that categorize TAZ-CAMTA1's binding partners (Fig. 7A). This analysis largely revealed that Hippo signaling processes may regulate TAZ-CAMTA1. We next identified the TEAD transcription factors as interacting partners of TAZ-CAMTA1, which is consistent with our observations that the TEAD transcription factors are required for TAZ-CAMTA1-induced tumorigenicity and that the C-terminally truncated TAZ in the TAZ-CAMTA1 fusion still retains the full TEAD-binding region. Paradoxically, we also identified LATS1 and the 14-3-3 complex as interacting partners of TAZ-CAMTA1 (Fig. 7B). Thus, the enhanced nuclear localization of TAZ-CAMTA1 does not preclude interactions with the upstream kinase that normally phosphorylates TAZ (LATS1) or the protein that normally binds TAZ in a phosphorylation-dependent manner (14-3-3). Of note, the truncated TAZ in TAZ-CAMTA1 still retains multiple LATS phosphorylation sites including the 14-3-3-binding site.
Figure 7.

TAZ-CAMTA1 is still susceptible to regulation by the Hippo pathway. (A) Gene ontology plot of TAZ-CAMTA1-binding proteins identified by mass spectrometry. (B) Spectral counts of Hippo pathway-related proteins identified in anti-FLAG immunoprecipitates from 293T cells expressing either empty vector (EV) or 2×-FLAG-TAZ-CAMTA1 (TC). (C–E) Immunostaining of transfected FLAG-tagged TAZ-CAMTA1 in 293A cells (C) or 293A LATS1/2 KO cells (D). 293A LATS1/2 KO (81.3% ± 6.4%, n = 6) cells had significantly more TAZ-CAMTA1 localized to the nucleus than 293A cells (47.8% ± 8.3%, n = 6). (**) P < 0.01, unpaired t-test. (F–H) Immunostaining of transfected FLAG-tagged TAZ-CAMTA1 or TAZ-CAMTA1S3A in 293T cells. TAZ-CAMTA1 (70.3% ± 4.4%, n = 10) was significantly more localized to the cytoplasm than TAZ-CAMTA1S3A (35.0% ± 4.9%, n = 10). (****) P < 0.0001, unpaired t-test. (I,J,K) qPCR for expression of Amotl2 (I), Ankrd1 (J), and Ctgf (K) in MS1 cells stably expressing empty vector (Ctrl), TAZ-CAMTA1 (TC), or TAZ-CAMTA1S3A (TC3A). Cells expressing TAZ-CAMTA1S3A had significantly increased expression of Amotl2 (Ctrl: 1.0 ± 0.1, n = 3; TC: 1.4 ± 0.1, n = 3; TC3A: 4.3 ± 0.1, n = 3), Ankrd1 (Ctrl: 1.0 ± 0.1, n = 3; TC: 0.9 ± 0.0, n = 3; TC3A: 2.3 ± 0.1, n = 3), and Ctgf (Ctrl: 1.0 ± 0.3, n = 3; TC: 1.0 ± 0.0, n = 3; TC3A: 1.5 ± 0.0, n = 3). (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001, one-way ANOVA with post-hoc Tukey test. (L–N) Immunostaining of doxycycline-induced FLAG-tagged TAZ-CAMTA1 in MS1 cells left untreated or treated with 1 µM latrunculin B (LatB) for 1 h. LatB-treated MS1 cells (45.5% ± 6.3%, n = 10) had more TAZ-CAMTA1 localized to the cytoplasm than untreated cells (5.7% ± 1.8%, n = 10). (****) P < 0.0001, unpaired t-test. (O) qPCR for expression of Cyr61 in MS1 cells without (−Dox) or with TAZ-CAMTA1 induction (+Dox), and untreated or treated with 5 µM simvastatin for 24 h. TAZ-CAMTA1-expressing cells had significantly reduced Cyr61 (untreated: 3.6 ± 0.2, n = 3, treated: 1.9 ± 0.1, n =3, [**] P < 0.01) after treatment with simvastatin. P-values were obtained by an unpaired t-test of the +Dox group treated or untreated. (P–R) Representative images showing soft agar growth of TAZ-CAMTA1-induced MS1 cells (TCiEC) in the absence or presence of 1 µM simvastatin (+Sim). Simvastatin significantly reduced the colony formation of TCiEC cells (Ctrl: 0.7 colonies ± 0.3, n = 3; TCiEC: 322.7 colonies ± 55.4, n = 3; TCiEC + Sim: 2.0 colonies ± 1.2, n = 3). (***) P < 0.001, one-way ANOVA with post-hoc Tukey test. All data are mean ± SEM. Scale bars, 25 µm.
To test whether TAZ-CAMTA1 is still susceptible to regulation by Hippo signaling, we expressed TAZ-CAMTA1 in 293A cells or 293A cells in which LATS1/2 had been deleted (Hansen et al. 2015). We observed increased nuclear localization of TAZ-CAMTA1 in the LATS1/2 KO cells as compared with wild-type cells (Fig. 7C–E). Consistent with this finding, mutating the three LATS phosphorylation sites in TAZ-CAMTA1 (TAZ-CAMTA1S3A) also resulted in increased nuclear localization (Fig. 7F–H). We also observed increased up-regulation of the canonical YAP/TAZ target genes Amotl2, Ankrd1, and Ctgf in MS1 cells stably expressing TAZ-CAMTA1S3A as compared with MS1 cells expressing empty vector or TAZ-CAMTA1, suggesting that abolishing Hippo-responsive phosphorylation increases the activity of TAZ-CAMTA1 (Fig. 7I–K). Taken together, these findings suggest that TAZ-CAMTA1 may still be susceptible to regulation by Hippo signaling. To further test this proposition, we treated TAZ-CAMTA1-expressing MS1 cells with latrunculin B (LatB), an F-actin inhibitor that is known to activate Hippo signaling and YAP/TAZ phosphorylation (Zhao et al. 2012; Chen et al. 2015; Moroishi et al. 2016; Mana-Capelli and McCollum 2018). Indeed, LatB treatment increased the cytoplasmic localization of TAZ-CAMTA1 (Fig. 7L–N). We conclude that, despite its gain-of-function activity, TAZ-CAMTA1 is still susceptible to regulation by Hippo signaling.
Because the localization of TAZ-CAMTA1 can still be regulated by Hippo signaling, we investigated whether other known mechanisms of increasing YAP/TAZ phosphorylation could sequester TAZ-CAMTA1 in the cytoplasm and thus offer a potential avenue for inhibiting its oncogenic activity. Previous publications have linked the mevalonate pathway to YAP/TAZ by showing that statins can drive YAP/TAZ phosphorylation and cytoplasmic sequestration, possibly by regulating the geranylgeranylation of Rho (Sorrentino et al. 2014; Wang et al. 2014; Mi et al. 2015). We therefore treated TAZ-CAMTA1-expressing MS1 cells with simvastatin. Consistent with our model, simvastatin significantly decreased the expression of TAZ-CAMTA1 target genes such as Cyr61 and Amotl2 (Fig. 7O; Supplemental Fig. S5H). Furthermore, simvastatin also abrogated the ability of TAZ-CAMTA1-expressing MS1 cells to grow in soft agar. Whereas TAZ-CAMTA1-expressing MS1 cells formed many colonies on soft agar, those treated with simvastatin formed few or no colonies (Fig. 7P–R). Therefore, therapies that are known to inhibit YAP/TAZ activity also may inhibit TAZ-CAMTA1-induced endothelial cell transformation.
Discussion
The TAZ-CAMTA1 fusion protein has been identified as an important and distinctive biomarker in EHE (Errani et al. 2011; Tanas et al. 2011; Shibuya et al. 2015). Our work shows that expression of this fusion protein in mammalian endothelial cells is sufficient to cause pulmonary EHE-like tumors and lethality. Furthermore, continued expression of this fusion protein is required for growth and maintenance of these tumors. Because EHE shows low mutational burden (Seligson et al. 2019; Rosenbaum et al. 2020) and the vast majority of cases exhibit the TAZ-CAMTA1 fusion protein (Errani et al. 2011; Tanas et al. 2011), targeting of TAZ-CAMTA1, whether directly or through its downstream transcriptional program, is likely to be highly effective for the vast majority of EHE patients. Beyond EHE, our study provides the first demonstration that a single genetic anomaly found in TAZ, a key effector of the Hippo pathway, causes its clinically relevant tumor type.
Because expression of an activated TAZ in vitro and in vivo phenocopies TAZ-CAMTA1 overexpression, we argue that the key function of these TAZ fusion proteins is to activate TAZ. Furthermore, the identification in EHE of rare and interchangeable C-terminal fusion protein partners that replace CAMTA1, such as ACTL6A and MAML2 (Suurmeijer et al. 2020), reinforces the importance of the role of TAZ in driving EHE. Hippo-regulated phosphorylation inhibits TAZ activity by inducing cytoplasmic sequestration (through a 14-3-3 binding site) and proteasomal degradation (through a β-TrCP-mediated phosphodegron site) (Lei et al. 2008; Liu et al. 2010). Indeed, we demonstrate that both enhanced nuclear localization and loss of proteasomal degradation likely contribute to the oncogenic activity of TAZ-CAMTA1. Previous molecular characterization of TAZ-CAMTA1 and other YAP/TAZ fusions suggested that the strong nuclear localization signal of CAMTA1 (and other fusion partners) prevents the fusion proteins from being regulated by the Hippo pathway and therefore results in the constitutive nuclear localization and activation of TAZ-CAMTA1 (Tanas et al. 2016; Pajtler et al. 2019; Szulzewsky et al. 2020). Our previous finding that nuclear localization of endogenous YAP by mutating its single 14-3-3-binding site does not cause tumorigenesis in vivo due to a compensatory mechanism that increases YAP degradation (Chen et al. 2015) shows that nuclear localization alone is unlikely to explain the oncogenic activity of YAP/TAZ fusions. Thus, the YAP/TAZ gene fusions likely activate YAP/TAZ by disrupting multiple mechanisms of regulation.
In contrast to previous reports (Tanas et al. 2016; Pajtler et al. 2019; Szulzewsky et al. 2020), we found that TAZ-CAMTA1 can still be regulated and inhibited by the Hippo pathway. The TAZ-CAMTA1 fusion protein not only retains multiple Hippo-responsive phosphorylation sites but also physically interacts with the Hippo pathway upstream kinase (LATS1) and its phosphorylation-dependent binding partners (the 14-3-3 proteins). We further showed that LATS1/2 regulate the subcellular localization of TAZ-CAMTA1 and that pharmacological intervention that is known to affect YAP/TAZ activity, such as statin treatment, could inhibit the activity of the TAZ-CAMTA1 fusion protein. Indeed, a retrospective analysis of patients with EHE showed that statin use was associated with increased survival, although the underlying mechanism was not investigated (Subramaniam et al. 2020). Together, these findings suggest that mechanisms that activate Hippo signaling and/or inhibit YAP/TAZ activity may provide a new avenue of therapy to inhibit TAZ-CAMTA1 activity in patients with EHE.
In addition to the therapeutic potential of targeting TAZ-CAMTA1 itself, we also demonstrate that eliminating TAZ-CAMTA1's transcriptional activity by inhibiting the TEAD family of transcription factors can inhibit the growth of EHE-like tumors. This also raises the possibility of therapeutic inhibition of the downstream target genes of TAZ-CAMTA1. Our scRNA-seq and RNA-seq analyses revealed a variety of up-regulated and down-regulated processes that can be manipulated by FDA-approved drugs. Interestingly, drugs targeting several of these processes have already shown some clinical benefit. For example, we identified Vegfa to be transcriptionally up-regulated in MS1 cells expressing TAZ-CAMTA1, and VEGF inhibitors have been shown to be beneficial in patients with EHE (Telli et al. 2020). Additionally, an mTOR-activated gene signature was observed in the proliferative endothelial cell population, and the mTOR inhibitor sirolimus has benefited some patients (Stacchiotti et al. 2016). Future studies aimed at validating and disrupting downstream target genes of TAZ-CAMTA1 may reveal useful therapeutic strategies for patients with EHE.
In conclusion, we demonstrate that a single genetic anomaly, TAZ-CAMTA1, is sufficient to drive the formation and progression of EHE-like vascular tumors. We show that the tumorigenic activity of TAZ-CAMTA1 is largely due to aberrant activation of TAZ target genes, as both entities activate similar downstream pathways and result in the formation of EHE-like vascular tumors. The findings of Seavey et al. (2021) reinforce our conclusions by demonstrating that the TAZ-CAMTA1 gene fusion is sufficient to result in EHE and that EHE from human patients indeed exhibits activation of YAP/TAZ target genes. Furthermore, we provide proof of concept for targeting the TAZ-CAMTA1 fusion protein, showing that its activity can be inhibited by Hippo pathway activation or by inhibiting its binding partners such as the TEAD transcription factors. That this gene fusion drives such a dramatic effect likely reveals that endothelial cells are exquisitely sensitive to YAP/TAZ activation, which is consistent with the important role of this pathway in endothelial cell development and homeostasis (Kim et al. 2017; Sakabe et al. 2017; Neto et al. 2018). Because transformed endothelial cells are so dependent on continued TAZ-CAMTA1 expression, monotherapy targeting the fusion protein may be highly effective for patients with EHE.
Materials and methods
Mice
All mice were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. All transgenic mice were maintained in a C57BL/6 and 129/Sv mixed background. TRE-H2B-GFP (Tumbar et al. 2004) and Cdh5-tTA (Sun et al. 2005) mice were from the Jackson Laboratory. NU(NCr)-Foxn1nu mice were from Charles River. TRE-TEAD2DN mice have been described previously (Liu-Chittenden et al. 2012).
To create the tetracycline-responsive TRE-TAZ-CAMTA1 transgenic mouse, a FLAG-tagged TAZ-CAMTA1 cDNA (Tanas et al. 2016) was cloned into the pTRE2 vector (BD Biosciences) that we had modified to contain an upstream sequence of rat insulin intron. The TRE-TAZ-CAMTA1 transgene was excised from the vector and injected into fertilized C57BL/6 mouse eggs by the University of Texas Southwestern Transgenic Core. Three founders were identified and crossed to the Cdh5-tTA transgenic line (Sun et al. 2005); two showed similar phenotypes, while one showed no phenotype and no expression of the transgene. One founder was used throughout the study and expression of the transgene was confirmed by immunofluorescence and Western blotting. TRE-TAZS4A mice were generated in the same way. Eight founders were identified and four of the founders showed similar phenotypes. One of the founders was mated to Cdh5-tTA mice for the results generated in this study. Genotyping primers are listed in Supplemental Table S3.
After crossing TRE-TAZ-CAMTA1 mice to Cdh5-tTA mice, pregnant mothers were treated with 1.5 mg/mL doxycycline in their drinking water with 1.25% sucrose. On the first day that pups were observed, the doxycycline water was replaced with normal water. The resulting mice were maintained on normal drinking water for the rest of their life span. TRE-H2B-GFP;TRE-TAZ-CAMTA1;Cdh5-tTA mice and TRE-TEAD2DN;TRE-TAZ-CAMTA1;Cdh5-tTA mice were treated in the same way. For reversibility experiments in Figure 2, mice were given water containing doxycycline (0.5 mg/mL for the survival study or 1.5 mg/mL with 1.25% sucrose for histological experiments) after 40 d of life.
Drosophila
FLAG-tagged TAZ, TAZS89A, TAZ-CAMTA1, and TAZ-CAMTA1S51A were cloned into the pUASTattb vector (Bischof et al. 2007) and confirmed by DNA sequencing. The resulting constructs were injected into the 51C landing site. The resulting flies were crossed to GMR-GAL4 (Freeman 1996) flies or GMR-GAL4 flies combined with UAS-SdRNAi (Bloomington Drosophila Stock Center, 29352).
Histology
After euthanasia, mice were subjected to transcardial perfusion with PBS + heparin (Sigma-Aldrich) and 4% paraformaldehyde. Tissue samples were collected and fixed in 4% PFA in 1× PBS overnight and embedded in paraffin. Sections were stained with hematoxylin-eosin for analysis. For immunohistochemistry, antibody detection was performed using the Elite ABC kit and DAB substrate according to the manufacturer's instructions (Vector Laboratories) and counterstained with hematoxylin (Thermo Fisher Scientific). Primary antibodies used were rat anti-FLAG (Biolegend 637319), anti-CD31 (Dianova DIA-310), anti-endomucin (Santa Cruz Biotechnology sc-65495), rabbit anti-S100a6 (Novus Biologicals NBP1-89388), anti-ERG (Abcam ab133264), anti-CD34 (Abcam ab81289), anti-Ki67 (Leica Biosystems NCLKi67p), anti-cytokeratin, wide spectrum screening (Agilent Z0622), anti-CRYAB (Invitrogen PA1-16950), and mouse anti-PDGFA (Santa Cruz Biotechnology sc-9974). Alexa488-, Alexa568-, and Alexa647-conjugated secondary antibodies (Molecular Probes) were used for immunofluorescence.
Quantifications
For quantifications of the tumor phenotypes in Figure 2, the results of multiple fields of view across a single lung section for each mouse were averaged and reported. For Ki67 quantifications, endothelial cells within multiple, independent, affected, noncapillary vessels were used and averaged across each sample. Similar vessels in control were used. All immunohistochemistry and H&E images were taken with a Zeiss Axioskop 2, and immunofluorescence images were taken with a Zeiss LSM 880.
Cell lines
MS1 (MILE-SVEN-1; source: mouse, C57BL/6, CRL2279) (Arbiser et al. 1997), MCF10A (source: human, female, CRL10317), and 293T (source: human, CRL3216) cells were obtained from ATCC. 293A (source: human) and 293A LATS1/2 KO (source: human) cells were a gift from Kun-Liang Guan (Hansen et al. 2015). All cell lines except MCF10A, which was cultured as previously described (Liu et al. 2010), were cultured in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1× penicillin/streptomycin (Sigma-Aldrich). All cells were cultured at 37°C with 5% CO2 in a humidified environment.
Plasmids
pBabeNeo-2xFLAG-TAZ-CAMTA1 and pBabeNeo-2xFLAG-TAZ-CAMTA1 S51A were gifts from Brian Rubin (Cleveland Clinic) (Tanas et al. 2016). pTRE2-TAZ-CAMTA1 was generated by inserting the TAZ-CAMTA1 cDNA into the HindIII cut site of the pTRE2 plasmid using the In-Fusion HD cloning kit (Takara). TAZ-CAMTA1 was cloned into the BamHI/XhoI sites of pcDNA3.1 (+) (Thermo Fisher) for transient expression, used in Figure 7. TAZ-CAMTA1S3A was generated by site-directed mutagenesis. pCDNA3.1-HA-TAZ was a gift from Kun-Liang Guan (Addgene plasmid 32839) (Lei et al. 2008). A 2xFLAG tag was added to replace the HA tag of TAZ, and TAZS4A was generated by site-directed mutagenesis. For generation of pUASTattb (Bischof et al. 2007) constructs, TAZ-CAMTA1 and TAZ-CAMTA1S51A were cut from pBabeNeo and cloned into the BamHI/XhoI cut sites of pUASTattb. TAZS89A was generated by site-directed mutagenesis, and TAZ and TAZS89A were given a 2xFLAG tag and cloned into the same sites of pUASTattb. All primers used for cloning are available in Supplemental Table S1. The integrity of all constructs was confirmed by DNA sequencing at the University of Texas Southwestern McDermott Sanger Sequencing Core.
Lentivirus production and stable cell generation
TAZ-CAMTA1, TAZ-CAMTA1S3A, TAZ, and TAZS4A cDNAs were cloned into the NheI/BamHI cut sites of pCDH-CMV-MCS-EF1-Puro (CD510B-1) from Systems Biosciences for constitutive expression. TAZ-CAMTA1 was cloned into the NheI/BamHI cut sites of the doxycycline-inducible all-in-one lentivirus construct pCW57-MCS1-2A-MCS2 (Barger et al. 2019). Primers used for cloning are listed in Supplemental Table S1. For virus production, the appropriate plasmid and the helper plasmids (psPAX2 and pMD2.G) were transfected into 293T cells cultured at 50% confluence in each well of a six-well plate dish using FuGene HD (Promega) according to the manufacturer's instructions. After 24 h, media was replaced. Viral supernatants were collected 76 h after transfection and filtered through a 0.45-µm filter. Viral supernatants were added to MS1 cells with 10 µg/mL polybrene (Santa Cruz Biotechnology). Pooled stable cells were selected with 1 µg/mL puromycin (Thermo Fisher Scientific). psPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259) were gifts from Didier Trono. pCW57.MCS1-2A-MCS2 was a gift from Adam Karpf (Addgene plasmid 71782).
Xenografts
MS1 cells transduced with the doxycycline-inducible TAZ-CAMTA1 lentivirus were injected, after selection, at a concentration of 106 cells subcutaneously into the flanks of 5-wk-old, male Nu/J mice (Jackson Laboratories). Control mice were given 2% sucrose in their drinking water, and experimental mice were given 2 mg/mL doxycycline + 2% sucrose to induce TAZ-CAMTA1 expression in the xenografts. Tumor volumes were measured weekly using calipers, and volumes were obtained from the formula 0.5 × length × width2, where width was the smaller dimension.
Cell imaging experiments
MS1 cells expressing doxycycline-inducible TAZ-CAMTA1 were seeded onto Lab-Tek II removable four-well chamber slides (Thermo Scientific). To induce TAZ-CAMTA1 expression, 2 µg/mL doxycycline were added to the appropriate chambers. After 24 h of seeding, cells were treated with vehicle or the respective drugs. Cells were treated with 1 µM latrunculin B for 1 h (Sigma Aldrich). The primary antibodies used were mouse anti-FLAG M2 antibody (Sigma-Aldrich F1804), Alexa488-conjugated anti-mouse (Molecular Probes), and Alexa fluor 647 phalloidin (Thermo Fisher A22287). For localization quantifications, cells were considered to have either majority nuclear or majority cytoplasmic localization based on immunostaining. The percentages of cells showing nuclear or cytoplasmic staining were averaged over multiple fields of view (n).
For transient transfections in Figure 7, 293A, 293A LATS1/2 KO, and 293T cells were seeded and transfected with pcDNA3.1 expressing TAZ-CAMTA1 or TAZ-CAMTA1S3A according to the manufacturer's instructions with FuGene HD (Promega). After 48 h of transfection, cells were fixed with 4% PFA in PBS and stained with the appropriate antibodies. Localizations were quantified as above. The individual chambers were removed and slides were imaged with an LSM 880 (Zeiss). Images were postprocessed with Fiji (Fiji is just ImageJ)
Coimmunoprecipitation and mass spectrometry
293T cells were grown on 10-cm plates and transfected according to the manufacturer's instructions by Effectene (Qiagen) with pcDNA3.1, pcDNA3.1 TAZ-CAMTA1, or pcDNA3.1 TAZ-CAMTA1S51A. After 48 h, cells were lysed with NP-40 lysis buffer, and FLAG-tagged TAZ-CAMTA1 was enriched through the use of anti-FLAG M2 affinity gel (Millipore Sigma). Expression of TAZ-CAMTA1 and enrichment by anti-FLAG beads was confirmed by Western blotting. Lysates were run a couple of inches on a Mini-Protean precast gel (Bio-Rad) and stained with Coomassie blue (Bio-Rad). A single 10-mm band of protein was cut out from the gel and submitted to the Proteomics Core at the University of Texas Southwestern for mass spectrometry. The samples were run on an Orbitrap Fusion Lumos instrument and proteins were discovered using Proteome Discoverer 2.2. The results are available in Supplemental Data File 3. No key differences were noted between TAZ-CAMTA1 and TAZ-CAMTA1S51A, and the results of TAZ-CAMTA1 versus empty vector are presented in Figure 7.
Western blots
Cells were lysed in RIPA buffer (Abcam) mixed with halt protease and phosphatase cocktail (Thermo Scientific) or lysed directly into SDS loading buffer. We used antibodies against FLAG (Sigma F1804), GAPDH (Millipore MAB374), and actin (Millipore MAB1501). Bands were visualized with secondary antibodies conjugated to horseradish peroxidase (Bio-Rad). ECL Western blotting detection reagents (Pierce) were used, and images were taken with a Bio-Rad ChemiDoc MP imaging system. Images were postprocessed and quantified in Fiji (Fiji is just ImageJ).
For protein stability experiments, TAZ- or TAZ-CAMTA1-expressing MCF10A cells were subjected to 40 µg/mL cycloheximide (CHX; Sigma Aldrich) or were treated for 24 h with 20 µM PS-341 (bortezomib, Selleck Chemicals).
Soft agar
Anchorage-independent growth was assayed by plating 1 × 105 cells or 5 × 105 cells per well in noble agar in six-well plates (Fisher Scientific). To set up these experiments, 2 mL of autoclaved 0.8% noble agar (Thermo Scientific) in 1× PBS was added and allowed to cool in each well. Then, cells were suspended in autoclaved UltraPure low melting point agarose (Fisher Scientific) and diluted to 0.35% in complete culture medium and added on top of the noble agar. After hardening, complete DMEM was added on top of the agarose. In experiments with drug treatments, drug or vehicle mixed into DMEM was added to each well. Experiments were set up in triplicate, and colonies were allowed to grow for 2 wk. For images, colonies were stained with 1 mg/mL MTT (Invitrogen). Colony numbers were counted using the “Find Maximum” function with Fiji.
RNA extraction and RT-qPCR
Total RNA was extracted using the Qiagen RNeasy Plus mini kit. cDNA synthesis was performed with 1µg of total RNA using the Bio-Rad iScript cDNA synthesis kit. cDNA was mixed with Bio-Rad iQ SYBR Green Supermix and run in triplicate on a Bio-Rad CFX96 real-time system. Primers used are listed in Supplemental Table S2. Expression levels given are normalized to Gapdh and then normalized to the average of the Ctrl sample.
RNA-seq
RNA was extracted from MS1 cells transduced with lentiviruses coding for control, TAZS4A, or TAZ-CAMTA1 expression with the Qiagen RNeasy Plus mini kit. RNA was DNase-treated with the Invitrogen Turbo DNA-free kit, and then libraries were prepared with the Illumina TruSeq stranded total RNA LT sample preparation kit. Poly-A RNA was purified and fragmented before cDNA synthesis. Adapters were ligated, and then samples were PCR-amplified and purified with Ampure XP beads. Samples were normalized, pooled, and run on an Illumina HiSeq 2500 using SBS v3 reagents. Library preparation and sequencing were performed by the University of Texas Southwestern Next-Generation Sequencing Core. Sequencing reads were mapped using TopHat2 to the mm10 mouse genome. Count data were generated using featureCounts, and differential gene expression analysis was carried out by the edgeR R package. Volcano plots and overlap analysis were generated in GraphPad Prism 7 using a list of genes comparing TAZ-CAMTA1- or TAZS4A-expressing MS1 cells with control cells with FDR < 0.01. GSEA (Subramanian et al. 2005) data were generated using the oncogenic and hallmarks gene sets database available in the Broad GSEA4.0.2 using the GSEA PreRanked software. The entire list of genes expressed by TAZS4A- or TAZ-CAMTA1-expressing MS1 cells was ranked in order by log(FC) compared with control. Heat maps were generated with Heatmapper. Correlation analysis and gene overlap were determined from the GeneOverlap R package. The RNA-seq data have been deposited in the GEO repository and are available at GSE156801.
Single-cell isolation and library preparation
Pooled lungs were isolated from three 5-wk-old TRE-H2B;TRE-TAZ-CAMTA1;Cdh5-tTA mice that had been induced (switched to normal water from 1.5 mg/mL doxycycline water) at birth. Lungs were digested in 0.2% collagenase 1 (Thermo Fisher) and treated with ACK lysis buffer before being mixed with the viability stain 7-AAD (BioLegend) and subjected to flow-activated cell sorting at the Moody Foundation Flow Cytometry Core on a FACS ARIA II SORP using FACSDiva software. Approximately 400,000 cells were obtained, and a viability of 80% was estimated with a Countess Automated Cell Counter (Thermo Scientific).
Samples were processed using the 10X genomics platform by the University of Texas Southwestern Next-Generation Sequencing Core. The appropriate volume of cells was loaded with single-cell 3′ gel beads into a Next GEM Chip G and run on the Chromium controller. GEM emulsions were incubated and then broken. Silane magnetic beads were used to clean up the GEM reaction mixture. Read 1 primer sequence was added during incubation and full-length, barcoded cDNA were then amplified by PCR after cleanup. Sample size was checked on the Agilent Tapestation 4200 using the DNAHS 5000 tape. Concentration was determined by the Qubit 4.0 fluorimeter (Thermo Scientific) using the DNA HS assay. Samples were enzymatically fragmented and underwent size selection before library construction. During library preparation, Read 2 primer sequence, sample index, and both Illumina adapter sequences were added. Subsequently, samples were cleaned up using Ampure XP beads and postlibrary preparation quality control was performed using the DNA 1000 tape on the Agilent Tapestation 4200. Final concentration was ascertained using the Qubit DNA HS assay. Samples were loaded at 1.6 pM and run on the Illumina NextSeq500 High-Output Flowcell using V2.5 chemistry. Run configuration was 28 × 98 × 8.
Single-cell data analysis
The digital expression matrix was obtained from the Cell Ranger pipeline. To analyze the data, we performed cell quality control and unsupervised clustering with the R package Seurat (Satija et al. 2015). Cells with <200 or >4000 features and >5% mitochondrial expression were excluded. The filtered data were normalized, the data were scaled, and variable genes were identified and used to generate principal components to group the cells. Based on the results of a Jack Straw plot, 30 principal components were used in a t-SNE analysis to reduce the dimensionality to two. Marker gene expression, identified by the Seurat FindAllMarkers function, was used to define the cells present in each cluster in conjunction with previously published scRNA-seq data of endothelial cells (Goveia et al. 2020). Differential expression was conducted by Seurat using the FindMarkers function, and the results were put into GraphPad Prism 7 to generate volcano plots and into the Broad GSEA4.0.2 software to generate GSEA results (Subramanian et al. 2005). Heat maps were generated by the Seurat DoHeatMap function along with the R package ggplot2. The scRNA-seq data have been deposited in the GEO repository and are available at GSE156803.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7. All data are presented as mean ± S.E.M. To calculate P-values, unpaired t-tests, χ2 tests, and one-way ANOVAs were used. For survival curves, the log-rank (Mantel-Cox) test was used to calculate P-values. P-values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Brian Rubin and Dr. Kun-Liang Guan for sharing reagents, and Dr. Ondine Cleaver, Dr. David McFadden, Dr. Hao Zhu, Dr. James Amatruda, and Dr. Gary Hon for advice on the project. We thank Lauren Scarborough, Maximino Villanueva, and Christopher Carl for technical assistance with mouse care, plasmid preparation, and tissue processing. We thank Dr. John Hulleman of the University of Texas Southwestern National Eye Institute Visual Science Core (P30 EY030413) for help with lentivirus production. We also thank the McDermott Sequencing Core, the Next-Generation Sequencing Core, the Bioinformatics Laboratory, the Moody Foundation Flow Cytometry Core, the Animal Resource Center, the Proteomics Core Facility, the Histo Pathology Core, and the Transgenic Core Facility at University of Texas Southwestern for their assistance with this work. This work was supported in part by National Institutes of Health grants EY015708 (to D.P.) and HL144793 (to M.D.). J.H.D. is supported in part by the Medical Scientist Training Program. D.P. is an investigator of the Howard Hughes Medical Institute.
Author contributions: J.H.D. and D.P. conceived the project, and D.P. supervised the research. J.H.D., Y.Z., M.D., and D.P. designed experiments. J.H.D., Y.Z., J.C., L.W., and M.D. conducted experiments and/or performed data analyses. J.C., B.-K.W., D.R., and M.D. contributed reagents, expertise, analytic tools, and/or grant support. J.H.D., Y.Z., M.D., and D.P. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348221.120.
Competing interest statement
The authors declare no competing interests.
References
- Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL, Chen HW, Pathan N, Krausz T, Dickson BC, et al. 2013. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 52: 775–784. 10.1002/gcc.22073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CR, Chen HW, Zhang L, Sung YS, Panicek D, Agaram NP, Dickson BC, Krausz T, Fletcher CD. 2014. ZFP36-FOSB fusion defines a subset of epithelioid hemangioma with atypical features. Genes Chromosomes Cancer 53: 951–959. 10.1002/gcc.22206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, et al. 1997. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci 94: 861–866. 10.1073/pnas.94.3.861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiser JL, Larsson H, Claesson-Welsh L, Bai X, LaMontagne K, Weiss SW, Soker S, Flynn E, Brown LF. 2000. Overexpression of VEGF 121 in immortalized endothelial cells causes conversion to slowly growing angiosarcoma and high level expression of the VEGF receptors VEGFR-1 and VEGFR-2 in vivo. Am J Pathol 156: 1469–1476. 10.1016/S0002-9440(10)65015-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger CJ, Branick C, Chee L, Karpf AR. 2019. Pan-cancer analyses reveal genomic features of FOXM1 overexpression in cancer. Cancers 11: 251. 10.3390/cancers11020251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. 2007. An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. Proc Natl Acad Sci 104: 3312–3317. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi KS, David KK, Huang B, Yabuta N, Nojima H, et al. 2015. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev 29: 1285–1297. 10.1101/gad.264234.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, et al. 2011. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147: 759–772. 10.1016/j.cell.2011.09.048 [DOI] [PubMed] [Google Scholar]
- Corrin B, Manners B, Millard M, Weaver L. 1979. Histogenesis of the so-called ‘intravascular bronchioloalveolar tumour’. J Pathol 128: 163–167. 10.1002/path.1711280308 [DOI] [PubMed] [Google Scholar]
- Doyle LA, Fletcher CD, Hornick JL. 2016. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol 40: 94–102. 10.1097/PAS.0000000000000511 [DOI] [PubMed] [Google Scholar]
- Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, Healey JH, Antonescu CR. 2011. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50: 644–653. 10.1002/gcc.20886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M. 1996. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell 87: 651–660. 10.1016/S0092-8674(00)81385-9 [DOI] [PubMed] [Google Scholar]
- Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, Geldhof V, de Rooij L, Kalucka J, Sokol L, et al. 2020. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37: 21–36.e13. 10.1016/j.ccell.2019.12.001 [DOI] [PubMed] [Google Scholar]
- Groeschl RT, Miura JT, Oshima K, Gamblin TC, Turaga KK. 2014. Does histology predict outcome for malignant vascular tumors of the liver? J Surg Oncol 109: 483–486. 10.1002/jso.23517 [DOI] [PubMed] [Google Scholar]
- Hansen CG, Ng YL, Lam WL, Plouffe SW, Guan KL. 2015. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res 25: 1299–1313. 10.1038/cr.2015.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, Halder G. 2014. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 13: 63–79. 10.1038/nrd4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YH, Kim J, Park DY, Bae H, Lee DH, Kim KH, Hong SP, Jang SP, Kubota Y, et al. 2017. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest 127: 3441–3461. 10.1172/JCI93825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JH, Guan KL. 2018. Interplay between YAP/TAZ and metabolism. Cell Metab 28: 196–206. 10.1016/j.cmet.2018.07.010 [DOI] [PubMed] [Google Scholar]
- Lee MY, da Silva B, Ramirez DC, Maki RG. 2019. Novel HMGA2-YAP1 fusion gene in aggressive angiomyxoma. BMJ Case Rep 12: e227475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. 2008. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 28: 2426–2436. 10.1128/MCB.01874-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, Li T, Chan SW, Lim CJ, Hong W, et al. 2010. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J Biol Chem 285: 37159–37169. 10.1074/jbc.M110.152942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. 2012. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26: 1300–1305. 10.1101/gad.192856.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Grueter CE, Song K, Qin S, Qi X, Kong YM, Shelton JM, Richardson JA, Zhang CL, Bassel-Duby R, et al. 2014. Ataxia and Purkinje cell degeneration in mice lacking the CAMTA1 transcription factor. Proc Natl Acad Sci 111: 11521–11526. 10.1073/pnas.1411251111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mana-Capelli S, McCollum D. 2018. Angiomotins stimulate LATS kinase autophosphorylation and act as scaffolds that promote Hippo signaling. J Biol Chem 293: 18230–18241. 10.1074/jbc.RA118.004187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AI, Shirali AS, Aragón R, Ma F, Hernandez G, Vaughn DA, Mack JJ, Lim TY, Sunshine H, Zhao P, et al. 2018. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell 23: 210–225.e6. 10.1016/j.stem.2018.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W, Lin Q, Childress C, Sudol M, Robishaw J, Berlot CH, Shabahang M, Yang W. 2015. Geranylgeranylation signals to the Hippo pathway for breast cancer cell proliferation and migration. Oncogene 34: 3095–3106. 10.1038/onc.2014.251 [DOI] [PubMed] [Google Scholar]
- Moroishi T, Hayashi T, Pan WW, Fujita Y, Holt MV, Qin J, Carson DA, Guan KL. 2016. The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 167: 1525–1539.e17. 10.1016/j.cell.2016.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto F, Klaus-Bergmann A, Ong YT, Alt S, Vion AC, Szymborska A, Carvalho JR, Hollfinger I, Bartels-Klein E, Franco CA, et al. 2018. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife 7: e31037. 10.7554/eLife.31037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, et al. 2015. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27: 728–743. 10.1016/j.ccell.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Wei Y, Okonechnikov K, Silva PBG, Vouri M, Zhang L, Brabetz S, Sieber L, Gulley M, Mauermann M, et al. 2019. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat Commun 10: 3914. 10.1038/s41467-019-11884-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagopoulos I, Lobmaier I, Gorunova L, Heim S. 2019. Fusion of the genes WWTR1 and FOSB in pseudomyogenic hemangioendothelioma. Cancer Genomics Proteomics 16: 293–298. 10.21873/cgp.20134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum E, Jadeja B, Xu B, Zhang L, Agaram NP, Travis W, Singer S, Tap WD, Antonescu CR. 2020. Prognostic stratification of clinical and molecular epithelioid hemangioendothelioma subsets. Mod Pathol 33: 591–602. 10.1038/s41379-019-0368-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rude MK, Watson R, Crippin JS. 2014. Recurrent hepatic epithelioid hemangioendothelioma after orthotopic liver transplantation. Hepatology 59: 2050–2052. 10.1002/hep.26891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe M, Fan J, Odaka Y, Liu N, Hassan A, Duan X, Stump P, Byerly L, Donaldson M, Hao J, et al. 2017. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc Natl Acad Sci 114: 10918–10923. 10.1073/pnas.1704030114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardaro A, Bardoscia L, Petruzzelli MF, Portaluri M. 2014. Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev 8: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A. 2015. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33: 495–502. 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seavey CN, Pobbati AV, Hallett A, Ma S, Reynolds JP, Kanai R, Lamar JM, Rubin BP. 2021. WWTR1(TAZ)-CAMTA1 gene fusion is sufficient to dysregulate YAP/TAZ signaling and drive epithelioid hemangioendothelioma tumorigenesis. Genes Dev (this issue). 10.1101/gad.348220.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Kiyono T, Ryo E, Ogawa R, Wakai S, Ichikawa H, Suzuki K, Arai S, Tsuta K, Ishida M, et al. 2019. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J Clin Invest 129: 3827–3832. 10.1172/JCI126185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seligson ND, Awasthi A, Millis SZ, Turpin BK, Meyer CF, Grand'Maison A, Liebner DA, Hays JL, Chen JL. 2019. Common secondary genomic variants associated with advanced epithelioid hemangioendothelioma. JAMA Netw Open 2: e1912416. 10.1001/jamanetworkopen.2019.12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya R, Matsuyama A, Shiba E, Harada H, Yabuki K, Hisaoka M. 2015. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology 67: 827–835. 10.1111/his.12713 [DOI] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, et al. 2014. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 16: 357–366. 10.1038/ncb2936 [DOI] [PubMed] [Google Scholar]
- Stacchiotti S, Provenzano S, Dagrada G, Negri T, Brich S, Basso U, Brunello A, Grosso F, Galli L, Palassini E, et al. 2016. Sirolimus in advanced epithelioid hemangioendothelioma: a retrospective case-series analysis from the Italian Rare Cancer Network database. Ann Surg Oncol 23: 2735–2744. 10.1245/s10434-016-5331-z [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam A, Zheng J, Yalamanchili S, Conley AP, Ratan R, Somaiah N, Livingston JA, Zarzour MA, Araujo DM, Benjamin RS, et al. 2020. Modulation of YAP/ TAZ by statins to improve survival in epithelioid hemangioendothelioma (EHE). J Clin Oncol 38: e23527. 10.1200/JCO.2020.38.15_suppl.e23527 [DOI] [Google Scholar]
- Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, et al. 2005. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci 102: 128–133. 10.1073/pnas.0403198102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurmeijer AJH, Dickson BC, Swanson D, Sung YS, Zhang L, Antonescu CR. 2020. Variant WWTR1 gene fusions in epithelioid hemangioendothelioma—a genetic subset associated with cardiac involvement. Genes Chromosomes Cancer 59: 389–395. 10.1002/gcc.22839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulzewsky F, Arora S, Hoellerbauer P, King C, Nathan E, Chan M, Cimino PJ, Ozawa T, Kawauchi D, Pajtler KW, et al. 2020. Comparison of tumor-associated YAP1 fusions identifies a recurrent set of functions critical for oncogenesis. Genes Dev 34: 1051–1064. 10.1101/gad.338681.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, Flanagan J, Luo Y, Fenwick K, Natrajan R, et al. 2011. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med 3: a82. [DOI] [PubMed] [Google Scholar]
- Tanas MR, Ma S, Jadaan FO, Ng CK, Weigelt B, Reis-Filho JS, Rubin BP. 2016. Mechanism of action of a WWTR1(TAZ)-CAMTA1 fusion oncoprotein. Oncogene 35: 929–938. 10.1038/onc.2015.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telli TA, Okten IN, Tuylu TB, Demircan NC, Arikan R, Alan O, Ercelep O, Ones T, Yildirim AT, Dane F, et al. 2020. VEGF-VEGFR pathway seems to be the best target in hepatic epithelioid hemangioendothelioma: a case series with review of the literature. Curr Probl Cancer 44: 100568. 10.1016/j.currproblcancer.2020.100568 [DOI] [PubMed] [Google Scholar]
- Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, Judson RN, Thway K, Nadal G, Selfe JL, et al. 2014. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 26: 273–287. 10.1016/j.ccr.2014.05.029 [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. 2004. Defining the epithelial stem cell niche in skin. Science 303: 359–363. 10.1126/science.1092436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi T, Naito H, Suehiro JI, Lin Y, Kawaji H, Iba T, Kouno T, Ishikawa-Kato S, Furuno M, Takara K, et al. 2018. CD157 marks tissue-resident endothelial stem cells with homeostatic and regenerative properties. Cell Stem Cell 22: 384–397.e6. 10.1016/j.stem.2018.01.010 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y, et al. 2014. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci 111: E89–E98. 10.1073/pnas.1319190110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SW, Enzinger FM. 1982. Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer 50: 970–981. [DOI] [PubMed] [Google Scholar]
- Weldon-Linne CM, Victor TA, Christ ML, Fry WA. 1981. Angiogenic nature of the ‘intravascular bronchioloalveolar tumor’ of the lung: an electron microscopic study. Arch Pathol Lab Med 105: 174–179. [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26: 54–68. 10.1101/gad.173435.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Pan D. 2019. The Hippo signaling pathway in development and disease. Dev Cell 50: 264–282. 10.1016/j.devcel.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.