

In this review, Pilley et al. examine the impact of different p53 mutations and focus on how heterogeneity of p53 status can affect relationships between cells within a tumor.
Keywords: cancer, cell competition, cell-cell interactions, mutant p53
Abstract
p53 is an important tumor suppressor, and the complexities of p53 function in regulating cancer cell behaviour are well established. Many cancers lose or express mutant forms of p53, with evidence that the type of alteration affecting p53 may differentially impact cancer development and progression. It is also clear that in addition to cell-autonomous functions, p53 status also affects the way cancer cells interact with each other. In this review, we briefly examine the impact of different p53 mutations and focus on how heterogeneity of p53 status can affect relationships between cells within a tumor.
Genome-wide analyses have shown that TP53 is the most frequently compromised gene in human cancer (Kandoth et al. 2013). In the 40 yr since p53 was discovered, its function has been studied intensively, yet new activities are still being found. Foremost, p53 is a transcription factor with the ability to control the expression of a large number of coding and noncoding RNA (Beckerman and Prives 2010; Grossi et al. 2016; Sullivan et al. 2018). Additionally, p53 interacts with cytoplasmic and mitochondrial proteins to directly modulate their activity (Ho et al. 2020). p53 sits as a central node in the cell's stress detection pathways, integrating signals from numerous sources, including DNA damage, oncogenic stress, hypoxia, and metabolic stress, to determine cell fate according to the type, severity, and duration of the stress. The induction of a p53 response can help cells deal with stress by supporting adaptation and survival or induce a permanent withdrawal of proliferative capacity through the induction of differentiation, senescence, or cell death (Kruiswijk et al. 2015). In either case, the induction of a p53 response can retard the evolution of a fully malignant cell. However, it is clear that there is still much to be learned about the exact mechanisms through which p53 fulfills its role as our most important defense against cancer development.
Mutant p53
While some cancer-associated somatic mutations in TP53 result in the loss of protein expression, many cancers express missense mutations that lead to the expression of full-length p53 proteins carrying a single amino acid substitution. Li-Fraumeni syndrome (LFS) patients carry germline p53 mutations and are consequently highly likely to develop a variety of different cancers at a young age (Bougeard et al. 2015). Most (∼80%) somatic missense mutations occur in the central DNA binding domain (DBD) of p53 and are concentrated at certain “hot spot” residues, including: 175, 245, 248, 273, and 282 (Bouaoun et al. 2016). These DBD p53 mutations have been classified as either affecting the conformation or DNA contacting ability of the protein, but most mutants show a reduction in both structural stability and sequence-specific DNA binding (Bullock et al. 2000), and it is not clear whether these classifications are functionally relevant (Sabapathy and Lane 2018). Missense mutations in the N-terminal domain and C-terminal oligomerization domain (OD), which flank the DBD of p53, are also found in human cancers, although somatic alterations in this region are less frequent. Interestingly, the R337H OD alteration is found in a significant number of LFS families in southern Brazil (Pinto and Zambetti 2020).
Cancer-derived p53 missense mutants are impaired for most wild-type (WT) p53 functions. While this loss of function can clearly contribute to tumor development, the high incidence of missense mutation compared with nonsense mutation or gene deletion has raised the possibility that there is a selective advantage to tumors in maintaining the expression of mutant p53 proteins. Studies comparing the consequences of loss of p53 versus expression of mutant p53 have revealed a number of mechanisms that could explain the selective advantage of mutant p53 expression during cancer development. These can be broadly grouped into dominant-negative effects on coexpressed WT p53 or independent gain of oncogenic functions (Fig. 1).
Figure 1.

Acquisition and impact of mutant p53 status. Expression of a mutant p53 protein is associated with more aggressive behavior in many cancer types, and mutant p53 status may be acquired through several mechanisms. Following point mutation in TP53, cancer cells can become genetically homozygous or hemizygous through loss of the WT allele (loss of heterozygosity). However, in cells that retain WT p53, the dominant-negative activity of mutant p53 can inhibit the WT protein and render the cell functionally mutant for p53. Finally, the adoption of a pseudomutant p53 conformation by the WT protein can allow mutant p53 behavior in cells without a TP53 mutation. Additional gain-of-function activities of mutant p53, beyond loss of WT activities, are associated with progression to metastasis in several cancer types.
Despite the vast literature identifying mutations in p53 as contributors to cancer development, a surprising study recently suggested that, in some contexts, mutant p53 could function as a tumor suppressor (Kadosh et al. 2020). Using a mouse model of intestinal cancer driven by the deletion of CKIa or mutation in Apc, mutant p53 was shown to have the expected oncogenic effect in the distal tissues of the intestine. However, in the proximal intestinal tissues of the same mice, the presence of mutant p53 was tumor-suppressive, a response that was shown to result from differences in the local microbiome. These results were quite unexpected, as such clear regional differences between mutant and loss of p53 have not been noted in other studies. The critical factor necessary to support mutant p53's oncogenic activity was found to be microbiota-derived gallic acid, leading to the possibility that changes in gallic acid levels between different mouse colonies could influence mutant p53-driven tumorigenesis. Nevertheless, while this tumor suppressor role for mutant p53 clearly warrants further investigation, almost all of our understanding of mutant p53 function to date addresses its protumorigenic activities.
The dominant-negative effect
p53 functions as a tetramer, assembled through the C-terminal OD. This region is retained by the common cancer-associated point mutants of p53, allowing the formation of functionally compromised heterotetramers containing both WT and mutant proteins. A dominant-negative (DN) ability of mutant p53 to restrain the activity of WT p53 could contribute to tumor development when both are expressed in the same cell (Fig. 2). In established tumors, estimates of the frequency of a loss of heterozygosity (LOH) in TP53 (the deletion of the WT TP53 allele after mutation in the other) vary between cancers. However, a recent analysis of >10,000 tumors reported that >90% of tumors with TP53 mutations did not retain a WT allele (Donehower et al. 2019). The frequent loss of the WT allele may indicate that the DN activity of mutant p53 proteins is not sufficient to fully inhibit WT p53 activity and allow for the development of tumors (Alexandrova et al. 2017). Furthermore, an analysis of several tumor types failed to show any difference in the rate of LOH in tumors expressing mutant p53 with DN capacity compared with those with p53 mutant proteins unable to tetramerize and so lacking DN function, or in the survival of patients harboring these tumors (Shahbandi and Jackson 2019). On the other hand, analysis of cancers arising in LFS patients—with a germline mutation in one TP53 allele—showed loss of the WT allele in almost all cases with a p53-null mutation but in only around a third of tumors in LFS patients carrying a germline p53 mutation in the DBD (Varley et al. 1997; Varley 2003; Malkin 2011). LFS patients carrying a potentially DN p53 allele also showed an earlier tumor onset than those with functionally null p53 mutations (Bougeard et al. 2015).
Figure 2.
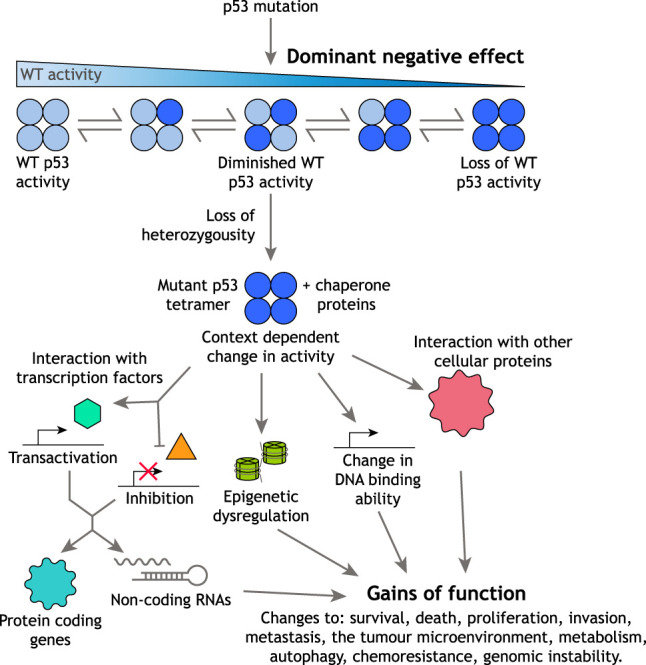
Mechanisms of mutant p53 action. The acquisition of p53 mutation can lead to the formation of heterotetramers made up of WT and mutant monomers. The presence of mutant subunits in a tetramer can reduce WT p53 activity and, through this dominant-negative activity, allow for tumor development. However, many tumors lose the remaining wild-type allele, leading to the expression of only mutant protein. Mutant p53 is frequently stabilized in cancer cells, in part as a consequence of complex with chaperone proteins, allowing it to mediate a variety of new activities (gains of function) via a number of different mechanisms.
To seek clarity around the role of DN functions of mutant p53, several experimental models have been used. Although the DN activity of mutant p53 has been demonstrated repeatedly over the past 30 yr, many of the early studies used systems that overexpressed the mutant protein and may not reflect a physiologically relevant situation (Gencel-Augusto and Lozano 2020). Nevertheless, more recent analyses of cells or mice expressing endogenous mutant and WT p53 have demonstrated DN activity of the mutant protein under some, but not all, conditions (Gencel-Augusto and Lozano 2020). In mouse models, a substantial proportion of tumors arising in Tp53+/− mice retain a functional WT allele (Venkatachalam et al. 1998). While it is possible that aging (Feng et al. 2007) and the acquisition of other alterations (such as defects in the pathways necessary to activate p53) weaken the p53 response in tumors that retain WT p53, it seems likely that complete loss of WT p53 function is not necessary for tumor development, so limiting a requirement for any DN effect. Tp53+/mutant mice, in which the p53 mutant has potential DN activity, show extended survival compared with p53-null mice, again suggesting that the expression of mutant p53 cannot completely disable the tumor restraining functions of the WT protein (Lang et al. 2004). This concept is further supported by the analysis of the consequences of loss of the p53 regulator MDM2 in mice. Expression of MDM2 during embryogenesis is essential to restrain p53 activity, but the lethality of Mdm2 loss can be rescued by concomitant deletion of both Tp53 WT alleles. However, deletion of Mdm2 in a Tp53+/mutant mouse also led to embryonic lethality, indicating that WT p53 is still active (Lang et al. 2004).
While these mouse experiments show that the expression of mutant p53 cannot completely inactivate WT p53 function, several models show that some mutant p53s have a partial ability to restrain WT p53. Several studies have reported that the presence of a mutant p53 allele can result in a decrease in WT p53 functions, mostly in response to various forms of DNA damage (Lang et al. 2004; Wijnhoven et al. 2007; Lee and Sabapathy 2008). Importantly, p53 protein can only be detected in tumor samples and not in normal tissue of Tp53+/mutant mice (Lang et al. 2004; Olive et al. 2004), suggesting that there are some tumor-specific changes required for mutant p53 to become stable and therefore able to exert a DN effect. Several mechanisms may underlie the stabilization of mutant p53 in cancer cells, including: the presence of tumor-specific stress signals that might normally function to stabilize WT p53, the inability of most mutant p53 proteins to induce the transcription of MDM2, and the ability of mutant p53 proteins to complex with members of the heat shock protein family, induced by the unfolded protein response often active in tumors, which stabilize mutant p53 proteins by preventing interaction with MDM2 (Wawrzynow et al. 2018; Mantovani et al. 2019). Furthermore, by disrupting the autophagic machinery in the cell, mutant p53 protects itself from degradation, adding to its stability (D'Orazi et al. 2020). Taken together, it appears that, in most cases, mutant p53 has only a partial ability to limit WT p53 function and that this may only become relevant in certain tissues or in response to stress such as DNA damage (Gencel-Augusto and Lozano 2020). A more in-depth analysis of studies that have or have not found evidence for the DN effect can be found in this recent review (Gencel-Augusto and Lozano 2020).
Oncogenic gains of function
In addition to inhibiting the function of WT p53, mutant p53 proteins can also acquire novel gains of function (GOFs) that contribute to tumor development independently of WT p53. The most compelling evidence for mutant p53 GOFs is seen by comparing mice expressing mutant p53 or no p53. While p53-null mice develop mostly lymphomas and soft tissue sarcomas (Donehower et al. 1992), mice expressing mutant p53 show a different tumor spectrum; developing more epithelial and endothelial tumors (Olive et al. 2004) and showing increased metastasis (Lang et al. 2004). Similarly, mice expressing a humanized version of mutant p53 show worse survival than mice without p53 (Hanel et al. 2013). A positive role for mutant p53 in maintaining malignancies has also been shown in cell culture systems in vitro (Bossi et al. 2006; Hui et al. 2006) and in vivo, where inactivation of mutant p53 caused tumor shrinkage and improved survival in lymphomas (Alexandrova et al. 2015) and tumor shrinkage in dextran sulphate sodium (DSS)-induced colorectal cancer (Schulz-Heddergott et al. 2018).
Further experimental studies have revealed a variety of cell-autonomous functions of mutant p53s, including cell survival, chemoresistance, metabolic rewiring, inhibition of autophagy and cell death, resistance to proteotoxic stress, increased rates of proliferation, genomic instability, and enhanced migratory and invasive capacity. These activities are discussed in detail in several recent reviews (Freed-Pastor and Prives 2012; Muller and Vousden 2014; Kim and Lozano 2018; Sabapathy and Lane 2018; Stein et al. 2019b; Zhang et al. 2020a; Zhu et al. 2020). In general, these functions reflect the acquisition or alteration of DNA and protein binding abilities of mutant p53s. While tumor-associated p53 mutant proteins lose most, but not necessarily all, of their ability to bind to the consensus DNA binding sequences recognized by WT p53 (Ludwig et al. 1996; Kato et al. 2003), they can acquire the ability to bind different promoters and so directly regulate new transcriptional programs (Dell'Orso et al. 2011). However, most of the GOF activities displayed by different mutant p53 proteins are thought to manifest through their interactions with other proteins. Mutant p53 proteins have been found to interact with a number of different transcription factors—such as NF-Y, SREBPs, ETS1/2, and NF-κB—and either potentiate or inhibit their activity (Kim and Lozano 2018). In particular, mutant p53s bind to and suppress the activities of the family member transcription factors p63 and p73 (Strano 2016). These mutant p53/transcription factor interactions lead to changes in expression of both protein coding genes and a range of different noncoding RNAs, including miRNAs and lncRNAs (Zhang et al. 2020a), all of which can contribute to the GOF phenotype. The activity of mutant p53s is further expanded by the ability to bind and modulate the activity of nontranscription factor proteins, resulting in the impairment of the DNA damage response, increased rates of glycolysis and lipid production (Zhu et al. 2020), and increased histone methylation (Chen et al. 2019). More generally, mutant p53s can bind to a variety of proteins controlling the epigenome, including those that regulate chromatin structure, histone modifications, and splicing, resulting in genome-wide dysregulation (Escobar-Hoyos et al. 2020; Zhang et al. 2020a). It should be noted, however, that the majority of the outcomes of mutant p53 expression are highly mutant- and cell type-specific, suggesting that the particular external stress and the composition of the tumor microenvironment (TME) may influence mutant p53 GOFs (Fig. 2; Amelio and Melino 2020).
Resolving the roles of mp53 DN activities and GOFs
There is considerable debate about the importance of DN and GOF activities of mutant p53 (Stein et al. 2020). An analysis of genome-wide RNAi and CRISPR-Cas9 survival screens of cancer cell lines did not reveal the dependence of cancer cells on mutant p53 expression, although this study did show that many p53 point mutants acquire a DN ability to overcome WT p53 activation and allow proliferation and survival of cells in culture (Giacomelli et al. 2018). Similarly, engineering of AML cell lines to express various combinations of WT and mutant p53 provided evidence for DN but not GOF activities. However, in vivo hematopoietic stem and progenitor cells (HSPCs) null for p53 outcompeted HSPCs expressing one mutant p53 and one WT allele in response to sublethal radiation, indicating the limitations of the DN effect (Boettcher et al. 2019). In a system lacking WT p53, an analysis of >10,000 p53 mutants found evidence for GOFs, with cells expressing mutant p53s gaining an in vivo advantage over cells without p53 (Kotler et al. 2018). Furthermore, expression of mutant p53 enhances the reprogramming and transformation of somatic cells (Sarig et al. 2010) and drives abnormal self-renewal in acute myeloid leukemia beyond that seen following p53 loss, again supporting a role for mutant p53 beyond loss of WT function (Loizou et al. 2019).
Collectively, these studies leave a confusing picture. There is good evidence for both GOFs and DN activities, but neither are consistently observed in all models, and it is clear that the manifestation of these phenotypes will be context- or tumor type-specific. It is worth considering that the temporal dynamics of LOH in TP53 in cancers are not well established, meaning DN activity may be important during tumor initiation but not in the later stages of tumor progression, at least in some cancers. While it may be possible for GOFs to occur in the presence of the WT protein, in certain contexts, GOFs cannot be identified unless LOH has occurred (Nakayama et al. 2020). On the other hand, GOFs may become more important in driving the metastatic progression of established malignancies.
Adding to the complexity are intriguing studies showing that mutant p53s can form prion-like aggregates that also capture p53 family members and may underlie both DN and GOF activities. When internalized by otherwise healthy cells, mutant p53 aggregates coaggregate with and consequently inhibit WT p53 protein activity, suggesting that mutant p53 status can spread between cells (de Oliveira et al. 2020; Navalkar et al. 2020).
WT p53 in tumorigenesis—the importance of structure
Whereas mutations in p53 are a common occurrence in cancers, it is clear that tumors can develop while retaining WT p53. The identification of mechanisms through which p53 promotes cell survival in the face of mild stresses, such as nonlethal DNA damage or transient nutrient starvation, provides for the possibility that WT p53 functions may help to support some stages of tumor development. Such activities explain an early paradoxical observation that WT p53 activity could support the development of carcinogen-induced skin papillomas in mice (Kemp et al. 1993) and several subsequent studies showing that retention of WT p53 can promote resistance to therapy (Ablain et al. 2016; Webster et al. 2020). Furthermore, WT p53 in tumors has been shown to maintain cell survival in response to the depletion of nutrients such as glucose (Jones et al. 2005), serine (Maddocks et al. 2016), or glutamine (Tajan et al. 2018) by promoting the metabolic plasticity required to deal with the stress. Some tumor-derived p53 mutants retain the ability to deal with these stresses, suggesting that a retention of some WT p53 activities by mutant p53 can benefit tumors (Tran et al. 2017; Humpton et al. 2018).
Interestingly, expression of a p53 protein with WT sequence does not always ensure the retention of WT p53 structure or function. WT p53 can adopt a “mutant” conformation under some conditions, such as defects in chaperones, leading WT p53-expressing cells to display mutant p53-like functions such as increased invasion (Trinidad et al. 2013) and gene expression patterns (Benor et al. 2020). The expression of this “pseudomutant” phenotype may explain how some cancers develop despite the retention of WT p53 and suggest that some plasticity with respect to WT and mutant p53 activity may be highly beneficial during tumorigenesis.
Notwithstanding the potential for some WT p53 functions to support cancer development, there is abundant evidence to support the therapeutic benefit of WT p53 activation in many cancers (Lozano 2019). Consequently, an approach that allows for the reactivation of WT p53 function in tumors expressing mutant p53 proteins is an attractive therapeutic strategy. A variety of different compounds have been tested for their ability to restore the WT conformation and consequently the function of mutant p53 proteins (Muller and Vousden 2014). These include APR-246, a prodrug reported to restore WT conformation by binding specific cysteine residues, which is currently in clinical trials. However, APR-246 and its related compounds also show a variety of p53-independent activities that could underlie therapeutic efficacy (Bykov et al. 2018; Eriksson et al. 2019).
Cell-nonautonomous roles of p53
It has become clear that, to fully understand cancer, the tumor cells need to be considered in the wider context of the TME. Cancer cells (of which there may be various subclones) make up only a fraction of a solid tumor. The rest of the tumor mass comprises the stroma, consisting of a variable proportion of different immune cells, fibroblasts, adipocytes, blood vessels, and nerves. This composition has led to the description of tumors as “pseudoorgans” (Lyssiotis and Kimmelman 2017). While competition between these cells for space and resources imposes challenges on tumor cells, surrounding stromal cells can also be reprogrammed by cancers to provide support and enhance malignant development. Most previous studies on p53 have focused on cell-autonomous functions of WT and mutant p53, but it is becoming clear that the complex interaction between cancer cells and their surroundings is also strongly influenced by the p53 status of each compartment.
WT p53 in cell-nonautonomous tumor suppression
Several studies have shown that loss of WT p53 in normal stromal cells enhances tumorigenesis (Kiaris et al. 2005; Guo et al. 2013, 2017), demonstrating a tumor-suppressive role for p53 beyond the cancer cells themselves. This activity is linked to the ability of WT p53 to activate expression of various secreted factors that can limit oncogenic progression (Fischer 2017). Examples include an ability of p53 in fibroblasts to exert a tumor-suppressive effect on the surrounding tissue by suppressing the expression of SDF-1/CXCL12, a chemokine that increases cell invasiveness (Moskovits et al. 2006) and the release of factors from p53-expressing hepatic stellate cells that promote macrophage polarization toward the tumor-suppressive M1 type (Lujambio et al. 2013). The induction of senescence in stromal cells following activation of p53 is associated with the release of cytokines and other factors related to the “senescence-activated secretory phenotype” (SASP). The consequences of SASP induction can be both tumor-promoting and tumor-limiting (Coppé et al. 2010), and the promalignant activities of SASP have been shown to be reduced by p53 (Coppé et al. 2008). Further evidence is now developing to show a function of WT p53 in other types of stromal cells, such as immune cells (Blagih et al. 2020a), with variable effects on tumorigenesis.
Although stromal cells can provide tumor-limiting functions, it is well established that cancer-associated fibroblasts (CAFs) exhibit activities that promote tumor growth and progression (Kalluri and Zeisberg 2006). Interestingly, cancer cells can dampen the tumor-limiting responses driven by p53 in surrounding fibroblasts by inhibiting their ability to activate p53 (Bar et al. 2009). In a similar model, p53 suppression or mutation in tumor cells was also seen to reduce p53 activity in cocultured fibroblasts and also stimulated fibroblast proliferation and their tumor-supporting functions via miRNAs found in exosomes released by the tumor cells (Yoshii et al. 2019). Loss of p53 in fibroblasts has previously been associated with their transformation into tumor-promoting CAFs (Addadi et al. 2010). Furthermore, p53 was found to be critical in supporting the expression of genes that distinguish CAFs from normal fibroblasts, including those of secreted proteins that increase cancer cell migration and invasion in vitro and growth in vivo (Arandkar et al. 2018). The investigators explain that tumor cells acquire the ability to inhibit the canonical tumor-suppressive functions of p53 in normal fibroblasts, and in CAFs, the p53 activity becomes “rewired” to support their tumor-promoting functions (Arandkar et al. 2018). Further investigations into how and why the functions of WT p53 switch from tumor-suppressive to tumor-promoting are required, although it is possible that the “pseudomutant” phenomenon may be responsible. WT p53 has also been shown to promote chemoresistance through a cell-nonautonomous manner by enabling cells to enter into senescence. While mutant p53 cells die due to their inability to arrest proliferation, WT p53 senescent cells secrete growth-stimulating cytokines in an autocrine and paracrine manner, allowing these tumors to relapse faster after the withdrawal of chemotherapy (Jackson et al. 2012).
The retention of WT p53 in cancer cells can also affect the tumor-stroma interaction, most clearly by supporting the activation of an anti-tumor immune response (Bezzi et al. 2018; Wellenstein et al. 2019; Blagih et al. 2020b). In these studies, loss of p53 in cancer cells was shown to increase expression of cytokines and factors that blunt various arms of the immune response to the tumor and enhance metastasis. Finally, p53 has also been shown to restrain the adrenergic transdifferentiation of neurons in murine oral tumors by promoting the release of vesicles containing the microRNA miR-34a. These changes in innervation resulting from p53 loss contributed to cancer progression in mice and were correlated with a significant worse prognosis in human cancers (Amit et al. 2020).
Cell-nonautonomous functions of mutant p53
As with WT p53, the presence of mutant p53 in a cell changes the expression of a variety of secreted proteins in a context- and mutation-dependent manner. The profile of molecules secreted by a tumor cell depends on the tumor genetics (Paltridge et al. 2013) but also varies widely based on the cancer type (Robinson et al. 2019). The impact of mutant p53 on the cancer cells secretome and consequent interactions between the tumor and stroma have been discussed in detail in several previous reviews (Cordani et al. 2016; Stein et al. 2019a; Blagih et al. 2020a; Pavlakis and Stiewe 2020; Pavlakis et al. 2020) and is briefly reviewed here.
Changes in the proteins that are secreted by cancer cells can have a direct effect on their behaviors through autocrine or paracrine mechanisms, and the induced expression of a variety of p53 mutants including R175H, R248Q, R248W, R249S, R273H, and R282W in H1299 cells led to the expression of a proinvasive secretome that increased the invasiveness of cells with no p53 expression (Neilsen et al. 2011). Mutant p53 was also shown to enhance exosome-mediated Hsp90a secretion (Zhang et al. 2020b), a protein that promoted invasion and migration of cancer cells and was previously shown to increase the efficiency of metastasis in mice (Wang et al. 2009). In addition to the up-regulation of specific secreted factors, mutant p53 has also been found to amplify total cell secretion by inducing the transcription of miR-30d, which is able to influence the structure of the Golgi apparatus. The mutant p53-dependent increase in secretion enhanced tumorigenesis and metastasis in vivo (Capaci et al. 2020).
A number of studies have identified how mutant p53 modulates the response of the NF-κB pathway to inflammatory signals, affecting the cellular secretions and consequently tumor cell motility and survival. For example, expression of various p53 mutants (including R175H, M237I, R273H, and R280K) supported invasive behavior and cancer cell survival in response to the inflammatory cytokine TNFα by promoting the secretion of proinvasive molecules such as MMP9 and CXCL10. Interestingly, these cells also showed mutant p53-dependent secretion of the lymphocyte-attracting chemokines CX3CL1 and LTB, suggesting that the increased motility was accompanied by an induction of a potentially tumor-limiting immune response (Di Minin et al. 2014). Similarly, several different mutant p53 proteins (D281G, H179L, R175H, and R273H) were shown to cooperate with the NF-κB pathway to promote CXC chemokine expression to increase cell motility over p53-null cells (Yeudall et al. 2012). The mutant p53-induced production of cytokines has also been shown in several studies to prevent the anti-tumor immune response and drive a protumoringenic inflammatory environment (Agupitan et al. 2020).
Another area of growing interest is how mutant p53 affects the production of exosomes by cancer cells can impact tumor progression. Exosomes are small extracellular vesicles that can contain nucleic acids, proteins, lipids, and metabolites, allowing communication between cells and tissues (Kalluri 2016). Exosomes secreted by mutant p53-expressing colon cancer cells can reprogram adjacent macrophages into a “cancer-promoting” state, making them more able to support tumor growth and metastasis than macrophages exposed to WT p53 cancer cells.(Cooks et al. 2018). Enhanced production of exosomes by mutant p53-expressing cancer cells also leads to an increase in fibroblast mobility and educates fibroblasts to deposit and remodel the extracellular matrix (ECM) into a state more conducive to cancer cell invasion (Novo et al. 2018).
Mutant p53 had previously been shown to play a role in modifying the structure of the ECM through the retention of the WT ability to repress the expression of TIMP3, an inhibitor of the matrix metalloproteinases (Loging and Reisman 1999), the activation of which results in the degradation of the surrounding ECM, consequently enhancing metastasis and invasion (Kessenbrock et al. 2010). Furthermore, MMP9 is expressed at much higher levels in colorectal cancers with p53 mutations than in those without (Rahnamoun et al. 2017). In nonsmall cell lung cancer, interactions between mutant p53 R273H HIF1α result in the up-regulation of ECM proteins VIIa1 collagen and laminin-γ2, the expression of which is associated with worse prognosis (Amelio et al. 2018). Another investigation found mutant p53 in tumor cells can have an indirect effect on the ECM by affecting fibroblast secretions. Mutant p53-expressing pancreatic cancer cells induced the secretion of perlecan in adjacent fibroblasts, an activity that was required for invasion of both mp53- and p53-null cancer cells into matrices (Vennin et al. 2019). The ability of mutant p53 to affect the structure of the ECM may have a feedback effect on the tumor cells, since mutant p53 is known to affect the trafficking of receptors that interact with the ECM, such as integrins (Muller et al. 2009), and become stabilized in response to changes in matrix stiffness (Ingallina et al. 2018).
Tumor-stromal interactions involve cross-talk between multiple different cell types in the TME, where an effect on one stromal compartment can have a subsequent impact on another. Not surprisingly, the p53 status of the tumor can influence these complex interaction networks. For example, tumor cells can induce fibroblasts to secrete IFNβ, which represses the invasive activity of the cancer cells and has potential to modulate the immune response to the tumor. The response to CAF-derived IFNβ is modulated by mutant p53 expression in the cancer cells, leading to increased invasive capacity (Madar et al. 2013).
Heterogeneity in the impact of mutant p53 on the stroma
There is a growing appreciation that different p53 mutants may have different activities that support and promote cancer development (Hanel et al. 2013; Giacomelli et al. 2018; Sabapathy and Lane 2018), an observation that also extends to the functions of p53 in controlling the stromal interaction. The ability to drive expression of secreted proteins that enhance malignancy, such as CXC chemokines, proinflammatory cytokines, and extracellular matrix-related proteins, is stronger in the “DNA contact” mutants R248Q and R273H than “conformational” mutants R175H and H179R, with some evidence that these two groups of p53 mutants function through different pathways (Buganim et al. 2010). In contrast, expression of the G245S mutant did not alter the expression of protumorigenic factors (Solomon et al. 2012). Similarly, the ability of mutant p53 to traffic Hsp90a into exosomes was more robust for the DNA contact mutants (R248W, R273H, and R280K) than conformational mutants (R175H and Y220C) (Zhang et al. 2020b). This function of mutant p53 contributes to enhanced invasion, although, interestingly, the conformational mutants such as R175H are as effective in driving increased invasive capacity as the DNA-contacting mutant R273H (Zhang et al. 2020b). Similarly, the ability of cancer cells to induce changes in macrophages was also shown to be dependent on the type of p53 mutation in the tumor cells, with R273H and R249S, but not p53 V157F and R175H, able to induce a tumor-promoting shift in the profile of M2 macrophages (Cooks et al. 2018).
Cell competition
The accumulation of postzygotic changes in DNA ensures that no two cells in an individual are likely to be genetically identical (Forsberg et al. 2017). In tumors, this genetic heterogeneity is further amplified by increased levels of DNA damage and enhanced still further by factors such as epigenetic, metabolic, and positional diversity (Martincorena and Campbell 2015). Evolutionary pressures driven by tumor progression and therapeutic intervention ensure that the complexity within the cancer cell population also changes over time (Caswell and Swanton 2017). Consequently, tumors are composed of ever-changing subpopulations of cells that differ from each other, establishing a situation where cells can compete with each other for survival and space.
First identified in Drosophila, cell competition describes the ability of cells to compare their fitness levels with that of their neighbours. During cell competition, cells carrying mutations that do not directly affect their viability in a tissue or tumor become disadvantaged when surrounded by fitter “winner” cells and are actively eliminated (Bowling et al. 2019). Various mechanisms can determine how differences in fitness levels between cells are sensed, and the elimination of the less fit “loser” cells can occur through a variety of different processes, including senescence, apoptosis, extrusion from an epithelial layer, or entosis. Finally, winner cells can compensate for the elimination of the loser cells by increasing proliferation or cell volume (Bowling et al. 2019; Baker 2020).
While much cell competition research has been conducted in nontumor developmental scenarios, there is clear potential for a tumor-suppressive role for cell competition in maintaining tissue homeostasis, ensuring that normal patterns of growth and development are not overrun by damaged or malignant cells. Several examples have been identified in flies and mammals, including “epithelial defense against cancer,” the process in which transformed cells are identified and eliminated in epithelia cell layers (Tanimura and Fujita 2020). However, cell competition can also be hijacked by tumor cells to allow them to outcompete normal cells in a process known as “supercompetition.” The complexity of competition between cells expands as tumors evolve, with likely ongoing competition between less and more aggressive clones. Emerging evidence also suggests the potential for cancer cells to compete with surrounding stromal cells. Given the clear potential for cell competition in modulating tumor development, it is not surprising that there is now a growing body of research identifying a role for p53 in this process.
A role for p53 in the losers during development and in normal tissue
During embryogenesis, the high mutation rate results in an increased chance that faulty cells may arise (Ju et al. 2017). Cells carrying defects such as abnormal ploidy or loss of genes crucial to patterning can persist if all the cells in the embryo are subject to the same defect. However, if the defective cells are surrounded by WT cells, then they will be eliminated by cell competition (Sancho et al. 2013), preventing them from propagating and forming part of the germline. Several studies have shown that p53 activity can track with the elimination of loser cells during development (Fig. 3A). An unbiased search for genes that controlled cell competition in mice showed that cells lacking p53 were able to outcompete cells retaining WT p53 in the embryo (Dejosez et al. 2013). Similarly, elimination of embryonic stem cells mutant for the BMP receptor BMPR1A depends on p53 in the losing cells. In this case, p53 was shown to signal a reduction in mTOR activity, leading to reduced fitness (Bowling et al. 2018). Consistently, cells with mildly increased p53 levels driven by reduced activity of MDM2 and MDM4, the key regulators of p53 stability and activity, were outcompeted by WT cells in mosaic embryos (Zhang et al. 2017). Interestingly, the fate of the p53-driven loser cells could be apoptosis (Bowling et al. 2018) or reduced proliferation (Zhang et al. 2017), suggesting that the response to p53 may differ depending on context.
Figure 3.

A role for p53 in cell competition. (A) Several studies have shown that WT p53 is required in aberrant cells that are outcompeted by surrounding cells containing WT p53. Loss of p53 allows the expansion of the aberrant clone. (B) In other contexts, cells expressing mutant p53 protein are outcompeted by WT p53-expressing cells and eliminated from epithelial layers. A similar loser phenotype is displayed by mutant KRAS-expressing cells. However, in the context of a mutant KRAS-expressing epithelial layer, cells with a concurrent mutation in TP53 are not outcompeted.
A role for p53 as a mediator of reduced fitness has also been seen in damaged or mutant cells, where limited activation of p53 (insufficient to promote cell-autonomous cell death) plays an important role in marking cells as losers (Fig. 3A). For example, in the mouse hematopoietic stem cell niche, mildly damaged cells are outcompeted by healthy cells and enter senescence, a response that is dependent on p53 in the damaged, loser cells (Bondar and Medzhitov 2010; Marusyk et al. 2010). The elimination of tetraploid cells by diploid cells during embryogenesis is also dependent on p53 (Horii et al. 2015). Similarly, epithelial cells from normal canine kidney (MDCK cells) depleted of the tumor suppressor gene SCRIB (orthologous to the Drosophila gene Scribble) are outcompeted by WT cells as a consequence of direct, mechanically induced competition. Again, elevated levels of p53 were found to be inducing elimination of the loser cells, in this instance via a ROCK-p38 mechanical stress sensing pathway (Wagstaff et al. 2016).
Cell competition in suppressing and enhancing tumorigenesis
Mutations in oncogenes and tumor suppressors can have varied effects on cell fitness, with some oncogenic changes increasing competitive advantage while others lower fitness. In several cases, loss of tumor suppressors or gain of activated oncogenes can promote elimination of cells by competition. A number of genes identified in Drosophila to be critical for maintaining competitive cell fitness, such as scrib and lgl, are orthologs of human tumor suppressor genes (Menéndez et al. 2010), and in Drosophila, as in humans, mutations in these genes can promote the development of neoplasia. The loss of fitness associated with these mutations and the elimination of cells carrying these defects by surrounding normal cells points to an effective additional tumor suppressor function of cell competition. Similarly, in mammalian systems, cells acquiring oncogenic alterations such as KRAS, SRC, or YES activation can be outcompeted by WT cells, while loss of tumor suppressor proteins SCRIBBLE or VPRBP confers loser status to cells when in competition with WT neighbors (Tanimura and Fujita 2020), consistent with a tumor-suppressive role of competition. In vivo, the removal of oncogenic KRAS-expressing cells from WT tissue has also been shown in mouse skin and intestinal epithelium (Brown et al. 2017; Kon et al. 2017).
However, not all oncogenic alterations result in competitive disadvantage. One of the most studied regulators of competition is MYC, a family of proteins that is misregulated and contributes to the development of many cancers. In both flies and humans, overexpression of MYC results in the acquisition of a “superfit” phenotype, enabling these cells to outcompete surrounding normal cells by inducing their elimination (Paglia et al. 2020). Subsequent studies showed that loss of the tumor suppressors APC and HIPPO can also induce this “superfit” status (Vermeulen et al. 2013; Mamada et al. 2015), although these effects may reflect the ability of these alterations to drive the downstream activation of MYC. Interestingly, in humans, high MYC tumor cells also correlate with the induction of cell death in surrounding stromal cells (Di Giacomo et al. 2017).
Interestingly, the tumor-suppressive cell competition response can be overridden by the simultaneous acquisition of oncogenic alterations, as described below. This feature is reminiscent of the oncogene cooperation in the transformation of primary cells, described some 40 yr ago (Land et al. 1983). Taken together, it seems likely that cells of increasing aggressiveness will be selected, in part, for their ability to outcompete normal or less aggressive neighbors within the shifting heterogeneous tumor mass.
A role for p53 in tumor cell competition
Selection for mutations in p53 can be seen to occur at different stages of tumor progression in different cancers (Rivlin et al. 2011), indicating that their effect on cell competition could be varied and context-dependent. Furthermore, the details of the consequences of loss of WT p53 activity or acquisition of the expression of a mutant p53 protein on cell competition have not been clarified. Such considerations are complicated by the observation that in Drosophila, WT p53 is required for Myc-overexpressing cells to become supercompetitors (De La Cova et al. 2014), an activity that might predict loss of p53 to be tumor-suppressive. However, a similar requirement for p53 to support the supercompetitor phenotype in mammalian cells has not been described.
More easily understood is a role for cancer-associated mutations or loss of p53 to lead to a failure to eliminate loser cells, as seen during development and in normal tissue. Indeed, the role of p53 in allowing elimination of defective cells during development has prompted the suggestion that the persistence of these defective cells in p53-null mice, where competitive elimination of these cells by WT cells cannot occur, explains the development of lymphomas and soft tissue sarcomas so early on in their lives (Bowling et al. 2019). However, in cancers, most p53 mutations occur within the context of a WT individual, conditions in which cell competition will play a role in regulating tumor progression. Recent large scale sequencing studies have shown that apparently normal tissue harbors large numbers of cells carrying known oncogenic alterations, the frequency of which increases with age (Martincorena et al. 2015, 2018). How these potentially malignant cells are held in check is unknown, but expansion of these mutant clones in older individuals suggests that these cells may have a competitive advantage over surrounding WT cells. Intriguingly, the second most common alteration seen in these quasi-normal cells in the esophagus was mutation in p53 (Martincorena et al. 2018). In contrast, in some tissues, mutant p53 clones appear to accumulate only later in cancer development. Overall, it would seem that conditions for competition between normal and tumor cells of different p53 status may occur at any point throughout the course of tumor progression, although it is likely that the magnitude and mechanisms of these effects will depend on the cell type and tissue context.
The mouse epidermis has proven to be a useful model in which to study the role of p53 in cell competition. Upon division, daughter keratinocytes in the basal layer of the epithelial membranes can either retain their ability to proliferate or become terminally differentiated and migrate to the subrabasal layers. The mosaic introduction of mutant p53-R245W (equivalent to R248W in humans) into basal epithelial cells in mouse skin reduced the generation of differentiated daughter cells following keratinocyte division, maintaining a pool of proliferating cells that continued to expand at the expense of their WT neighbors. Interestingly, the resultant increased epidermal thickness and higher cell density in the basal layer of the epidermis was slowly restored to normal once the entire basal layer had been populated with mutant cells, suggesting the imbalance in cell fate had returned to normal (Murai et al. 2018). In response to UV irradiation, mutant p53 clones initially showed a more rapid expansion than WT clones, although continued UV exposure caused the mutant p53 population to regress, possibly because the acquisition of further mutations by WT cells allowed them to outcompete the mutant p53-expressing cells (Murai et al. 2018). Interestingly, other studies have suggested that the expansion of mutant p53-expressing clones of keratinocytes is restrained in normal epithelium but released in response to UV exposure, which allows the mutant p53-expressing clones to expand (Brash et al. 2005).
In the mouse intestine, KRAS activation or APC loss, introduced via low level recombination into Lgr5+ stem cells in the base of crypts, gives cells a competitive advantage over WT progenitor cells; in this case, the advantage of KRAS-expressing cells is seemingly in contrast to the loser phenotype seen following KRAS activation in other contexts as described above. However, acquisition of the p53 mutation R172H (equivalent to R175H in humans) was only seen to confer an advantage to cells after the induction of colitis by the administration of DSS, potentially reflecting the ability of p53 mutant cells to deal with colitis-associated reactive oxygen species (ROS) (Vermeulen et al. 2013).
A similar role of ROS has been seen in murine esophageal epithelium, where exposure to low levels of ionizing radiation (IR) drives the differentiation of cells due to an increase in oxidative stress. In this system, expression of mutant p53 made cells more resistant to ROS, thus protecting them against irradiation-induced differentiation and allowing for the expansion of the mutant, and potentially tumorigenic, clones (Fernandez-Antoran et al. 2019). The loser phenotype of normal cells in this context was rescued with antioxidant treatment, supporting the previous suggestion that mutant p53 increases cell fitness by helping cells to cope with increased ROS (Vermeulen et al. 2013).
Despite the broadly consistent ability of mutation in p53 to promote advantage in cell competition, studies on cultured cells have revealed that this can be context-dependent. p53-R273H-expressing MDCK cells were extruded from a monolayer by surrounding cells that were either WT or null for p53. Similar results were obtained from experiments performed with mouse intestinal organoids. Interestingly, however, if p53-R273H was sporadically induced within a monolayer of oncogenic RAS-expressing MDCK cells, there was no evidence of cell death or extrusion, suggesting that oncogenic RAS allowed the p53-R273H-expressing cells to escape the effects of the competition (Watanabe et al. 2018). Previously, the same lab reported that oncogenic RAS-expressing cells were extruded from WT epithelia (Hogan et al. 2009), indicating that epithelia possess a strong intrinsic tumor-suppressive mechanism protecting them from acquiring individual oncogenic events that can be overridden by certain combinations of alterations, such as KRAS activation and p53 mutation. Such considerations are likely to underlie the observation that activation of KRAS can promote loser or winner behavior in different experimental systems (Fig. 3B).
To date, analysis of the effect of mutation in p53 on interactions with surrounding WT cells has focused on p53 point mutations that give rise to mutant protein expression. As described earlier, these mutants generally lose WT p53 function but also have a potential to acquire dominant-negative or gain-of-function activities. Whether any p53 mutants show GOFs that support a “superfit” cell phenotype and how this might be modulated by the transformed state of tumor cells remains to be identified. The functions of WT or mutant p53 required to control cell competition are also not yet established, although the ability of various forms of p53 to control metabolite production, cytokine secretion, and the mechanical properties of the cell may all contribute to this activity.
Does mutant p53 affect competitiveness between tumor cells?
While interactions between WT and mutant p53-expressing cells are the most likely to occur during tumor development, it is also possible that tumors contain a mixture of cells that harbor different p53 mutations or with different levels of p53 expression. Immunohistological evidence shows heterogeneous levels of mutant p53 within tumors; for example, the Human Protein Atlas contains multiple images of tumor sections stained for p53, with clear evidence for profoundly different levels of p53 in individual cells in the tumor (https://www.proteinatlas.org/ENSG00000141510-TP53/pathology). The reasons for this heterogeneity are unclear, but there is evidence to suggest that mutant p53 expression patterns can be affected at both the gene and protein level.
p53 mutations acquired during tumorigenesis could lead to the formation of subclones with different p53 statuses within a tumor, resulting in areas with WT p53 and areas with no or mutant p53. Subclonal p53 mutations have been observed at varying frequencies in different cancer types, including lung (Jamal-Hanjani et al. 2017), skin (Albibas et al. 2018), and chronic lymphocytic leukemia (Nadeu et al. 2016), although a study of nine major cancer types showed that ∼95% of tumors are clonal for p53 mutations (McGranahan et al. 2015). These studies suggest that subclonality has a limited role as a driver of mutant p53 heterogeneity in tumors, although they may underestimate heterogeneity as a consequence of limited size of the tumor sample and sequencing depth (McGranahan et al. 2015). Furthermore, some tumors arise with more than one p53 mutation, potentially leading to a mixture of cells expressing different p53 mutants (Donehower et al. 2019; Gorelick et al. 2020). However, heterogeneity of mutant p53 expression in tumors is not necessarily a reflection of genetic differences in TP53 in different subclones of cells. A study of human breast cancers showed that heterogeneity in mutant p53 protein levels was found in the majority of tumors (Bouchalova et al. 2014). Interestingly, variation in the mutant p53 levels was found to be dispersed randomly throughout the tumors and not localized to distinct areas, suggesting subclonal expansion to be an unlikely explanation.
Various explanations for this heterogeneous expression of mutant p53 have been put forward. Heterogeneity in the expression of genes that affect the proteasomal degradation of mutant p53, such as MDM2 or TRIM71, may provide a genetic explanation for heterogenous mutant p53 expression (Koga et al. 2001), or changes in other systems that control the stability of mutant p53, such as association with chaperones or autophagy (as described above). Tumor cell differentiation has been proposed to lead to a drop in the transcription of TP53, causing a reduction in mp53 protein in some cells, and mp53 staining patterns may differ depending on the presence of the second WT p53 allele (Xue et al. 2019). IFNβ, which is expressed by stromal cells, can lead to the destabilization of mutant, but not wild-type, p53 mRNA, potentially leading to heterogeneity of mutant p53 expression (Madar et al. 2013). The stability of mutant p53 protein is also influenced by the stiffness of the surrounding extracellular matrix, through an ability of RHOA to prevent ubiquitination and degradation of mutant p53. Both genetic inhibition and a reduction extracellular matrix stiffness resulted in increased degradation of mutant p53 (Ingallina et al. 2018), leading to a situation where tumors carrying clonal p53 mutations can contain mixtures of cells that express high or low levels of mutant p53 protein, depending on local matrix availability. Similarly, tumors clonal for WT p53 may nevertheless contain mixtures of cells with WT and “pseudo”-mutant p53, where WT p53 adopts a mutant p53 conformation. An analysis of human breast tumors without TP53 mutations found that some of them had transcriptional profiles that more closely resembled mutant p53 tumors than other WT p53 tumors. Furthermore, patients with pseudomutant p53 tumors show survival rates more similar to patients with mutant p53 tumors (Benor et al. 2020). Taken together, there is clear potential for tumors to contain mixtures of cells that express high and low (or no) levels of mutant p53 (Fig. 4).
Figure 4.

Heterogeneous p53 status in tumors. While some cancers retain a WT TP53 gene, mutations in TP53 occur during the development of many cancers, leading to expansion of clones of tumor cells that express no p53 or a mutant p53 protein among the WT p53-expressing cells. In some tumors, this heterogeneity of p53 expression persists, but most cancers tend to become clonal for cells carrying the TP53 mutation, leading to genetic homozygosity with respect to TP53. However, signals that drive WT p53 into a pseudomutant conformation, or changes in adhesion characteristics within the tumor that result in loss of mutant p53 protein expression, can result in heterogeneity in p53 expression within a tumor that is clonal for the TP53 gene.
While the impact of contacts between tumor cells that differ in p53 status has not been widely considered, emerging evidence suggests these interactions may have a role during oncogenesis. In mouse epithelium, cells expressing the p53-R273H mutant were eliminated from p53-null epithelia, as they were from p53 WT epithelia, clearly indicating that mutant p53 shows functions that are different from loss of p53 and, in this case, lead to the retention of the loser phenotype (Watanabe et al. 2018). An interesting form of cell competition results from entosis, a process by which winning cells can engulf surrounding loser cells (Overholtzer et al. 2007). In this case, cancer cells expressing mutant p53 were winners when competing with p53-null cells. Mixtures of isogenic p53-null and p53 R273H-expressing human carcinoma cells showed that p53-null cells were significantly more likely to be engulfed by mutant p53 cells. Further investigation revealed that mutant p53 cells were better at surviving the replication stress imposed by engulfing a cell, due to their ability to activate CHK1 and so persist through subsequent abnormal cell divisions such as tripolar mitosis. The resulting genomic instability in the daughters of engulfing cells led the authors to postulate that this process may act as a driver of intratumoral genetic heterogeneity. Interestingly, the presence of “cell-in-cell” structures was found to be highest in tumors with heterogeneous p53 expression and correlate with poor disease outcome and recurrence (Mackay et al. 2018). Parenthetically, engulfment of surrounding cells may also provide metabolic support for cancer cells under nutrient-limiting conditions (Fais and Overholtzer 2018).
Conclusions
p53 is well established as a critical node in the cell's stress detection network, and it is therefore not surprising that p53 is implicated in determining cell fate in response to stresses imposed by other cells. The molecular mechanisms of mutant p53 action are still being debated, with the evidence for increased cancer-promoting capacity balanced by recent evidence that, in some tissues, mutant p53 may even function to suppress tumorigenesis (Kadosh et al. 2020). In addition, more attention is being given to how p53 mutations affect a cell's response to the presence of other cells within their surrounding environment at all stages of cancer development. The multitude of mutations detected in apparently normal human epithelia attests to the general robustness of tumor-suppressive processes, and there is a growing appreciation of the importance of interactions between cells in controlling or enhancing malignant progression. Various functions of WT and mutant p53 are clearly implicated in determining how cancer cells affect and respond to their environment, although much remains to be learned. Different activities of WT p53 and various cancer-associated p53 mutations, as well as heterogeneity in the levels of mutant p53 within tumors, provides added complexity. Despite these challenges, our appreciation of these functions of p53 can only serve to open up more avenues for treatment and improve our targeting of therapeutics.
Acknowledgments
This work was funded by Cancer Research UK grant C596/A26855 and Biotechnology and Biological Sciences Research Council grant BB/S008284/1. The work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001557), the United Kingdom Medical Research Council (FC001557), and the Wellcome Trust (FC001557).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.347542.120.
Freely available online through the Genes & Development Open Access option.
Competing interest statement
K.H.V. is on the board of directors and a shareholder of Bristol Myers Squibb; a shareholder of GRAIL, Inc.; and on the science advisory board (with stock options) of PMV Pharma, RAZE Therapeutics, and Volastra Therapeutics, Inc. She is also on the Scientific Advisory Board of Ludwig Cancer Research. K.H.V. is a cofounder and consultant of Faeth Therapeutics, funded by Khosla Ventures. She has been in receipt of research funding from Astex Pharmaceuticals and AstraZeneca and contributed to Cancer Research UK Cancer Research Technology filing of patent application WO/2017/144877.
References
- Ablain J, Poirot B, Esnault C, Lehmann-Che J, de Thé H. 2016. P53 as an effector or inhibitor of therapy response. Cold Spring Harb Perspect Med 6: a026260. 10.1101/cshperspect.a026260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addadi Y, Moskovits N, Granot D, Lozano G, Carmi Y, Apte RN, Neeman M, Oren M. 2010. P53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res 70: 9650–9658. 10.1158/0008-5472.CAN-10-1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agupitan AD, Neeson P, Williams S, Howitt J, Haupt S, Haupt Y. 2020. p53: a guardian of immunity becomes its saboteur through mutation. Int J Mol Sci 21: 3452. 10.3390/ijms21103452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albibas AA, Rose-Zerilli MJJ, Lai C, Pengelly RJ, Lockett GA, Theaker J, Ennis S, Holloway JW, Healy E. 2018. Subclonal evolution of cancer-related gene mutations in p53 immunopositive patches in human skin. J Invest Dermatol 138: 189–198. 10.1016/j.jid.2017.07.844 [DOI] [PubMed] [Google Scholar]
- Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, Lozano G, Dobbelstein M, Moll UM. 2015. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523: 352–356. 10.1038/nature14430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova EM, Mirza SA, Xu S, Schulz-Heddergott R, Marchenko ND, Moll UM. 2017. P53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis 8: e2661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio I, Melino G. 2020. Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov 6: 16. 10.1038/s41420-020-0251-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, Marini A, Antonov AA, Le Quesne J, Baena Acevedo JD, et al. 2018. P53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci 115: E10869–E10878. 10.1073/pnas.1808314115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, Anfossi S, Osman AA, Cai Y, Wang R, et al. 2020. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 578: 449–454. 10.1038/s41586-020-1996-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arandkar S, Furth N, Elisha Y, Nataraj NB, Van Der Kuip H, Yarden Y, Aulitzky W, Ulitsky I, Geiger B, Oren M. 2018. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc Natl Acad Sci 115: 6410–6415. 10.1073/pnas.1719076115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NE. 2020. Emerging mechanisms of cell competition. Nat Rev Genet 21: 683–697. 10.1038/s41576-020-0262-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar J, Feniger-Barish R, Lukashchuk N, Shaham H, Moskovits N, Goldfinger N, Simansky D, Perlman M, Papa M, Yosepovich A, et al. 2009. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene 28: 933–936. 10.1038/onc.2008.445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman R, Prives C. 2010. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2: a000935. 10.1101/cshperspect.a000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benor G, Fuks G, Chin SF, Rueda OM, Mukherjee S, Arandkar S, Aylon Y, Caldas C, Domany E, Oren M. 2020. Transcriptional profiling reveals a subset of human breast tumors that retain wt TP53 but display mutant p53-associated features. Mol Oncol 14: 1640–1652. 10.1002/1878-0261.12736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, Mitchell C, Ng C, Katon J, Lunardi A, et al. 2018. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med 24: 165–175. 10.1038/nm.4463 [DOI] [PubMed] [Google Scholar]
- Blagih J, Buck MD, Vousden KH. 2020a. p53, cancer and the immune response. J Cell Sci 133: jcs237453. 10.1242/jcs.237453 [DOI] [PubMed] [Google Scholar]
- Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, Hock AK, Walton JB, Morton JP, Gronroos E, et al. 2020b. Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep 30: 481–496.e6. 10.1016/j.celrep.2019.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, Giacomelli AO, Wong W, Kim J, Chao S, et al. 2019. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365: 599–604. 10.1126/science.aax3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar T, Medzhitov R. 2010. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 6: 309–322. 10.1016/j.stem.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. 2006. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 25: 304–309. 10.1038/sj.onc.1209026 [DOI] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. 2016. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 37: 865–876. 10.1002/humu.23035 [DOI] [PubMed] [Google Scholar]
- Bouchalova P, Nenutil R, Muller P, Hrstka R, Appleyard MV, Murray K, Jordan LB, Purdie CA, Quinlan P, Thompson AM, et al. 2014. Mutant p53 accumulation in human breast cancer is not an intrinsic property or dependent on structural or functional disruption but is regulated by exogenous stress and receptor status. J Pathol 233: 238–246. 10.1002/path.4356 [DOI] [PubMed] [Google Scholar]
- Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, Gauthier-Villars M, Stoppa-Lyonnet D, Consolino E, Brugières L, et al. 2015. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 33: 2345–2352. 10.1200/JCO.2014.59.5728 [DOI] [PubMed] [Google Scholar]
- Bowling S, Di-Gregorio A, Sancho M, Pozzi S, Aarts M, Signore M, Schneider M, Martinez Barbera JP, Gil J, Rodríguez TA. 2018. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat Commun 9: 1763. 10.1038/s41467-018-04167-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling S, Lawlor K, Rodríguez TA. 2019. Cell competition: the winners and losers of fitness selection. Development 146: dev167486. 10.1242/dev.167486 [DOI] [PubMed] [Google Scholar]
- Brash DE, Zhang W, Grossman D, Takeuchi S. 2005. Colonization of adjacent stem cell compartments by mutant keratinocytes. Semin Cancer Biol 15: 97–102. 10.1016/j.semcancer.2004.08.006 [DOI] [PubMed] [Google Scholar]
- Brown S, Pineda CM, Xin T, Boucher J, Suozzi KC, Park S, Matte-Martone C, Gonzalez DG, Rytlewski J, Beronja S, et al. 2017. Correction of aberrant growth preserves tissue homeostasis. Nature 548: 334–337. 10.1038/nature23304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Solomon H, Rais Y, Kistner D, Nachmany I, Brait M, Madar S, Goldstein I, Kalo E, Adam N, et al. 2010. P53 regulates the Ras circuit to inhibit the expression of a cancer-related gene signature By various molecular pathways. Cancer Res 70: 2274–2284. 10.1158/0008-5472.CAN-09-2661 [DOI] [PubMed] [Google Scholar]
- Bullock AN, Henckel J, Fersht AR. 2000. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19: 1245–1256. 10.1038/sj.onc.1203434 [DOI] [PubMed] [Google Scholar]
- Bykov VJN, Eriksson S, Bianchi J, Wiman K. 2018. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 18: 89–102. 10.1038/nrc.2017.109 [DOI] [PubMed] [Google Scholar]
- Capaci V, Bascetta L, Fantuz M, Beznoussenko GV, Sommaggio R, Cancila V, Bisso A, Campaner E, Mironov AA, Wiśniewski JR, et al. 2020. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat Commun 11: 3945. 10.1038/s41467-020-17596-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell DR, Swanton C. 2017. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 15: 133. 10.1186/s12916-017-0900-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang Q, Yu H, Capitano ML, Vemula S, Nabinger SC, Gao R, Yao C, Kobayashi M, Geng Z, et al. 2019. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun 10: 5649. 10.1038/s41467-019-13542-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooks T, Pateras IS, Jenkins LM, Patel KM, Robles AI, Morris J, Forshew T, Appella E, Gorgoulis VG, Harris CC. 2018. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat Commun 9: 771. 10.1038/s41467-018-03224-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: e301. 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis 5: 99–118. 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordani M, Pacchiana R, Butera G, D'Orazi G, Scarpa A, Donadelli M. 2016. Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: involvement in cancer invasion and metastasis. Cancer Lett 376: 303–309. 10.1016/j.canlet.2016.03.046 [DOI] [PubMed] [Google Scholar]
- Dejosez M, Ura H, Brandt VL, Zwaka TP. 2013. Safeguards for cell cooperation in mouse embryogenesis shown by genome-wide cheater screen. Science 341: 1511–1514. 10.1126/science.1241628 [DOI] [PubMed] [Google Scholar]
- De La Cova C, Senoo-Matsuda N, Ziosi M, Wu DC, Bellosta P, Quinzii CM, Johnston LA. 2014. Supercompetitor status of Drosophila Myc cells requires p53 as a fitness sensor to reprogram metabolism and promote viability. Cell Metab 19: 470–483. 10.1016/j.cmet.2014.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Orso S, Fontemaggi G, Stambolsky P, Goeman F, Voellenkle C, Levrero M, Strano S, Rotter V, Oren M, Blandino G. 2011. ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. Omi A J Integr Biol 15: 305–312. 10.1089/omi.2010.0084 [DOI] [PubMed] [Google Scholar]
- de Oliveira GAP, Petronilho EC, Pedrote MM, Marques MA, Vieira TCRG, Cino EA, Silva JL. 2020. The status of p53 oligomeric and aggregation states in cancer. Biomolecules 10: 548. 10.3390/biom10040548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giacomo S, Sollazzo M, De Biase D, Ragazzi M, Bellosta P, Pession A, Grifoni D. 2017. Human cancer cells signal their competitive fitness through MYC activity. Sci Rep 7: 12568. 10.1038/s41598-017-13002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Minin G, Bellazzo A, DalFerro M, Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R, Sommaggio R, et al. 2014. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol Cell 56: 617–629. 10.1016/j.molcel.2014.10.013 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221. 10.1038/356215a0 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, Li X, Babur O, Hsu TK, Lichtarge O, et al. 2019. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep 28: 1370–1384.e5. 10.1016/j.celrep.2019.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Orazi G, Cordani M, Cirone M. 2020. Oncogenic pathways activated by pro-inflammatory cytokines promote mutant p53 stability: clue for novel anticancer therapies. Cell Mol Life Sci 78: 1853–1860. 10.1007/s00018-020-03677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson SE, Ceder S, Bykov VJN, Wiman KG. 2019. P53 as a hub in cellular redox regulation and therapeutic target in cancer. J Mol Cell Biol 11: 330–341. 10.1093/jmcb/mjz005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, Apken LH, Hobbs GA, Luo R, Lecomte N, et al. 2020. Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic cancer. Cancer Cell 38: 198–211.e8. 10.1016/j.ccell.2020.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fais S, Overholtzer M. 2018. Cell-in-cell phenomena in cancer. Nat Rev Cancer 18: 758–766. 10.1038/s41568-018-0073-9 [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. 2007. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci 104: 16633–16638. 10.1073/pnas.0708043104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, Jones PH. 2019. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell 25: 329–341.e6. 10.1016/j.stem.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M. 2017. Census and evaluation of p53 target genes. Oncogene 36: 3943–3956. 10.1038/onc.2016.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Gisselsson D, Dumanski JP. 2017. Mosaicism in health and disease-clones picking up speed. Nat Rev Genet 18: 128–142. 10.1038/nrg.2016.145 [DOI] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. 2012. Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286. 10.1101/gad.190678.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gencel-Augusto J, Lozano G. 2020. P53 tetramerization: at the center of the dominant-negative effect of mutant p53. Genes Dev 34: 1128–1146. 10.1101/gad.340976.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, Howard TP, Takeda DY, Ly SH, Kim E, et al. 2018. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 50: 1381–1387. 10.1038/s41588-018-0204-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick AN, Sánchez-Rivera FJ, Cai Y, Bielski CM, Biederstedt E, Jonsson P, Richards AL, Vasan N, Penson AV, Friedman ND, et al. 2020. Phase and context shape the function of composite oncogenic mutations. Nature 582: 100–103. 10.1038/s41586-020-2315-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossi E, Sánchez Y, Huarte M. 2016. Expanding the p53 regulatory network: lncRNAs take up the challenge. Biochim Biophys Acta Gene Regul Mech 1859: 200–208. 10.1016/j.bbagrm.2015.07.011 [DOI] [PubMed] [Google Scholar]
- Guo G, Marrero L, Rodriguez P, Del Valle L, Ochoa A, Cui Y. 2013. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res 73: 1668–1675. 10.1158/0008-5472.CAN-12-3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Yu M, Xiao W, Celis E, Cui Y. 2017. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res 77: 2292–2305. 10.1158/0008-5472.CAN-16-2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanel W, Marchenko N, Xu S, Xiaofeng Yu S, Weng W, Moll UM. 2013. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ 20: 898–909. 10.1038/cdd.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T, Tan BX, Lane D. 2020. How the other half lives: what p53 does when it is not being a transcription factor. Int J Mol Sci 21: 13. 10.3390/ijms21010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan C, Dupré-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, Piddini E, Baena-López LA, Vincent JP, Itoh Y, et al. 2009. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol 11: 460–467. 10.1038/ncb1853 [DOI] [PubMed] [Google Scholar]
- Horii T, Yamamoto M, Morita S, Kimura M, Nagao Y, Hatada I. 2015. P53 suppresses tetraploid development in mice. Sci Rep 5: 8907. 10.1038/srep08907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. 2006. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 25: 7305–7310. 10.1038/sj.onc.1209735 [DOI] [PubMed] [Google Scholar]
- Humpton TJ, Hock AK, Maddocks ODK, Vousden KH. 2018. P53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer Metab 6: 18. 10.1186/s40170-018-0191-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingallina E, Sorrentino G, Bertolio R, Lisek K, Zannini A, Azzolin L, Severino LU, Scaini D, Mano M, Mantovani F, et al. 2018. Mechanical cues control mutant p53 stability through a mevalonate–RhoA axis. Nat Cell Biol 20: 28–35. 10.1038/s41556-017-0009-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JG, Pant V, Li Q, Chang LL, Quintás-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al. 2012. P53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21: 793–806. 10.1016/j.ccr.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. 2017. Tracking the evolution of non-small-cell lung cancer. N Engl J Med 376: 2109–2121. 10.1056/NEJMoa1616288 [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18: 283–293. 10.1016/j.molcel.2005.03.027 [DOI] [PubMed] [Google Scholar]
- Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, Wedge DC, Davies HR, Ramakrishna M, Fullam A, et al. 2017. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543: 714–718. 10.1038/nature21703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadosh E, Snir-Alkalay I, Venkatachalam A, May S, Lasry A, Elyada E, Zinger A, Shaham M, Vaalani G, Mernberger M, et al. 2020. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 586: 133–138. 10.1038/s41586-020-2541-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R. 2016. The biology and function of exosomes in cancer. J Clin Invest 126: 1208–1215. 10.1172/JCI81135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. 2006. Fibroblasts in cancer. Nat Rev Cancer 6: 392–401. 10.1038/nrc1877 [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Han S, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. 2003. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci 100: 8424–8429. 10.1073/pnas.1431692100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ, Donehower LA, Bradley A, Balmain A. 1993. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74: 813–822. 10.1016/0092-8674(93)90461-X [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. 2010. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141: 52–67. 10.1016/j.cell.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaris H, Chatzistamou I, Trimis G, Frangou-Plemmenou M, Pafiti-Kondi A, Kalofoutis A. 2005. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res 65: 1627–1630. 10.1158/0008-5472.CAN-04-3791 [DOI] [PubMed] [Google Scholar]
- Kim MP, Lozano G. 2018. Mutant p53 partners in crime. Cell Death Differ 25: 161–168. 10.1038/cdd.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga T, Hashimoto S, Sugio K, Yoshino I, Nakagawa K, Yonemitsu Y, Sugimachi K, Sueishi K. 2001. Heterogeneous distribution of P53 immunoreactivity in human lung adenocarcinoma correlates with MDM2 protein expression, rather than with p53 gene mutation. Int J Cancer 95: 232–239. [DOI] [PubMed] [Google Scholar]
- Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, Kajita M, Ishikawa S, Yamauchi H, Yako Y, et al. 2017. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol 19: 530–541. 10.1038/ncb3509 [DOI] [PubMed] [Google Scholar]
- Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, Hopf TA, Marks DS, Oren M, Segal E. 2018. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell 71: 178–190.e8. 10.1016/j.molcel.2018.06.012 [DOI] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. 2015. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16: 393–405. 10.1038/nrm4007 [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. 1983. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304: 596–602. 10.1038/304596a0 [DOI] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh Y-A, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119: 861–872. 10.1016/j.cell.2004.11.006 [DOI] [PubMed] [Google Scholar]
- Lee MK, Sabapathy K. 2008. The R246S hot-spot p53 mutant exerts dominant-negative effects in embryonic stem cells in vitro and in vivo. J Cell Sci 121: 1899–1906. 10.1242/jcs.022822 [DOI] [PubMed] [Google Scholar]
- Loging WT, Reisman D. 1999. Inhibition of the putative tumor suppressor gene TIMP-3 by tumor-derived p53 mutants and wild type p53. Oncogene 18: 7608–7615. 10.1038/sj.onc.1203135 [DOI] [PubMed] [Google Scholar]
- Loizou E, Banito A, Livshits G, Ho YJ, Koche RP, Sánchez-Rivera FJ, Mayle A, Chen CC, Kinalis S, Bagger FO, et al. 2019. A gain-of-function p53-mutant oncogene promotes cell fate plasticity and myeloid leukemia through the pluripotency factor foxh1. Cancer Discov 9: 962–979. 10.1158/2159-8290.CD-18-1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano G. 2019. Restoring p53 in cancer: the promises and the challenges. J Mol Cell Biol 11: 615–619. 10.1093/jmcb/mjz063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig RL, Bates S, Vousden KH. 1996. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol 16: 4952–4960. 10.1128/MCB.16.9.4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153: 449–460. 10.1016/j.cell.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, Kimmelman AC. 2017. Metabolic interactions in the tumor microenvironment. Trends Cell Biol 27: 863–875. 10.1016/j.tcb.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay HL, Moore D, Hall C, Birkbak NJ, Jamal-Hanjani M, Karim SA, Phatak VM, Piñon L, Morton JP, Swanton C, et al. 2018. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat Commun 9: 3070. 10.1038/s41467-018-05368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar S, Harel E, Goldstein I, Stein Y, Kogan-Sakin I, Kamer I, Solomon H, Dekel E, Tal P, Goldfinger N, et al. 2013. Mutant p53 attenuates the anti-tumorigenic activity of fibroblasts-secreted interferon β. PLoS One 8: e61353. 10.1371/journal.pone.0061353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Labuschagne CF, Adams PD, Vousden KH. 2016. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol Cell 61: 210–221. 10.1016/j.molcel.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D. 2011. Li-Fraumeni syndrome. Genes Cancer 2: 475–484. 10.1177/1947601911413466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamada H, Sato T, Ota M, Sasaki H. 2015. Cell competition in mouse NIH3T3 embryonic fibroblasts is controlled by the activity of Tead family proteins and Myc. J Cell Sci 128: 790–803. 10.1242/jcs.163675 [DOI] [PubMed] [Google Scholar]
- Mantovani F, Collavin L, Del Sal G. 2019. Mutant p53 as a guardian of the cancer cell. Cell Death Differ 26: 199–212. 10.1038/s41418-018-0246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Campbell PJ. 2015. Somatic mutation in cancer and normal cells. Science 349: 1483–1489. 10.1126/science.aab4082 [DOI] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. 2015. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348: 880–886. 10.1126/science.aaa6806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. 2018. Somatic mutant clones colonize the human esophagus with age. Science 362: 911–917. 10.1126/science.aau3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. 2010. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol 8: e1000324. 10.1371/journal.pbio.1000324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Favero F, De Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. 2015. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 7: 283ra54. 10.1126/scitranslmed.aaa1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez J, Pérez-Garijo A, Calleja M, Morata G. 2010. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci 107: 14651–14656. 10.1073/pnas.1009376107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovits N, Kalinkovich A, Bar J, Lapidot T, Oren M. 2006. P53 attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res 66: 10671–10676. 10.1158/0008-5472.CAN-06-2323 [DOI] [PubMed] [Google Scholar]
- Muller PAJ, Vousden KH. 2014. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25: 304–317. 10.1016/j.ccr.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PAJ, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. 2009. Mutant p53 drives invasion by promoting integrin recycling. Cell 139: 1327–1341. 10.1016/j.cell.2009.11.026 [DOI] [PubMed] [Google Scholar]
- Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, Nagy T, Cagan A, Goulding D, Klein AM, et al. 2018. Epidermal tissue adapts to restrain progenitors carrying clonal p53 mutations. Cell Stem Cell 23: 687–699.e8. 10.1016/j.stem.2018.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, Jares P, Navarro A, Martín-García D, Beà S, et al. 2016. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 127: 2122–2130. 10.1182/blood-2015-07-659144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Hong CP, Oshima H, Sakai E, Kim SJ, Oshima M. 2020. Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties. Nat Commun 11: 2333. 10.1038/s41467-020-16245-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navalkar A, Ghosh S, Pandey S, Paul A, Datta D, Maji SK. 2020. Prion-like p53 amyloids in cancer. Biochemistry 59: 146–155. 10.1021/acs.biochem.9b00796 [DOI] [PubMed] [Google Scholar]
- Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, Lane DP, Callen DF. 2011. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget 2: 1203–1217. 10.18632/oncotarget.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo D, Heath N, Mitchell L, Caligiuri G, MacFarlane A, Reijmer D, Charlton L, Knight J, Calka M, McGhee E, et al. 2018. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat Commun 9: 5069. 10.1038/s41467-018-07339-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119: 847–860. 10.1016/j.cell.2004.11.004 [DOI] [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, Brugge JS. 2007. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131: 966–979. 10.1016/j.cell.2007.10.040 [DOI] [PubMed] [Google Scholar]
- Paglia S, Sollazzo M, Di Giacomo S, Strocchi S, Grifoni D. 2020. Exploring MYC relevance to cancer biology from the perspective of cell competition. Semin Cancer Biol 63: 49–59. 10.1016/j.semcancer.2019.05.009 [DOI] [PubMed] [Google Scholar]
- Paltridge JL, Belle L, Khew-Goodall Y. 2013. The secretome in cancer progression. Biochim Biophys Acta 1834: 2233–2241. 10.1016/j.bbapap.2013.03.014 [DOI] [PubMed] [Google Scholar]
- Pavlakis E, Stiewe T. 2020. p53's extended reach: the mutant p53 secretome. Biomolecules 10: 307. 10.3390/biom10020307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlakis E, Neumann M, Stiewe T. 2020. Extracellular vesicles: messengers of p53 in tumor–stroma communication and cancer metastasis. Int J Mol Sci 21: 9648. 10.3390/ijms21249648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto EM, Zambetti GP. 2020. What 20 years of research has taught us about the TP53 p.R337H mutation. Cancer 126: 4678–4686. 10.1002/cncr.33143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnamoun H, Lu H, Duttke SH, Benner C, Glass CK, Lauberth SM. 2017. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun 8: 754. 10.1038/s41467-017-01117-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin N, Brosh R, Oren M, Rotter V. 2011. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes and Cancer 2: 466–474. 10.1177/1947601911408889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Feizi A, Uhlén M, Nielsen J. 2019. A systematic investigation of the malignant functions and diagnostic potential of the cancer secretome. Cell Rep 26: 2622–2635.e5. 10.1016/j.celrep.2019.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K, Lane DP. 2018. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol 15: 13–30. 10.1038/nrclinonc.2017.151 [DOI] [PubMed] [Google Scholar]
- Sancho M, Di-Gregorio A, George N, Pozzi S, Sánchez JM, Pernaute B, Rodríguez TA. 2013. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell 26: 19–30. 10.1016/j.devcel.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarig R, Rivlin N, Brosh R, Bornstein C, Kamer I, Ezra O, Molchadsky A, Goldfinger N, Brenner O, Rotter V. 2010. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med 207: 2127–2140. 10.1084/jem.20100797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Heddergott R, Stark N, Edmunds SJ, Li J, Conradi LC, Bohnenberger H, Ceteci F, Greten FR, Dobbelstein M, Moll UM. 2018. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits Stat3-mediated tumor growth and invasion. Cancer Cell 34: 298–314.e7. 10.1016/j.ccell.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbandi A, Jackson JG. 2019. Analysis across multiple tumor types provides no evidence that mutant p53 exerts dominant negative activity. npj Precis Oncol 3: 1–3. 10.1038/s41698-018-0074-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon H, Buganim Y, Kogan-Sakin I, Pomeraniec L, Assia Y, Madar S, Goldstein I, Brosh R, Kalo E, Beatus T, et al. 2012. Various p53 mutant proteins differently regulate the ras circuit to induce a cancer-related gene signature. J Cell Sci 125: 3144–3152. 10.1242/jcs.099663 [DOI] [PubMed] [Google Scholar]
- Stein Y, Aloni-Grinstein R, Rotter V. 2019a. Mutant p53—a potential player in shaping the tumor-stroma crosstalk. J Mol Cell Biol 11: 600–604. 10.1093/jmcb/mjz071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein Y, Rotter V, Aloni-Grinstein R. 2019b. Gain-of-function mutant p53: all the roads lead to tumorigenesis. Int J Mol Sci 20: 6197. 10.3390/ijms20246197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein Y, Aloni-Grinstein R, Rotter V. 2020. Mutant p53 oncogenicity: dominant-negative or gain-of-function? Carcinogenesis 41: 1635–1647. 10.1093/carcin/bgaa117 [DOI] [PubMed] [Google Scholar]
- Strano S. 2016. Oncogenic intra-p53 family member interactions in human cancers. Front Oncol 6: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. 2018. Mechanisms of transcriptional regulation by p53. Cell Death Differ 25: 133–143. 10.1038/cdd.2017.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD, Vousden KH. 2018. A role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab 28: 721–736.e6. 10.1016/j.cmet.2018.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N, Fujita Y. 2020. Epithelial defense against cancer (EDAC). Semin Cancer Biol 63: 44–48. 10.1016/j.semcancer.2019.05.011 [DOI] [PubMed] [Google Scholar]
- Tran TQ, Lowman XH, Reid MA, Mendez-Dorantes C, Pan M, Yang Y, Kong M. 2017. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 36: 1991–2001. 10.1038/onc.2016.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad AG, Muller PAJ, Cuellar J, Klejnot M, Nobis M, Valpuesta JM, Vousden KH. 2013. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol Cell 50: 805–817. 10.1016/j.molcel.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley JM. 2003. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat 21: 313–320. 10.1002/humu.10185 [DOI] [PubMed] [Google Scholar]
- Varley JM, Thorncroft M, McGown G, Appleby J, Kelsey AM, Tricker KJ, Evans D, Birch JM. 1997. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene 14: 865–871. 10.1038/sj.onc.1201041 [DOI] [PubMed] [Google Scholar]
- Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. 1998. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 17: 4657–4667. 10.1093/emboj/17.16.4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennin C, Mélénec P, Rouet R, Nobis M, Cazet AS, Murphy KJ, Herrmann D, Reed DA, Lucas MC, Warren SC, et al. 2019. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun 10: 3637. 10.1038/s41467-019-10968-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, Kemp R, Tavare S, Winton DJ. 2013. Defining stem cell dynamics in models of intestinal tumor initiation. Science 342: 995–998. 10.1126/science.1243148 [DOI] [PubMed] [Google Scholar]
- Wagstaff L, Goschorska M, Kozyrska K, Duclos G, Kucinski I, Chessel A, Hampton-O'Neil L, Bradshaw CR, Allen GE, Rawlins EL, et al. 2016. Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun 7: 11373. 10.1038/ncomms11373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Song X, Zhuo W, Fu Y, Shi H, Liang Y, Tong M, Chang G, Luo Y. 2009. The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc Natl Acad Sci 106: 21288–21293. 10.1073/pnas.0908151106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Ishibashi K, Mano H, Kitamoto S, Sato N, Hoshiba K, Kato M, Matsuzawa F, Takeuchi Y, Shirai T, et al. 2018. Mutant p53-expressing cells undergo necroptosis via cell competition with the neighboring normal epithelial cells. Cell Rep 23: 3721–3729. 10.1016/j.celrep.2018.05.081 [DOI] [PubMed] [Google Scholar]
- Wawrzynow B, Zylicz A, Zylicz M. 2018. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim Biophys Acta Rev Cancer 1869: 161–174. 10.1016/j.bbcan.2017.12.004 [DOI] [PubMed] [Google Scholar]
- Webster MR, Fane ME, Alicea GM, Basu S, Kossenkov AV, Marino GE, Douglass SM, Kaur A, Ecker BL, Gnanapradeepan K, et al. 2020. Paradoxical role for wild-type p53 in driving therapy resistance in melanoma. Mol Cell 77: 633–644.e5. 10.1016/j.molcel.2019.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, Henneman L, Kas SM, Prekovic S, Hau CS, et al. 2019. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572: 538–542. 10.1038/s41586-019-1450-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnhoven SWP, Speksnijder EN, Liu X, Zwart E, VanOostrom CTM, Beems RB, Hoogervorst EM, Schaap MM, Attardi LD, Jacks T, et al. 2007. Dominant-negative but not gain-of-function effects of a p53.R270H mutation in mouse epithelium tissue after DNA damage. Cancer Res 67: 4648–4656. 10.1158/0008-5472.CAN-06-4681 [DOI] [PubMed] [Google Scholar]
- Xue Y, San Luis B, Lane DP. 2019. Intratumour heterogeneity of p53 expression; causes and consequences. J Pathol 249: 274–285. 10.1002/path.5328 [DOI] [PubMed] [Google Scholar]
- Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi MY, Chapman CG, Wang H, Black E, Bulysheva AA, Deb SP, et al. 2012. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 33: 442–451. 10.1093/carcin/bgr270 [DOI] [PubMed] [Google Scholar]
- Yoshii S, Hayashi Y, Iijima H, Inoue T, Kimura K, Sakatani A, Nagai K, Fujinaga T, Hiyama S, Kodama T, et al. 2019. Exosomal microRNAs derived from colon cancer cells promote tumor progression by suppressing fibroblast TP53 expression. Cancer Sci 110: 2396–2407. 10.1111/cas.14084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Xie Y, Zhou Y, Xiang C, Chen L, Zhang C, Hou X, Chen J, Zong H, Liu G. 2017. p53 pathway is involved in cell competition during mouse embryogenesis. Proc Natl Acad Sci 114: 498–503. 10.1073/pnas.1617414114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Xu D, Zhang T, Hu W, Feng Z. 2020a. Gain-of-function mutant p53 in cancer progression and therapy. J Mol Cell Biol 12: 674–687. 10.1093/jmcb/mjaa040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang C, Ma B, Xu M, Xu S, Liu J, Tian Y, Fu Y, Luo Y. 2020b. Mutant p53 drives cancer metastasis via RCP-mediated Hsp90α secretion. Cell Rep 32: 107879. 10.1016/j.celrep.2020.107879 [DOI] [PubMed] [Google Scholar]
- Zhu G, Pan C, Bei JX, Li B, Liang C, Xu Y, Fu X. 2020. Mutant p53 in cancer progression and targeted therapies. Front Oncol 10: 595187. 10.3389/fonc.2020.595187 [DOI] [PMC free article] [PubMed] [Google Scholar]