

ABSTRACT
Background
Identifying sex‐related differences is critical for enhancing our understanding of factors that may impact prognosis and advance treatments in Huntington's disease (HD).
Objectives
To investigate if sex‐related differences exist in clinical HD.
Methods
Longitudinal study of the Enroll‐HD database. Manifest HD patients were included in the analysis (N = 8401). Linear mixed models were used to assess motor, behavioral, and cognitive functioning over a series of four annual visits, and compared male and female HD gene carriers.
Results
HD patients showed significant sex‐dependent differences in motor, cognitive, and behavioral symptoms. Both sexes had worsened motor symptoms over the course of four visits, but there was a significant disparity between sexes, with females consistently presenting with more symptoms than males. For behavioral symptoms, specifically depressive symptoms, females had significantly more depressive symptoms, although self‐reported symptoms in both sexes became less severe throughout time.
Conclusions
Our analyses suggest that women have worse symptoms than men during the course of HD.
Keywords: Huntington's disease, sex‐differences, enroll‐HD
Huntington's disease (HD) is a neurodegenerative disease caused by a CAG repeat expansion on the huntingtin gene (HTT). HD is characterized by a triad of symptoms that include motor, cognitive and behavioral changes, progressing to worsening function and eventual death. 1 The motor dysfunction associated with HD involves both an onset of involuntary movements, usually chorea, and the deterioration of normal movements. Cognitive and psychiatric symptoms often precede the motor symptoms. While variable, the psychiatric manifestations include major depressive disorder, anxiety, and irritability among others. 1 , 2
Neurodegenerative diseases are influenced by a multitude of variables. Sex, an unmodifiable risk factor, has been recognized as a potential contributing factor in the pathophysiology of neurodegenerative diseases that can influence both prognosis and treatment. 3 , 4 , 5 Previous investigations have identified a sex‐dependent bias in the risk, symptomatology, and progression of neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, multiple sclerosis and HD, possibly due to subtle sex‐related biological differences in brain structure and organization. 4 , 5 However, the mechanisms associated with sex‐dependent effects on the central nervous system are not fully understood, and elucidating their relationship in the development and progression of neurodegenerative diseases is of great value to clinicians and patients. For instance, sex‐differences in HD may affect disease prognosis and guide the management and treatment of symptoms.
The majority of evidence of sex‐related differences in HD comes from studies that examined instability in CAG repeat length in male intergenerational transmission. Large expansions occur almost exclusively through paternal transmission, while offspring of affected mothers are more likely to show no change or contractions in CAG length. 6 Unexpectedly, in maternal transmissions, repeat‐length changes were also dependent upon the sex of the offspring, with a tendency for expansion in male offspring and contraction in female offspring. 4 , 7 Despite the well‐known CAG repeat instability in male intergenerational transmission, little is known about sex‐related differences in the natural history of HD. There is clinical relevance to understanding sex differences, particularly in relation to disease outcomes. Such differences could play an important role in symptom manifestation, disease progression, and potentially, response to treatment. Therefore, the current study was designed to investigate if sex‐related differences exist in clinical HD. Taking advantage of the Enroll‐HD database, we specifically aimed to define whether: (1) there is a difference between male and female HD gene carriers at the age of clinical diagnosis; (2) sex is a significant predictor of disease severity and/or progression; (3) sex is associated with disease phenotype (ie, presence of specific motor, cognitive or behavioral symptoms); and (4) the CAG repeat length varied between males and females.
Methods
This study used information from the Enroll‐HD database, a longitudinal observational study on HD with study sites in North America, Latin America, Europe, Australia, and New Zealand. The Enroll‐HD study, in addition to other goals, aims to improve understanding of the dynamic phenotypic presentation of HD and disease mechanisms. 8
We used the periodic dataset containing Enroll‐HD participants which met the criteria for inclusion as of December 14, 2018 (PDS4, N = 15,301 participants). The Enroll‐HD assesses participants annually and manifest HD gene carriers were included in our analysis. Manifest patients were defined by the gene expansion for HD (larger CAG allele ≥36) and a clinical diagnosis of HD based on motor signs certainty, i.e., set to 4 at the Diagnostic Confidence Level (DCL) from the Unified Huntington's Disease Rating Scale (UHDRS).
Because biology and clinical phenomenology in HD might be influenced by the CAG length and age, we calculated the ‘CAG age product’ (CAP) score, which was used as a proxy for cumulative disease burden. The CAP has been defined as:
where AGE is the current age of the individual, CAG is the repeat length, and L and K are constants. L is an estimate of the lower limit of the CAG expansion at which phenotypic expression of the effects of mutant HTT could be observed, and K is a normalizing constant. We used the CAP proposed by Warner & Sampaio (2016), in which L = 30 and K = 6.27, and the CAP will be equal to 100 at the subject's expected age of motor symptoms onset. 9
First, we performed cross‐sectional analyses in order to investigate differences between male and female HD gene carriers at visit 4. Associations between dichotomous variables were assessed with the Pearson chi‐square test. All continuous variables were tested to assess whether they follow a Gaussian distribution using the Shapiro–Wilk normality test. Two groups were compared using the Mann–Whitney U test since data were determined to not follow a normal distribution. For these analyses, we used data from manifest HD gene carriers who completed the study visit number 4 (baseline plus three annual visits).
Then, longitudinal data on motor, behavioral, and cognitive functioning over a series of up to four visits (baseline plus three annual visits) were analyzed using a linear mixed model to study the sex‐differences (adjusted for the CAP score). The P values for post‐hoc comparisons at each visit time were adjusted for multiple testing. Motor symptoms were assessed by the UHDRS ‐ Total Motor Score (TMS). Cognitive performance was evaluated by the scores in the verbal fluency test (VFT), the Symbol Digit Modalities Test (SMDT), and the Stroop Interference Test (SIT). The Problem Behaviors Assessment—Short Version (PBA‐s) was used to evaluate behavioral symptoms.
Results
The participants' selection process is summarized in Fig. 1. A total of 2145 manifest HD gene carriers completed at least four study visits and were included in our cross‐sectional analyses (N = 1097 female and 1048 male subjects, Table 1). There were no differences between male and female individuals in CAG repeat length (Table 1). Although males were found to be older than females at the time of the study visit and at the clinical diagnosis of HD (Table 1), these differences were not significant after controlling for the CAP score (data not shown). History of alcohol abuse was more common in men while smoking was common among women (Table 1). Medical history of depression was also more common in women than men with HD (Table 1).
FIG. 1.
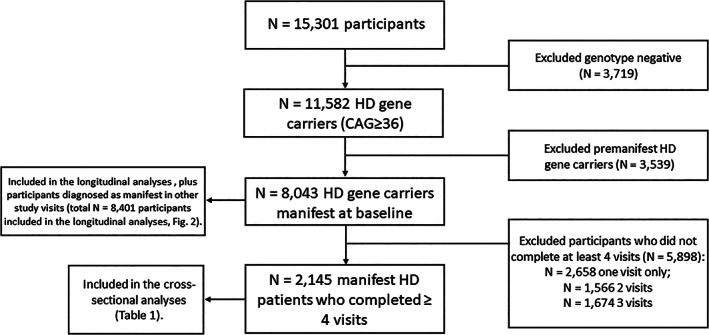
Flowchart showing participants' selection. This study evaluated enroll‐HD participants that meet the criteria for inclusion into the dataset as of December 14, 2018 (PDS4).
TABLE 1.
Cross‐sectional analyses comparing female and male patients with Huntington's disease (HD) at visit 4. Only CAG ≥36 and manifest HD subjects from enroll‐HD which meet the criteria for inclusion into the dataset as of December 14, 2018 (PDS4) and who completed at least four study visits were included in these analyses (N = 2145)
| Sex | |||
|---|---|---|---|
| Female (N = 1097) | Male (N = 1048) | P value | |
| Age in years at visit 4 [mean ± SD (median)] | 54.4 ± 12.1 (55) | 55.6 ± 12.2 (56) | 0.028 a b |
| Days relative to baseline date of Enroll‐HD at visit 4 [mean ± SD (median)] | 1098 ± 128 (1099) | 1098 ± 129 (1099) | 0.951 a |
| Age of clinical HD diagnosis [mean ± SD (median)] | 47.5 ± 11.8 (48) | 48.9 ± 12.0 (50) | 0.017 a b |
| CAG repeats [mean ± SD (median)] | 43.9 ± 3.6 (43) | 43.8 ± 3.8 (43) | 0.075 a |
| Medical history of: | |||
| Alcohol abuse (%) | 32.0 | 42.4 | <0.001 c |
| Smoking (%) | 23.4 | 18.3 | 0.013 c |
| Drugs abuse (%) | 2.2 | 3.2 | 0.320 c |
| Depression (%) | 80.9 | 71.4 | <0.001 c |
| Irritability (%) | 73.5 | 74.4 | 0.880 c |
| Violent/aggressive behavior (%) | 41.9 | 45.6 | 0.228 c |
| Perseverative/obsessive behavior (%) | 59.6 | 61.4 | 0.711 c |
| Apathy (%) | 71.5 | 68.0 | 0.131 c |
| Psychosis (%) | 14.0 | 12.2 | 0.281 c |
| Significant cognitive impairment (%) | 64.9 | 61.5 | 0.225 c |
| Previous suicidal ideation (%) | 34.4 | 31.5 | 0.171 c |
Medical history data were collected at the Enroll‐HD baseline visit and verified again at every annual visit.
Mann‐Whitney test.
The differences are not significant when controlling for the CAP score.
Pearson chi‐square test. Significant values are highlighted in bold.
SD, standard deviation.
For the longitudinal analyses, we included all subjects with a diagnosis of manifest HD at any of the four analyzed study visits (baseline plus three annual visits, N = 8401 participants). Figure 2 shows the scores obtained in the motor, cognitive, and behavioral scales over the four visits. Manifest HD patients showed significant sex‐dependent differences in motor symptoms (P < 0.0001 for all visits). While both sexes showed gradual motor worsening over the course of four annual visits, female patients with HD consistently presented with more severe symptoms than males (Fig. 2A).
FIG. 2.
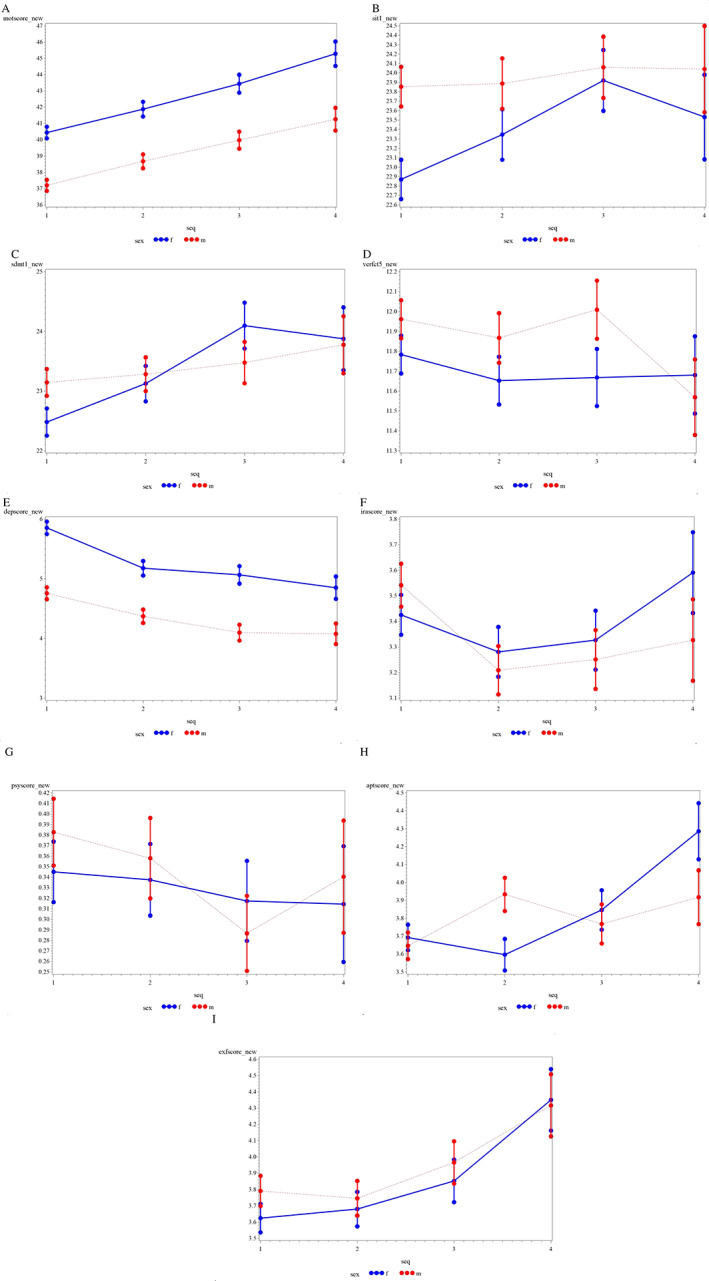
Differences between male (in red) and female (in blue) patients with HD. The graphs show motor, cognitive, and behavioral symptoms progression over the period of four annual visits. (A) the unified Huntington's disease rating scale‐Total motor score (motscore_new). (B) Stroop interference test (sit1_new). (C) Symbol digit modalities test (sdmt1_new). (D) Verbal fluency test (verfct5_new). (E) Problem behaviors assessment ‐ short version (PBA‐s), Depression score (depscore_new). (F) PBA‐s, irritability/aggression score (irascore_new) (G) PBA‐s, psychosis score (psyscore_new). (H) PBA‐s, apathy score (aptscore_new). (I) PBA‐s, executive function score (exfscore_new).
Regarding cognitive assessment, men tended to perform better than women [VFT, visits 1–3; SMDT, visits 1–2; and SIT, visits 1–4], and the differences were statistically significant for the SIT visit 1 (P = 0.021) (Fig. 2B,D). Interestingly, female patients performed better over time on the SIT and SDMT (Fig. 2B,C). This may be due to repeated testing effects, as patients are repetitively exposed to the test and they may learn and perform better on each visit. However, if that is the case, an explanation is needed to determine why male patients were not able to improve their scores significantly over time. On the contrary, a worsening of the scores for the VFT was seen in males (Fig. 2D).
When comparing the subscales of the PBA‐s, a significant difference between sexes was found for the depression sub‐score (P < 0.0001 for visit 1 to 3, and P = 0.007 for visit 4). Females presented with worse symptoms than males, although both groups progressed similarly through follow‐ups. Interestingly, the depression sub‐scores decreased over time, which could be explained by less insight into the disease or reduced capacity to comment about the symptoms in patients with advancing HD (Fig. 2E). No differences between sexes were found for irritability/aggression and psychosis sub‐scores of the PBA‐s (Fig. 2F,G). Regarding the apathy sub‐score, although men scored significantly worse than women at visit 2 (P = 0.003), female HD gene carriers progressed worse over time (Fig. 2H). Finally, male and female HD gene carriers presented similar scores in the executive function component of the PBA‐s, with both sexes worsening over the course of the four visits (Fig. 2I).
Discussion
Studying sex differences in HD adds valuable information on the natural history of the disease. Herein, we assessed clinical outcomes linked to the natural history of HD over a period of 4 years. We found that female HD gene carriers present with worse motor, cognitive, and depressive symptoms than males, although symptom progression over the years was similar between the two sexes.
Sex‐differences are often not highlighted in the literature in HD, possibly because of the HD autosomal inheritance pattern, which is considered sex‐independent. 10 However, sex may play an important role in disease progression and clinical phenotype. Our results corroborate previous analysis of sex‐differences in HD, which showed that the presence of depression and history of depression is more prevalent in women than in men, while the history of alcohol abuse is more prevalent in men than in women. 10 , 11 Our findings also confirm data from the PREDICT‐HD study, which reported the female sex as a significant predictor of increased depressive symptoms severity, 12 and from the REGISTRY study, which reported that the rate of HD progression is faster in women than in men. 11 Not only the motor dysfunction was worse in women than in men, but also the motor domain affected function in women more than in men, thus suggesting that the motor symptoms of HD have a significantly stronger impact on functional abilities in women. 13
Other studies, however, have failed to show any sex‐related effect on HD progression or clinical phenotype. An elegant study conducted by the Huntington Study Group revealed that sex did not affect overall function, motor, cognitive, and behavior in a cohort of 960 patients with HD followed prospectively for a mean of 18.3 months. 14 In addition, contrary to our data, sex was not described as a significant predictor of depressive symptoms, anxiety, or general distress in HD. 15
We observed that depressive symptoms became less severe over time in both male and female HD patients. Our results corroborate previous reports in which the proportion of HD patients endorsing significant depression diminished with disease progression. 16 , 17 It is not clear, however, if patients indeed become less depressive over the course of HD or if they fail to recognize and/or report such symptoms. It has been well documented that anosognosia can be a major component of HD and has implications on management and treatment, particularly of symptoms such as depression. 18 Another explanation could be due to an adaptation to the disease process and its outcomes. 16 , 17
We found no differences between male and female HD gene carriers regarding the time of clinical diagnosis of HD when controlling for the CAP score. Our results corroborate a previous report of no sex‐differences in the age of HD diagnosis. 19 However, these results are controversial, since another study has reported that females have an earlier age of onset than males. 20 Noteworthy, that the age of HD clinical onset is highly influenced by the CAG repeat length, and in the latter study, the comparisons were not controlled for the patients' CAG length or CAP score. 20 In addition to the CAG repeat length, other genetic factors might play a significant role in determining age at onset in HD. Sex‐specific differences in genotypes and allele frequencies may influence age at onset differently in male and female patients. For example, a study reported that a coding variant in PPARGC1A was associated with an earlier motor onset in male but not female HD gene carriers. 21 In addition, males with the apolipoprotein E genotype ε2ε3 have significantly earlier onset than females of the same genotype. 4 These findings suggest that subtle differences in the course of the neurodegeneration in HD may allow interacting genes to exert sex‐specific effects upon the age at disease onset. It is worth mentioning that there were no significant sex differences for age at death in individuals with HD. 22
We are aware of the limitations of our study. Additional studies including variables such as brain structure, genetic analysis, and biomarkers, among others, with a longer follow‐up, would be of great value to increase our understanding of the pathophysiology of HD and evaluate potential sex differences. In addition, our analysis did not control for potential confounders, such as age at disease onset and affected parent. Future studies including different populations and controlling for potential confounders are needed to confirm our findings. The strengths of our study include the longitudinal assessment of different clinical outcomes linked to the natural history of HD, and the high number of participants, as they belong to a large‐scale multicenter study.
In conclusion, this study increases awareness of different clinical outcomes for female and male patients with HD. Future research in humans is needed to characterize hormonal factors and other variables pertaining to disease onset as the manifest of clinically apparent symptoms appear to be the result of a multifactorial pathogenesis.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the first draft, B. Review and Critique.
S.H.: 1C, 3A.
L.Z.: 2A, 2B.
J.P.: 3A.
J.W.F.: 1A, 1B.
N.P.R.: 1A, B, 1C, 2A, 2B, 3B.
E.F.S.: 1A, 1B, 2C, 3B.
Disclosures
Ethical Compliance Statement
This study has been approved by the UTHealth Committee for the Protection of Human Subjects (IRB # HSC‐MS‐18‐0903). The authors confirm that patient consent was not required for this work, as we performed retrospective research from the Enroll‐HD database, whose participants signed a consent form agreeing that their data would be used for studies such as the current one. The Enroll‐HD is an observational study/registry that provides de‐identified data about patients with HD. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest
This study did not receive specific funding. The Enroll‐HD is a CHDI Foundation Project. There are no conflicts of interest to report.
Financial Disclosures for the Previous 12 Months
EFS receives research funding from Roche/Genetech, Vaccinex, Cures Within Reach, HDSA, Uniqure and CHDI Foundation.
Acknowledgments
We would like to thank the CHDI Foundation for granting us access to the Enroll‐HD periodic datasets.
References
- 1. Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10(4):204–216. [DOI] [PubMed] [Google Scholar]
- 2. Paoli RA, Botturi A, Ciammola A, et al. Neuropsychiatric burden in Huntington's disease. Brain Sci 2017;7(6):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bruzelius E, Scarpa J, Zhao Y, Basu S, Faghmous JH, Baum A. Huntington's disease in the United States: variation by demographic and socioeconomic factors. Mov Disord 2019;34(6):858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meoni S, Macerollo A, Moro E. Sex differences in movement disorders. Nat Rev Neurol 2020;16(2):84–96. [DOI] [PubMed] [Google Scholar]
- 5. Ullah MF, Ahmad A, Bhat SH, Abu‐Duhier FM, Barreto GE, Ashraf GM. Impact of sex differences and gender specificity on behavioral characteristics and pathophysiology of neurodegenerative disorders. Neurosci Biobehav Rev 2019;102:95–105. [DOI] [PubMed] [Google Scholar]
- 6. Aziz NA, van Belzen MJ, Coops ID, Belfroid RD, Roos RA. Parent‐of‐origin differences of mutant HTT CAG repeat instability in Huntington's disease. Eur J Med Genet 2011;54(4):e413–e418. [DOI] [PubMed] [Google Scholar]
- 7. Wheeler VC, Persichetti F, McNeil SM, et al. Factors associated with HD CAG repeat instability in Huntington disease. J Med Genet 2007;44(11):695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. CHDI Foundation . A user guide to the clinical datasets and biosamples available from Enroll‐HD. 2018. p. 1‐8.
- 9. Warner JH, Sampaio C. Modeling variability in the progression of Huntington's disease a novel modeling approach applied to structural imaging markers from TRACK‐HD. CPT Pharmacometrics Syst Pharmacol 2016;5(8):437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zielonka D. Factors contributing to clinical picture and progression of Huntington's disease. Neural Regen Res 2018;13(8):1364–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zielonka D, Marinus J, Roos RA, et al. The influence of gender on phenotype and disease progression in patients with Huntington's disease. Parkinsonism Relat Disord 2013;19(2):192–197. [DOI] [PubMed] [Google Scholar]
- 12. Epping EA, Mills JA, Beglinger LJ, et al. Characterization of depression in prodromal Huntington disease in the neurobiological predictors of HD (PREDICT‐HD) study. J Psychiatr Res 2013;47(10):1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zielonka D, Ren M, De Michele G, et al. The contribution of gender differences in motor, behavioral and cognitive features to functional capacity, independence and quality of life in patients with Huntington's disease. Parkinsonism Relat Disord 2018;49:42–47. [DOI] [PubMed] [Google Scholar]
- 14. Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington's disease. Huntington Study Group. Neurology 2000;54(2):452–458. [DOI] [PubMed] [Google Scholar]
- 15. Dale M, Maltby J, Shimozaki S, Cramp R, Rickards H. Disease stage, but not sex, predicts depression and psychological distress in Huntington's disease: a European population study. J Psychosom Res 2016;80:17–22. [DOI] [PubMed] [Google Scholar]
- 16. Epping EA, Paulsen JS. Depression in the early stages of Huntington disease. Neurodegener Dis Manag 2011;1(5):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paulsen JS. Depression and stages of Huntington's disease. J Neuropsychiatr 2005;17(4):496–502. [DOI] [PubMed] [Google Scholar]
- 18. Tranel D, Paulsen J, Hoth K. Anosognosia in Huntington's Disease. New York: Oxford University Press; 2010:147–158. [Google Scholar]
- 19. Glidden AM, Luebbe EA, Elson MJ, et al. Patient‐reported impact of symptoms in Huntington disease: PRISM‐HD. Neurology 2020;94(19):e2045–e2053. [DOI] [PubMed] [Google Scholar]
- 20. Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington's disease based on age at onset. J Neurol Neurosurg Psychiatry 1999;66(1):52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weydt P, Soyal SM, Landwehrmeyer GB, Patsch W. A single nucleotide polymorphism in the coding region of PGC‐1α is a male‐specific modifier of Huntington disease age‐at‐onset in a large european cohort. BMC Neurol 2014;14(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Solberg OK, Filkukova P, Frich JC, Feragen KJB. Age at death and causes of death in patients with Huntington disease in Norway in 1986‐2015. J Huntingtons Dis 2018;7(1):77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]