

Abstract
Introduction
To evaluate the usefulness of the 2018 NIA‐AA (National Institute on Aging and Alzheimer's Association) research framework in a longitudinal memory clinic study with different clinical outcomes and underlying disorders.
Methods
We included 420 patients with mild cognitive impairment or subjective cognitive impairment. During the follow up, 27% of the patients converted to dementia, with the majority converting to Alzheimer's disease (AD) or mixed dementia. Based on the baseline values of the cerebrospinal fluid biomarkers, the patients were classified into one of the eight possible ATN groups (amyloid beta [Aβ] aggregation [A], tau aggregation reflecting neurofibrillary tangles [T], and neurodegeneration [N]).
Results
The majority of the patients converting to AD and mixed dementia were in ATN groups positive for A (71%). The A+T+N+ group was highly overrepresented among converters to AD and mixed dementia. Patients converting to dementias other than AD or mixed dementia were evenly distributed across the ATN groups
Discussion
Our findings provide support for the usefulness of the ATN system to detect incipient AD or mixed dementia.
Keywords: Alzheimer's disease, cerebrospinal fluid biomarkers, dementia, diagnosis, frontotemporal dementia, Lewy body dementia, mild cognitive impairment, mixed dementia, subcortical vascular dementia
1. INTRODUCTION
The recent research framework from the National Institute on Aging and Alzheimer's Association (NIA‐AA) concludes that Alzheimer's disease (AD) is defined by its underlying pathologic processes rather than its clinical presentation. 1 This suggests that the ATN classification system, which reflects these processes, is suitable for use in observational and interventional studies. 1 The ATN classification system employs, based on the measurements that are available, markers originating from cerebrospinal fluid (CSF), positron emission tomography (PET), and/or structural magnetic resonance imaging (MRI) to detect signs of amyloid beta (Aβ) aggregation (A), tau aggregation reflecting neurofibrillary tangles (T), and neurodegeneration (N). The NIA‐AA research framework is an update of the NIA‐AA guidelines from 2011, 2 emphasizing the usefulness of AD biomarkers for staging of the disease, building on increased support for the view that the biomarkers deviate progressively reflecting a slow continuous deterioration during the course of the disease. 3
Although the individual markers in the ATN system have been firmly established to reflect the pathological processes in AD, only a few studies have evaluated the ATN system in relation to clinical outcome. The A+T+N+ group has been linked to clinical or cognitive deterioration in the general population, 4 and studies in memory clinic populations found that the A+T+N+ group was more prevalent when the clinical symptoms were severe or when the risk of future conversion to AD was elevated. 5 , 6 , 7
A biological definition of AD using the ATN system could simplify the diagnostic process and result in early identification of individuals with the potential to benefit from disease‐modifying therapies. This addresses one commonly stated reason for the so‐far negative results from recent AD trials, namely that the therapies were given too late in the disease process. 8 Although promising, the ATN construct needs further validation, preferable in the general population with long‐term follow‐up or in clinical populations reflecting the inflow of patients with cognitive complaints. The objective of the present study was therefore to apply the ATN system in a naturalistic memory clinic sample with different clinical outcomes and different underlying disorders.
2. MATERIAL AND METHODS
2.1. The Gothenburg MCI study
The Gothenburg MCI (mild cognitive impairment) study is a mono‐center, clinically based longitudinal study that aims to identify neurodegenerative and vascular disorders prior to the development of dementia. 9 The Gothenburg MCI study was approved by the local ethics committee in Gothenburg (approval number: L091‐99, 1999; T479‐11, 2011). Participants gave their consent for participating in the study, which was directed in accordance with the Helsinki Declaration. Inclusion required self‐ or informant‐reported progressive cognitive impairment for >6 months, age ≥50 and ≤79 years, and Mini Mental State Examination (MMSE) score >18. Exclusion criteria were acute/unstable somatic disease, severe psychiatric disorder, or substance abuse. (For an overview of the Gothenburg MCI study, please see reference 10.) Vascular burden was classified as two or more vascular risk factors (arterial hypertension, symptomatic arterial hypotension, congestive heart failure, angina pectoris, cardiac dysrhythmia, myocardial infarction, transient ischemic attack, stroke [also silent], hyperlipidemia, diabetes mellitus, and claudicatio intermittens) and the presence of not insignificant cerebral white matter changes (WMCs) on MRI.
2.2. Global Deterioration Scale (GDS)DS classification
The patients' degree of cognitive decline was staged according to the Global Deterioration Scale (GDS). 11 The GDS classifications were made using the following instruments: Variables 13‐20 of the Stepwise Comparative Status Analysis (STEP) 12 (ie, memory disturbance, disorientation, reduced abstract thinking, visuospatial disturbance, poverty of language, sensory aphasia, visual agnosia, and apraxia); IFlex, which is a short form of the Executive Interview (EXIT) 13 (ie, number‐letter task, word fluency, anomalous sentence repetition, interference task, Luria hand sequences, and counting task); Min Mental State Examination (MMSE) 14 ; and Clinical Dementia Rating (CDR). 15 The CDR sum of boxes assessment was based on information from both the patient and an informant. The guidelines for the classification were as follows: For GDS 2 (subjective cognitive impairment [SCI]) participants should have MMSE ≥28, CDR ≤0.5, I‐FLEX <3, and no positive outcomes on variables 13‐20 of STEP; GDS 3 (MCI) corresponds to MMSE ≥26, CDR >0.5, I‐FLEX ≤3, and one or fewer positive outcomes on variables 13‐20 of STEP; and for GDS 4 (mild dementia) participants should have MMSE ≤25, CDR >1.0, STEP >1, and I‐FLEX >3. When the guidelines were not applicable, a consensus decision among the physicians at the clinic was made to determine the appropriate GDS score.
RESEARCH IN CONTEXT
Systematic review: We analyzed articles that have referenced the 2018 NIA‐AA (National Institute on Aging and Alzheimer's Association) research framework, with particular emphasis on studies applying and evaluating the ATN system (amyloid beta [Aβ] aggregation [A], tau aggregation reflecting neurofibrillary tangles [T], and neurodegeneration [N]) on a clinical or general population. We additionally searched PubMed using the term “ATN system” for any articles that we have missed.
Interpretation: Our findings validate the use of the ATN system in a memory clinic sample with different clinical outcomes and different underlying disorders. However, the ATN system does not seem to cluster patients subsequently converting to forms of dementia other than Alzheimer's disease (AD).
Future directions: Although the group fulfilling the criteria for biological AD were highly represented among the patients converting to AD or mixed dementia, 35% did not develop manifest dementia during the course of the study. Longer follow‐up times are needed to ascertain that biological defined AD will also manifest itself clinically.
Participants classified as GDS 4 were further classified according to the following etiological dementia criteria: AD according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) criteria 16 and subcortical vascular dementia (SVD) according to the Erkinjuntti criteria, 17 frontotemporal dementia (FTD) according to, 18 and Lewy body dementia (LBD) according to. 19 For mixed dementia, AD criteria had to be fulfilled as well as moderate/severe WMC (Fazekas score ≥2) on MRI or alternatively, mild WMC in combination with a marked frontosubcortical‐dysexecutive syndrome. If no diagnostic criteria could be fulfilled, the patient was classified as unspecified dementia. The clinician who set the dementia diagnoses had access to clinical symptomatology and MRI images but was blinded to volumetric data, neuropsychological test results, and CSF biomarker data. The guidelines and diagnostic procedures have been described in detail previously. 9
2.3. The study population
In the present study, we included all participants in the Gothenburg MCI study that were classified as either SCI or MCI at baseline and had at least one follow‐up examination. This resulted in a study population (N = 420) of 226 patients with MCI and 194 patients with SCI at baseline. At subsequent follow‐ups, 115 of the patients with SCI or MCI at baseline converted to dementia (53 converted to AD, 34 to mixed dementia, 16 to SVD, two to LBD, two to FTD, and eight to unspecified dementia). Of the remaining patients, 117 MCI and 173 SCI patients remained stable and 19 MCI and 20 SCI patients recovered (MCI/SCI at baseline and healthy at follow‐up examination or MCI at baseline and SCI at follow‐up) during the study. The presented follow‐up times are the time from baseline until conversion for MCI patients subsequently progressing to dementia, and the time until last follow‐up examination for other groups. Patients converting to FTD, LBD, and unspecified dementia were grouped together as “Other dementias.”
2.4. Cerebrospinal fluid and genetic analysis
CSF samples were collected by lumbar puncture, which was performed in the morning to exclude influence from possible diurnal fluctuations in biomarker levels. Twenty milliliters of CSF was collected in a polypropylene tube and immediately transported to the local laboratory for centrifugation at 2000 × g at room temperature for 10 minutes. The supernatant was collected, gently mixed to avoid possible gradient effects, and aliquoted in 0.5 mL aliquots in screw‐cap polypropylene tubes that were stored at −80°C, without being thawed and refrozen, pending biochemical analyses. CSF levels of total (T)‐tau, phosphorylated (P)‐tau181, and Aβ amino acids 1 to 42 (Aβ42) were determined using sandwich enzyme‐linked immunosorbent assays (INNOTEST hTau Ag, INNOTEST PHOSPHO‐TAU(181P), and INNOTEST β‐AMYLOID(1‐42), respectively) from Innogenetics, Gent. 20 CSF P‐tau levels for 16 study participants were analyzed using the xMAP AlzBio3 kit (Innogenetics, Ghent, Belgium) and were used together with enzyme‐linked immunosorbent assay (ELISA) results after applying a correction factor, as described previously. 21 The CSF samples were processed at the Clinical Neurochemical Laboratory at Sahlgrenska University Hospital as part of clinical routine on multiple occasions during the course of the study. Still, the analytical variability was low. 22
Apolipoprotein E (APOE) ε4 status was analyzed using solid‐phase minisequencing. 9
2.5. Neuropsychological assessment
The Gothenburg MCI study includes a comprehensive neuropsychological test battery, which has previously been described in detail. 23 The following tests were included in the present study: Trail Making Test A and B, Rey Auditory Verbal Learning Test Delayed Recall, Visual Object and Space Perception Silhouettes subtest, Rey Complex Figure copy and recall, Wechsler Adult Intelligence Scale digit span, Word fluency F‐A‐S, Parallel Serial Mental Operations, and Stroop Test Victoria version. Not all study participants completed the neuropsychological test battery. The test scores available were: A+T+N+: 88 (91%), A+T‐N‐: 33 (97%), A‐T+N+: 84 (94%), A‐T+N‐: 31 (91%), A‐T‐N+: 22 (92%), A‐T‐N‐: 124 (91%). In addition, the Stroop test was included in the study at a later date and therefore had a lower completion rate. Study participants with available Stroop test score were: A+T+N+: 74 (76%), A+T‐N‐: 27 (79%), A‐T+N+: 56 (63%), A‐T+N‐: 23 (68%), A‐T‐N+: 13 (54%), A‐T‐N‐: 105 (77%).
2.6. Neuroimaging
Baseline MRI data from a 1.5 T scanner (Siemens Symphony, Erlangen, Germany) was available for a subset of participants (135 MCI and 103 SCI patients). Volumetry was performed on T1 three‐dimensional (3D) Inversion recovery/Gradient echo (IR/GR) images (repetition time 1610 ms, echo time 2.38 ms, flip angle 15°, coronal slices, field of view 250*203 mm, slice thickness 1 mm, pixel spacing 0.49*0.49 mm, matrix size 512*416). WMC volumes were obtained using the automated segmentation software FreeSurfer version 5.3.0.
2.7. ATN classification
All patients were classified according to the ATN classification system signifying the presence or absence of amyloid deposition (A), tau aggregation (T), and neurodegeneration (N). 1 We used CSF biomarker data for the classification of ATN groups; as described by, 1 we used Aβ42 for the classification of A, P‐tau for the classification of T, and T‐tau for the classification of N. More specifically: for A, CSF Aβ42 ≤482 ng/L was required for a positive classification; for T, CSF P‐tau ≥52 ng/L was classified as T+; and for N, CSF T‐tau ≥320 ng/L was classified as N+. Cut‐off values were derived from. 24 Based on these criteria, the study participants were then divided into one of the eight possible ATN groups.
2.8. Statistical analyses
Demographic differences between the A‐T‐N‐ group versus the other ATN groups combined were analyzed using the independent samples t test (age, MMSE, and years of education) and χ² (APOE ε4 status and sex). Differences between A‐T‐N‐ and other ATN groups in WMC volumes and cognitive tests were evaluated using analysis of covariance (ANCOVA) with age as a covariate (age and years of education for neuropsychological tests), or χ² for vascular burden. Differences between A‐T‐N‐ and other ATN groups in follow‐up status were analyzed using χ². Because of their small size, the A+T+N‐ and A+T‐N+ groups were not analyzed in terms of biomarker levels, cognitive tests, or follow‐up status. Predictive values were calculated using cross‐tabulation. All analyses were performed using IBM SPSS software (version 24.0).
3. RESULTS
Demographic data of the ATN groups are presented in Table 1. Although there were no differences regarding years of education and MMSE score, the A‐T‐N‐ group was younger and less likely to carry an APOE ε4 allele compared to most of the other groups.
TABLE 1.
Demographic data for ATN groups
| A+T+N+ | A+T+N‐ | A+T‐N‐ | A+T‐N+ | A‐T+N+ | A‐T+N‐ | A‐T‐N+ | A‐T‐N‐ | |
|---|---|---|---|---|---|---|---|---|
| N (%) | 97 (23) | 4 (1) | 34 (8) | 3 (1) | 87 (20) | 34 (8) | 24 (6) | 137 (33) |
| Baseline cognitive status: | ||||||||
| MCI N (%) | 72 (74) | 4 (100) | 18 (53) | 2 (67) | 49 (56) | 13 (38) | 10 (42) | 58 (42) |
| SCI N (%) | 25 (26) | 0 | 16 (47) | 1 (33) | 38 (44) | 21 (62) | 14 (58) | 79 (58) |
| Sex (% female) | 57 | 25 | 53 | 33 | 59 | 53 | 67 | 57 |
| Age (years) | 68.1 ± 6.9** | 68.3 ± 6.0 | 63.6 ± 8.4 | 70.3 ± 7.2* | 65.7 ± 7.3** | 61.3 ± 5.9 | 66.4 ± 8.6* | 61.1 ± 7.5 |
| Education (years) | 12.0 ± 3.3 | 12.0 ± 3.3 | 12.2 ± 3.9 | 11.0 ± 4.2 | 12.1 ± 3.6 | 12.8 ± 3.3 | 13.1 ± 2.9 | 13.2 ± 3.4 |
| MMSE | 28.0 ± 1.5 | 27.0 ± 1.0 | 28.5 ± 1.7 | 28.3 ± 0.6 | 28.6 ± 1.3 | 29.1 ± 1.3 | 28.3 ± 1.6 | 28.8 ± 1.2 |
| APOE ε4% (1 allele/2 alleles) | 49 / 26** | 25 / 50** | 36 / 21** | 100 / 0* | 45 / 5* | 12 / 9 | 38 / 0 | 29 / 3 |
Data are presented as the mean ± SD. The A‐T‐N‐ group was compared to other ATN groups. APOE ε4% (1 allele/2 alleles): percentage of study participant with one or two APOE ε4 alleles.
P‐value <.05 versus A‐T‐N‐.
P‐value <.001 versus A‐T‐N‐.
Table 2 shows that the A+T+N+ group performed worse on most neuropsychological tests at baseline compared to the A‐T‐N‐ group. Cognitive deficits were not as pronounced for the other groups, with the exception for the A+T‐N‐ group, which was characterized by poor performance on memory tests. Vascular burden was similar in the ATN groups, whereas the A+T+N+ and A‐T+N+ groups had smaller WMC volumes compared to the A‐T‐N‐ group.
TABLE 2.
Baseline levels of CSF biomarkers, cognitive tests, and vascular factors
| A+T+N+ | A+T‐N‐ | A‐T+N+ | A‐T+N‐ | A‐T‐N+ | A‐T‐N‐ | |
|---|---|---|---|---|---|---|
| N | 97 (23) | 34 (8) | 87 (20) | 34 (8) | 24 (6) | 137 (33) |
| CSF markers: | ||||||
| Aβ42, (ng/L) | 365 ± 74 | 382 ± 84 | 751 ± 197 | 750 ± 133 | 763 ± 228 | 707 ± 150 |
| P‐tau (ng/L) | 89 ± 32 | 35 ± 8 | 71 ± 18 | 69 ± 40 | 46 ± 4 | 38 ± 7 |
| T‐tau (ng/L) | 676 ± 354 | 192 ± 64 | 519 ± 202 | 267 ± 57 | 365 ± 50 | 208 ± 59 |
| Neuropsychological tests: | ||||||
| RAVLT DR | 4.5 ± 4.0** | 7.2 ± 3.2* | 6.8 ± 3.6* | 8.6 ± 3.2 | 7.3 ± 4.7 | 8.9 ± 3.4 |
| TMT A | 49 ± 20* | 41 ± 15 | 43 ± 15 | 38 ± 16 | 49 ± 18 | 37 ± 13 |
| TMT B | 133 ± 69* | 108 ± 57 | 104 ± 48 | 82 ± 27 | 111 ± 60 | 90 ± 44 |
| WAIS digit span | 12.8 ± 3 | 13.4 ± 4 | 12.9 ± 4 | 13.4 ± 3 | 12.3 ± 3 | 13.6 ± 3 |
| RCF recall | 9.0 ± 7** | 13.3 ± 7* | 14.1 ± 7 | 18.5 ± 7 | 14.5 ± 7 | 17.4 ± 7 |
| RCF copy | 30 ± 6* | 32 ± 4 | 32 ± 5 | 33 ± 3 | 30 ± 6* | 32 ± 3 |
| F‐A‐S test | 38 ± 12 | 37 ± 15 | 39 ± 13 | 41 ± 14 | 35 ± 11 | 43 ± 13 |
| VOSP silhouettes | 19 ± 5* | 20 ± 4 | 20 ± 4 | 22 ± 4 | 20 ± 5 | 21 ± 4 |
| Stroop | 36 ± 17* | 33 ± 17 | 31 ± 9 | 28 ± 8 | 33 ± 11 | 28 ± 10 |
| Vascular factors: | ||||||
| WMC (cm3) | 3.6±3.5* | 8.3±8.9 | 4.4±5.0* | 4.3±3.0 | 4.8±4.0 | 6.7±6.9 |
| Vascular burden (%) | 31 | 30 | 26 | 31 | 35 | 28 |
Data are presented as the mean ± SD. ATN groups were compared to the A‐T‐N‐ group.
F‐A‐S test, Word fluency F‐A‐S test; RAVLT DR, Rey Auditory Verbal Learning Test Delayed Recall; RCF, Rey Complex Figure copy and recall; Stroop, Stroop Test Victoria version; TMT, Trail Making Test; Vascular burden, percentage of study participants with at least two vascular risk factors and not insignificant WMC; VOSP silhouettes, Visual Object and Space Perception Silhouettes subtest; WAIS, Wechsler Adult Intelligence Scale digit span; WMC volume, white matter changes volume.
P‐value <.05 versus A‐T‐N‐.
P‐value <.001 versus A‐T‐N‐.
Results from the follow up examinations are presented in Table 3. The follow‐up status differed between the groups, as <10% of participants in the A‐T+N‐ and A‐T‐N‐ group had converted to dementia compared to more than half in the A+T+N+ group. Of note, most converters in groups with N+ tended to convert within 3 years from the baseline examination. Figure 1 displays follow‐up status in ATN groups. Figure 2 displays ATN groups in the participants converting to dementia.
TABLE 3.
Follow‐up status in the ATN groups
| A+T+N+ | A+T‐N‐ | A‐T+N+ | A‐T+N‐ | A‐T‐N+ | A‐T‐N‐ | |
|---|---|---|---|---|---|---|
| N | 97 | 34 | 87 | 34 | 24 | 137 |
| Follow‐up time (months) | 31.6 ± 19* | 31.0 ± 27* | 34.5 ± 27 | 32.5 ± 28 | 39.0 ± 28 | 36.2 ± 29 |
| Follow‐up status: | ||||||
| Recovered N (%) | 2 (2)† | 0† | 4 (4)* | 6 (18) | 2 (8) | 20 (15) |
| Stable N (%) | 32 (33)† | 25 (74) | 62 (70) | 27 (79) | 16 (67) | 106 (77) |
| Converting N (%) | 63 (65) | 9 (26) | 21 (26) | 1 (3) | 6 (25) | 11 (8) |
| AD N (%) | 34 (54)† | 2 (22) | 11 (52)† | 1 (100) | 1 (17) | 3 (27) |
| Mixed dementia N (%) | 21 (33)† | 3 (33)† | 7 (33)† | 0 | 2 (33)† | 0 |
| SVD N (%) | 4 (6) | 2 (22) | 2 (10) | 0 | 3 (50)* | 3 (27) |
| Other dementia | 4 (6) | 2 (22) | 1 (5) | 0 | 0 | 4 (36) |
| Conversion within 3 years: | ||||||
| AD and Mix N (%) | 39 (61) | 1 (11)† | 14 (67)† | 1 (100) | 2 (33) | 2 (18) |
| SVD and Other N (%) | 5 (8) | 3 (33) | 2 (10) | 2 (33) | 3 (27) | |
| Conversion after 3 years: | ||||||
| AD & Mix N (%) | 16 (25) | 4 (44) | 4 (19) | 1 (17) | 1 (9) | |
| SVD & Other N (%) | 3 (5) | 1 (11) | 1 (5) | 1 (17) | 4 (36) | |
Follow‐up time was defined as the time until conversion or to last follow‐up and is presented as the mean ± SD. ATN groups were compared to the A‐T‐N‐ group. Furthermore, in each ATN group, converters were divided in two groups depending on whether the conversion occurred within or after 3 years from baseline. Then, these groups were further divided based on whether the conversion was to AD/mixed dementia or SVD/other dementia. Between‐group differences were analyzed using χ². Percentage values in follow‐up status and conversion within/after 3 years represent distribution within each ATN group. Percentage values of dementia diseases in follow‐up status represent proportion of converting patients in the individual ATN group.
P‐value <.05 versus A‐T‐N‐.
P‐value <.05 Proportion of converters within/after >3 years from baseline compared to A‐T‐N‐.
FIGURE 1.
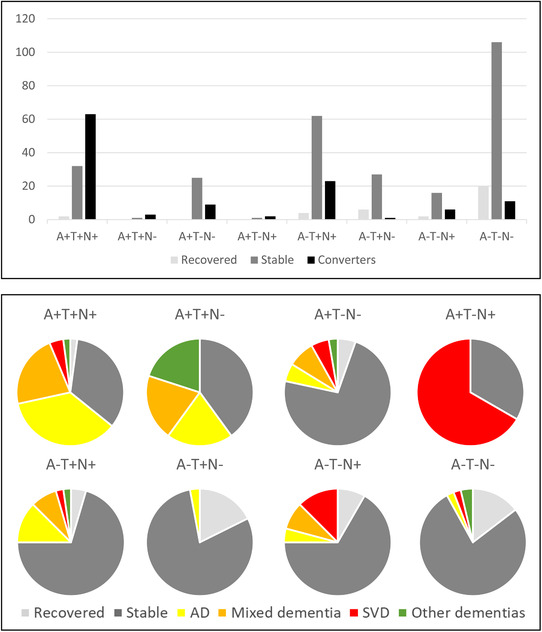
Follow‐up status in ATN groups. The top panel displays the number of patients who had recovered, been stable, and converted to dementia in each ATN group. The bottom panel displays the proportion of patients in the ATN groups who had recovered, been stable, or converted to a specific dementia diagnosis
FIGURE 2.
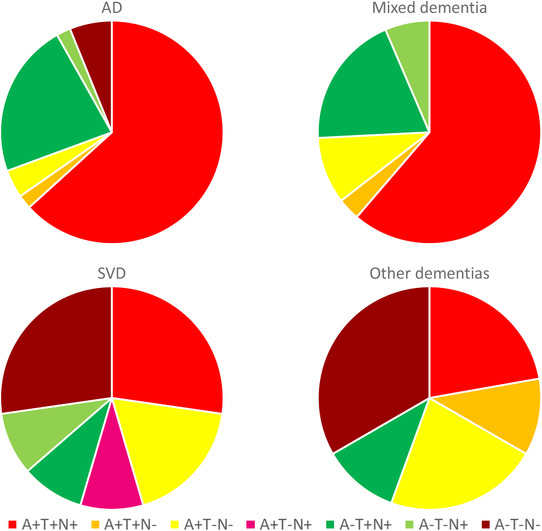
Baseline ATN group status in patients who converted to a specific dementia diagnosis at follow‐up
Table 4 displays predictive values for the individual components of the ATN system and their combination in A+T+N+. All individual components were useful for the prediction of AD and mixed dementia, with A+ being the best individual marker and the combination (A+T+N+) having the highest predictive value. The components of the ATN system were less useful for the prediction of SVD and other dementia.
TABLE 4.
Predictive values for components of the ATN system
| Sens. | Spec. | +LR | ‐LR | PPV | NPV | |
|---|---|---|---|---|---|---|
| AD and Mixed dementia: | ||||||
| A+ | 71 | 80 | 3.6 (2.7‐4.6) | 0.4 (0.3‐0.5) | 50 (44‐57) | 91 (87‐93) |
| T+ | 87 | 56 | 2.0 (1.7‐2.3) | 0.2 (0.1‐0.4) | 36 (33‐40) | 94 (90‐96) |
| N+ | 87 | 61 | 2.3 (1.9‐2.7) | 0.2 (0.1‐0.4) | 39 (35‐43) | 94(91‐97) |
| A+T+N+ | 63 | 89 | 5.7 (4.0‐8.1) | 0.4 (0.3‐0.6) | 62 (53‐70) | 89 (86‐92) |
| SVD and Other dementias: | ||||||
| A+ | 54 | 80 | 2.7 (1.8‐4.0) | 0.6 (0.4‐0.9) | 20 (14‐27) | 95 (93‐97) |
| T+ | 43 | 56 | 1.0 (0.6‐1.5) | 1.0 (0.7‐1.4) | 8 (5‐12) | 91 (88‐94) |
| N+ | 57 | 61 | 1.5 (1.0‐2.1) | 0.7 (0.5‐1.1) | 12 (9‐16) | 94 (91‐96) |
| A+T+N+ | 29 | 89 | 2.6 (1.3‐5.0) | 0.8 (0.6‐1.0) | 19 (11‐31) | 93 (91‐94) |
+LR, Positive likelihood ratio;–LR, Negative likelihood ratio; NPV, Negative predictive value; PPV, Positive predictive value; Sens., Sensitivity %; Spec., Specificity %. Likelihood ratios and predictive values are given with 95% confidence intervals in brackets.
4. DISCUSSION
We applied the ATN system retrospectively to the Gothenburg MCI study and found that 23% of the study population were classified as A+T+N+. However, this group accounted for a majority of the patients subsequently converting to dementia, with 89% of converters converting to either AD or mixed dementia. In contrast, only a few participants in the A‐T+N‐ and A‐T‐N‐ groups converted to dementia.
The accumulation of Aβ deposits has been found to start early in adult life. 3 A strict biological definition of AD using the ATN system could therefore enable researchers to undertake intervention studies already in the presymptomatic stages of AD. Nevertheless, it is still unclear whether all patients fulfilling the criteria for biologically defined AD will develop clinically manifest AD and, if so, in which time frame. Previous reports suggest that there is a group of individuals that exhibit all the pathological hallmarks of AD without obvious signs of cognitive deterioration, 25 suggesting that biologically defined AD is more prevalent than the traditional definition. Indeed, the results of a recent population study supported this notion, reporting that biologically defined AD was up to three times more prevalent than clinically defined AD depending on the age group studied. 26 In our study, 35% of the patients in the A+T+N+ group did not convert to dementia during follow‐up. This does not necessarily invalidate the ATN concept, but could be secondary to insufficient length of the follow‐up time (mean 32 months in the A+T+N+ group). Therefore, although a longer follow‐up time would have been preferable, is clear that a proportion of individuals with A+T+N+ will not convert to dementia in a relatively long time. This might in turn provide an opportunity for early identification of individuals with the potential to benefit from disease modifying therapies.
It should be noted that 23% of our patients were classified as A+T+N+, which is higher than that reported in most previous studies. Two population‐based studies found prevalence levels between 8% and 10%, 27 , 28 and two clinically based studies observed a prevalence of A+T+N+ of around 20%. 5 , 6 The prevalence of A+T+N+ in the clinically based study by Altomare et al. was only marginally lower than that found in our study, but their study included relatively young patients and also patients with dementia at baseline, who showed the highest prevalence of A+T+N+ (35%). 5 Although this difference in the composition of study participants highlights the high prevalence of A+T+N+ in the present study, our results concur with the previous findings of increased prevalence of A+T+N+ with increasing age and more severe clinical symptoms. In addition, our findings are in line with a previous study from the Gothenburg MCI study, evaluating the NIA‐AA guidelines from 2011 on a smaller data set, finding that patients positive for amyloid and either t‐tau or P‐tau were most likely to convert to AD. 29 The distribution of SCI patients differed between the groups, with the highest prevalence was seen in the A‐T+N‐, A‐T‐N+, and A‐T‐N‐ groups. These groups also had the lowest rate of conversion to dementia, which is consistent with the view that SCI has a lower conversion rate than MCI, 30 and also with studies finding that Aβ42 is the best individual predictor for clinical progression in SCI. 31
Examination of the baseline neuropsychological test performance showed that the A+T+N+ group clearly stood out compared to the A‐T‐N‐ group. Most tests results were impaired in the A+T+N+ group, including tests for memory, visuospatial, and executive functions. The neuropsychological profiles in the other ATN groups were not as conspicuous, likely reflecting a lower rate of conversion. Cardiovascular risk factors not only increase the risk for AD, but are also together with cerebrovascular disease important confounders in clinical diagnostics. 32 , 33 Although we found no difference in vascular burden between the ATN groups, WMC volume was smaller in the A+T+N+ and A‐T+N+ groups. This could reflect that the number of converters to SVD was low in these groups, but the mechanisms underlying the higher WMC volumes seen in the other groups are unclear, as these groups also had few converters to SVD. Taken together, the number of converters to non‐AD dementias was low, and we found no accumulation of SVD or other dementias in any of the ATN groups. Thus, we conclude that the ATN system is specific for AD and not suitable for detection of subsequent conversion to other dementias.
A previously addressed limitation of the ATN system is the use and interpretation of the intermediate ATN groups (A+T‐N+, A+T‐N‐, A‐T+N+, A‐T+N‐). We found that the A+T+N‐ and A+T‐N+ groups together accounted for <2% of the study population, raising questions about the validity of these groups. Although the categorization is highly dependent on the cut‐off points applied, previous studies have also found low prevalence of these groups. 27 , 28 Although different variants of T and N in the A+ groups are believed to reflect different time points in the preclinical stages of AD, as outlined in the dynamic biomarker model, 34 the A‐ groups does not as easily fit into the model. The low number of converters to AD in the A‐/N‐ groups is in line with results of a previous study. 35 We found, however, that the A‐T+N+ group is not only quite prevalent, but also accounts for a large proportion of patients converting to AD and mixed dementia, second only to the A+T+N+ group. This raises questions regarding this group. A‐ with either T+, N+, or both, is commonly classified as Suspected Non‐Alzheimer Pathology (SNAP), and although there have been discussions about whether SNAP should be regarded as an early manifestation of AD, 36 the current interpretation of the A‐T+ groups is that they do not reflect the AD pathologic change. 1 Instead, it has been suggested that A‐T+ may reflect the primary age‐related tauopathy (PART). 37 Although this merits further study, the relatively young age of the patients in our A‐T+N+ group is not fully consistent with PART, but rather favors the view that tauopathy may have a more prominent role in the pathophysiology of AD. 38 Another interesting aspect of the A‐T+N+ group is the high number of patients with one or more APOE ε4 alleles. ApoE binds to Aβ and the ε4 variant has been linked to Aβ deposition, which is believed to be the reason for the increased risk of AD for APOE ε4 patients. 39 However, a recent study supports the idea that that APOE ε4 is involved also in tau‐mediated neurodegeneration independent of Aβ, 40 which could explain the findings in the A‐T+N+ group.
5. LIMITATIONS
A consequence of evaluating a system with eight study groups is the difficulty of having enough power to analyze all groups, especially with regard to longitudinal outcome. Despite including 420 memory clinic patients at baseline, the low frequencies of some ATN groups precluded meaningful statistical analysis of these groups. Consequently, we believe it could be beneficial to cluster some groups to improve the statistical power, as has previously been suggested. 5
Another drawback is the diagnostic accuracy of dementia. Evaluating a biological definition of AD should ideally be compared with a definite diagnosis obtained through neuropathological examination. Furthermore, although the diagnostic system in the Gothenburg MCI study is carefully constructed, the possibility cannot be excluded that some of the dementia patients were misclassified. This risk of misclassification is likely greatest in the spectrum of AD and mixed dementia. Reflecting this, we opted to group AD and mixed dementia together in some analyses.
6. CONCLUSION
The high frequency of converters to AD and mixed dementia in the A+T+N+ group and the low number of converters in the A‐T‐N‐ group validate the ATN system in SCI and MCI patients with different underlying disorders studied at a single memory clinic. The even distribution of patients converting to SVD or other dementia suggests that the ATN system is useful only in AD.
CONFLICTS OF INTEREST
The authors have no competing interests to report.
FUNDING
This work was financed by grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement [Grant number ALFGBG‐784831, ALFGBG‐720661, ALFGBG‐238501]. Additional support was received from the Sahlgrenska University Hospital, the Swedish Research Council, Swedish Brain Power, the Swedish Dementia Foundation, the Swedish Alzheimer Foundation, Stiftelsen Psykiatriska forskningsfonden, and Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse. The funding sources were not involved in the drafting of this manuscript.
ACKNOWLEDGMENTS
We would like to thank Marie Johansson and Eva Bringman for administrating the study, Erik Olsson and Niklas Klasson for help with the MRI analyses, and the staff at the memory clinic in Mölndal and the staff at the Neurochemical Laboratory for data collection and analyses.
Eckerström C, Svensson J, Kettunen P, Jonsson M, Eckerström M. Evaluation of the ATN model in a longitudinal memory clinic sample with different underlying disorders. Alzheimer's Dement. 2021;13:e12031. 10.1002/dad2.12031
REFERENCES
- 1. Jack CR Jr., Bennett DA, Blennow K, et al. NIA‐AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR Jr., Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer's disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack CR Jr., Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321:2316‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Altomare D, de Wilde A, Ossenkoppele R, et al. Applying the ATN scheme in a memory clinic population: The ABIDE project. Neurology. 2019;93:e1635‐e1646. [DOI] [PubMed] [Google Scholar]
- 6. Hwang J, Jeong JH, Yoon SJ, et al. Clinical and biomarker characteristics according to clinical spectrum of Alzheimer's disease (AD) in the validation cohort of Korean Brain Aging Study for the Early Diagnosis and Prediction of AD. J Clin Med. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Maurik IS, Vos SJ, Bos I, et al. Biomarker‐based prognosis for people with mild cognitive impairment (ABIDE): a modelling study. Lancet Neurol. 2019;18:1034‐1044. [DOI] [PubMed] [Google Scholar]
- 8. Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17:1060‐1065. [DOI] [PubMed] [Google Scholar]
- 9. Wallin A, Nordlund A, Jonsson M, et al. The Gothenburg MCI study: design and distribution of Alzheimer's disease and subcortical vascular disease diagnoses from baseline to 6‐year follow‐up. J Cereb Blood Flow Metab. 2016;36:114‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wallin A, Nordlund A, Jonsson M, et al. Alzheimer's disease–subcortical vascular disease spectrum in a hospital‐based setting: Overview of results from the Gothenburg MCI and dementia studies. J Cereb Blood Flow Metab. 2016;36:95‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Auer S, Reisberg B. The GDS/FAST staging system. Int Psychogeriatr. 1997;9(Suppl 1):167‐171. [DOI] [PubMed] [Google Scholar]
- 12. Wallin A, Edman A, Blennow K, et al. Stepwise comparative status analysis (STEP): a tool for identification of regional brain syndromes in dementia. J Geriatr Psychiatry Neurol. 1996;9:185‐199. [DOI] [PubMed] [Google Scholar]
- 13. Royall DR, Mahurin RK, Gray KF. Bedside assessment of executive cognitive impairment: the executive interview. J Am Geriatr Soc. 1992;40:1221‐1226. [DOI] [PubMed] [Google Scholar]
- 14. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 15. Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9( Suppl 1):173‐176; discussion 7‐8. [DOI] [PubMed] [Google Scholar]
- 16. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 17. Erkinjuntti T, Inzitari D, Pantoni L, et al. Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl. 2000;59:23‐30. [DOI] [PubMed] [Google Scholar]
- 18. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546‐1654. [DOI] [PubMed] [Google Scholar]
- 19. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53:902‐905. [DOI] [PubMed] [Google Scholar]
- 20. Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid beta‐amyloid(1‐42) in Alzheimer disease: differences between early‐ and late‐onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673‐680. [DOI] [PubMed] [Google Scholar]
- 21. Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta‐amyloid(1‐42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336‐345. [DOI] [PubMed] [Google Scholar]
- 22. Mattsson N, Andreasson U, Persson S, et al. The Alzheimer's Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7:386‐395 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nordlund A, Rolstad S, Hellstrom P, Sjogren M, Hansen S, Wallin A. The Goteborg MCI study: mild cognitive impairment is a heterogeneous condition. J Neurol Neurosurg Psychiatry. 2005;76:1485‐1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385‐393. [DOI] [PubMed] [Google Scholar]
- 25. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun. 2014;2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jack CR Jr., Therneau TM, Weigand SD, et al. Prevalence of Biologically vs Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging‐Alzheimer's Association Research Framework. JAMA Neurol. 2019;76(10):1174‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR Jr., Wiste HJ, Weigand SD, et al. Age‐specific and sex‐specific prevalence of cerebral beta‐amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50‐95 years: a cross‐sectional study. Lancet Neurol. 2017;16:435‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soldan A, Pettigrew C, Fagan AM, et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology. 2019;92:e1567‐e1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eckerstrom M, Gothlin M, Rolstad S, et al. Longitudinal evaluation of criteria for subjective cognitive decline and preclinical Alzheimer's disease in a memory clinic sample. Alzheimers Dement. 2017;8:96‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rabin LA, Smart CM, Amariglio RE. Subjective Cognitive Decline in Preclinical Alzheimer's Disease. Annu Rev Clin Psychol. 2017;13:369‐396. [DOI] [PubMed] [Google Scholar]
- 31. van Harten AC, Visser PJ, Pijnenburg YA, et al. Cerebrospinal fluid Abeta42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimers Dement. 2013;9:481‐487. [DOI] [PubMed] [Google Scholar]
- 32. Jokinen H, Koikkalainen J, Laakso HM, et al. Global burden of small vessel disease‐related brain changes on MRI predicts cognitive and functional decline. Stroke. 2020;51(1):170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nordlund A, Gothlin M, Wallin A. Vascular disease, Alzheimer's disease biomarkers and cognition in mild cognitive impairment: additive or synergetic effects? Dement Geriatr Cogn Disord. 2011;32:250‐256. [DOI] [PubMed] [Google Scholar]
- 34. Jack CR Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ekman U, Ferreira D, Westman E. The A/T/N biomarker scheme and patterns of brain atrophy assessed in mild cognitive impairment. Sci Rep. 2018;8:8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dani M, Brooks DJ, Edison P. Suspected non‐Alzheimer's pathology ‐ Is it non‐Alzheimer's or non‐amyloid? Ageing Res Rev. 2017;36:20‐31. [DOI] [PubMed] [Google Scholar]
- 37. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kametani F, Hasegawa M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease. Front Neurosci. 2018;12:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81:740‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau‐mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]