

Abstract
Aims/Introduction
To investigate the changes in the gut microbiome in the second trimester of pregnancy associated with later‐diagnosed gestational diabetes mellitus (GDM) and their relationship with fasting serum levels of metabolites, especially glucose.
Materials and Methods
We carried out a case–control study with 110 GDM patients and 220 healthy pregnant women who provided fecal samples for 16S ribosomal ribonucleic acid sequencing in the second trimester of pregnancy.
Results
Our results showed that GDM patients had lower α‐diversity that was significantly associated with glycemic traits. Principal coordinates analysis showed significantly different microbial communities, as within GDM patients, seven genera within the phylum Firmicutes and two within the phylum Actinobacteria were significantly decreased, and four genera within phylum Bacteroidetes were increased. In addition, microbiota co‐occurrence network analysis was carried out, and decreased genera within the phylum Firmicutes in GDM patients showed a significant negative correlation with oral glucose tolerance test values. Finally, microbial gene functions related to glycan biosynthesis and metabolism were found to be enriched in GDM patients.
Conclusions
Our results show the relationship between changed gut microbiota composition in the second trimester of pregnancy before the diagnosis of GDM and fasting serum levels of metabolites, which might inform the diagnosis, prevention and treatment of GDM.
Keywords: Gestational diabetes mellitus, Gut microbiome, Oral glucose tolerance test
A significantly lower α‐diversity and changed microbial communities were identified in the gestational diabetes mellitus group compared with the normal pregnancy group. Changed microbiome composition is associated with fasting serum levels of metabolites, especially glucose. Microbial gene functions related to glycan biosynthesis and metabolism are found to be enriched in gestational diabetes mellitus patients.

Introduction
Insulin resistance (IR) and immune tolerance are increased during pregnancy, along with the development of pregnancy complications in susceptible populations 1 . Gestational diabetes mellitus (GDM), one of the most common pregnancy complications, is defined as glucose intolerance that is first detected during pregnancy and affects 1–16.5% of pregnancies 2 . GDM increases the risk of hypertension, fetal macrosomia, neonatal jaundice and hypoglycemia. There are also some long‐term adverse effects of GDM on women and their children, mainly increased risk of developing metabolic disorders, including obesity and type 2 diabetes 3 , 4 .
Emerging evidence has shown the influence of the gut microbiome on human metabolic health and IR, and previous studies have shown different fecal microbial communities between healthy individuals and patients with obesity and type 2 diabetes 5 , 6 . Qin et al. 6 carried out a two‐stage metagenome‐wide association study, and identified decreased butyrate‐producing bacteria and increased opportunistic pathogens in type 2 diabetes patients. In addition, some recent studies have focused on the association of gut microbiome changes during pregnancy with pregnancy complications. Koren et al. 7 found reduced richness and increased abundance of Proteobacteria and Actinobacteria between the first and third trimesters. A study based on 16S ribosomal ribonucleic acid (16SrRNA) gene sequencing showed a correlation between altered levels of bacteria, including Ruminococcaceae, Lachnospiraceae, Collinsella and Coprococcus, in the gut microbiome and circulating metabolic hormones in overweight and obese pregnant women 8 . During pregnancy, being overweight or obese might increase the risk of IR and GDM 9 . In 2017, Kuang et al. 10 carried out a metagenome‐wide association study, and found a significantly limited gene count, lower Shannon index and enriched Parabacteroides distasonis, Klebsiellavariicola and so on in GDM patients. Decreased α‐diversity in the gut microbiome and development of GDM have also been reported in other studies 11 . However, the findings of the alterations in microbiota composition, and their relationship with glucose concentration and IR during pregnancy have been inconsistent between studies 11 , 12 , 13 .
Previous studies mainly focused on the effect of GDM on gut microbiome changes during late pregnancy and postpartum. Hence, we aimed to investigate the potential differences in the abundance and composition of the gut microbiome before GDM development, and their correlation with different fasting serum levels of metabolites, including glucose, fatty acids and lipids, between women with GDM and those with a healthy pregnancy. In the present study, the gut microbiota composition of 110 GDM patients and 220 healthy pregnant women were analyzed through 16SrRNA gene sequencing of fecal samples collected at 22 and 24 weeks before diagnosis of GDM to explore the association between gut microbiome composition and subsequent diagnosis of GDM. In addition, the results might inform the diagnosis, prevention and treatment of GDM through early gut microbiome interventions.
Methods
Study population and sampling
This work was approved by the Medicine Ethics Committee of Nanjing Maternity and Child Health Care Hospital (reference number: [2017] 003), and all participants provided informed consent. Maternal fecal samples were obtained from pregnant women between 22 and 24 weeks of pregnancy at the Nanjing Maternity and Child Health Care Hospital (Nanjing, Jiangsu Province, China) between 2017 and 2018. A standardized 75‐goral glucose tolerance test (OGTT) between 25 and 26 weeks of pregnancy was administered to all participants. Women with GDM were diagnosed by qualified doctors if one or more of the following glucose criteria were met: fasting ≥5.1 mmol/L, 1 h ≥10.0 mmol/L or 2 h ≥8.5 mmol/L 14 . Using a case–control design, eligible cases were identified and then matched with healthy controls for prepregnancy body mass index, parity and maternal age at a ratio of 1:2. The exclusion criteria were as follows: (i) prepregnancy diabetes or other prediagnosed metabolic diseases; (ii) diagnosed with GDM based on fasting glucose levels at enrollment; (iii) treatment with antibiotics within 3 months before sample collection; and (iv) chronic diseases requiring medication. Finally, a total of 330 pregnant women (110 women with GDM and 220 healthy pregnant women) participated in the present study. All pregnant women in this study were unrelated ethnic Han Chinese.
Demographic information and clinical records of the study population were extracted from a structured questionnaire and the Hospital Information Systems, respectively. Fecal samples were collected in a sterile and uniform manner, frozen in −20°C freezers immediately and stored at −80°C until deoxyribonucleic acid (DNA) extraction.
DNA extraction and 16SrRNA sequencing
All bacterial DNA was obtained from fecal samples of 180–220 mg in a sterile environment using a QIAamp Fast DNA Stool Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions 15 . The variable regions 3 and 4 (V3–V4) of the 16SrRNA gene were amplified used the primers 338F (5′‐ACTCCTACGGGAGGCAGCAG‐3′) and 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′), and then sequenced using a MiSeq platform (Illumina Inc., San Diego, CA, USA).
16SrRNA analysis
Sequence quality control was carried out with QIIME 2 v.2019.10 (https://qiime2.org). The dada2 plugin was used to denoise sequences, and operational taxonomic units (OTUs) were obtained at 100% sequence homology, a method that improves distinctive taxonomic classification at single nucleotide accuracy. All representative reads were classified in the lowest taxonomic rank by a trained feature classifier using the Greengenes database as the training reference (the Greengenes database v13_8 99% OTUs). The α‐diversity (Shannon index and observed OTUs) and β‐diversity (weighted unifrac distance) were calculated with QIIME2, with all samples rarefied to the lowest sequence depth 37,747. Principle coordinates analysis (PCoA) was used to visualize β‐diversity, and permutational manova in R package “vegan’” (R Foundation for Statistical Computing, Vienna, Austria) was used to calculate the variance between groups. High‐dimensional microbiome biomarkers at the genus level were identified by linear discriminant analysis (LDA) effect size (LEfSe; logarithmic LDA scores >2.0). The predicted metagenome of the 16SrRNA OTU data was obtained by PICRUSt (http://huttenhower.sph.harvard.edu/galaxy) and categorized by function according to Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa Laboratories, Kyoto University, Kyoto, Japan). The co‐occurrence network of the 100 most abundant genera, which accounted for >99.8% of the whole abundance, was calculated by weighted gene co‐expression network analysis (WGCNA). Soft‐thresholding powers were defined by the pickSoftThreshold function in the R package “WGCNA”. Highly co‐expressed genera were grouped into one module. The network was visualized by Cytoscape 3.7.1 (http://cytoscape.org/).
Statistical analysis
All statistical analyses were carried out in R v3.4.3 (R Foundation for Statistical Computing). Categorical variables were compared between groups with χ2‐tests. Comparisons of continuous clinical variables were carried out by t‐tests. The relative abundance of taxa at the phylum level, α‐diversity indexes and PCoA2 indexes between groups were compared with Mann–Whitney tests. Logistic regression analysis was carried out to explore the association of the α‐diversity index with GDM, with and without adjusting for other factors. Linear regression was used to estimate the association of clinical data with the PCoA 2 index. Spearman’s correlation analysis between clinical data and genera biomarkers was carried out, and false discovery rate correction was used for multiple comparisons. A two‐tailed P value <0.05 was considered significant in all analyses.
Results
Description of the study population
The selected characteristics of the 110 GDM patients and 220 healthy controls are shown in Table 1. Prepregnancy body mass index, parity and maternal age were matched between the two groups (P = 0.877, P = 1.000 and P = 0.996, respectively). As expected, markers of glucose, including fasting glucose and OGTT values, were higher in the GDM patients than in the healthy controls (P < 0.001). Similarly, total protein, triglycerides, fructosamine, diastolic blood pressure and glycated hemoglobin were higher in the GDM patients (P = 0.012, P < 0.001, P = 0.031, P = 0.038 and P = 0.016, respectively). However, the characteristics of body mass index and blood biochemical indexes measured at enrollment (between 22 and 24 weeks of pregnancy), including blood urea nitrogen, creatinine, uric acid, albumin, cholesterol, alanine aminotransferase, aspartate transaminase, alkaline phosphatase, lactic dehydrogenase, γ‐glutamyltranspeptidase, total bilirubin, direct bilirubin and total bile acid in the GDM patients were not significantly different from those in the healthy controls.
Table 1.
Clinical variables of gestational diabetes mellitus patients and healthy controls in the second trimester of pregnancy
| Characters | Control (n = 220) | GDM (n = 110) | P‐value |
|---|---|---|---|
| Descriptive measurements | |||
| Prepregnancy BMI | |||
| BMI <18.5 | 40 (18.2%) | 18 (16.4%) | 0.877 |
| 18.5 ≤ BMI < 25 | 150 (68.2%) | 78 (70.9%) | |
| BMI ≥ 25 | 30 (13.6%) | 14 (12.7%) | |
| Parity | |||
| No | 150 (68.2%) | 75 (68.2%) | 1.000 |
| Yes | 70 (31.8%) | 35 (31.8%) | |
| Maternal age | |||
| <30 | 97 (44.1%) | 48 (43.6%) | 0.996 |
| 30–35 | 85 (38.6%) | 43 (39.1%) | |
| ≥35 | 38 (17.3%) | 19 (17.3%) | |
| Educational levels | |||
| High school or lower | 20 (9.1%) | 5 (4.5%) | 0.089 |
| College | 156 (70.9%) | 90 (81.8%) | |
| Master’s degree or above | 44 (20.0%) | 15 (13.6%) | |
| Prepregnancy BMI | 21.4 (3.1) | 21.4 (3.2) | 0.889 |
| BMI ~24 weeks | 23.6 (3.2) | 23.8 (3.3) | 0.604 |
| BMI_difference | 2.2 (1.4) | 2.4 (1.1) | 0.359 |
| SBP (mmHg) | 108.3 (11.8) | 110.4 (12.6) | 0.146 |
| DBP (mmHg) | 65.9 (8.4) | 67.9 (8.3) | 0.038 |
| Biochemistry | |||
| GLU (mmol/L) | 4.3 (0.3) | 4.5 (0.3) | <0.001 |
| OGTT 0 h (mmol/L) | 4.2 (0.3) | 4.4 (0.4) | <0.001 |
| OGTT 1 h (mmol/L) | 7.4 (1.3) | 9.6 (1.2) | <0.001 |
| OGTT 2 h (mmol/L) | 6.5 (1.0) | 8.7 (1.0) | <0.001 |
| BUN (mmol/L) | 2.8 (0.6) | 2.8 (0.8) | 0.533 |
| CREA (μmol/L) | 38.0 (5.4) | 36.9 (5.5) | 0.067 |
| UA (μmol/L) | 242.5 (45.4) | 247.7 (55.0) | 0.394 |
| TP (g/L) | 65.2 (3.3) | 66.1 (3.3) | 0.012 |
| ALB (g/L) | 38.6 (2.2) | 38.8 (2.2) | 0.439 |
| CHOL (mmol/L) | 5.8 (0.9) | 5.8 (1.0) | 0.941 |
| TG (mmol/L) | 2.2 (0.8) | 2.5 (0.9) | <0.001 |
| ALT (U/L) | 17.1 (10.8) | 16.8 (8.9) | 0.810 |
| AST (U/L) | 17.7 (5.9) | 18.1 (5.2) | 0.624 |
| ALP (U/L) | 46.2 (10.3) | 46.6 (12.5) | 0.715 |
| LDH (U/L) | 163.2 (26) | 166.2 (36.6) | 0.386 |
| GGT (U/L) | 16.6 (8.7) | 16.3 (11.3) | 0.800 |
| TBIL (μmol/L) | 7.8 (2.4) | 7.8 (2.4) | 0.909 |
| DBIL (μmol/L) | 2.3 (0.8) | 2.2 (0.7) | 0.603 |
| TBA (μmol/L) | 1.7 (1.4) | 1.6 (1.2) | 0.440 |
| FMN (mmol/L) | 1.8 (0.2) | 1.9 (0.5) | 0.031 |
| HbAlc (%) | 4.9 (0.2) | 5.0 (0.3) | 0.016 |
The characteristics of body mass index (BMI) and blood biochemistry indexes, except for oral glucose tolerance test (OGTT) values, were measured at enrollment (between 22 and 24 weeks of pregnancy). Data presented are the mean ± standard deviation or n (%). The t‐test and χ2‐test were used to compare the control and gestational diabetes mellitus (GDM) groups. ALB, albumin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate transaminase; BUN, blood urea nitrogen; CHOL, cholesterol; CREA, creatinine; DBIL, direct bilirubin; DBP, diastolic blood pressure; FMN, fructosamine; GGT, γ‐glutamyltranspeptidase; GLU, fasting glucose; HbAlc, glycated hemoglobin; LDH, lactic dehydrogenase; OGTT 0 h, glucose at time 0 min; OGTT 1 h, glucose at time 60 min; OGTT 2 h, glucose at time 120 min; SBP, systolic blood pressure; TBA, total bile acid; TBIL, total bilirubin; TGs, triglycerides; TP, total protein; UA, uric acid.
Microbiome profile of the study population
At the phylum level, Firmicutes, Bacteroidetes and Actinobacteria were the dominant taxa across all samples (Figure 1). However, the GDM patients showed a greater abundance of Bacteroidetes (P = 0.033), and lower abundances of Firmicutes (P = 0.007) and Actinobacteria (P = 0.014) than the healthy controls (Figure 1a).
Figure 1.
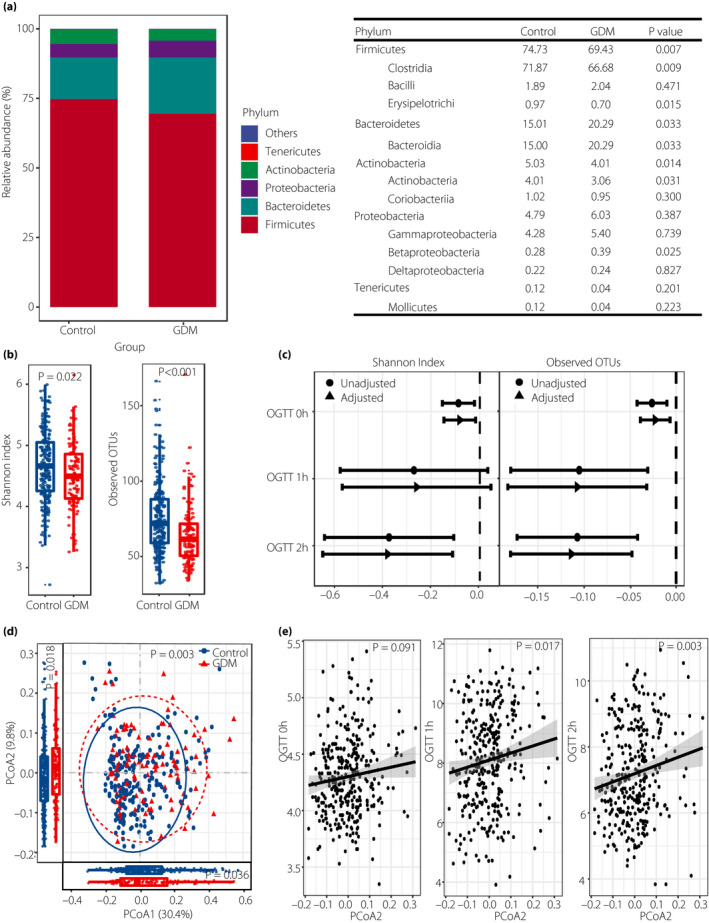
Microbiome profile of the study population. (a) Relative abundance of dominant taxa in the gut microbiome between the gestational diabetes mellitus (GDM) patients and the healthy controls. (b) α‐Diversity indexes of the fecal microbiome between the GDM patients and the healthy controls. (c) Oral glucose tolerance test (OGTT) values were regressed on α‐diversity measures (per 1 increase in Shannon index or 10 in observed operational taxonomic units [OTUs]) with and without adjustment for prepregnancy body mass index, parity and maternal age. (d) Principle coordinates analysis (PCoA) plot based on weighted unifrac distance between the GDM patients and the healthy controls. Permutational manova was used to calculate similarity between two groups. (e) Linear regression analysis of PCoA2 index and OGTT values with adjustment for prepregnancy body mass index, parity and maternal age. OGTT 0 h, blood glucose level at time 0 min; OGTT 1 h, blood glucose level at time 60 min; OGTT 2 h, blood glucose level at time 120 min.
Through α‐diversity analysis, the GDM patients were shown to have significantly lower Shannon indexes (P = 0.022) and observed OTUs (P = 8.117e‐06) than the healthy controls in the second trimester of pregnancy (Figure 1b). When glycemic traits were regressed against α‐diversity measures, we found significant associations between the Shannon index and glucose level, represented by the results of the OGTT, including OGTT 0 h and OGTT 2 h values (β = −0.08, 95% confidence interval [CI] −0.15, −0.02; β = −0.38, 95% CI −0.65, −0.11, respectively), as well as between observed OTUs and OGTT 0 h, OGTT 1 h and OGTT 2 h (β = −0.02, 95% CI −0.04, −0.01; β = −0.11, 95% CI −0.18, −0.03; β = −0.11, 95% CI −0.18, −0.05, respectively; Figure 1c and Table S1).
We also plotted a PCoA to compare the similarity between groups, and found a significant difference in weighted unifrac distances (P = 0.003) between the GDM patients and the healthy controls (Figure 1d). The PCoA1 and PCoA2 indexes also showed significant differences between the two groups (P = 0.036 and P = 0.018). In addition, PCoA2 was significantly associated with OGTT 1 h and OGTT 2 h (P = 0.017 and P = 0.003, respectively; Figure 1e).
Differences in dominant genera and their association with fasting serum levels of metabolites between the two groups
LEfSe was used to identify the most variable features at the genus level between the GDM patients and the healthy controls. In total, we found 20 genera that showed differences between the two groups. Seven genera within the phylum Firmicutes and two within the phylum Actinobacteria were identified as significantly lower in the GDM group, and four genera within the phylum Bacteroidetes were higher in the GDM group. These results were consistent with the aforementioned results at the phylum level. The results showed that the genera Bacteroides, Dialister and Campylobacter, and an unassigned genus of Enterococcaceae were taxonomic biomarkers of GDM, whereas the genera Gemmiger and Bifidobacterium, and unassigned genera of Clostridiales and Ruminococcaceae were markers of normal glucose during pregnancy (Figure 2a).
Figure 2.
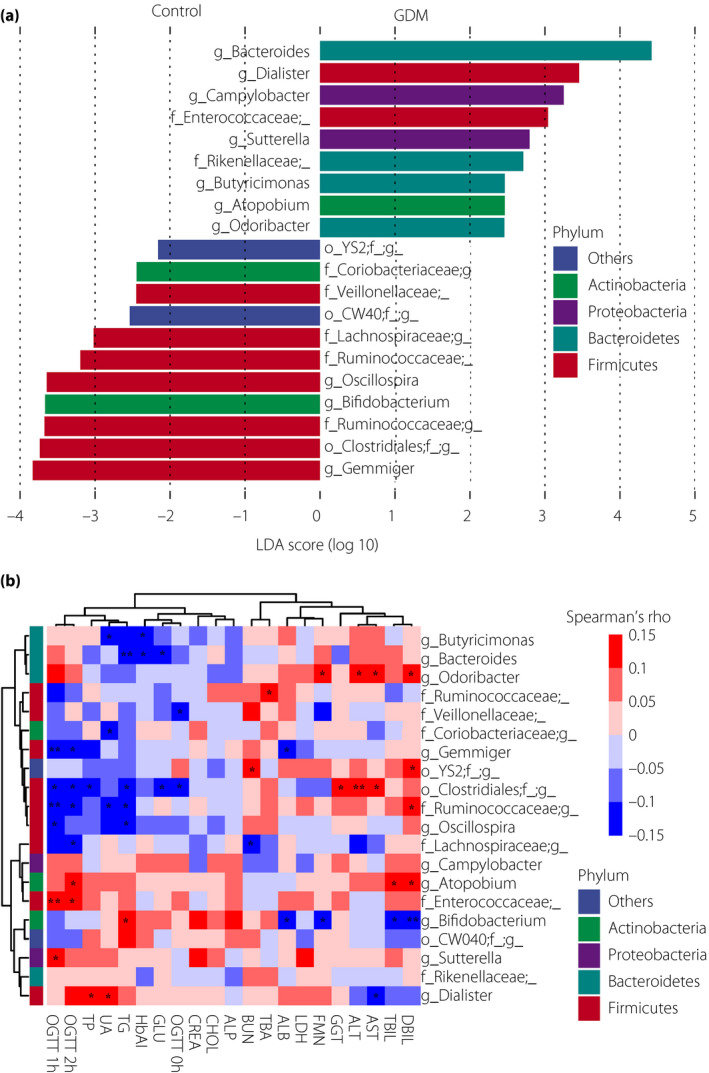
Differences in dominant genera and their association with blood biochemical traits between the gestational diabetes mellitus (GDM) patients and the healthy controls. (a) Differences in dominant genera between the GDM patients and the healthy controls. Linear discriminant analysis effect size was carried out with an absolute value of linear discriminant analysis (LDA) score (log10) >2.0 as the threshold. (b) Spearman’s correlation of different dominant genera and blood biochemical traits (red for positive correlations and blue for negative correlations). *P < 0.05, **P < 0.01. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate transaminase; BMI, body mass index; BUN, blood urea nitrogen; CHOL, cholesterol; CREA, creatinine; DBIL, direct bilirubin; DBP, diastolic blood pressure; FMN, fructosamine; GGT, γ‐glutamyltranspeptidase; GLU, fasting glucose; LDH, lactic dehydrogenase; OGTT 0 h, glucose at time 0 min; OGTT 1 h, glucose at time 60 min; OGTT 2 h, glucose at time 120 min; SBP, systolic blood pressure; TBA, total bile acid; TBIL, total bilirubin; TP, total protein; UA, uric acid.
We used Spearman’s correlation analysis to identify whether the different dominant genera were associated with fasting serum levels of metabolites in the second trimester of pregnancy (Figure 2b). Genera whose abundances were decreased in GDM patients, including Gemmiger, Oscillospira and unassigned genera of Clostridiales, Ruminococcaceae and Lachnospiraceae within the phylum Firmicutes, were significantly negatively correlated with at least one OGTT value (P < 0.05). However, an unassigned genera of Enterococcaceae within the phylum Firmicutes, Atopobium within the phylum Actinobacteria and Sutterella within the phylum Proteobacteria were significantly positively associated with OGTT 1 h or OGTT 2 h values (P < 0.05).
Genera co‐occurrence network and module–trait associations
A network of co‐occurring genera was created, and weighted gene co‐expression network analysis was used to identify modules in the network (Figure 3b). The entire list of the 100 most abundant genera and the organization of their modules is available in Table S2. Phylogenetically related OTUs were preferentially clustered in the same module, although other OTUs from different taxa are also found in each module.
Figure 3.
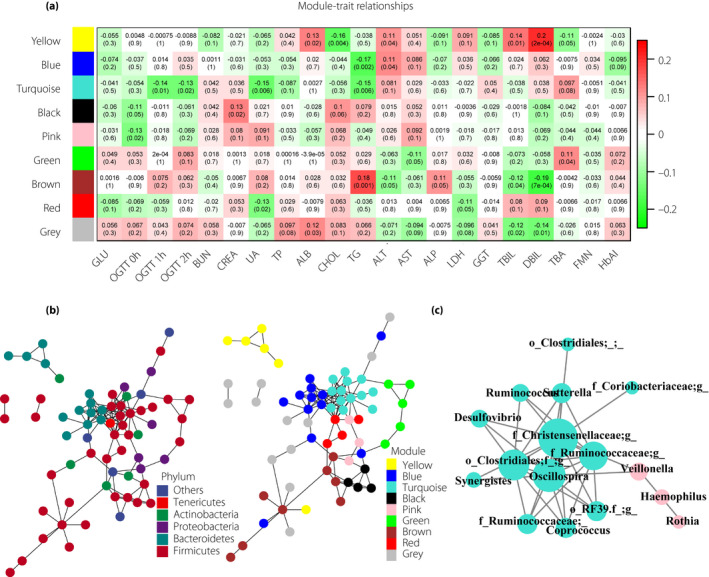
Genera co‐occurrence network and module–trait associations. (a) Module–trait associations. The correlation value and the corresponding P‐value between genera modules and biochemical traits are shown in each cell of the matrix (red for positive correlations and green for negative correlations). (b) Genera co‐occurrence network where observed operational taxonomic units (nodes) are colored according to corresponding phyla or module colors. (c) Detailed network of the turquoise module and pink module. Node size is proportional to the degree of unweighted connectivity. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate transaminase; BUN, blood urea nitrogen; CHOL, cholesterol; CREA, creatinine; DBIL, direct bilirubin; FMN, fructosamine; GGT, γ‐glutamyltranspeptidase; GLU, fasting glucose; HbAlc, glycated hemoglobin; LDH, lactic dehydrogenase; OGTT 0 h, glucose at time 0 min; OGTT 1 h, glucose at time 60 min; OGTT 2 h, glucose at time 120 min; TBA, total bile acid; TBIL, total bilirubin; TG, triglycerides; TP, total protein; UA, uric acid.
The module–trait associations were in accordance with previous results of trait–taxa relationships (Figure 3a). The turquoise module, mostly composed of genera within the phylum Firmicutes, was negatively associated with OGTT values. In this module, five genera were the biomarkers found to be significantly decreased in the GDM group by LEfSe analysis. In addition, the pink module, representing the genera Veillonella, Haemophilus and Rothia, was negatively associated with OGTT 0 h values. We also found significant associations between modules and other biochemical traits. However, both modules, especially the pink module, also contained other OTUs from different taxa, highlighting the importance of the combined effects of different members of the gut microbiome on biochemical traits (Figure 3c).
Association of abundant Kyoto Encyclopedia of Genes and Genomes pathways of microbial genes between the two groups
To determine the importance of dysbiosis in GDM patients between 22 and 24 weeks of pregnancy, the relative proportions of the predicted microbial genetic functional pathways between the two groups were compared based on the 16SrRNA data. Statistical hypothesis tests highlighted significantly differentially (P < 0.05) abundant pathways of microbial genes that discriminate between the GDM patients and the healthy controls, including microbial gene functions related to three pathways in glycan biosynthesis and metabolism, four pathways in cellular processes and signaling, and two pathways in the metabolism of cofactors and vitamins that were enriched in GDM patients (Figure 4).
Figure 4.
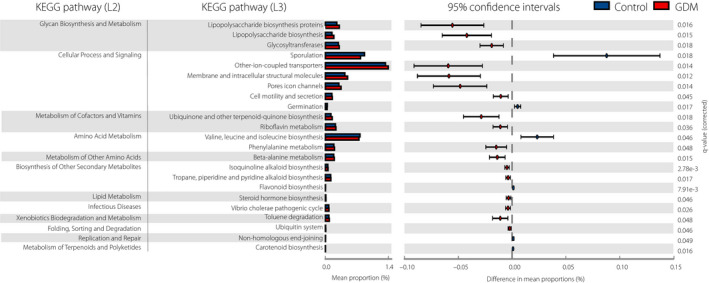
PICRUSt analysis results of predicted functional pathways of the gut microbiome of gestational diabetes mellitus patients and healthy controls. Two‐sided Welch’s t‐test and false discovery rate correction were used to identify the differentially abundant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (P < 0.05).
Discussion
In a case–control study of 330 pregnant women (110 women with GDM and 220 normal pregnant women), we identified the association of gut microbiome composition with subsequently diagnosed GDM and fasting serum levels of metabolites, especially OGTT values. Furthermore, microbial genetic functional pathways were predicted, and glycan biosynthesis and metabolism were found to be enriched in GDM patients.
The differences in taxonomic compositions associated with metabolic disease, including obesity, type 2 diabetes and GDM, have not been consistent across different studies 12 , 13 , 16 , 17 , 18 , 19 . However, a lower abundance of Firmicutes was reported in type 2 diabetes and GDM patients 6 , 19 . In the present study, the gut microbiome composition of the entire study population was dominated by the phyla Firmicutes and Bacteroidetes; and an increased abundance of Bacteroidetes, and decreased abundances of Firmicutes and Actinobacteria were detected in the GDM group.
Decreased richness of the gut microbiome has been associated with elevated IR and pro‐inflammatory markers 20 . IR and inflammation are two characteristics of the metabolic environment, and worsen with advancing gestation in GDM patients 21 , 22 . Decreased microbial richness and diversity of the gut microbiome in late pregnancy and in GDM patients compared with individuals with a healthy pregnancy were reported in previous studies 7 , 11 , 12 . The present study also showed a significantly lower Shannon index and observed OTUs in GDM patients compared with the same parameters in healthy controls, and these parameters were significantly associated with OGTT values. These results further showed the association between decreased α‐diversity and GDM, and suggested a relationship between these variables and elevated plasma OGTT values.
Bacterial community structure analyses showed that the gut microbiota composition of the GDM patients was significantly distinct from that of the healthy controls, and PCoA2 was significantly associated with plasma OGTT values. We further detected four genera within the phylum Bacteroidetes (specifically, Bacteroides, Butyricimonas, Odoribacter and an unassigned genera of Rikenellaceae), two genera within the phylum Proteobacteria (specifically, Campylobacter and Sutterella) and genus Atopobium within the phylum Actinobacteriaas taxonomic biomarkers of GDM. Within Firmicutes, two genera, Dialister and an unassigned genera of Enterococcaceae, were biomarkers of GDM, whereas the genera Gemmiger, Oscillospira, unassigned genera of Clostridiales, Ruminococcaceae, Lachnospiraceae and Veillonellaceae were markers of normal glucose during pregnancy.
The GDM‐enriched bacteria detected in the present study were related to gut flora dysbiosis reported in previous studies. Bacteroides was associated with a high‐fat animal‐based diet and overweight pregnant woman 23 , 24 . Rikenellaceae, Butyricimonas and Odoribacter are also butyrate producers, and Ferrocino et al. 13 reported a direct association between Butyricimonas and IR in overweight GDM patients. Obesity and type 2 diabetes characterized by a pro‐inflammatory state might be associated with Gram‐negative bacteria that produce inflammatory lipopolysaccharide 25 . It is well known that Enterococcaceae, Campylobacter and Sutterella are bacteria with pro‐inflammatory capacity. Some species of Enterococcaceae and Campylobacter are associated with intestinal inflammatory disease 26 , 27 , and Sutterella is positively associated with lipopolysaccharide biosynthesis during the third trimester of pregnancy 13 . Thus, the present study proposed that changed composition of functional bacterial populations, including increased abundance of butyrate‐producing bacteria and pathogenic bacteria, was associated with later‐diagnosed GDM.
In the present study, bacterial populations mostly composed of genera within the phylum Firmicutes (specifically, Gemmiger, Oscillospira, unassigned genera of Clostridiales, Ruminococcaceae and Lachnospiraceae) were enriched in normoglycemic pregnant women and were significantly negatively correlated with one or more OGTT values. However, Atopobium, Sutterella and an unassigned genera of Enterococcaceae, which were increased in GDM patients, were significantly positively associated with plasma glucose levels. Relationships between bacterial abundance and plasma OGTT values were also reported in previous studies 6 , 13 . Although OGTT is the gold standard for the diagnosis of GDM, it requires a blood test and is time‐consuming 28 . These discriminatory bacteria, which correlate with plasma OGTT values, might provide new strategies for the diagnosis, prevention and treatment of GDM. In addition, we found that microbial gene functions related to glycan biosynthesis and metabolism pathways, cellular processes and signaling, and metabolism of cofactors and vitamins were enriched in GDM patients. These results strengthened the association between changed gut microbiome composition and dysmetabolism 29 , 30 .
There were still some limitations in the present study. First, we obtained fecal samples only at one time point, and the interval between fecal sampling and OGTT was short, which makes our study lack prediction power, but instead provides supplementary information for the prevention and treatment of GDM through early gut microbiome interventions. Although we excluded those patients diagnosed with GDM at enrollment based on fasting glucose levels, some of the patients might have been missed, because OGTT was not carried out at that time point. Other limitations include the relatively small sample size and lack of dietary questionnaires. Further prospective studies are required to explore the possible association between microbiota imbalance and long‐term metabolic disease in GDM mothers and their offspring.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 | Association of α‐diversity indices and oral glucose tolerance test results.
Table S2 | Weighted gene co‐expression network analysis modules and the list of genera in each module.
Acknowledgment
This work was funded by National Natural Science Foundation of China, grant number 81571458 and 81100436; National Key Research and Development Program of China, grant number 2016YFC1000303; 333 Training Fund Project of Jiangsu Province, grant number (2016) III‐0087; Six talent peaks project in Jiangsu Province, grant number WSW‐121; Nanjing Medical Science and Technique Development Foundation, grant number QRX17162; Nanjing Science and Technology Development Project, grant number 201911040; and Nanjing Medical Science and Technology Development Fund, grant number ZKX19044.
J Diabetes Investig. 2021
Contributor Information
Yankai Xia, Email: yankaixia@njmu.edu.cn.
Zhonghua Shi, Email: Jesse_1982@163.com, Email: yankaixia@njmu.edu.cn.
References
- 1. Angueira AR, Ludvik AE, Reddy TE, et al. New insights into gestational glucose metabolism: lessons learned from 21st century approaches. Diabetes 2015; 64: 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kjos SL, Buchanan TA. Gestational diabetes mellitus. N Engl J Med 1999; 341: 1749–1756. [DOI] [PubMed] [Google Scholar]
- 3. Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care 2002; 25: 1862–1868. [DOI] [PubMed] [Google Scholar]
- 4. Allalou A, Nalla A, Prentice KJ, et al. A predictive metabolic signature for the transition from gestational diabetes mellitus to type 2 diabetes. Diabetes 2016; 65: 2529–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karlsson F, Tremaroli V, Nielsen J, et al. Assessing the human gut microbiota in metabolic diseases. Diabetes 2013; 62: 3341–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin J, Li Y, Cai Z, et al. A metagenome‐wide association study of gut microbiota in type 2 diabetes. Nature 2012; 490: 55–60. [DOI] [PubMed] [Google Scholar]
- 7. Koren O, Goodrich JK, Cullender TC, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012; 150: 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomez‐Arango LF, Barrett HL, McIntyre HD, et al. Connections between the gut microbiome and metabolic hormones in early pregnancy in overweight and obese women. Diabetes 2016; 65: 2214–2223. [DOI] [PubMed] [Google Scholar]
- 9. Chu SY, Callaghan WM, Kim SY, et al. Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care 2007; 30: 2070–2076. [DOI] [PubMed] [Google Scholar]
- 10. Kuang YS, Lu JH, Li SH, et al. Connections between the human gut microbiome and gestational diabetes mellitus. Gigascience 2017; 6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J, Zheng J, Shi W, et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut 2018; 67: 1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crusell MKW, Hansen TH, Nielsen T, et al. Gestational diabetes is associated with change in the gut microbiota composition in third trimester of pregnancy and postpartum. Microbiome 2018; 6: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferrocino I, Ponzo V, Gambino R, et al. Changes in the gut microbiota composition during pregnancy in patients with gestational diabetes mellitus (GDM). Sci Rep 2018; 8: 12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu WW, Yang HX. Diagnosis of gestational diabetes mellitus in China. Diabetes Care 2013; 36: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mirsepasi H, Persson S, Struve C, et al. Microbial diversity in fecal samples depends on DNA extraction method: easyMag DNA extraction compared to QIAamp DNA stool mini kit extraction. BMC Res Notes 2014; 7: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ley RE, Turnbaugh PJ, Klein S, et al. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444: 1022–1023. [DOI] [PubMed] [Google Scholar]
- 17. Verdam FJ, Fuentes S, de Jonge C, et al. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity 2013; 21: E607–E615. [DOI] [PubMed] [Google Scholar]
- 18. Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013; 498: 99–103. [DOI] [PubMed] [Google Scholar]
- 19. Fugmann M, Breier M, Rottenkolber M, et al. The stool microbiota of insulin resistant women with recent gestational diabetes, a high risk group for type 2 diabetes. Sci Rep 2015; 5: 13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500: 541–546. [DOI] [PubMed] [Google Scholar]
- 21. Friedman JE. Obesity and gestational diabetes mellitus pathways for programming in mouse, monkey, and man‐where do we go next? The 2014 Norbert Freinkel Award Lecture. Diabetes Care 2015; 38: 1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barbour LA, McCurdy CE, Hernandez TL, et al. Chronically increased S6K1 is associated with impaired IRS1 signaling in skeletal muscle of GDM women with impaired glucose tolerance postpartum. J Clin Endocrinol Metab 2011; 96: 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014; 505: 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collado MC, Isolauri E, Laitinen K, et al. Distinct composition of gut microbiota during pregnancy in overweight and normal‐weight women. Am J Clin Nutr 2008; 88: 894–899. [DOI] [PubMed] [Google Scholar]
- 25. Allin KH, Nielsen T, Pedersen O. Mechanisms in endocrinology: gut microbiota in patients with type 2 diabetes mellitus. Eur J Endocrinol 2015; 172: R167–R177. [DOI] [PubMed] [Google Scholar]
- 26. Brooks PT, Brakel KA, Bell JA, et al. Transplanted human fecal microbiota enhanced Guillain Barre syndrome autoantibody responses after Campylobacter jejuni infection in C57BL/6 mice. Microbiome 2017; 5: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakamoto N, Sasaki N, Aoki R, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol 2019; 4: 492–503. [DOI] [PubMed] [Google Scholar]
- 28. Olabi B, Bhopal R. Diagnosis of diabetes using the oral glucose tolerance test. BMJ 2009; 339: b4354. [DOI] [PubMed] [Google Scholar]
- 29. Friedman JE. Developmental programming of obesity and diabetes in mouse, monkey, and man in 2018: where are we headed? Diabetes 2018; 67: 2137–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Natividad JM, Agus A, Planchais J, et al. Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab 2018; 28, 737–749 e734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Association of α‐diversity indices and oral glucose tolerance test results.
Table S2 | Weighted gene co‐expression network analysis modules and the list of genera in each module.