

Abstract
Operando X-ray absorption spectroscopy (XAS) and X-ray diffraction (XRD) were performed on a Co/TiO2 Fischer–Tropsch synthesis (FTS) catalyst at 16 bar for (at least) 48 h time-on-stream in both a synchrotron facility and a laboratory-based X-ray diffractometer. Cobalt carbide formation was observed earlier during FTS with operando XAS than with XRD. This apparent discrepancy is due to the higher sensitivity of XAS to a short-range order. Interestingly, in both cases, the product formation does not noticeably change when cobalt carbide formation is detected. This suggests that cobalt carbide formation is not a major deactivation mechanism, as is often suggested for FTS. Moreover, no cobalt oxide formation was detected by XAS or XRD. In other words, one of the classical proposals invoked to explain Co/TiO2 catalyst deactivation could not be supported by our operando X-ray characterization data obtained at close to industrially relevant reaction conditions. Furthermore, a bimodal cobalt particle distribution was observed by high-angle annular dark-field scanning transmission electron microscopy and energy-dispersive X-ray analysis, while product formation remained relatively stable. The bimodal distribution is most probably due to the mobility and migration of the cobalt nanoparticles during FTS conditions.
Keywords: catalyst stability, catalyst deactivation, X-ray absorption spectroscopy, operando spectroscopy, Fischer−Tropsch synthesis, Co/TiO2
1. Introduction
Cobalt nanoparticles (NPs) supported on TiO2 comprise one of the most industrially applied Fischer–Tropsch synthesis (FTS) catalysts. This is a process used to synthesize, for example, ultra-clean diesel from crude oil alternative feedstocks, such as biomass, municipal waste, and natural gas.1 The application of FTS in the chemical industry is experiencing a clear revival due to increased environmental awareness, an ever-growing demand for (increasingly cleaner) transportation fuels, and the environmental impact from burning fossil carbon.2−7 FTS is a versatile production route for the conversion of synthesis gas, a mixture of CO and H2, in different ratios and with different catalyst materials depending on the desired product.5,8−10 A catalyst in the form of cobalt NPs supported on TiO2 (Co/TiO2) exhibits the desirable capability to form long-chain hydrocarbons, or synthetic diesel, with the added benefit of zero sulfur content.11 As is inevitable for many industrial processes, catalyst materials deactivate over time. This also holds for FTS.4,12−17 Catalyst deactivation is a parameter that greatly influences a process’s economic viability, yet it is often not well-understood and, most importantly, is less studied under relevant conditions by the academic community.18−20 One of the main reasons for this is the challenge to downscale industrial processes and related reaction conditions to a level that allows detailed laboratory-based spectroscopic investigations of mechanisms that only take place at those conditions (i.e., mainly high pressure). The proposed deactivation mechanisms for Co-based FTS catalysts are related to the conversion of the active metallic phase into an inert phase via, for example, Co re-oxidation or carburization,1,14,21−23 the formation of support-Co phases that form through strong metal–support interactions,18,24−26 the loss of active surface area due to crystalline growth,1,12,13,27 and finally by fouling via, for example, hydrocarbon deposition in the form of coke species.17,28 In particular, the effect of cobalt carbide (Co2C) phase on the FTS reaction has long been debated. It was long thought to be an inactive phase, but more recent studies have demonstrated that it can indeed be active for lower hydrocarbon products, and the interface between Co2C and Co may actually be beneficial.29−36
X-ray techniques provide a wide range of characterization possibilities, and synchrotron radiation-based deactivation studies have been performed on Co/TiO2 FTS catalysts,20,37,38 with many of the studies being performed under ex situ conditions.15 A common synchrotron-based technique to study these systems is X-ray absorption spectroscopy (XAS), as it is a powerful tool to gain fundamental insights into the chemical and structural changes taking place in FTS catalysts during catalytic activity and deactivation.39 In previous work, we have studied the FTS reaction by use of operando scanning X-ray transmission microscopy.38 Here, we showed that laboratory-based deactivation studies that comprise a few days of FTS actually capture a genesis period of the catalyst before it reaches its actual steady state.10,38,40 By the use of single-particle soft X-ray microscopy, we showed that there is the formation of a hydrocarbon layer in the pores of micrometer-sized Co/TiO2 catalyst particles through which the reactants subsequently diffuse in steady-state operation. Another recent study has shown the applicability of some of the first laboratory-based in situ X-ray absorption near-edge structure (XANES) studies on FTS catalysis under moderate pressure (5 bar).35 Yet for the vast majority of studies, hard X-ray synchrotron radiation is still desired, as the substantially higher photon flux allows for the detection of more subtle changes in the extended X-ray absorption fine structure (EXAFS) such as those expected in catalyst activation and deactivation. A more recent operando synchrotron-based study (primarily using XAS) presented data at 30 bar for over 300 h during FTS for a Co–Ni–Re/γ-Al2O3 catalyst.20 In this study, the initial 60 h was followed in real time, with a subsequent 250 h gap where no synchrotron data were recorded, likely due to the scarcity in measurement time. However, their study elegantly pointed toward a pathway for conducting long-term experiments at a synchrotron radiation source under meaningful reaction conditions.
To the best of our knowledge, no study has investigated the influence of process conditions approaching those of true industrial ones, that is, the combination of measuring operando spectroscopy, at relevant pressures (i.e., 1–20 bar), and with realistic (i.e., industrial) reaction times of multiple days of time-on-stream (TOS) for the Co/TiO2 catalyst.38,41,42 A program under development at the Stanford Synchrotron Radiation Lightsource (SSRL) at the SLAC National Accelerator Laboratory is exploring the potential of enabling long-term studies of, for example, catalyst deactivation, and the results presented here are the first results from this program. As such, in this work, we report the results of a pilot study for the use of a user-friendly long-term synchrotron operando XAS experimental arrangement. In combination with operando X-ray diffraction (XRD), as recently developed in the Utrecht University laboratories, we are using the Co/TiO2 showcase to increase our knowledge of the effect of Co2C formation during FTS under relevant conditions of temperature and pressure. This work lays the foundation for future studies utilizing both synchrotron- and lab-based X-ray radiation to study long-term catalyst deactivation processes under conditions close to those employed in chemical industries.
2. Materials and Methods
2.1. Catalyst Preparation
An industrially relevant 10 wt % Co/TiO2 catalyst was prepared by incipient wetness impregnation of an aqueous solution of Co(NO3)2·6H2O (99.99% Sigma-Aldrich) on a P25 TiO2 support material (Evonic, SABET: 9 m2·g–1, PV: 0.06 cm3·g–1). More specifically, 2.6 g of TiO2 support was dried under vacuum for 5 h at room temperature. Subsequently, this support was impregnated with a saturated Co precursor solution (pH = 4) until a cobalt metal loading of ∼10 wt % Co was reached. The impregnated material was dried overnight at 60 °C in a static air atmosphere and subsequently calcined at 450 °C in a N2 atmosphere (100 mL·min–1, Linde, ≥99.999%) for 4 h. The Co loading was determined to be 10.2 wt % by use of inductively coupled plasma–optical emission spectroscopy using a Spectro Arcos instrument.
2.2. Catalyst Characterization
The fresh 10.2 wt % Co/TiO2 catalyst was characterized by use of N2 physisorption, XRD, XAS, and scanning transmission electron microscopy–energy dispersion X-ray (STEM–EDX) analysis. The details of this fresh catalyst characterization can be found in the Supporting Information (Section S1: Catalyst Characterization, Figures S1–S6, and Catalytic Testing, Figure S7).
2.3. Operando XAS
Operando XAS measurements were performed using a home-built cell16,43,44 and an experimental setup at beamline 2-2 at SSRL at the SLAC National Accelerator Laboratory in Menlo Park, California, USA, which is optimized for the safe operations of high-pressure (up to 80 bar) He, H2, and CO. Beamline 2-2 incorporates a water-cooled double-bounce Si(220) monochromator. A 0.5 mm vertical beam size was used, optimized for energy resolution, and the horizontal size adjusted to match the catalyst bed size. The XAS data were collected in transmission mode using nitrogen-filled ionization chambers, calibrated at 7709.0 eV for the Co K-edge, and detuned 40% for harmonic rejection. A cobalt foil was placed between the It and Iref ion chambers for energy alignment.
Two operando XAS experiments were performed, the first lasted for 160 h and the second for 47 h, and different methodologies were used for each (more information can be found in the Supporting Information Section S3). In both experiments, the XAS measurements were performed in transmission and therefore required dilution of the catalyst bed with an inert, low atomic number diluent for optimum transmission. In the first experiment, the powdered catalyst was diluted with an inert MCM-41 material (Sigma-Aldrich) in a ratio of 1:2 by mass, pressed and sieved to a mesh fraction of 120–180 μm. The inert/catalyst blend was loaded into a quartz capillary (1.0 mm OD × 0.98 mm ID, Hilgenberg GmbH, 1471501) and held in place with quartz wool. The effluent gas was characterized by a mass spectrometer (Hiden Analytical QGA). While the overall experiment ran successfully for 6 days, unfortunately the process was not stable as some pressure variations were observed, suggesting that some small leaks had developed (Figure S9). Stable operations were obtained for the first 50 h, and then there were several pressure variations over the subsequent time. Thus, although we were successful in conducting a high-pressure FTS long-term deactivation experiment at the beamline, given that there were some technical difficulties encountered, this experiment was treated as preliminary and will not be further discussed in detail. However, there was one major observation from this experiment that is discussed later on and in Section S6.
In the second experiment, conducted for 47 h, the powdered catalyst was diluted with inert diamond powder (microcrystalline powder, ∼1 μm, Sigma-Aldrich) in a ratio of 1:3 by mass and then fed into a borosilicate capillary (1.0 mm OD × 0.96 mm ID, Hilgenberg GmbH, 1471501) and held in place with quartz wool. The length of the catalyst bed was 5 mm (sieve fraction of 120–180 μm). The capillary was glued into an aluminum frame with two component epoxy (Devcon, 14310), creating the seal between the capillary and the gas flow path. The aluminum frame was mounted in the in situ cell.44 A graphite ferrule was used to hold a thermocouple in place in the gas stream, fed into the quartz capillary, pressed against the quartz wool ensuring correct temperature measurement, and catalyst bed stability. The flow system was leak-checked with He at 20 bar, giving sufficient safety room for the operando XAS experiment operated at 16 bar. The catalyst was reduced at ambient pressure at 400 °C under a flow of 100% H2 at a flow rate of 5 mL·min–1 with a ramp rate of 10 °C·min–1. The cell was then cooled to 220 °C and pressurized to 16 bar at a ratio of 1:1 H2:CO at a total flow rate of 5 mL·min–1. The effluent gas was characterized by a mass spectrometer (Hiden Analytical QGA). The extent (i.e., 47 h) and conditions of this second experiment matched those of the separately conducted operando XRD experiment, allowing for a direct comparison of the results.
The XAS spectra of reference samples [Co3O4, CoO, CoTiO3, and Co2C diluted with cellulose and pressed into pellets, face-centered cubic (fcc)-Co, and hexagonal closest packed (hcp)-Co foils] were measured ex situ at room temperature. At all times, a Co foil was placed between the second and third ion chambers for energy calibration.
The raw EXAFS data were energy-calibrated, merged, and normalized using the Athena interface of the Demeter software package.45 The EXAFS data were extracted in k-space and Fourier-transformed on the k-range of 3.0–12.0 Å–1 and analyzed on a R-range between 1.0 and 2.7 Å for metallic Co and k-range of 3.0–11.7 Å–1 and R-range of 1.0–3.0 Å for carburized Co, respectively. Details of the extensive data analysis are given in the Supporting Information Section S3.3.
2.4. Operando XRD
The operando XRD, conducted for 48 h, measurements were carried out in a similar cell design46 to the XAS measurements using the same catalyst and dilution by diamond powder, as described above for the second XAS measurements. The length of the catalyst bed was 20 mm (sieve fraction of 75–125 μm), which was loaded into the borosilicate capillary. The cell design was adapted to fit a Bruker D8 Discovery XRD with a Mo Kα (0.7107 Å) source. The X-rays were focused on the capillary (OD 1 mm, wall thickness 0.01 mm) using a Göbel mirror. Photons were detected using an energy-dispersive Lynxeye XE-T detector, making it possible to filter the Kβ radiation from the signal. The setup, previously developed at Utrecht University, is comprehensively described elsewhere in the open literature.46 The XRD diffraction patterns were recorded from 8 to 39° 2Θ, with a 0.03° increment and a 6.26 s dwell time per data point.
The products were analyzed on-line by a Thermo Scientific TRACE 1300 gas chromatograph (GC, by Interscience) equipped with two FID and two TCD detectors. FID-1 detects alkanes and alkenes from C1–5 (Al2O3/Na2SO4 column). These are assigned to the specific product with a calibration gas. FID-2 detects C5+, oxygenates, and aromatics, and these are not calibrated but are used to estimate the relative amount of the C5+ products which are not waxes (these accumulate slowly in the transfer lines) (Lowox column). TCD-1 detects the permanent gases (Molsieve 5A column), and TCD-2 is used to separate He and H2 (ShinCarbon column).
The obtained XRD patterns were binned per three scans to increase the signal-to-noise (S/N) ratio and exported by Brucker EVA software.47 The exported files were further analyzed in Origin v9.1. (normalization and base line subtraction in the case of the diamond diluted catalyst) and by Rietveld Quantitative Phase Analysis (Rietveld QPA) in TOPAS v5.0.
2.5. Scanning Transmission Electron Microscopy
The catalyst powder samples (both fresh and spent) were ground using a mortar and pestle. The resulting fine powder was placed on top of a 3 mm holey carbon-coated Cu mesh TEM grid. The sample was analyzed by an aberration-corrected FEI Titan 80–300 kV operated at 200 kV.
3. Results and Discussion
Operando XAS and XRD measurements (and other supporting characterization techniques, where relevant) have been used to study the main cobalt-mediated activation and the potential deactivation mechanisms postulated for FTS in the literature. That is, Co carbide formation, Co particle growth, and cobalt oxide formation1,12,23 were studied, and these structural changes were correlated to the changes in catalyst performance.
3.1. Activation and Catalytic Activity of the Co/TiO2 FTS Catalyst
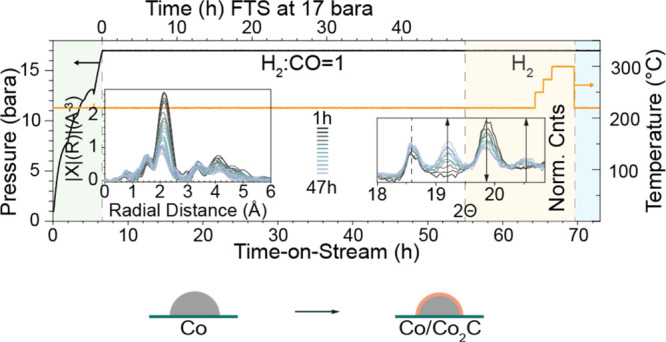
Before each experiment, the as-synthesized Co/TiO2 catalyst is activated for FTS in situ by reduction in pure H2 (5 mL·min–1) at 400 °C at 1 bar. The cobalt in the fresh calcined catalyst is present as well-dispersed approximately 20 nm diameter particles of the Co3O4 phase on the titania support as shown by high-angle annular dark-field (HAADF) images in STEM, accompanied by EDX mapping, Figure 1 and Table S1 for quantitative analysis of the Co/Ti ratio, XRD (Figures S2 and S3), and Co K-edge XANES (Figure S5b). During activation (reduction), Co3O4 goes through a CoO phase prior to the formation of metallic cobalt (Figures S4–S6).41,46,48 At the end of the reduction procedure, the cobalt is present as metallic Co NPs of approximately 15–20 nm diameter, as determined by XRD using Rietveld refinement, and these NPs comprised a mixture of both hcp and fcc phases (Figure S4). The catalytic activity of the 10 wt % Co/TiO2 FTS catalyst was measured by on-line GC, while measuring XRD (Figures 2 and S8), and with mass spectrometry (MS) during XAS (Figures S9 and S10) under operando conditions at 15 and 16 bar (XRD and XAS, respectively) of pressure and at a ratio of 1:1 H2/CO.
Figure 1.
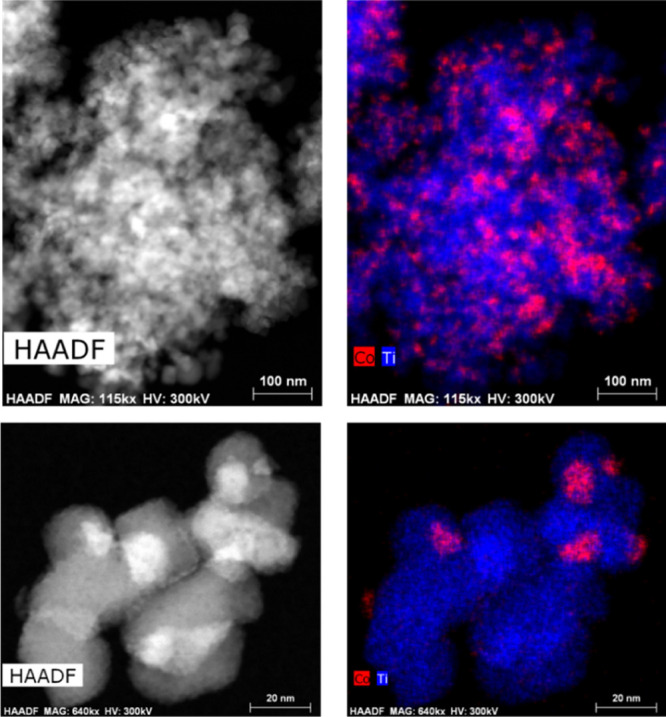
Fresh 10 wt % Co/TiO2 HAADF-STEM images (left) and EDX chemical mapping. Cobalt is shown in red and titanium in blue (right).
Figure 2.

Operando activity of the 10 wt % Co/TiO2 catalyst diluted with diamond powder, as measured by GC during 48 h while simultaneously measuring XRD. The gray dotted line in each panel indicates the start of carbide formation as determined by XRD. (a) The sum of the different products formed from C1 to C5. (b) The selectivity toward those different products showing a preference toward methane. (c) Comparison of the production of olefins and paraffins. Note that C5+ products were not quantified in this experiment. For additional catalytic testing with full product analysis and alpha numbers, see Figure S7.
The activity measurements of the 48 h FTS experiment, in which XRD data were simultaneously measured, are shown in Figure 2. Figure 2a shows the summed amounts of the C1–C5 FTS products, and Figure 2b shows the selectivity toward these C1–C5 FTS products. Figures S9 and S10 of the Supporting Information show the MS profiles of the reaction products measured during the operando XAS measurements (both 47 h and 6 day). These data show that in both operando XAS experiments, the catalyst is active and produces higher molecular weight hydrocarbons. During the initial 10–15 h (Figure 2a–c of the operando XRD experiment) of FTS TOS, there is a pronounced induction period during which the production of all products decreases with TOS. Figure 2a shows that the production of some products decreases more severely in this period than others. As such, the selectivity of the Co/TiO2 FTS catalyst changes during this induction period from longer chain hydrocarbons to shorter chain hydrocarbons as can be seen in Figure 2b. More specifically, the selectivity of C1 (i.e., methane) increases, the selectivity of C2 (ethane or ethene) remains relatively stable, and the selectivity of C3+ products decreases, with C3 products decreasing more strongly than C4 and C5. Subsequent to this induction period in which there are large changes in the activity, there is a period in which changes in activity become less pronounced. The absolute activity of C1 products is stable, as can be seen in Figure 2a, while the selectivity toward C1 products gently increases due to the loss in activity toward higher hydrocarbons (C3<). Figure 1c plots the reactivity data in terms of paraffins and olefins. Generally, more olefins are formed than paraffins, and the decrease in olefin production yields to a stable production or increase in the production of paraffins (ethane) with time. The dashed line within each panel in Figure 2 indicates the start of the carbide formation, determined by XRD (see the following section). There is no obvious deactivation after the start of this formation, at least for the lower molecular weight hydrocarbons.36
3.2. Cobalt Carbide Formation as Probed with Operando XRD
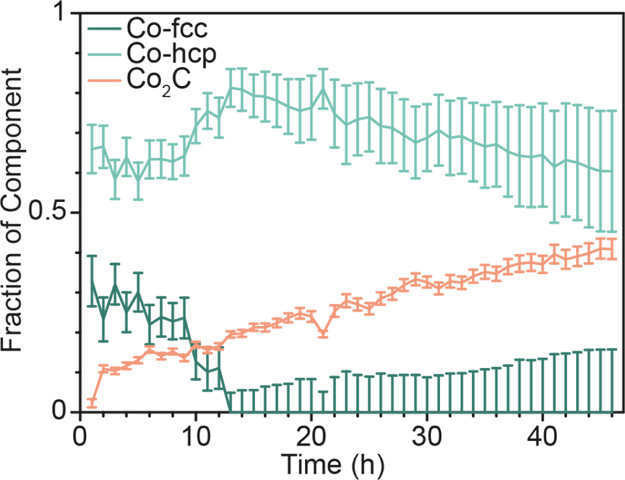
Figure 3 shows the diffractograms collected during lab-based operando XRD measurements of the 10 wt % Co/TiO2 catalyst. The XRD pattern was recorded approximately every 2 h, and there are clear changes noted with increasing TOS. Figure 3a shows the diffractograms over the whole measured region (8–39° 2Θ) and the inset shows the region with the most significant change where a new peak at 19.25° 2Θ emerges. This new peak is assigned to the emergence of a cobalt carbide phase, Co2C, and emerges along with one at 20.56° 2Θ. Unfortunately, the dilution of the XRD sample with diamond dust, which was done to duplicate the conditions of the XAS measurements, results in an intense diffraction peak from the diamond at 19.80° 2Θ, which interferes with the detection of several of the relevant peaks in XRD. This created difficulties with the quantification of the emergence of the Co2C phase (Figure 3b). Thus, the operando XRD experiment was repeated with an undiluted catalyst bed. These results are shown in Figure 4 (the GC activity measurements can be found in the Supporting Information, Figure S8). By performing this experiment without the diluent, it is both possible to distinguish the Co-fcc crystalline phase and hence to quantify the formation of the Co2C phase (Figure 4b). In addition to the increase of the Co2C phase, there is a corresponding decrease in the Co-fcc phase (Figure 4a (insets) and b). It has previously been noted that carburization can form at the expense of the fcc phase of cobalt.49−55
Figure 3.

(a) Operando XRD data of the 10 wt % Co/TiO2 diluted with diamond powder with TOS during 2 days of FTS at 15 bar. (b) Evolution of key phases as a function of TOS. The diffractograms are normalized to the first TiO2 peak at 11.55° 2Θ and background-subtracted. The intense peak at 19.80° 2Θ is from the diamond powder used for the dilution of the sample. Peaks indicated in the inset shown in (a) correspond to the species indicated in (b), where the peak area of Co2C (pink square) is shown with TOS (peak area from y = 0).
Figure 4.

(a) Operando XRD data of the undiluted 10 wt % Co/TiO2 with TOS during 2 days of FTS at 15 bar. (b) Evolution of key phases as a function of TOS. Peaks indicated in the diffractograms shown in the inset of (a) correspond to the species indicated in (b), where the peak areas of Co-fcc (green circle) and Co2C (pink square) are shown with TOS. It can be seen, from the insets in (a), that the Co-fcc phase is decreasing over the whole range, as the corresponding peaks at 22.9 and 38.6° 2Θ are disappearing as well.
3.3. Cobalt Carbide Formation as Probed with Operando XAS
The time-resolved XRD data during the FTS reaction in the previous section provide insights regarding the ordered longer-range crystalline structural transformation of the metallic Co NPs to the Co2C phase. Operando XAS, on the other hand, is more sensitive to the short-range structure around the cobalt during the transformation. Figure 5 shows the series of operando XAS Co K-edge spectra recorded during the FTS experiment at 16 bar, measured for 2 days. Spectra were recorded every 20 min and then every three spectra were merged resulting in hourly data, as plotted. The Co K-edge XANES spectra are plotted in Figure 5a and show that during the operando measurements, significant and continuous changes take place. This is particularly visible in the Co pre-edge region (at approximately 7713 eV), where a significant change occurs commensurate with a change in the white line (at approximately 7730 eV), Figure 5a insets. Figure 5b shows the corresponding magnitude of the Fourier transform (in R-space) of the XAS spectra shown in Figure 5a, together with the reference spectrum of bulk Co (a metallic Co foil, Figures S11–S13). The corresponding k2·χ(x) data are shown in Figure S14a. The panel in Figure 5c shows the magnitude of the Fourier transform (in R-space) of the reference materials and the catalyst spectra at both the start and end of the 2 day experiment. These spectral changes are consistent with the continuous transformation of the metallic cobalt in the fresh reduced catalyst to an increased amount of Co2C phase as the FTS reaction proceeds, as is now discussed.
Figure 5.

(a) Operando XAS Co K-edge spectra recorded for Co/TiO2 during the FTS experiment at 16 bar, measured for 2 days at a ratio of 1:1 H2:CO at a temperature of 220 °C with a total flow rate of 5 mL·min–1. (b) Corresponding magnitude of the Fourier transform (in R-space) and (c) the magnitude and imaginary parts of the Fourier transform (in R-space) of the reference materials and the catalyst spectra at both the start and end of the reaction.
These time-evolution XAS data are analyzed using two different methods: (i) multivariate analysis and clustering of the EXAFS data and (ii) linear combination fitting of the Co K-edge XANES spectra.
In order to obtain spectroscopic fingerprints of the different (temporal) phases of the experiment with an improved S/N ratio in comparison to individual spectra, principal component analysis (PCA) and clustering were performed for the recorded time series. PCA was used to reduce the original, 378-dimensional, data space to three dimensions based on the inspection of the cumulative variance, the eigenspectra, and the loadings (see the Supporting Information, Figure S16). Here, 378 is the number of energy points of the recorded spectra. After PCA, every spectrum was represented by a data point in the three-dimensional PC space, in which k-means clustering was performed, effectively pooling data points (i.e., spectra) based on their Euclidian distance in the PC space, that is, based on spectral similarity. This approach is advantageous as it allows for a more efficient clustering of the data in terms of computational costs (much lower data dimensionality), removes noise (captured and removed by higher PCs), and, in the case of time series, allows visualizing the time behavior of the data points in the score plot (see Figure S28 and the discussion there). The result of the clustering allowed us to establish distinct time periods over the total time of the experiment for which an average spectrum could be determined at the expense of a higher time resolution. Note that any significant changes in the spectral features of the recorded spectra (expressed by a change in cluster index between two subsequent spectra) will still be captured at the time resolution used to record individual spectra, while slower, gradual changes will be expressed via an “intermediate cluster” containing a few spectra with features common to both neighboring clusters (see Figure 6a). In other words, the total time period consisting of 47 spectra was binned into 5 time periods (i.e., “clusters”) plus one reference using an adaptive binning that was exclusively based on the degree of spectral similarity. With this reduced number of spectra that show an improved S/N ratio (due to the averaging of all spectra of a cluster) and are characteristic for the distinct temporal phases of the catalytic reaction, it becomes possible to perform a detailed analysis of the EXAFS indicative for each of those process periods.
Figure 6.

PCA and clustering of the operando XAS data shown in Figure 5. With this method, the large number of spectra is downsized to five different clusters and a reference with TOS indicated in (a) (spectra offset for clarity). (b,c) A zoom-in of the areas with the most spectral changes. (d) The time frame of the different clusters with TOS. The colored boxes represent the cluster and the number represents the time in hours. The different clusters were fitted with the spectra of post-reduction, post-H2 treatment (post-H2), and the Co2C reference, and the linear combination result with the residual is shown in (e). With TOS, the amount of Co2C phase is increasing and metallic Co is decreasing.
Figure 6 shows the result of the multivariate analysis applied to the operando XAS data, as shown in Figure 5. More specifically, PCA and clustering were applied to cluster together spectra with similar spectral features in a way that uses no a priori knowledge of the system.
In our case, this procedure results in six clusters that capture the changes of the XAS spectra with TOS. Figure 6a shows these six different clusters (one reference and clusters 1–5 with TOS), and Figure 6b,c shows the white line region, and Co pre-edge features, respectively. Figure 6d represents a map of the clusters in time, where the numbers represent the TOS in hours, starting from the top downward (the first pink square represents the metallic Co reference) and the colors represent the corresponding clusters shown in Figure 6a. Thus, instead of analyzing a plethora of spectra, we only have five different spectra to analyze, and these capture the changes taking place in the entire data set. Each of the clusters can now be individually fit by, for example, least-squares linear combination fitting (LSLC, as done in Figure 6e) with metallic Co and Co2C references to yield the ratio of different Co phases in each spectrum. As our XRD data shows, and as is described in the literature,49,50,54 the initial reduction of a Co-based FTS catalyst results in NPs with intermixed Co-fcc and Co-hcp phases. With carburization during catalysis, the carbide is believed to form first by the conversion of Co-fcc (Figures 3 and 4) and followed by the conversion of Co-hcp.56 With decarburization in an H2 environment, Co2C is converted to a phase that mainly contains Co-hcp.49,52
By assessing the operando-clustered XAS data (Figure 6) and comparing them with the operando XRD data of the same catalyst under the same reaction conditions (Figures 3 and 4), we gain additional information. Although it is difficult to distinguish between Co-fcc and Co-hcp by XAS for these NPs (Section S3.3.3), we suggest that this was a valid approach, especially with the knowledge gained from XRD. Thus, the reduced catalyst at the start of the FTS reaction was composed of a mixture of Co NPs with both hcp and Co-fcc phases present.
As can be seen from Figure 6d,e, the first change occurs in the first 11 h of the reaction, where initial carbide formation takes place and still both metallic Co-phases are present (cluster 1). In the next 12 h (cluster 2), the formation of Co2C proceeds first via the decrease of the more Co-fcc characteristic phase as was observed from the XRD results. After 24 h, with increasing TOS, the carbide formation continues to grow at the expense of the metallic Co until at approximately, at 48 h, there is 37.7% (±1.5) Co2C and 62.3% (±2.3) metallic Co remaining. Note that cluster 3 shows a lower content of carbide than cluster 2 (see Sections 3.4 and S5.5 and Figures S26 and S27 for the explanation).
The time evolution of the XANES data were also analyzed by linear combination fitting using the entire XAS data set, and the results are shown in Figure 7. This method shows that Co-fcc gradually disappears within the first 11 h TOS. This corresponds well with the first cluster from the above applied method. The amount of carbide phase gradually grows with TOS at the expense of the overall metallic Co phase, ending up with ∼40% (±2.5) Co2C and ∼60% (±15.2) metallic cobalt. This analysis method agrees well with the clustering method. With TOS, the uncertainty of the fit increases, which is likely due to the intergrowth of the different cobalt phases present, combined with the effect of using bulk phases to fit XANES data of NPs.54,57 This is also in line with van Deelen et al.(57) Stacking faults appear by the intergrowth of the metallic Co0 structures and break up long-range order needed for fitting.54 For thoroughness, we show (Figures S16–S18) that the results of linear combination fitting with different combinations of cobalt references, from which we conclude that the fit in Figure 7 is the most reliable fit.
Figure 7.
Linear combination fitting of the normalized Co K-edge XANES with Co-hcp/fcc and Co2C references over the whole FTS range. In the end state, at 47 h, there is 60.4% metallic Co and 40.6% Co2C. With increasing TOS, the error becomes larger, probably due to the intergrowth of the different cobalt fractions.
In order to further understand the changes occurring during the 2 day experiment, each of the XAS spectra in Figure 5 were processed using the Cauchy wavelet transform (CCWT) algorithm.58 Wavelet transforms (WT) are becoming more common in EXAFS analysis in the catalysis community due to its ability to aid in interpretation and analysis of complex EXAFS spectra.59,60 The WT of an EXAFS spectrum can be used to identify the presence, or lack thereof, of a scattering path,59 or can be used to deconvolute overlapping scattering paths from atomically different neighboring atoms.60 The latter are often difficult to identify, and model, using conventional Fourier transform EXAFS. The ability to identify unique scattering paths in the WT is due to the k-dependency of the back-scattering amplitude for each element. In the WT map, this Z-dependence can be observed, in the simplest manner, as a peak that is shifted to lower or higher k depending on a lower or higher Z element, respectively. There are advanced methods of interpretation of WTs that also allow for the identification of multiple scattering paths,61 but this is outside of the scope of the analysis presented here.
The resulting plots (of every 5 h) are shown in Figure S20. The intensities of the plots are all normalized to the same scale. In this manner, the gradual transformation from the metallic Co phase to the Co2C phase is clearly visualized. The intense scattering path of the nearest-neighbor Co–Co single-scattering path, CCWT signature across all k at R ≈ 2.25 Å, in metallic Co gradually decreases in intensity while shifting to higher R with time as a weak signal at k ≈ 3–8 Å–1 and R ≈ 1.75 Å appears. This decrease in the intensity of the Co–Co scattering path with the addition of a short-R scattering component is indicative of the multiple Co–Co paths and the Co–C path in the Co2C structure. The weaker intensity of the Co–Co paths in Co2C has previously been noted.33 Decreased intensity can also be observed in the CCWT plots at higher R values, approximately 3–4 Å, matching the new (destructively interfering) scattering paths of the Co2C species.
3.4. Comparing XRD and XAS for Cobalt Carbide Formation
Given the information obtained from the analysis of XRD, the clustering of the XAS data, and the linear combination fitting of the XAS data during the 47 h FTS reaction, it is interesting to compare these data together. Such a comparison is shown in Figure 8. Here, the result of the above-mentioned characterization is plotted within one figure with TOS. The Co-fcc and the Co2C results from the XRD are plotted on the right axis and both XAS analysis methods on the left axis. Both the XRD and XAS data show a linear increase in carbide formation with TOS. However, from these combined data, it can be seen that the carbide is detected at a later TOS (10 h) with XRD than with XAS (2 h), as would be expected given the difference in length scale/degree of ordering that is probed, combined with the relative sensitivity of each method. The time frames of the clusters are indicated by the colored background and the fraction of carbide with pink triangles. The linear combination fitting of the XAS data is shown by the half-filled squares. Except for cluster 3 (see Section S5.5), the indicated fraction of carbide from the clusters follows the linear combination fitting well. This is also an indication that the clustering is a valid method to downsize large data sets.
Figure 8.
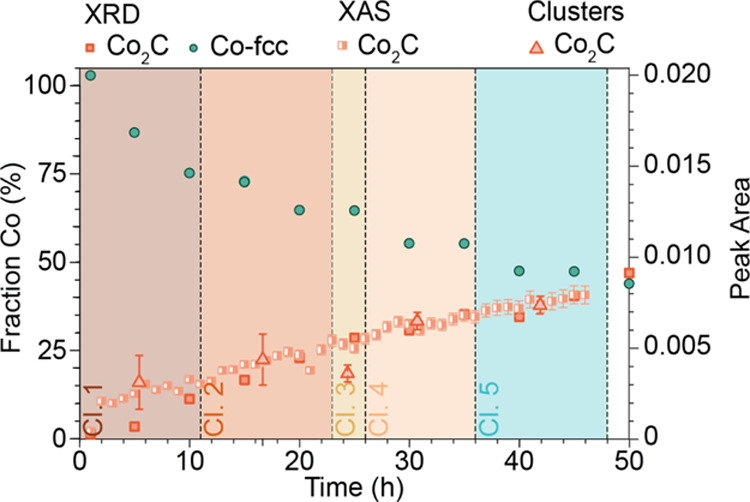
Combined results of the operando XRD and XAS measurements for the Co/TiO2 FTS catalyst. The XAS data shows the fraction of Co2C as a function of time. Both from LCF (half-filled rectangles) and from the clustering (triangles), the end state results in approximately 40% Co2C and 60% metallic Co. The XRD data show the same results, with only a delay in the detection of the carbide, as can be seen from the lower initial amount (before 10 h TOS). In addition to the formation of cobalt carbide, XRD also gives information on the Co-fcc, which decreases with TOS.
3.5. Evaluation of Potential Cobalt Oxide Formation
As it has been discussed in the literature that the formation of oxidized Co can occur during the FTS reaction,1 we closely analyzed the XRD and XAS data in an attempt to identify any oxidized cobalt phase. Figure S21 shows diffractograms of the fresh, reduced, and spent Co/TiO2 catalyst samples. From close examination of these data, it was not possible to assign any of the peaks to the formation of CoO or Co3O4 in the operando XRD analysis of the catalyst sample after approximately 2 days of FTS TOS. Moreover, neither the detailed XAS modeling nor the EXAFS modeling of the clusters, combined with the LSLC fitting of the clusters, leads us to conclude that there is measurable cobalt oxide formation during 2 days of FTS under the 1:1 CO/H2, 15 bar (XRD), 220 °C conditions used here. Here, it must be noted that we also did not observe catalyst deactivation, and we operated under low conversion conditions; thus, the partial pressure of the byproduct water would be low.
3.6. Decarburization after FTS
At the end of the 47 h TOS, the FTS reaction CO flow was stopped, resulting in a pure hydrogen feed, and the temperature was ramped in stages to 300 °C at 16 bar. This was followed by a reduction in temperature to 220 °C, with the gas feed switched to pure CO at 16 bar (Figure S22). This experiment was aimed at studying the stability of the cobalt carbide phase and then the ease of reforming it. The XANES, the results of the PCA and clustering, and the LCF of the Co K-edge XANES during the decarburization and re-carburization are shown in Figures S22–S26 in Section S5. It can be seen that the carbide is quite stable and only begins to decompose above 275 °C (Figure S25). From the clustering (Figure S26, clusters 6 and 7), it can be seen that the carbide gradually decomposes, and metallic cobalt is forming into mostly Co-hcp. However, the error within the fitting is large, due to the NP effect, caused by the intergrowth of different cobalt phases, and fitting with bulk material is significant here.54 At the end of decarburization, there is still a small amount of carbide present.
3.7. Characterization of the Spent Co/TiO2 Catalyst by Electron Microscopy
At the end of the whole operando XAS experiment, the sample was studied by STEM–EDX, and the results are summarized in Figure 9 (see also Section S6 for additional images). Co is observed as discrete NPs on the titania support, as expected. While it is not trivial to determine an accurate particle size analysis due to the similarity of the atomic number of Co and Ti, this analysis was conducted on several regions of the catalyst using the chemical maps. We note that this analysis is not sensitive to any small sub-nanometer clusters. Two such particle size histograms are shown in Figure S29. In one image, the average size was 3.8 ± 1.5 nm, and the other was 2.5 ± 0.7 nm. From analyzing many different images, it was concluded that the average particle size was ca. 3 nm. However, there was a wide distribution, and occasionally large agglomerates, >5 nm, were observed (some as large as 30 nm). This leads us to suggest that there might be a dual particle size distribution. Moreover, these data indicate that there is some mobility of the Co during this experiment (the XRD of the initial reduced catalyst indicated that the size of the Co NPs was 15–20 nm and the EXAFS analysis was also consistent with large NPs). This is in line with previous work done by Cats et al., where they found a bimodal cobalt distribution due to the mobility of the NPs over micrometer range in an ex situ sample.18 They also performed a 10 h 2D in situ TXM (nano-tomographic transmission X-ray microscopy) experiment at a synchrotron facility in order to track the migration. However, they conclude that the migration is a rather slow process as they were not able to visualize it within their time. Additionally, some studies on Co/SiO2 or Co/Al2O3 mention the mobility or migration over the surface of the support.62,63 We performed ex situ XRD of the sealed sample after the XAS experiment (Figure S31). However, there were no defined metallic cobalt peaks found in the X-ray diffractograms due to the diamond peak (at 19.80° 2Θ), but the broad band at lower angles could indicate the small particle distribution observed using STEM–EDX. Given the history of the sample (not only reaction but also decarburization and re-carburization), it is unfortunately not possible to be definitive regarding at what stage the smaller cobalt particle formation took place. This observation will be the subject of further study.
Figure 9.

HAADF-STEM image of the spent Co/TiO2 FTS catalyst (left) and the EDX imaging of the Co and Ti. Cobalt is shown in red and titanium in blue (right).
Even though the sample had undergone decarburization and partial re-carburization at the end of the 47 h FTS experiment, the STEM–EDX was used to probe for evidence of coke formation on the supported Co NPs. The carbon (assumed to be coke species) was found to be non-uniformly distributed. There were regions where the coke was closely coordinated to Co particles and Co particles with no detectable coke near them (Figure S30). Additionally, there were regions where the coke appeared to be finely dispersed on the titania, which could also be an indication of the fine dispersion of small cobalt particles distributed over the surface of the TiO2 support. Thus, it appears that in addition to some cobalt agglomeration and redispersion, there was evidence of coke deposition on the catalyst even after decarburization.
3.8. of Co/TiO2 Catalyst Diluted with MCM-41
In the operando XAS experiment that was conducted continuously for 6 days, performed with a 1:1 CO/H2 ratio and at 14 bar and 220 °C (Figure S9), the catalyst was diluted with a MCM-41 silica material. This dilution was used to achieve a mass fraction of catalyst in the X-ray beam that would give optimum absorption at the Co K-edge, and the assumption was that the silica would act as an inert diluent. While in this experiment we were unsuccessful in maintaining a stable pressure, there was an unexpected observation when the spent catalyst was studied by STEM–EDX. As shown in Figure S32, in addition to the expected Co NPs on the titania, the most frequent observation was that of Co metal NPs on the MCM-41 silica that has been in close proximity to the titania. Figure S33 provides additional elemental analysis of the catalyst material. This implies that under the reaction conditions used for this experiment, the cobalt was mobile and migrated over microscopic scales from the titania to the silica. This was somewhat unexpected, however, it is in line with the previous work mentioned earlier where it has been demonstrated that the cobalt is mobile.18,62,63 This observation may be correlated with the particle dispersion observed in the sample where diamond was used as a diluent. However, we again note that we operated under non-standard FTS conditions (1:1 CO/H2, with Co2C formation). This observation will also be the subject of future research.
Given that the sample was only studied by STEM at the end of the 6 day experiment, it is unknown when or by what mechanism the Co migrated. However, the operando XAS data from this experiment are different from those of the diamond-dust dilution experiment, and the formation of the Co2C phase was slow and difficult to quantify. Furthermore, we were not able to maintain stable pressure and there was migration of the Co from the TiO2 support to the MCM-41 support. This speaks to some of the complexities of conducting FTS catalysis at a remote location and that there are some aspects of this well-studied catalytic reaction that are still not fundamentally understood.
4. Conclusions
Via the combination of operando X-ray spectroscopy and diffraction methods, we were able to follow the state of Co within a Co/TiO2 catalyst material under realistic FTS conditions and—most notably—observed cobalt carburization without a decrease in measured catalyst activity and without cobalt oxide formation. We have performed operando XAS and XRD on a 10 wt % Co/TiO2 FTS catalyst at 15 bar, with a 1:1 ratio of H2/CO and for an extended period of time. The first 2 days of FTS measured by XAS and XRD were compared, and it was found that the carburization process of Co/TiO2 occurred at the cost of metallic cobalt (i.e., the Co-fcc crystal phase). After 2 days, both XRD and XAS showed that the intergrown metallic cobalt phases in the catalyst was converted into a mixed phase of metallic cobalt (i.e., the Co-hcp crystal phase) and cobalt carbide (Co2C). No formation of any cobalt oxide phase was found during FTS conditions. Furthermore, no significant catalyst deactivation was measured. As the consensus in FTS literature is that metallic cobalt is the active FTS phase and no clear correlation between carbidization and deactivation could be found, more research should be done as to the location of this carbide. Possible explanations could be that there are factors compensating for a loss in activity due to carbide formation, such as increased activity due to the formation of smaller particles or higher activity of the increasing Co-hcp phase. Another explanation is that mainly bulk carbide is formed, which is backed by the recent research showing that subsurface carbide is more stable than surface carbide.64
After the FTS reaction, when a decarburization procedure was applied, the sample was left in a mostly Co-hcp crystal structure. HAADF-STEM–EDX was performed on the spent catalyst, and a bimodal particle size distribution with particles <5 nm and particles 20−30 nm were observed, most probably due to migration of cobalt. STEM–EDX was performed ex situ after FTS and the decarburization procedure, and as such, it is hard to tell when this migration took place and the bimodal particle size distribution was formed. Further study is necessary. Finally, we found that cobalt migrates from the TiO2 support to the SiO2 phase that was tested for dilution in an FTS experiment lasting 6 days. These combined conclusions lead us to the final point that it is highly important to follow up with long-term operando characterization studies in order to gain information on the complexity of the FTS catalyst. This study shows that it is feasible to conduct longer term catalyst deactivation experiments at a synchrotron facility. Indeed, as a result of the success of this pilot experiment (for 6 days and 2 days), we are planning on making this capability available to catalyst researchers and use the learnings from this study to develop a user-friendly capability at SSRL.
Acknowledgments
The authors thank Shell and the Netherlands Research Council (NWO) for a TA-CHIPP grant. Shell is also thanked for the catalytic testing experiments. Further financial support for this work came from the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation program funded by the Ministry of Education, Culture and Science of the government of the Netherlands. Erik Maris (Utrecht University, UU) is acknowledged for his help during the synchrotron measurements. Part of this work was also performed at Stanford Synchrotron Radiation Lightsource (SSRL) of SLAC National Accelerator Laboratory and use of the SSRL is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515 and by Co-ACCESS supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c04695.
List of characterization methods, extra information on the undiluted XRD experiment, information on potential cobalt oxide formation, decarburization and recarburization, HAAFD-STEM–EDX mapping, and extra information on operando XAS (PDF)
Author Present Address
# Institute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany.
Author Contributions
I.K.v.R., A.S.H., and C.V. contributed equally to this work. The paper was written through contributions of all the authors. All the authors have given approval to the final version of the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Tsakoumis N. E.; Rønning M.; Borg Ø.; Rytter E.; Holmen A. Deactivation of Cobalt Based Fischer-Tropsch Catalysts: A Review. Catal. Today 2010, 154, 162–182. 10.1016/j.cattod.2010.02.077. [DOI] [Google Scholar]
- Xie J.; Paalanen P. P.; van Deelen T. W.; Weckhuysen B. M.; Louwerse M. J.; de Jong K. P. Promoted Cobalt Metal Catalysts Suitable for the Production of Lower Olefins from Natural Gas. Nat. Commun. 2019, 10, 167. 10.1038/s41467-018-08019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S. G.; Ciobîcă I. M.; Hensen E. J. M.; van Santen R. A. Site Regeneration in the Fischer–Tropsch Synthesis Reaction: A Synchronized CO Dissociation and C–C Coupling Pathway. Chem. Commun. 2011, 47, 9822–9824. 10.1039/c1cc11499a. [DOI] [PubMed] [Google Scholar]
- de Smit E.; Weckhuysen B. M. The Renaissance of Iron-Based Fischer-Tropsch Synthesis: On the Multifaceted Catalyst Deactivation Behaviour. Chem. Soc. Rev. 2008, 37, 2758–2781. 10.1039/b805427d. [DOI] [PubMed] [Google Scholar]
- Van Helden P.; Ciobîcă I. M.; Coetzer R. L. J. The Size-Dependent Site Composition of FCC Cobalt Nanocrystals. Catal. Today 2016, 261, 48–59. 10.1016/j.cattod.2015.07.052. [DOI] [Google Scholar]
- Van Steen E.; Claeys M.; Dry M. E.; Van De Loosdrecht J.; Viljoen E. L.; Visagie J. L. Stability of Nanocrystals: Thermodynamic Analysis of Oxidation and Re-Reduction of Cobalt in Water/Hydrogen Mixtures. J. Phys. Chem. B 2005, 109, 3575–3577. 10.1021/jp045136o. [DOI] [PubMed] [Google Scholar]
- Fischer N.; Van Steen E.; Claeys M. Structure Sensitivity of the Fischer-Tropsch Activity and Selectivity on Alumina Supported Cobalt Catalysts. J. Catal. 2013, 299, 67–80. 10.1016/j.jcat.2012.11.013. [DOI] [Google Scholar]
- Argyle M.; Bartholomew C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. 10.3390/catal5010145. [DOI] [Google Scholar]
- van der Eerden A. M. J.; Visser T.; Nijhuis T. A.; Ikeda Y.; Lepage M.; Koningsberger D. C.; Weckhuysen B. M. Atomic XAFS as a Tool to Probe the Electronic Properties of Supported Noble Metal Nanoclusters. J. Am. Chem. Soc. 2005, 127, 3272–3273. 10.1021/ja043107l. [DOI] [PubMed] [Google Scholar]
- Oosterbeek H. Bridging the Pressure and Material Gap in Heterogeneous Catalysis: Cobalt Fischer–Tropsch Catalysts from Surface Science to Industrial Application. Phys. Chem. Chem. Phys. 2007, 9, 3570–3576. 10.1039/b703003g. [DOI] [PubMed] [Google Scholar]
- Rytter E.; Tsakoumis N. E.; Holmen A. On the Selectivity to Higher Hydrocarbons in Co-Based Fischer-Tropsch Synthesis. Catal. Today 2016, 261, 3–16. 10.1016/j.cattod.2015.09.020. [DOI] [Google Scholar]
- Claeys M.; Dry M. E.; Van Steen E.; van Berge P. J.; Booyens S.; Crous R.; Van Helden P.; Labuschagne J.; Moodley D. J.; Saib A. M. Impact of Process Conditions on the Sintering Behavior of an Alumina-Supported Cobalt Fischer-Tropsch Catalyst Studied with an in Situ Magnetometer. ACS Catal. 2015, 5, 841–852. 10.1021/cs501810y. [DOI] [Google Scholar]
- Sadeqzadeh M.; Chambrey S.; Piché S.; Fongarland P.; Luck F.; Curulla-Ferré D.; Schweich D.; Bousquet J.; Khodakov A. Y. Deactivation of a Co/Al2O3 Fischer–Tropsch Catalyst by Water-Induced Sintering in Slurry Reactor: Modeling and Experimental Investigations. Catal. Today 2013, 215, 52–59. 10.1016/j.cattod.2013.03.022. [DOI] [Google Scholar]
- Schanke D.; Hilmen A. M.; Bergene E.; Kinnari K.; Rytter E.; Ådnanes E.; Holmen A. Reoxidation and Deactivation of Supported Cobalt Fischer–Tropsch Catalysts. Energy Fuels 1996, 10, 867–872. 10.1021/ef950197u. [DOI] [Google Scholar]
- van de Loosdrecht J.; Balzhinimaev B.; Dalmon J.-A.; Niemantsverdriet J. W.; Tsybulya S. V.; Saib A. M.; van Berge P. J.; Visagie J. L. Cobalt Fischer-Tropsch Synthesis: Deactivation by Oxidation?. Catal. Today 2007, 123, 293–302. 10.1016/j.cattod.2007.02.032. [DOI] [Google Scholar]
- Fischer N.; Clapham B.; Feltes T.; van Steen E.; Claeys M. Size-Dependent Phase Transformation of Catalytically Active Nanoparticles Captured in Situ. Angew. Chem., Int. Ed. 2014, 53, 1342–1345. 10.1002/anie.201306899. [DOI] [PubMed] [Google Scholar]
- Moodley D. J.; van de Loosdrecht J.; Saib A. M.; Overett M. J.; Datye A. K.; Niemantsverdriet J. W. Carbon Deposition as a Deactivation Mechanism of Cobalt-Based Fischer-Tropsch Synthesis Catalysts under Realistic Conditions. Appl. Catal., A 2009, 354, 102–110. 10.1016/j.apcata.2008.11.015. [DOI] [Google Scholar]
- Cats K. H.; Andrews J. C.; Stéphan O.; March K.; Karunakaran C.; Meirer F.; de Groot F. M. F.; Weckhuysen B. M. Active Phase Distribution Changes within a Catalyst Particle during Fischer-Tropsch Synthesis as Revealed by Multi-Scale Microscopy. Catal. Sci. Technol. 2016, 6, 4438–4449. 10.1039/c5cy01524c. [DOI] [Google Scholar]
- Creemer J. F.; Santagata F.; Morana B.; Mele L.; Alan T.; Iervolino E.; Pandraud G.; Sarro P. M.. An All-in-One Nanoreactor for High-Resolution Microscopy on Nanomaterials at High Pressures. Proceedings of the IEEE International Conference on Micro Electro Mechanical Systems, 2011; pp 1103–1106.
- Loewert M.; Serrer M.-A.; Carambia T.; Stehle M.; Zimina A.; Kalz K. F.; Lichtenberg H.; Saraçi E.; Pfeifer P.; Grunwaldt J.-D. Bridging the Gap Between Industry and Synchrotron: An Operando Study at 30 Bar over 300 h During Fischer–Tropsch Synthesis. React. Chem. Eng. 2020, 5, 1071–1082. 10.1039/c9re00493a. [DOI] [Google Scholar]
- van Berge P. J.; van de Loosdrecht J.; Barradas S.; Van Der Kraan A. M. Oxidation of Cobalt Based Fischer-Tropsch Catalysts as a Deactivation Mechanism. Catal. Today 2000, 58, 321–334. 10.1016/s0920-5861(00)00265-0. [DOI] [Google Scholar]
- Xiong J.; Ding Y.; Wang T.; Yan L.; Chen W.; Zhu H.; Lu Y. The Formation of Co2C Species in Activated Carbon Supported Cobalt-Based Catalysts and Its Impact on Fischer-Tropsch Reaction. Catal. Lett. 2005, 102, 265–269. 10.1007/s10562-005-5867-1. [DOI] [Google Scholar]
- Karaca H.; Hong J.; Fongarland P.; Roussel P.; Griboval-Constant A.; Lacroix M.; Hortmann K.; Safonova O. V.; Khodakov A. Y. In Situ XRD Investigation of the Evolution of Alumina-Supported Cobalt Catalysts under Realistic Conditions of Fischer-Tropsch Synthesis. Chem. Commun. 2010, 46, 788–790. 10.1039/b920110f. [DOI] [PubMed] [Google Scholar]
- Hernández Mejía C.; van Deelen T. W.; de Jong K. P. Activity Enhancement of Cobalt Catalysts by Tuning Metal-Support Interactions. Nat. Commun. 2018, 9, 4459. 10.1038/s41467-018-06903-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Xu Y.; Chen Y.; Li W.; Lin L.; Li M.; Deng Y.; Wang X.; Ge B.; Yang C.; Yao S.; Xie J.; Li Y.; Liu X.; Ma D. Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal-Support Interaction. Angew. Chem., Int. Ed. 2017, 56, 10761–10765. 10.1002/anie.201705002. [DOI] [PubMed] [Google Scholar]
- Prieto G.; De Mello M. I. S.; Concepción P.; Murciano R.; Pergher S. B. C.; Martínez A. Cobalt-Catalyzed Fischer-Tropsch Synthesis: Chemical Nature of the Oxide Support as a Performance Descriptor. ACS Catal. 2015, 5, 3323–3335. 10.1021/acscatal.5b00057. [DOI] [Google Scholar]
- Bezemer G. L.; Remans T. J.; van Bavel A. P.; Dugulan A. I. Direct Evidence of Water-Assisted Sintering of Cobalt on Carbon Nanofiber Catalysts during Simulated Fischer–Tropsch Conditions Revealed with in Situ Mössbauer Spectroscopy. J. Am. Chem. Soc. 2010, 132, 8540–8541. 10.1021/ja103002k. [DOI] [PubMed] [Google Scholar]
- Chen W.; Kimpel T. F.; Song Y.; Chiang F.-K.; Zijlstra B.; Pestman R.; Wang P.; Hensen E. J. M. Influence of Carbon Deposits on the Cobalt-Catalyzed Fischer-Tropsch Reaction: Evidence of a Two-Site Reaction Model. ACS Catal. 2018, 8, 1580–1590. 10.1021/acscatal.7b03639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller S.; Hofer L. J. E.; Anderson R. B.; Weller S.; Hofer L. J. E.; Anderson R. B. The Role of Bulk Cobalt Carbide in the Fischer-Tropsch Synthesis. J. Am. Chem. Soc. 1948, 70, 799–801. 10.1021/ja01182a108. [DOI] [Google Scholar]
- Mohandas J. C.; Gnanamani M. K.; Jacobs G.; Ma W.; Ji Y.; Khalid S.; Davis B. H. Fischer-Tropsch Synthesis: Characterization and Reaction Testing of Cobalt Carbide. ACS Catal. 2011, 1, 1581–1588. 10.1021/cs200236q. [DOI] [Google Scholar]
- Yang R.; Xia Z.; Zhao Z.; Sun F.; Du X.; Yu H.; Gu S.; Zhong L.; Zhao J.; Ding Y.; Jiang Z. Characterization of CoMn Catalyst by In Situ X-Ray Absorption Spectroscopy and Wavelet Analysis for Fischer–Tropsch to Olefins Reaction. J. Energy Chem. 2019, 32, 118–123. 10.1016/j.jechem.2018.07.005. [DOI] [Google Scholar]
- Pei Y.-P.; Liu J.-X.; Zhao Y.-H.; Ding Y.-J.; Liu T.; Dong W.-D.; Zhu H.-J.; Su H.-Y.; Yan L.; Li J.-L.; Li W.-X. High Alcohols Synthesis via Fischer-Tropsch Reaction at Cobalt Metal/Carbide Interface. ACS Catal. 2015, 5, 3620–3624. 10.1021/acscatal.5b00791. [DOI] [Google Scholar]
- Singh J. A.; Hoffman A. S.; Schumann J.; Boubnov A.; Asundi A. S.; Nathan S. S.; Nørskov J.; Bare S. R.; Bent S. F. Role of Co2C in ZnO-Promoted Co Catalysts for Alcohol Synthesis from Syngas. ChemCatChem 2019, 11, 799–809. 10.1002/cctc.201801724. [DOI] [Google Scholar]
- Jiao G.; Ding Y.; Zhu H.; Li X.; Li J.; Lin R.; Dong W.; Gong L.; Pei Y.; Lu Y. Effect of La2O3 Doping on Syntheses of C1–C18 Mixed Linear α-Alcohols from Syngas over the Co/AC Catalysts. Appl. Catal., A 2009, 364, 137–142. 10.1016/j.apcata.2009.05.040. [DOI] [Google Scholar]
- Moya-Cancino J. G.; Honkanen A.; van der Eerden A. M. J.; Schaink H.; Folkertsma L.; Ghiasi M.; Longo A.; Meirer F.; de Groot F. M. F.; Huotari S.; Weckhuysen B. M. Elucidating the K-Edge X-Ray Absorption Near-Edge Structure of Cobalt Carbide. ChemCatChem 2019, 11, 3042–3045. 10.1002/cctc.201900434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Wu D.; Yu F.; Yang R.; Zhang H.; Sun F.; Zhong L.; Jiang Z. In Situ XAFS Study on the Formation Process of Cobalt Carbide by Fischer-Tropsch Reaction. Phys. Chem. Chem. Phys. 2019, 21, 10791–10797. 10.1039/c9cp01298b. [DOI] [PubMed] [Google Scholar]
- Beale A. M.; Jacques S. D. M.; Di Michiel M.; Mosselmans J. F. W.; Price S. W. T.; Senecal P.; Vamvakeros A.; Paterson J. X-Ray Physico-Chemical Imaging During Activation of Cobalt-Based Fischer–Tropsch Synthesis Catalysts. Philos. Trans. R. Soc., A 2018, 376, 20170057. 10.1098/rsta.2017.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ravenhorst I. K.; Vogt C.; Oosterbeek H.; Bossers K. W.; Moya-Cancino J. G.; van Bavel A. P.; van der Eerden A. M. J.; Vine D.; de Groot F. M. F.; Meirer F.; Weckhuysen B. M. Capturing the Genesis of an Active Fischer-Tropsch Synthesis Catalyst with Operando X-ray Nanospectroscopy. Angew. Chem., Int. Ed. 2018, 57, 11957–11962. 10.1002/anie.201806354. [DOI] [PubMed] [Google Scholar]
- Herbert J. J.; Senecal P.; Martin D. J.; Bras W.; Beaumont S. K.; Beale A. M. X-Ray Spectroscopic and Scattering Methods Applied to the Characterisation of Cobalt-Based Fischer–Tropsch Synthesis Catalysts. Catal. Sci. Technol. 2016, 6, 5773–5791. 10.1039/c6cy00581k. [DOI] [Google Scholar]
- Kuipers E. W.; Vinkenburg I. H.; Oosterbeek H. Chain Length Dependence of α-Olefin Readsorption in Fischer-Tropsch Synthesis. J. Catal. 1995, 152, 137–146. 10.1006/jcat.1995.1068. [DOI] [Google Scholar]
- Rønning M.; Tsakoumis N. E.; Voronov A.; Johnsen R. E.; Norby P.; van Beek W.; Borg Ø.; Rytter E.; Holmen A. Combined XRD and XANES Studies of a Re-Promoted Co/γ-Al2O3 Catalyst at Fischer–Tropsch Synthesis Conditions. Catal. Today 2010, 155, 289–295. 10.1016/j.cattod.2009.10.010. [DOI] [Google Scholar]
- Handbook of Heterogeneous Catalysis; Ertl G., Knözinger H., Schüth F., Weitkamp J., Eds.; Wiley-VCH: Weinheim, 2008. [Google Scholar]
- Vogt C.; Groeneveld E.; Kamsma G.; Nachtegaal M.; Lu L.; Kiely C. J.; Berben P. H.; Meirer F.; Weckhuysen B. M. Unravelling Structure Sensitivity in CO2 Hydrogenation over Nickel. Nat. Catal. 2018, 1, 127–134. 10.1038/s41929-017-0016-y. [DOI] [Google Scholar]
- Hoffman A. S.; Singh J. A.; Bent S. F.; Bare S. R. IUCr. In Situ Observation of Phase Changes of a Silica-Supported Cobalt Catalyst for the Fischer–Tropsch Process by the Development of a Synchrotron-Compatible In Situ/Operando Powder X-Ray Diffraction Cell. J. Synchrotron Radiat. 2018, 25, 1673–1682. 10.1107/s1600577518013942. [DOI] [PubMed] [Google Scholar]
- Ravel B.; Newville M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. 10.1107/s0909049505012719. [DOI] [PubMed] [Google Scholar]
- Cats K. H.; Weckhuysen B. M. Combined Operando X-Ray Diffraction/Raman Spectroscopy of Catalytic Solids in the Laboratory: The Co/TiO2 Fischer-Tropsch Synthesis Catalyst Showcase. ChemCatChem 2016, 8, 1531–1542. 10.1002/cctc.201600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruker . EVA Software.
- Enache D. I.; Rebours B.; Roy-Auberger M.; Revel R. In Situ XRD Study of the Influence of Thermal Treatment on the Characteristics and the Catalytic Properties of Cobalt-Based Fischer-Tropsch Catalysts. J. Catal. 2002, 205, 346–353. 10.1006/jcat.2001.3462. [DOI] [Google Scholar]
- Karaca H.; Safonova O. V.; Chambrey S.; Fongarland P.; Roussel P.; Griboval-Constant A.; Lacroix M.; Khodakov A. Y. Structure and Catalytic Performance of Pt-Promoted Alumina-Supported Cobalt Catalysts under Realistic Conditions of Fischer-Tropsch Synthesis. J. Catal. 2011, 277, 14–26. 10.1016/j.jcat.2010.10.007. [DOI] [Google Scholar]
- Liu J.-X.; Su H.-Y.; Sun D.-P.; Zhang B.-Y.; Li W.-X. Crystallographic Dependence of CO Activation on Cobalt Catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. 10.1021/ja408521w. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zhang C.; Li Y.; Niemantsverdriet J. W.; Wagner J. B.; Hansen T. W. Environmental Transmission Electron Microscopy (ETEM) Studies of Single Iron Nanoparticle Carburization in Synthesis Gas. ACS Catal. 2017, 7, 4867–4875. 10.1021/acscatal.7b00946. [DOI] [Google Scholar]
- Tsakoumis N. E.; Dehghan R.; Johnsen R. E.; Voronov A.; Van Beek W.; Walmsley J. C.; Borg Ø.; Rytter E.; Chen D.; Rønning M.; Holmen A. A Combined In Situ XAS-XRPD-Raman Study of Fischer-Tropsch Synthesis over a Carbon Supported Co Catalyst. Catal. Today 2013, 205, 86–93. 10.1016/j.cattod.2012.08.041. [DOI] [Google Scholar]
- Tsakoumis N. E.; Dehghan-Niri R.; Rønning M.; Walmsley J. C.; Borg Ø.; Rytter E.; Holmen A. X-Ray Absorption, X-Ray Diffraction and Electron Microscopy Study of Spent Cobalt Based Catalyst in Semi-Commercial Scale Fischer-Tropsch Synthesis. Appl. Catal., A 2014, 479, 59–69. 10.1016/j.apcata.2014.03.035. [DOI] [Google Scholar]
- Sławiński W. A.; Zacharaki E.; FjellvaÅg H.; SjaÅstad A. O. Structural Arrangement in Close-Packed Cobalt Polytypes. Cryst. Growth Des. 2018, 18, 2316–2325. 10.1021/acs.cgd.7b01736. [DOI] [Google Scholar]
- Ducreux O.; Rebours B.; Lynch J.; Roy-Auberger M.; Bazin D. Microstructure of Supported Cobalt Fischer-Tropsch Catalysts. Oil Gas Sci. Technol. 2009, 64, 49–62. 10.2516/ogst:2008039. [DOI] [Google Scholar]
- Paterson J.; Peacock M.; Ferguson E.; Purves R.; Ojeda M. In Situ Diffraction of Fischer-Tropsch Catalysts: Cobalt Reduction and Carbide Formation. ChemCatChem 2017, 9, 3463–3469. 10.1002/cctc.201700754. [DOI] [Google Scholar]
- van Deelen T. W.; Yoshida H.; Oord R.; Zečević J.; Weckhuysen B. M.; de Jong K. P. Cobalt Nanocrystals on Carbon Nanotubes in the Fischer-Tropsch Synthesis: Impact of Support Oxidation. Appl. Catal., A 2020, 593, 117441. 10.1016/j.apcata.2020.117441. [DOI] [Google Scholar]
- Muñoz M.; Argoul P.; Farges F. Continuous Cauchy Wavelet Transform Analyses of EXAFS Spectra: A Qualitative Approach. Am. Mineral. 2003, 88, 694–700. 10.2138/am-2003-0423. [DOI] [Google Scholar]
- Xia Z.; Zhang H.; Shen K.; Qu Y.; Jiang Z. Wavelet Analysis of Extended X-Ray Absorption Fine Structure Data: Theory, Application. Phys. B 2018, 542, 12–19. 10.1016/j.physb.2018.04.039. [DOI] [Google Scholar]
- Timoshenko J.; Kuzmin A. Wavelet Data Analysis of EXAFS Spectra. Comput. Phys. Commun. 2009, 180, 920–925. 10.1016/j.cpc.2008.12.020. [DOI] [Google Scholar]
- Sushkevich V. L.; Safonova O. V.; Palagin D.; Newton M. A.; van Bokhoven J. A. Structure of Copper Sites in Zeolites Examined by Fourier and Wavelet Transform Analysis of EXAFS. Chem. Sci. 2020, 11, 5299–5312. 10.1039/d0sc01472a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnik P.; De Jongh P. E.; De Jong K. P. Control and Impact of the Nanoscale Distribution of Supported Cobalt Particles Used in Fischer-Tropsch Catalysis. J. Am. Chem. Soc. 2014, 136, 7333–7340. 10.1021/ja500436y. [DOI] [PubMed] [Google Scholar]
- Borg Ø.; Walmsley J. C.; Dehghan R.; Tanem B. S.; Blekkan E. A.; Eri S.; Rytter E.; Holmen A. Electron Microscopy Study of γ-Al2O3 Supported Cobalt Fischer-Tropsch Synthesis Catalysts. Catal. Lett. 2008, 126, 224–230. 10.1007/s10562-008-9650-y. [DOI] [Google Scholar]
- Jansen A. P. J.; Agrawal R.; Spanu L. Thermodynamics and Kinetics of Carbon Deposits on Cobalt: A Combined Density Functional Theory and Kinetic Monte Carlo Study. Phys. Chem. Chem. Phys. 2016, 18, 28515–28523. 10.1039/c6cp04719j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.