

ABSTRACT
Background
Determination of disease onset in Huntington's disease is made by clinical experience. The diagnostic confidence level is an assessment regarding the certainty about the clinical diagnosis based on motor signs. A level of 4 means the rater has ≥99% confidence motor abnormalities are unequivocal signs of disease. However, it does not state which motor abnormalities are signs of disease and how many must be present.
Objective
Our aim is to explore how accurate the diagnostic confidence level is in estimating disease onset using the Enroll‐HD data set. For clinical disease onset we use a cut‐off total motor score >5 of the Unified Huntington's Disease Rating Scale. This score is used in the TRACK‐HD study, with ≤5 indicating no substantial motor signs in premanifests.
Methods
At baseline premanifest participants who converted to manifest (converters) and non‐converters were compared for clinical symptoms and diagnostic confidence level. Clinical symptoms and diagnostic confidence levels were longitudinally displayed in converters.
Results
Of 3731 eligible participants, 455 were converters and 3276 non‐converters. Baseline diagnostic confidence levels were significantly higher in converters compared to non‐converters (P < 0.001). 232 (51%) converters displayed a baseline motor score >5 (mean = 6.7). Converters had significantly more baseline clinical symptoms, and higher disease burden compared to non‐converters (P < 0.001). Diagnostic confidence level before disease onset ranged between 1 and 3 in converters.
Conclusions
According to this data the diagnostic confidence level is not an accurate instrument to determine phenoconversion. With trials evaluating disease modifying therapies it is important to develop more reliable diagnostic criteria.
Keywords: Huntington's disease, disease onset, unified Huntington's disease rating scale, diagnostic confidence interval
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease characterized by involuntary movements, psychiatric disorders, and cognitive deterioration. HD results from an unstable and expanded CAG trinucleotide repeat in the Huntingtin (HTT) gene on chromosome 4. 1 A CAG repeat size of 36 or more is associated with HD. The age that symptoms and signs become manifest is called the age at onset (AO). The AO has a mean of 40 years with a tremendous spread from 2 to 80 years. 2 The importance of a precise determination of the AO plays at several levels. First, one wants to make an accurate clinical diagnosis in patient care, second, a precise AO is needed for participation in clinical trials, and third, the moment disease inhibiting drugs become available one needs to estimate when to start medication.
The clinical diagnosis of HD is predominantly made on the basis of characteristic motor signs often with a positive family history and/or confirmed by DNA analysis. The Unified Huntington's Disease Rating Scale (UHDRS) was designed for clinical follow‐up and research purposes. 3 The UHDRS contains a motor (total motor score [TMS]), psychiatric, psychological, and functional part. Based on the motor signs a 4 item assessment regarding the certainty about the clinical diagnosis was added, the diagnostic confidence level (DCL). By definition in this scale only a person with a DCL of 4 receives the formal diagnosis of manifest HD, meaning that the AO is determined at that time point. A DCL of 4 means that the motor abnormalities are unequivocal signs of HD (≥99% confidence of the rater) and not otherwise explained by another movement disorder. However, several remarks can be made; (1) the DCL does not state which motor abnormalities are unequivocal signs of HD; and (2) does not mention how many motor signs must be present. In other words, there is no cut‐off point on the TMS (range 0–124). An operational definition that guides the clinician is lacking. The TMS rates movement disorder motor abnormalities. This suggests that healthy persons, including premanifest persons, are supposed to have a TMS of 0. Otherwise, healthy persons including premanifest persons should have a movement disorder other than HD. In the TRACK‐HD study a defined cut‐off TMS of 5 or less in the UHDRS was chosen, indicating no substantial motor signs in premanifest gene carriers. 4 This allows Huntington's disease gene expansion carriers (HDGECs) to have some motor abnormalities without being called manifest yet. However, this cut‐off is not used in the current diagnostic criteria.
Early motor symptoms are variable in presentation and occurrence. Subtle motor changes may easily be missed during neurological examination 5 and recognition is dependent on the experience of the person who performs the clinical examination. All this makes it difficult to determine the exact moment of motor onset of HD.
Furthermore, cognitive and behavioral changes are frequently mentioned by HDGECs before the start of motor symptoms and signs. 6 , 7 The PREDICT‐HD and TRACK‐HD data show that not only subtle motor, but also cognitive and behavioral changes as well as brain atrophy on MRI are present in premanifest HDGECs up to more than 10 years from estimated HD diagnosis. 4 , 8 , 9 , 10 , 11 , 12 , 13 However, the findings of PREDICT‐HD are compromised by subjects in the premanifest cohort exhibiting UHDRS‐TMS scores ranging up to above 20. Significant cognitive changes in premanifest participants >10 years from predicted onset in TRACK‐HD were seen on the Stroop Word Reading test compared to controls. However, during 36‐month follow‐up no cognitive changes were seen in this group of participants.
These early neuropsychiatric symptoms contribute to a decline in daily functioning and are often the most distressing aspects of HD for patients, families, as well as their caregivers. 13 , 14 The current version of the UHDRS does not ask for a DCL rating that expands to non‐motor features. 3 , 15 , 16 This means the investigator is not able to rate the DCL as 4 based on all elements of the UHDRS, and scoring is restricted to the motor part.
Last, but not least, motor manifest patients often find it difficult to remember, because of a lack of disease insight or denial, when the first motor symptoms started. Sometimes the patient and their spouse do not recognize the movements or deny symptoms and signs of the disease. This makes it almost impossible to adequately estimate onset of disease.
Therefore, our aim is to find out how consistent the TMS >5 and DCL of 4 relate to one another in estimating disease onset using the Enroll‐HD data set. For clinical disease onset, we use the TRACK‐HD cut‐off score of TMS >5. Furthermore, we compare Enroll‐HD participants who converted from premanifest to manifest (converters) with those who are still premanifest (non‐converters) and show a follow‐up of DCL, behavioral, cognitive, and motor symptoms in the converters group.
Methods
Data were extracted from the Enroll‐HD database on October 31, 2018 (Periodic Dataset 4 (PDS4). Enroll‐HD is a global clinical research platform designed to facilitate clinical research in Huntington's disease. 17 Core data sets are collected annually on all research participants as part of this multi‐center longitudinal observational study of Huntington's disease. Data are monitored for quality and accuracy using a risk‐based monitoring approach. All sites are required to obtain and maintain local ethics committee approvals. This data set not only includes data gathered from the Enroll‐HD study, but also integrates data from REGISTRY 2 and 3, as well as ad hoc data (data, drawn from a variety of different sources, gathered before a participant's enrollment into Enroll‐HD). 18 , 19 All participants provided written informed consent for this observational study. The HTT CAG genotyping was carried out at the local genetic laboratories of the sites as well as at a central biorepository facility (Biorep). Data collection adhered to a standard protocol including electronic case report forms and used identical study protocols for the assessment and sampling of biomaterials. At baseline, HD mutation carriers are classified as manifest/motor‐manifest HD (carriers with clinical features that are regarded in the opinion of the investigator as diagnostic of HD) or premanifest/premotor‐manifest HD (carriers without clinical features regarded as diagnostic of HD). Participants were evaluated by a specialist with experience in HD. The study site raters are trained and certified annually to minimize inter‐rater and intra‐rater variability (https://hdtraining.enroll-hd.org/).
Participants and Clinical Assessments
Only participants who entered the database as premanifest were included in this study. The data set contains participants aged ≥18 years with CAG information. Clinical characteristics (motor, behavior, and cognitive) and age of onset were documented.
The time of diagnosis was documented by the clinician converting the participant from premanifest to manifest in the database. Participants with unknown category (manifest or premanifest) were excluded. The disease‐burden score is calculated from a formula (age × [CAG – 35.5]). This score gives an a posteriori estimate of an individual's lifetime exposure to mutant huntingtin, at any age, before and after motor onset. 20 To provide insight in which symptoms of the disease are most frequently reported at the onset of disease, motor and cognitive signs were scored using the UHDRS. 3 A UHDRS TMS >5 was used for clinical disease onset (maximum score 124). For cognition, we used the cognitive UHDRS composite score (UHDRS total corrected for letter fluency, symbol digit modalities test, trail making test, and Stroop subscores for word reading, color naming, and interference), with lower scores indicating lower performance. For behavior, we used the problem behavior assessment short form (PBAs) 21 or the behavior section of the UHDRS that consists of items evaluating several behavioral signs and symptoms (depressed mood, anxiety, suicidal ideation, irritability, aggressive behavior, apathy, perseverative/obsessive–compulsive behavior, delusions and hallucinations). In both assessments, participants are ranked on severity (0–4) with higher scores indicating higher degree of behavioral impairment. The item of “disoriented behavior” in the PBAs is not used in the behavior section of the UHDRS, whereas the item “low self‐esteem/guilt” is only used in the UHDRS. Therefore, these 2 items are not used for analysis. Perseverative behavior and obsessive–compulsive behavior were combined because the PBAs and the UHDRS use different sub‐items. The disease stage was obtained using the DCL that ranges from 0 to 4: 0 = normal; 1 = non‐specific motor abnormalities (<50% confidence); 2 = motor abnormalities that may be signs of HD (50%–89% confidence); 3 = motor abnormalities that are likely signs of HD (90%–98% confidence); and 4 = motor abnormalities that are unequivocal signs of HD (≥99% confidence). Participants with an unknown DCL at baseline were excluded. If the rater changed the DCL of a participant to 4 during follow‐up, the database does not automatically change the participant from premanifest to manifest. The rater has to do this manually.
Functionality was documented using the total functional capacity (TFC) scores with a maximum score of 13 (lower scores indicate a lower functional status). Comorbidities were documented according to the International Statistical Classification of Diseases 10th revision (ICD‐10) code. Participants were followed up on a yearly basis according to the Enroll‐HD protocol.
Statistical Analysis
Baseline characteristics such as age of onset, gender, and CAG repeat size (both alleles) were recorded/counted and the frequency of symptoms and signs in each group, converters and non‐converters, were calculated. Differences between the groups were tested with the t test for continuous variables and the χ2 or Fisher exact test (if necessary) for the categorical variables.
For the natural course of symptoms, data were gathered and visualized by plotting all data separately as well as looking at a trend. All available measurements of all participants were analyzed. All analyses were carried out using R. P values of ≤0.05 were considered as statistically significant.
Results
Baseline Analysis
The PDS4 consisted of 15,301 participants generating a total of 50,452 visits. Categorization of the participant (premanifest or manifest) was not available for ad hoc and Registry 2. Therefore, these data were not used for analysis. Of all 15,301 participants 3528 also participated in Registry 3.
A total of 3775 participants were premanifest at enrollment. Participants with unknown category (manifest or still premanifest) (n = 13) and unknown DCL at baseline (n = 9) were excluded. Twenty‐two participants who were categorized as premanifest with a DCL of 4 (motor abnormalities that are unequivocal signs of HD) at presentation were excluded from analysis. Of the remaining 3731 premanifest participants 455 (12.2%) became manifest (converters) during follow‐up and 3276 remained premanifest (non‐converters) (Fig. 1).
FIG. 1.
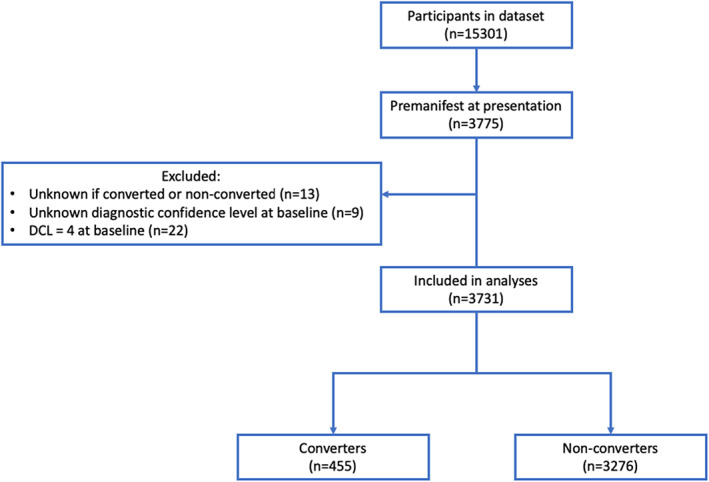
Flow chart of inclusion. DCL, diagnostic confidence interval.
The baseline characteristics of all participants, stratified by disease category, are given in Table 1. 232 (51%) converters displayed a baseline TMS >5 (mean = 6.7). The number of participants with a baseline TMS ≤5 in the converters (n = 223, 49.1%) was significantly lower than in the non‐converters (n = 2732, 83.7%) (P < 0.001). The baseline disease burden score was significantly higher in the converters (mean = 326.4; standard deviation [SD] = 80.8) compared to the non‐converters (mean = 246.9; SD = 80.6) (P < 0.001). The mean number of visits was significantly higher in the converters (mean = 3.31; SD = 1.31) compared to non‐converters (mean = 2.4; SD = 1.3) (P < 0.001). The years of visits in the converters ranged from a maximum of 7 years before and a maximum of 4 years after becoming manifest. The DCL (range = 0–4) at baseline was significantly higher in the converters compared to the non‐converters (P < 0.001) (Table 1 and Fig. 2).
TABLE 1.
Baseline characteristics participants stratified by non‐converters and converters
| Non‐converters n = 3276 | Converters n = 455 | P value | Missing | |
| Age at first visit, mean (SD) | 38.8 (11.9) | 44.0 (12.1) | <0.001 | 2/0 |
| Sex, male (%) | 1334 (40.7) | 189 (41.5) | 0.7394 | 0/0 |
| CAG repeats size high, mean (SD) | 42.3 (2.7) | 43.6 (3.3) | <0.001 | 0/0 |
| CAG repeats size low, mean (SD) | 18.3 (2.9) | 18.2 (2.8) | 0.5375 | 45/11 |
| Baseline disease burden score, mean (SD) | 246.93 (80.6) | 326.37 (80.8) | <0.001 | 2/0 |
| Number of visits, mean (SD) | 2.41 (1.3) | 3.31 (1.3) | <0.001 | 0/0 |
| Baseline motor score, mean (SD) | 2.6 (3.7) | 6.7 (6.6) | <0.001 | 11/1 |
| Baseline total motor score ≤5 (%) | 2732 (83.7) | 223 (49.1) | <0.001 | 11/1 |
| DCL at baseline, yes (%) | <0.001 | 0/0 | ||
| 0 = Normal | 2069 (63.2) | 120 (26.4) | ||
| 1 = Non‐specific motor abnormalities (<50% confidence) | 787 (24.0) | 151 (33.2) | ||
| 2 = Motor abnormalities that may be signs of HD (50%–89% confidence) | 282 (8.6) | 93 (20.4) | ||
| 3 = Motor abnormalities that are likely signs of HD (90%–98% confidence) | 138 (4.2) | 91 (20.0) | ||
| 4 = Motor abnormalities that are unequivocal signs of HD (≥99% confidence) | – | – | ||
| Baseline UHDRS total functional capacity, mean (SD) | 12.8 (0.9) | 12.4 (1.3) | <0.001 | 8/1 |
| Baseline UHDRS functional assessment, mean (SD) | 24.7 (1.3) | 24.3 (2.0) | <0.001 | 65/8 |
Data are mean (SD) or n (%). SD, standard deviation; UHDRS, Unified Huntington's Disease Rating Scale; DCL, Diagnostic Confidence Level.
FIG. 2.
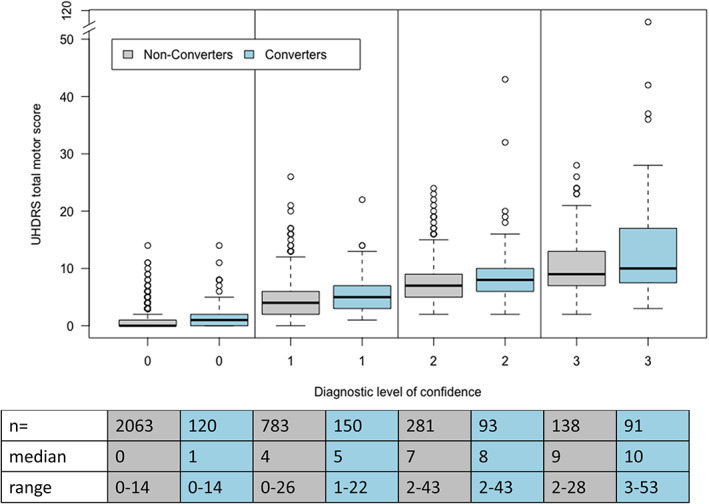
Unified Huntington Disease Rating Scale. Total motor score compared to diagnostic level of confidence at baseline in premanifest and converted HD patients; UHDRS, Unified Huntington's Disease Rating Scale.
The converters had significantly more neurological (P = 0.014) and psychiatric (P = 0.007) comorbid conditions compared to non‐converters (Table 2). Two converters had movement disorders compared to 26 non‐converters. The 2 converters had an essential tremor and an unspecified dystonia. Of the non‐converters, 3 had essential tremor, 1 had Parkinson's disease, 2 had myoclonus, 2 had other chorea, 1 had blepharospasms, and 17 had an unspecified movement disorder. In both groups, 5%–6% of the participants received physical therapy (before or) at baseline, around 4.5% received psychotherapy, 4%–6% psychological counseling, and 5%–7% relaxation therapy.
TABLE 2.
Baseline cognitive and behavioral characteristics participants stratified by non‐converters and converters
| Non‐converters n = 3276 | Converters n = 455 | P value | Missing | |
| Cognitive assessments | ||||
| SDMT total correct, mean (SD) | 50.3 (11.9) | 41.6 (12.4) | <0.001 | 197/44 |
| Verbal fluency test (category) total correct | 21.4 (5.7) | 19.1 (6.2) | <0.001 | 226/61 |
| Verbal fluency test (letters) total correct | 39.5 (12.7) | 34.0 (13.1) | <0.001 | 643/82 |
| Stroop color naming total correct | 73.1 (14.6) | 65.2 (15.3) | <0.001 | 213/48 |
| Stroop reading test total correct | 94.0(17.9) | 84.0 (19.8) | <0.001 | 210/49 |
| Stroop interference test total correct | 43.6 (11.1) | 38.0 (11.3) | <0.001 | 372/61 |
| Trailmaking test Part A total correct | 24.7 (2.7) | 25.0 (0.1) | <0.001 | 934/158 |
| Trailmaking test Part B total correct | 24.5 (3.8) | 24.6 (2.3) | 0.8344 | 936/163 |
| PBA‐s (or UHDRS‐Ba) | ||||
| Depression | ||||
| Depressed mood (3120/290) | 1309 (43.5) | 214 (53.2) | <0.001 | 268/53 |
| Suicidal ideation (3120/290) | 190 (6.3) | 40 (10.0) | 0.0064 | 268/53 |
| Anxiety (3119/290) | 1557 (51.8) | 208 (51.7) | 0.9886 | 269/53 |
| Irritability/aggression | ||||
| Irritability (3118/290) | 1276 (42.4) | 194 (48.4) | 0.0240 | 269/54 |
| Angry/aggressive (3118/290) | 570 (19.0) | 89 (22.2) | 0.1229 | 269/54 |
| Psychosis | ||||
| Delusions (3118/290) | 70 (2.3) | 13 (3.2) | 0.2688 | 270/53 |
| Hallucinations (3118/290) | 20 (0.7) | 4 (1.0) | 0.5175 | 269/54 |
| Apathy | ||||
| Apathy (3116/290) | 697 (23.2) | 149 (37.2) | <0.001 | 271/54 |
| Executive function | ||||
| Perseverative thinking/OCB (3116/290) | 422 (14.0) | 76 (18.9) | <0.001 | 271/54 |
| Comorbid conditions, yes (%) | ||||
| Neurologic | 443 (13.5) | 81 (17.8) | 0.0138 | |
| Psychiatric | 917 (28.0) | 155 (34.1) | 0.0073 |
Data are mean (SD) or n (%). aPBA‐s data were complemented with UHDRS‐B in case PBA‐s data were not available. The numbers of PBA‐s or UHDRS‐B are given (PBA‐s + UHDRS‐B). SD, standard deviation; UHDRS‐B, Unified Huntington Disease Rating Scale Behavioral part; SDMT, Symbol Digit Modality Test; PBA‐s, Problem Behavior Assessment‐short form; OCBs, Obessive Compulsive Behaviors.
Longitudinal Analysis
The TMS gradually increased before disease onset, with a majority of the participants displaying a score of >5, 3 years before disease onset (Fig. 3A). After disease onset, the TMS gradually increased per visit. The TFC score respectively remained 13 or just below until the participants became manifest and then slowly decreased. The severity of behavioral symptoms gradually increased in time and was always present before disease onset. Especially apathy and perseverative/obsessive–compulsive behavior increased, respectively, 6 and 4 years before disease‐onset (Fig. 3B and C). The DCL score from 4 years previous to disease‐onset was above 1 (Fig. 3D). Figure 4A and B show slight decrease in the total correct answers on the Stroop interference test and Symbol Digit Modality Test over time, but no significant decrease before disease onset.
FIG. 3.
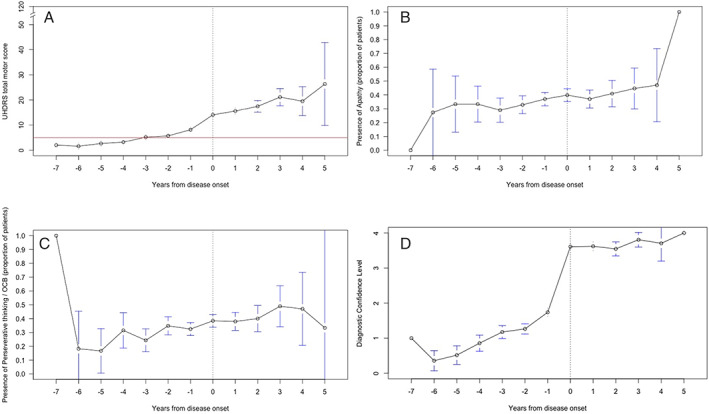
Unified Huntington Disease Rating Scale. Total motor score (A), apathy (B), perseverative/obsessive–compulsive behavior, (C) and diagnostic confidence level (D) in time in converted group. UHDRS, Unified Huntington's Disease Rating Scale; OCB, obsessive–compulsive behavior.
FIG. 4.
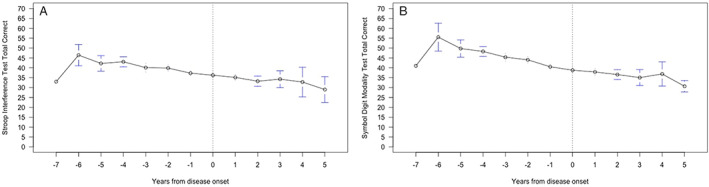
Stroop interference test total correct (A), Symbol Digit Modality Test total correct (B) in time in the converted group.
Longitudinally, converters show a higher TMS (>5), DCL (>1) (Fig. 5A and B) as well as more cognitive and behavioral problems (Data S1) compared to non‐converters from baseline to their last Enroll‐HD visit. The data shown are regardless onset of disease.
FIG. 5.
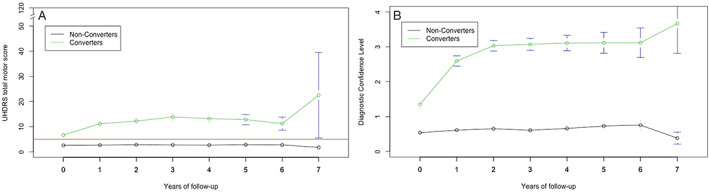
Total motor score (A) and diagnostic confidence level (B) in years of follow‐up in converted and non‐converted group.
Discussion
We are the first to analyze how consistent a TMS cut‐off score of 5 and the DCL of 4 relate to one another in estimating disease onset in Enroll‐HD participants. As expected, and in concordance with the results of the TRACK‐HD and PREDICT‐HD studies, 4 , 8 , 9 , 10 , 11 , 12 , 13 we found that HDGEC's closer to disease onset show more motor and psychiatric symptoms compared to those further away from onset. However, the data revealed that 232 (51%) converters had a mean TMS >5 at baseline. Although longitudinally the TMS fluctuated, it still remained >5 before disease onset. This means that, at that point, participants already had clear motor abnormalities. We found that 2 converters had a movement disorder, 1 had an essential tremor, and the other had a dystonia. This might partially explain the fact that the participants were not scored as manifest at that point in time despite a TMS >5. However, the TMS >5 of the other 230 participants can not otherwise be explained than motor abnormalities matching HD. Furthermore, our results show that the TMS already was >5 at least 3 years before disease onset.
In converters, the DCL in the 3 years previous to disease‐onset had a score of 1 or more and not the score 0. This means the raters had <50%–98% confidence the participant had signs and symptoms of HD at least 3 years before they rated the participant to be manifest.
Perhaps the raters struggle with the fact they observe motor abnormalities but do not want to distress the participants with their findings. From the clinician's point of view, this seems understandable, because these abnormalities do not interfere with daily life functioning, and there are currently no disease progression stabilizing treatments. However, from a scientific point of view, this is worrisome.
There are some limitations to this study. The Enroll‐HD and Registry data contain multiple instances of missing data, so we had to modify our statistical model accordingly. Ideally, all participants would have been tracked from the same moment in time from estimated disease onset, but this is not feasible. As a result, some participants were already closer to their disease onset than others when they were enrolled in this database. Furthermore, the data set contains fewer participants further from disease onset. Medication use might also induce or suppress motor abnormalities. Unfortunately, the Enroll‐HD data are registered in a way that makes it impossible to analyze the effect of medication use on motor scores.
Another limitation is that, in the Registry data, the behavior section of the UHDRS is used, whereas Enroll‐HD uses the PBAs. Although both assessments use the same ranking scale on both severity and frequency, they differ on 2 items. These 2 items “disoriented behavior” and “low self‐esteem/guilt” were omitted according to the advice of the Behavioral Working Group of the European Huntington's Disease Network (EHDN). Furthermore, subitems on perseverative and obsessive–compulsive behavior differ between the 2 questionnaires and were therefore combined to 1.
Unfortunately, it seems that even with premanifest follow‐up, it is impossible to estimate an accurate time of motor disease onset, let alone non‐motor onset. Despite cognitive and behavioral symptoms and signs frequently preceding motor symptoms, motor changes are still the landmark for clinical research on age of onset. 15 The Movement Disorder Society Task Force on Huntington's disease diagnostic categories suggests redefinition of the diagnostic criteria of genetically confirmed HD into pre‐symptomatic (no significant motor signs; DCL of 0 or 1 and no cognitive change), prodromal (DCL of 2 plus clear cognitive changes), and manifest HD (DCL of 3 plus minor neurocognitive disorder or DCL of 4). 22 , 23 This classification will make it possible to categorize HDGECs as prodromal. The prodromal stage is not used in Enroll‐HD. However, in our opinion, with these suggested diagnostic criteria, actual clinical disease onset remains unclear in many participants. What is meant with “no significant motor signs”? The new criteria still do not use cut‐off scores for motor symptoms (TMS).
In addition, biomarkers, such as brain MRI and spinal fluid examination, might be helpful to support early diagnosis. However, Van der Plas et al 24 recently showed that asymptomatic child and adolescent mutant HTT carriers already have abnormal brain development, possibly making this less useful.
We conclude that estimating disease onset in HD remains very difficult even in premanifest participants who are followed annually by trained and skilled HD physicians. The current diagnostic criteria give no guidance when an HDGEC has unequivocal signs of HD. The diagnosis is made by clinical experience and, currently there is no operational definition that guides the clinician. It also remains unclear when behavioral, cognitive, and functional changes may be attributed to manifest HD as there are no clear cutoff points of the used scales. We conclude that the DCL is not an accurate and reliable instrument to determine HD disease onset. With numerous trials evaluating disease modifying therapies, it is extremely important to develop more adequate diagnostic criteria, including non‐motor symptoms. With this in mind, it is also important to become upfront with our patients. If we want them to benefit from future therapies, we need to confront them regarding symptoms and signs. We believe it is time to define motor and non‐motor disease onset more clearly. First, we should use cut‐off scores for the TMS. Here, we used a cut‐off score of 5. Although this cut‐off score has been used in an important and leading HD study (TRACK‐HD), there is no scientific background why this value should be the cut‐off score. Therefore, further investigation on what cut‐off score is reliable and justifies motor disease onset is necessary. Perhaps the DCL could be categorized by using separate cut‐off points for the prodromal and manifest stages. Furthermore, we need cut‐off points in the PBA and cognitive tests used in Enroll‐HD to diagnose non‐motor disease onset. There is clear evidence that cognitive and behavioral changes may start several decades before motor‐onset and cannot be attributed to other causes.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
M.O.: 1A, 1B, 1C, 2C, 3A
B.T.A.dG.: 1A, 1B, 1C, 2A, 2B, 2C
E.K.B.: 1A, 3B
A.D.: 1A, 3B
S.J.T.: 1A, 3B
C.E‐F.: 1A, 3B
R.A.C.R.: 1A, 3B
C.E.M.dD‐S.: 1A, 3B
Disclosures
Ethical Compliance Statement
All Enroll‐HD sites are required to obtain and maintain local ethics committee approvals. Participants provided written informed consent for this observational study. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
No specific funding was received for this research. The authors declare that there is no conflict of interest.
Financial Disclosures for the Previous 12 Months
During the past year, S.J.T. has, through the offices of UCL Consultants Ltd, a wholly owned subsidiary of University College London, undertaken consultancy services for Alnylam Pharmaceuticals Inc., Annexon Inc., DDF Discovery Ltd, F. Hoffmann‐La Roche Ltd, Genentech, Novartis Pharma, Takeda Pharmaceuticals Ltd, Triplet Theraputics, and Vertex Pharmaceuticals Incorporated. A.D. serves, through the ICM contracts, as an advisor of Brainvectis, F. Hoffmann‐La Roche Ltd, and Triplet Therapeutics. R.A.C.R. is an adviser of UniQure at the time this study was conducted. The other authors declare they have no conflict of interest.
Supporting information
Data S1. Longitudinal cognitive and behavioral changes in converters and non‐converters.
Acknowledgments
Enroll‐HD is a longitudinal observational study for Huntington's disease families intended to accelerate progress toward therapeutics, it is sponsored by CHDI Foundation, a nonprofit biomedical research organization exclusively dedicated to developing therapeutics for HD. Enroll‐HD would not be possible without the vital contribution of the research participants and their families (https://www.enroll-hd.org/acknowledgments/).
References
- 1. The Huntington's Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72(6):971–983. [DOI] [PubMed] [Google Scholar]
- 2. Bruyn GW. Huntington's chorea. Historical, clinical and laboratory synopsis. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1968. [Google Scholar]
- 3. Huntington Study Group . Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord 1996;11(2):136–142. [DOI] [PubMed] [Google Scholar]
- 4. Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK‐HD study: cross‐sectional analysis of baseline data. Lancet Neurol 2009;8(9):791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Boo G, Tibben A, Hermans J, Maat A, Roos RA. Subtle involuntary movements are not reliable indicators of incipient Huntington's disease. Mov Disord 1998;13(1):96–99. [DOI] [PubMed] [Google Scholar]
- 6. Witjes‐Ane MN, Vegter‐van der Vlis M, van Vugt JP, Lanser JB, Hermans J, Zwinderman AH, et al. Cognitive and motor functioning in gene carriers for Huntington's disease: a baseline study. J Neuropsychiatry Clin Neurosci 2003;15(1):7–16. [DOI] [PubMed] [Google Scholar]
- 7. Witjes‐Ane MN, Zwinderman AH, Tibben A, van Ommen GJ, Roos RA. Behavioural complaints in participants who underwent predictive testing for Huntington's disease. J Med Genet 2002;39(11):857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the predict‐HD study. J Neurol Neurosurg Psychiatry 2008;79(8):874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biglan KM, Ross CA, Langbehn DR, et al. Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT‐HD study. Mov Disord 2009;24(12):1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paulsen JS, Long JD, Johnson HJ, Aylward EH, Ross CA, Williams JK, et al. Clinical and biomarker changes in premanifest Huntington disease show trial feasibility: a decade of the PREDICT‐HD study. Front Aging Neurosci 2014;6:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC, Predict HDIotHSG . Psychiatric symptoms in Huntington's disease before diagnosis: the predict‐HD study. Biol Psychiatry 2007;62(12):1341–1346. [DOI] [PubMed] [Google Scholar]
- 12. Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: the 12‐month longitudinal analysis. Lancet Neurol 2011;10(1):31–42. [DOI] [PubMed] [Google Scholar]
- 13. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early‐stage Huntington's disease in the TRACK‐HD study: analysis of 36‐month observational data. Lancet Neurol 2013;12(7):637–649. [DOI] [PubMed] [Google Scholar]
- 14. Hamilton JM, Salmon DP, Corey‐Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington's disease. J Neurol Neurosurg Psychiatry 2003;74(1):120–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Biglan KM, Zhang Y, Long JD, Geschwind M, Kang GA, Killoran A, et al. Refining the diagnosis of Huntington disease: the PREDICT‐HD study. Front Aging Neurosci 2013;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huntington Study Group . Unified Huntington's Disease Rating Scale(UHDRS). http://huntingtonstudygroup.org/tools-resources/uhdrs/.
- 17. Landwehrmeyer GB, Fitzer‐Attas CJ, Giuliano JD, Goncalves N, Anderson KE, Cardoso F, et al. Data analytics from enroll‐HD, a global clinical research platform for Huntington's Disease. Mov Disord Clin Pract 2017;4(2):212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Orth M, Handley OJ, Schwenke C, Dunnett SB, Craufurd D, Ho AK, et al. Observing Huntington's disease: the European Huntington's Disease Network's REGISTRY. PLoS Curr 2010;2:RRN1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Orth M, European Huntington's Disease N , Handley OJ, Schwenke C, Dunnett S, Wild EJ, et al. Observing Huntington's disease: the European Huntington's Disease Network's REGISTRY. J Neurol Neurosurg Psychiatry 2011;82(12):1409–1412. [DOI] [PubMed] [Google Scholar]
- 20. Penney JB Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington's disease. Ann Neurol 1997;41(5):689–692. [DOI] [PubMed] [Google Scholar]
- 21. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol 2001;14(4):219–226. [PubMed] [Google Scholar]
- 22. Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington's disease based on natural history. Mov Disord 2014;29(11):1335–1341. [DOI] [PubMed] [Google Scholar]
- 23. Ross CA, Reilmann R, Cardoso F, McCusker EA, Testa CM, Stout JC, et al. Movement Disorder Society task force viewpoint: Huntington's Disease diagnostic categories. Mov Disord Clin Pract 2019;6(7):541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van der Plas E, Langbehn DR, Conrad AL, Koscik TR, Tereshchenko A, Epping EA, et al. Abnormal brain development in child and adolescent carriers of mutant huntingtin. Neurology 2019;93:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Longitudinal cognitive and behavioral changes in converters and non‐converters.