

Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by persistent airway inflammation and fixed airflow obstruction. Patients with COPD have increased risk of lung cancer (LC), and the coexistence of both diseases is associated with poorer survival. However, the mechanisms predisposing patients with COPD to LC development and poor prognosis remain unclear. Gene expression profiles were downloaded from the Gene Expression Omnibus. Twenty‐two data sets were included (n = 876). We identified 133 DEGs and 145 DEGs in patients with COPD and LC compared with healthy controls, respectively. There were 1544 DEGs in patients with LC and coexisting COPD compared with COPD, and these DEGs are mainly involved in the cell cycle, DNA replication, p53 signalling and insulin signalling. The biological processes primarily associated with these DEGs are oxidation reduction and apoptosis. SPP1 was the only overlapping DEG that was up‐regulated in patients with COPD and/or LC, and this was validated by qPCR in an independent cohort. The area under the curve value for SPP1 was 0.893 (0.822–0.963) for the prediction of LC in patients with COPD. High expression of SPP1 in patients with LC was associated with shorter survival time. Up‐regulation of SPP1 may be associated with increased risk of LC in patients with COPD and therefore may have potential as a therapeutic target for LC in patients with COPD.
Keywords: bioinformatics, chronic obstructive pulmonary disease, lung cancer, meta‐analysis, SPP1
SPP1 is progressively up‐regulated in the lungs of patients with COPD, lung cancer (LC) and LC coexisting with COPD, as shown by bioinformatic analysis. Up‐regulation of SPP1 expression in COPD and its further up‐regulation in NSCLC coexisting with COPD were confirmed by qPCR. The area under the curve value for SPP1 was 0.893 (0.822–0.963) for the prediction of LC in patients with COPD. Thus, up‐regulation of SPP1 may be associated with increased risk of LC in patients with COPD.

Abbreviations
- AUC
area under the curve
- CI
confidence intervals
- COPD
chronic obstructive pulmonary disease
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- DEGs
differentially expressed genes
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- GO
gene ontology
- GSEA
Gene Set Enrichment Analysis
- IGF
insulin growth factor
- NSCLC
non‐small‐cell lung cancer
- OPN
osteopontin
- qPCR
quantitative real‐time polymerase chain reaction
- ROC
receiver operating characteristic curve
- SPP1
secreted phosphoprotein 1
Chronic obstructive pulmonary disease (COPD) and lung cancer (LC) are leading cause of morbidity and mortality worldwide. COPD is characterized by persistent airway inflammation and fixed airflow obstruction [1]. The overall prevalence of spirometry‐defined COPD is 8.6% among the general population aged 20 years or older, accounting for 99.9 million people with COPD in China [2]. It is currently the third leading cause of death worldwide and imposes huge economic burden on patients [3]. LC is the number one cause of cancer deaths worldwide with a low 5‐year overall survival rate [4, 5]. A previous study has reported that the prevalence of COPD was 40–70% of those diagnosed with LC [6, 7]. The incidence of these two diseases has been increasing in recent years, and this trend is estimated to continue. More importantly, both conditions often coexist such that patients with COPD have an increased risk of developing LC resulting in a poorer prognosis compared with LC alone [8].
The mechanisms underlying the coexistence of COPD and LC are not well understood. One of the main hypotheses is the common environmental risk factor for both diseases in particular cigarette smoke exposure. However, smokers with COPD are fourfold to sixfold as likely to develop LC as smokers without COPD [9], and the presence of emphysema in COPD predicts a higher LC risk adjusted for smoking status [10, 11]. Therefore, the presence of COPD itself is an independent risk factor for LC. In terms of prognoses, a series of studies have demonstrated that patients with non‐small‐cell lung cancer (NSCLC) coexisting with COPD were related to worse survival and complained of more symptoms [12, 13, 14, 15]. Potential mechanisms linking COPD and LC may be associated with shared pathogenic processes such as increased oxidative stress and DNA damage, repression of the DNA repair mechanisms, chronic exposure to pro‐inflammatory cytokines, repression of innate immunity and increased cellular proliferation [16]. Nevertheless, the molecular basis that predisposes patients with COPD to LC and is related to worse survival remains unclear. Therefore, it is important to identify potential pathological genes and pathways involved.
With the development of microarray technology, gene expression analysis using high‐throughput platforms has been widely applied to explore the differentially expressed genes (DEGs) within various diseases. A group of gene expression profiling studies on COPD and LC have been performed using microarray technology and identified certain DEGs associated with COPD and LC [17, 18, 19]. However, the DEGs identified with microarray in a single study depend on sample size, technology platform and sample types, etc., while the overlapping genes obtained from multiple microarray data sets might be more accurate and representative. There is no study addressing DEGs and pathways related to the coexistence of COPD and LC using meta‐analysis of combined data sets so far.
In the present study, we interrogated the Gene Expression Omnibus (GEO) database for multiple microarray expression data sets and compared mRNA expression among different disease groups in terms of COPD, LC and LC coexisting with COPD, aiming to identify molecular signatures, significant biological functions and pathways associated with the coexistence of COPD and LC. More importantly, results were further validated by quantitative real‐time polymerase chain reaction (qPCR) and analysed for the association with survival.
Material and methods
Data collection
All data sets in the current study were searched in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) by using key words as follows: [(COPD or chronic obstructive pulmonary disease or emphysema or chronic bronchitis or LC or lung squamous cell carcinoma or lung adenocarcinoma or NSCLC or lung or epithelial) AND (smok* or cigarette) AND human]. Original studies were further searched in PubMed to ensure the inclusion of all relevant data sets by using key words: [(COPD or chronic obstructive pulmonary disease or emphysema or chronic bronchitis or LC or lung squamous cell carcinoma or lung adenocarcinoma or NSCLC or lung or epithelial) AND (mRNA or messenger RNA or gene) AND (smok* or cigarette) AND human].
The inclusion criteria for data sets were as follows: (a) having screened for DEGs related to COPD and LC by using human biological samples; (b) available information of smoking history for each sample; (c) available results of pulmonary function test if there was no description of COPD diagnosis in study design; (d) available raw data or matrix file in GEO database; and (e) data sets in which samples were not completely overlapped with other data sets.
The inclusion criteria for patients were as follows: (a) available information for smoking history; (b) with data of lung function tests or information for the diagnosis of COPD; and (c) available raw data or matrix file of mRNA expression on airway epithelial cell sample or lung tissue sample. COPD was defined in patients with pulmonary function test of FEV1 (forced exhalation volume in one second) /FVC (forced vital capacity) < 0.7 or with doctor‐diagnosed COPD in the data set. LC was confirmed by pathological diagnosis. The exclusion criteria were as follows: (a) unavailable information for smoking history or non‐smoker; (b) overlapping samples in other data sets that have been included in current study; (c) other sample types such as blood; and (d) with respiratory diseases other than COPD and LC.
Identification of DEGs
Data sets were merged and analysed only when the experimental platforms and sample types were identical. Limma package in r software (version 3.5.1, https://www.r‐project.org/) was applied to perform the normalization of raw data or matrix file, and log2 conversion of fold change for each disease group. Genes with absolute fold change ≥ 1.5 in expression level and adjusted P value < 0.05 were considered as statistically significant DEGs when two groups were compared. Ggplot2 package in r software was used to perform the volcano plots of specific genes. Gplots package in r software was applied to perform the heatmap of top 50 DEGs (top 25 up‐regulated genes and top 25 down‐regulated genes). Venn diagram package in r software was applied to identify the overlapping DEGs.
Functional annotation analysis
Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov), an online tool, was used to analyse gene ontology (GO) of DEGs. In detail, GO depicts three complementary biological concepts including biological process, molecular function and cellular component. P < 0.05 was regarded as an accepted threshold criterion.
Gene Set Enrichment Analysis (GSEA)
Gene Set Enrichment Analysis is a computational method that determines whether an a priori defined set of genes shows statistically significant and concordant differences between two biological states [20]. GSEA was applied to perform Kyoto Encyclopedia of Genes and Genomes pathway analysis. Data of gene expression, phenotype, gene sets that compose GSEA were input to GSEA software (version 3.0) to run pathway analysis. The number of permutations was 1000. Gene sets in GSEA with false discovery rate (FDR) < 0.25 and nominal P < 0.05 were considered statistically significant.
The associations between key gene and clinical outcomes
Receiver operating characteristic curve (ROC) analysis was performed to investigate the role of key gene in the prediction of coexisting LC in COPD, and the area under the curve (AUC) value with 95% confidence intervals (CI) was calculated. The levels of key gene were compared by different GOLD grades and by different stages of LC in patients with COPD and in those with LC, respectively.
The Kaplan–Meier plotter database is capable of assessing the effect of 54 000 genes on survival in 21 cancer types, which includes survival analysis data of 3452 patients with LC (http://kmplot.com/analysis/index.php?p=background). Survival analysis of the selected key gene was performed by the Kaplan–Meier plotter database. Hazard ratios (HR) with 95% CI and log‑rank P values were calculated and displayed.
RNA extraction and qPCR validation
Lung tissue specimens were obtained from patients who underwent lobectomy or pneumonectomy for cancer in West China Hospital. After surgical resection, lung tissues were immediately frozen to −80 °C. Histologically normal tissues adjacent to the tumour were used as controls. This study was approved by the Clinical Trial and Biomedical Ethics Committee of West China Hospital of Sichuan University (No.2016‐120) and was conducted in accordance with the principles of Declaration of Helsinki. All patients volunteered to attend the study and signed an informed consent. Total RNA was extracted from lung tissues (nine non‐smoker controls, 15 smoker controls, 13 COPD, eight NSCLC and 16 NSCLC coexisting with COPD) according to the manufacturer's protocol using the E.Z.N.A. HP Total RNA Kit (OMEGA, GA, USA). iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA, USA) was applied to synthesize cDNA following the manufacturer's instructions. qPCR was performed in triplicate by using iQ™ SYBR Green Supermix (Bio‐Rad) according to the manufacturer's protocol. Relative expression level of gene was normalized via the GAPDH C t value (endogenous reference), applying a relative quantification method. The qPCR primers were as follows:
SPP1‐forward: 5′‐CAACAAATACCCAGATGCTGTGGC‐3′.
SPP1‐reverse: 5′‐GGACTTACTTGGAAGGGTCTGTGG‐3′.
GAPDH‐forward: 5′‐TGCACCACCAACTGCTTAGC‐3′.
GAPDH‐reverse: 5′‐GGCATGGACTGTGGTCATGAG‐3′.
Statistical analysis
Statistical analysis was performed with spss version 22 (IBM Corporation, Chicago, IL, USA). Levels of mRNA expression were expressed as median (interquartile range) or mean ± standard deviation according to data distributed type. Comparisons between two different groups were determined by Student's t‐test for parametric data or Mann–Whitney test for nonparametric data. One‐way analysis of variance was used for multiple comparisons. P < 0.05 was considered statistically significant.
Results
Data collection
There were 79 data sets that were found with two different types of tissue (airway epithelial cell and lung tissue) based on 18 platforms. According to the inclusion criteria, 22 data sets (n = 876) based on three platforms (GPL570, GPL96 and GPL1708) were finally included in the current study for analysis (Table 1). To control the impact of smoking status, only smokers in the disease groups were included for analysis. The median age and number of male|female of the patients are listed in Table S1.
Table 1.
Data sets included in the current study.
| Tissue type | Platform | Dataset | Sample size | |||
|---|---|---|---|---|---|---|
| HC | LC | COPD | LC + COPD | |||
| Lung tissue |
GPL570 [HG‐U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array |
GSE43580, GSE31210, GSE37768 | 17 | 226 | 0 | 0 |
|
GPL96 [HG‐U133A] Affymetrix Human Genome U133A Array |
GSE10072 | 151 | 42 | 0 | 0 | |
| Airway epithelial cell |
GPL570 [HG‐U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array |
366 | 0 | 91 | 0 | |
|
GPL96 [HG‐U133A] Affymetrix Human Genome U133A Array |
GSE994, GSE19027 | 26 | 21 | 0 | 0 | |
| GPL1708 [Agilent‐012391 Whole Human Genome Oligo Microarray G4112A] | GSE12428, GSE12472 | 0 | 0 | 36 | 36 | |
Identification of DEGs
There were 133 DEGs (72 up‐regulated and 61 down‐regulated genes) when comparing COPD patients with healthy controls (HC; Fig. 1A). A total of 429 DEGs (232 up‐regulated and 197 down‐regulated genes) in LC patients were identified compared with HCs using samples of airway epithelial cells (Fig. 1B).
Fig. 1.
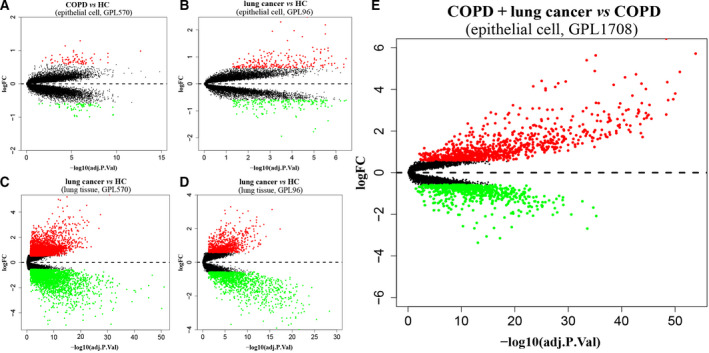
Volcano plots of DEGs. (A) COPD vs HC in epithelial cell of GPL570; (B) LC vs HC in epithelial cell of GPL96; (C) LC vs HC in lung tissue of GPL570; (D) LC vs HC in lung tissue of GPL96; (E) LC coexisting with COPD vs COPD in epithelial cell of GPL1708. Red and green dots denote up‐regulated and down‐regulated genes, respectively.
By using lung tissue samples, there were 6476 DEGs (3307 up‐regulated and 3169 down‐regulated genes) in GPL570 and 2261 DEGs (954 up‐regulated and 1307 down‐regulated genes) in GPL96 when comparing LC patients with HCs (Fig. 1C,D). A total of 145 DEGs were confirmed in patients with LC across all sample types and platforms (Fig. 2A). There were 1544 DEGs (889 up‐regulated and 655 down‐regulated genes) in LC patients coexisting with COPD compared with COPD patients (Fig. 1E), and a heatmap was clustered by top 50 DEGs as shown in Fig. 3. The secreted phosphoprotein 1 (SPP1) was the only overlapping DEG that was up‐regulated in patients with COPD, LC and LC coexisting with COPD (Fig. 2B).
Fig. 2.
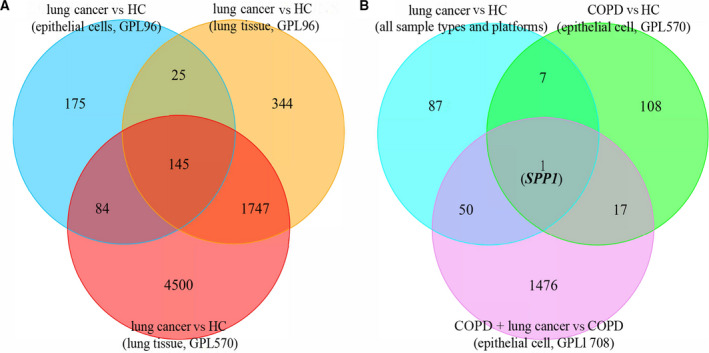
Venn diagrams of the DEGs. (A) The DEGs in LC vs HC across all sample types and platforms; (B) the overlapping DEG that was up‐regulated in patients with COPD, LC and LC coexisting with COPD.
Fig. 3.
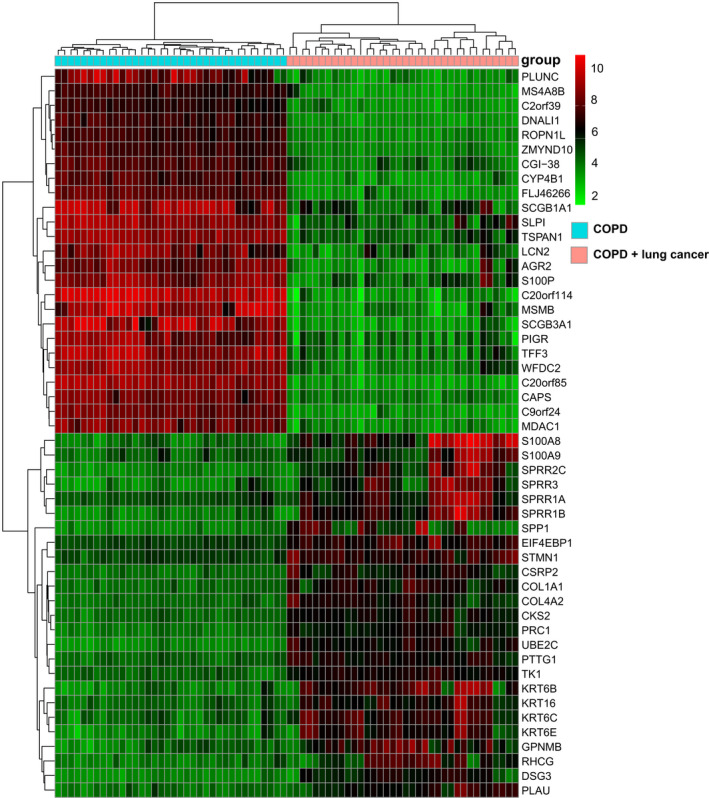
Heatmap of the top 50 DEGs (top 25 up‐regulated and 25 down‐regulated genes) in patients with LC and coexisting COPD vs COPD. Red indicates up‐regulation, and blue indicates down‐regulation.
Functional annotation analysis
Functional enrichment analysis was performed with 1544 DEGs identified in the comparison of LC coexisting with COPD and COPD alone. The biological process was primarily associated with oxidation reduction process, positive regulation of transcription from RNA polymerase II promoter and apoptotic process (Fig. 4A). The cellular component mainly located in extracellular exosome, cytoplasm and cytosol (Fig. 4B). The molecular function was mainly related to protein binding, poly(A) RNA binding and identical protein binding (Fig. 4C).
Fig. 4.
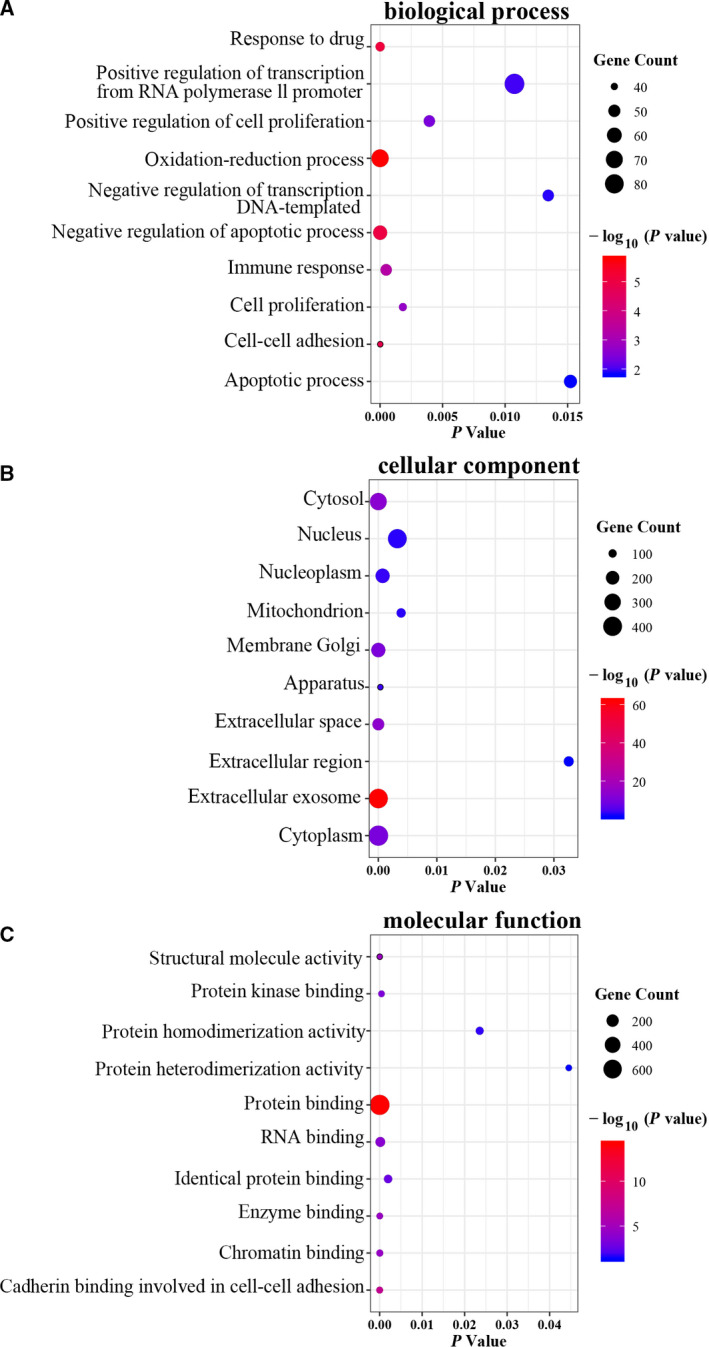
The top 10 significant enriched GO terms based on DEGs related to LC coexisting with COPD vs COPD by DAVID. (A) Biological process; (B) cellular component; (C) molecular function.
Gene Set Enrichment Analysis
The GSEA results showed that gene sets related to the groups of LC coexisting with COPD were mainly enriched in the pathways of cell cycle, P53 signalling pathway, DNA replication, pyrimidine metabolism, insulin signalling pathway, etc. (FDR < 0.25 and nominal P < 0.05; Table 2).
Table 2.
GSEA of genes related to LC coexisting with COPD vs COPD (top 10 ranked according to P value).
| Pathway name | NOM P value | FDR q value |
|---|---|---|
| Pyrimidine metabolism | < 0.001 | 0.035 |
| Cell cycle | < 0.001 | 0.026 |
| DNA replication | 0.002 | 0.054 |
| Pathways in cancer | 0.01 | 0.025 |
| Insulin signalling pathway | 0.01 | 0.04 |
| Fructose and mannose metabolism | 0.017 | 0.037 |
| p53 signalling pathway | 0.017 | 0.048 |
| RNA degradation | 0.017 | 0.045 |
| FC gamma r‐mediated phagocytosis | 0.018 | 0.051 |
| ECM–receptor interaction | 0.022 | 0.046 |
The associations between SPP1 and clinical outcomes
The diagnostic value of relative expression levels of SPP1 in the lungs for the prediction of coexisting LC in patients with COPD was evaluated by a ROC analysis. The AUC value with 95% CI for the relative expression levels of SPP1 in the lungs was 0.893 (0.822–0.963), and the cut‐off value for the relative expression levels of SPP1 in the lungs was 3.625 according to Youden's index with sensitivity of 80.56% and specificity of 77.78% (Fig. 5).
Fig. 5.
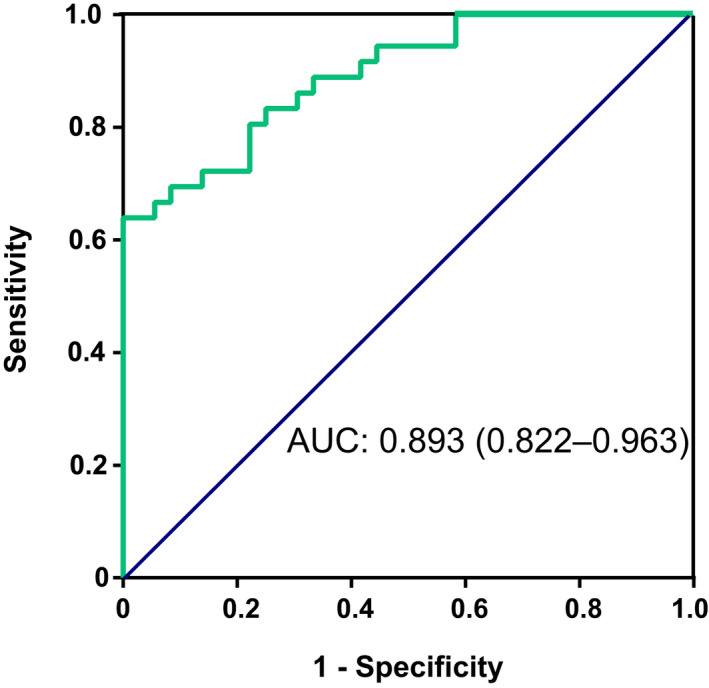
ROC analysis revealed that the relative expression levels of SPP1 in the lungs could predict coexisting LC in patients with COPD, and AUC value with 95% CI was 0.893 (95% CI: 0.822–0.963).
There were 91 patients with COPD across seven data sets in the bioinformatic analysis, in which 52 patients were reported with COPD severity. These patients were from the same platform (GPL570) with sample type of airway epithelial cells. There were 34 patients classified as GOLD stage 1, fifteen patients as GOLD stage 2 and three patients as GOLD stage 3, and the levels of SPP1 expression were not significantly different among the three groups (P = 0.811).
The presence of advanced LC (stages IIIB and IV) is not suitable for surgery, we found few patients with stage III and IV LC in the bioinformatic analysis. Data of 226 lung tissue samples reporting the stage of LC were retrieved from platform GPL570, in which 129 samples were from patients with stage I LC and 97 samples were from patients with stage II LC. The levels of SPP1 expression were statistically different between patients with stage I (11.74 ± 1.63) and stage II (12.16 ± 1.16; P = 0.030). Data of 42 lung tissues samples reporting the stage of LC were retrieved from platform GPL96, in which only two lung tissue samples were obtained from patients with stage IV. There was no significant difference in the levels of SPP1 expression among patients with stage I (n = 16), stage II (n = 17) and stage III (n = 7) LC (P = 0.690).
Survival analysis of the key gene SPP1 was performed in 1926 patients diagnosed with LC using the Kaplan–Meier plotter database. Elevated level of SPP1 expression was significantly associated with poor overall survival [HR = 1.32 (1.16–1.49); log‐rank P = 1.9 × 10−5] (Fig. 6).
Fig. 6.
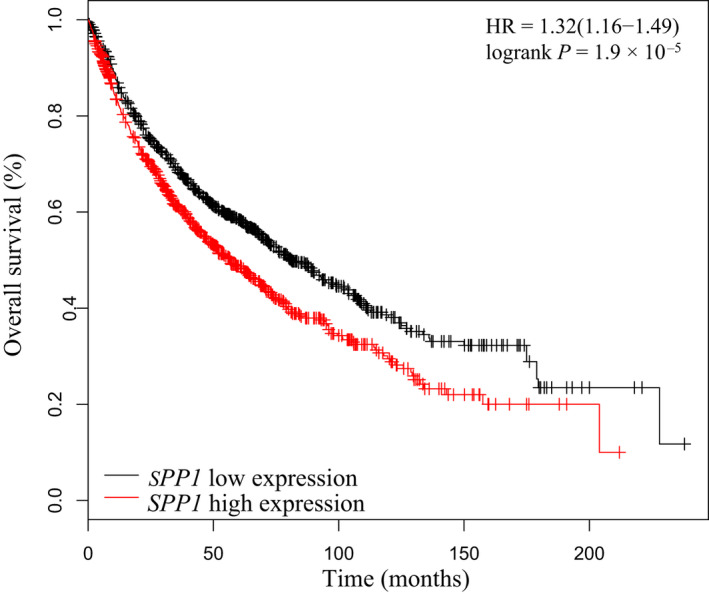
Survival analysis of SPP1 in LC by using the Kaplan–Meier plotter database.
Results of qPCR validation
To validate the results of bioinformatic analysis, the relative expression levels of gene SPP1 were assayed in lung tissues using qPCR. The results showed that levels of SPP1 expression in cancer tissues were higher than that in non‐smoker controls (P = 0.007) and smoker controls (P = 0.002). The expression level of SPP1 in the COPD group was significantly higher compared with either non‐smokers (P = 0.002) or smokers (P = 0.001), and the level was further up‐regulated with an enormous increase in the group of NSCLC coexisting with COPD (P < 0.001). In addition, levels of SPP1 expression were also significantly higher in the group of NSCLC and coexisting COPD than that in NSCLC group (P < 0.001; Fig. 7).
Fig. 7.
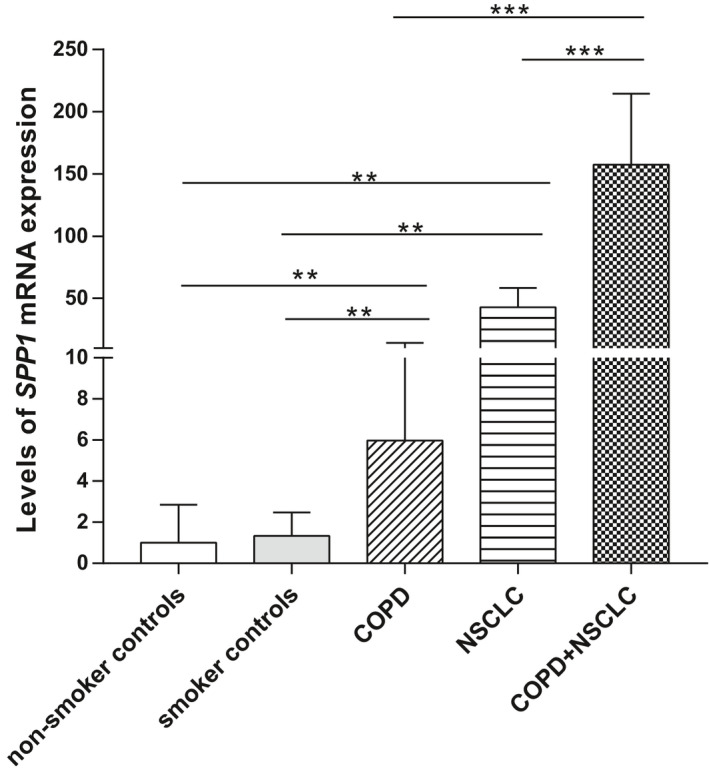
qPCR validation of SPP1 expression in a cancer cohort. Levels of SPP1 expression in lung tissues between non‐smoker controls (n = 9), smoker controls (n = 15), COPD (n = 13), NSCLC (n = 8) and NSCLC coexisting with COPD (n = 16) were compared by the Mann–Whitney test. Data are presented as median (interquartile range). **P < 0.01. ***P < 0.001.
Discussion
In this study, we firstly identified DEGs that were associated with COPD, LC and LC coexisting with COPD by bioinformatic analysis using multiple microarray expression data sets, in order to explore potential molecular mechanisms underlying the increased risk of LC in patients with COPD. Functional annotation analysis and GSEA were performed to investigate the potential roles of significant genes in the biological function and relevant pathways.
The most important finding of our study is the identification of the key gene SPP1 in the coexistence of COPD and LC. SPP1, also named osteopontin (OPN), is a phosphorylated acidic glycoprotein that functions as both an extracellular matrix molecule and a cytokine [21]. The overexpression of the SPP1 is associated with the growth and stage of LC, tumour angiogenesis and lymph node metastasis [22, 23, 24, 25, 26], and represents a more aggressive NSCLC type [23]. SPP1 could also mediate macrophage polarization and immune escape in lung adenocarcinoma [27]. Besides, the tumour‐derived SPP1 isoforms cooperate with both TRP53 and CCL2 and the FN1/SPP1‐ITGAV signalling chemoattracts tumour cells and inhibits their apoptosis, promoting the intrapulmonary metastasis of LC [28, 29]. Our study results of qPCR validation confirmed the up‐regulation of SPP1 in patients with NSCLC, indicating its potential role as oncogene.
On the other hand, SPP1 is associated with neutrophilic inflammation and emphysema [30]. Cigarette smoking can increase SPP1 expression in induced sputum, and its level is increased in induced sputum [31] and lung tissue [32] of patients with COPD. Hence, SPP1 may participate in the pathogenesis of COPD. By using bioinformatic analysis, we firstly found that SPP1 was the only gene that differentially expressed in the comparisons of COPD vs HCs, LC vs HCs, and LC coexisting with COPD vs COPD alone in the lungs. Although the comparisons were based on different platforms and sample types, it may suggest a progressive up‐regulation of SPP1 in COPD, LC and LC coexisting with COPD. We therefore conducted the qPCR validation, and the results confirmed that SPP1 gene expression was significantly up‐regulated in COPD and further up‐regulated to a considerably higher level in the group of NSCLC coexisting with COPD. This suggests that as an oncogene, SPP1 may participate in the predisposition of patients with COPD to LC development.
The above finding drives us to further investigate the clinical role of SPP1 in predicting coexisting LC in COPD. The high AUC value of 0.893 for SPP1 expression level indicated promising utility of SPP1 as a biomarker in the lung for the prediction of LC risk in patients with COPD, although the sample size was not large enough. A previous study using serum OPN to predict cancer risk in patients with COPD identified a rather small AUC value of 0.636 [33]. There are two forms of OPN, intracellular form (iOPN) and a secreted form (sOPN) [34], and sOPN may be produced by many cell types, including hepatocytes, cholangiocytes, hepatic stellate cells, macrophages, T lymphocytes and natural killer T cells [35]. A previous study demonstrated that the lungs produce plasma OPN [36]; however, plasma OPN may partly originate from the lungs in COPD and LC because many other cells are capable of producing OPN. Hence, detecting SPP1 gene or protein expression levels in local specimens such as lung tissue, BAL and sputum to predict coexisting LC in COPD is superior to plasma OPN and should be the direction of future research.
Previous studies have investigated the associations between SPP1 and clinical outcomes in both COPD and LC. Patients with frequent exacerbations of COPD had higher plasma OPN than the non‐frequent exacerbators [37]. We did not find the association between SPP1 and COPD severity, which was consistent with an early study [31]. Conflicting results exist in terms of cancer stage and SPP1 level based on blood assay [38, 39]. We did not find the difference in the lungs using our data, which may be attributed to the somehow limited number of patients especially the limited number of patients with advanced LC, and this needs to be confirmed in following study. However, in terms of role of SPP1 in clinical prognosis, the crucial role of SPP1 found by our study was manifested by its association with low overall survival in LC. High SPP1 expression in the tumour tissue was associated with inferior survival in a previous study with small sample size [24]. We confirmed its association with survival in LC by analysing data of 1926 patients in Kaplan–Meier plotter database. This indicates that the up‐regulation of SPP1 expression may also be involved in the poor prognosis in terms of survival in patients with LC and coexisting COPD. Therefore, SPP1 might be a therapeutic target to reduce the development of LC in patients with COPD and to improve survival time.
Several significant pathways were identified that related to the coexistence of LC with COPD. Cell cycle deregulation is a common feature of human cancer, and cancer cells frequently display unscheduled proliferation [40]. Cell cycle‐related and expression‐elevated protein in tumour overexpression is associated with cell proliferation and poor prognosis in NSCLC [41]. DNA replication occurs during the S (synthetic) phase of cell cycle [42] and may cause mutation and DNA damage [42]. Gene mutation has identified in squamous cell carcinoma and adenocarcinoma of lung [43, 44] and has led to resistance to chemotherapy, resulting in lower survival rates in small‐cell LC [45]. DNA damage is often associated with oxidative stress, resulting in cancer initiation [46]. Previous study has found that the insulin growth factor (IGF) pathway is activated in NSCLC [47]. Reduced levels of insulin‐like growth factor binding protein 3 in serum were associated with increased LC risk in current smokers independent of obesity [48]. Small molecule inhibitors targeting both the IR and IGF1R or blockading beta‐adrenergic receptor‐mediated insulin‐like growth factor receptor activation could reduce NSCLC cell proliferation and prevent LC [49, 50]. The p53 gene is the most frequently mutated gene in cancer [51], playing an important role in the regulation of tumour progression [52]. Meanwhile, p53 protein expression is significantly higher in patients with emphysema compared with healthy smokers or non‐smokers [53]. Our results further showed that the p53 signalling pathway was an important pathway related to the coexistence of COPD and LC. Thus, the p53 signalling pathway may participate in LC development in patients with COPD. Understanding of the pathways beyond significant molecules will provide mechanisms and research bases for the novel therapy, which needs to be addressed in future study.
Airway epithelial cells are extensively exposed to oxidants, causing both oxidative stress and inflammation, which contribute to COPD pathophysiology [54] and carcinogenesis [46]. We found that oxidation reduction was an important biological process related to patients with LC and coexisting COPD, which therefore may be involved in the increased risk of developing LC in patients with COPD. Apoptotic process was another important biological process linking the coexistence of COPD and LC in the current study. SPP1 has been shown to participate in regulating apoptotic process in LC [28, 29]. Therefore, the apoptosis pathway might be the mechanism or pathway by which SPP1 functions resulting in the development of LC in COPD and deserving further study.
There are some highlights in our study. It is the first study that investigated the molecular signatures and pathways underlying the coexistence of COPD and LC by integrating multiple mRNA microarray datasets and confirmed the DEGs in multi‐platforms using different tissue types from the lung, which provides more accurate and reliable results than a single study of microarray. Additionally, the results of bioinformatic analysis were validated by qPCR using lung tissues. Moreover, we identified the level of SPP1 in the lungs for the prediction of coexisting LC in COPD for the first time. Finally, we confirmed the prognostic role of SPP1 in the overall survival of patients with LC using data of 1926 patients from the Kaplan–Meier plotter database. The study also has certain limitations. We only used lung tissues from patients with NSCLC to reduce the effects of histological subtypes of LC on study results in validation study. However, squamous cell carcinoma was not classified, which is known to be more closely related to COPD, due to the limitation of sample size. The study results will be confirmed in future with larger sample size, and molecules and pathways involved in the upstream and downstream mechanism of SPP1 deserve further study.
Conclusion
In conclusion, our study identified the progressive up‐regulation of gene SPP1 in the lungs of patients with COPD, LC and LC coexisting with COPD by using meta‐analysis of multiple microarray data sets with bioinformatic approach. Validation study confirmed the up‐regulation of SPP1 expression in COPD and its further up‐regulation in NSCLC coexisting with COPD. Survival analysis showed that high expression of SPP1 was associated with shorter survival time. The current study might indicate the roles of SPP1 in the predisposition of patients with COPD to LC development. Lung SPP1 might be a biomarker that could predict the coexistence of LC in COPD and the poorer prognosis of patients with coexisting COPD and LC. SPP1 might be a therapeutic target to reduce the development of LC in patients with COPD and to improve survival time, which deserves further investigation.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
T‐WM and WX collected and analysed data and drafted the manuscript. L‐YD, X‐MC, CL, WH and YW collected the samples, performed qPCR and drafted the manuscript. BM designed the experiment. J‐JF designed the experiment and revised the manuscript.
Supporting information
Table S1. Demographic of the patients included for analysis.
Acknowledgement
We thank staffs in West China Biobanks, Department of Clinical Research Management and Prof. Long‐qi Chen in the Department of Cardiovascular and Thoracic Surgery of West China Hospital, Sichuan University, for providing specimens of lung tissue for this study. This study was funded by the Thousand Talents Program, Sichuan province, China (0082204151045), and the National Natural Science Foundation of China (No. 81870014).
Ti‐wei Miao and Wei Xiao, contributed equally to this work, should be considered as cofirst authors
Data Accessibility
The data used to support the findings of this study are available from the corresponding author upon request.
References
- 1. Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M, Fabbri LM et al. (2017) Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Eur Respir J 49, 1700214. [DOI] [PubMed] [Google Scholar]
- 2. Wang C, Xu J, Yang L, Xu Y, Zhang X, Bai C, Kang J, Ran P, Shen H, Wen F et al. (2018) Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross‐sectional study. Lancet 391, 1706–1717. [DOI] [PubMed] [Google Scholar]
- 3. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY et al. (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Didkowska J, Wojciechowska U, Manczuk M and Łobaszewski J (2016) Lung cancer epidemiology: contemporary and future challenges worldwide. Ann Transl Med 4, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong MCS, Lao XQ, Ho KF, Goggins WB and Shelly LA (2017) Incidence and mortality of lung cancer: global trends and association with socioeconomic status. Sci Rep 7, 14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Loganathan RS, Stover DE, Shi W and Venkatraman E (2006) Prevalence of COPD in women compared to men around the time of diagnosis of primary lung cancer. Chest 129, 1305–1312. [DOI] [PubMed] [Google Scholar]
- 7. Congleton J and Muers MF (1995) The incidence of airflow obstruction in bronchial carcinoma, its relation to breathlessness, and response to bronchodilator therapy. Respir Med 89, 291–296. [DOI] [PubMed] [Google Scholar]
- 8. Brett AS and Al‐Hasan MN (2019) COPD exacerbations ‐ a target for antibiotic stewardship. N Engl J Med 381, 174–175. [DOI] [PubMed] [Google Scholar]
- 9. Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P and Gamble GD (2009) COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J 34, 380–386. [DOI] [PubMed] [Google Scholar]
- 10. de Torres JP, Bastarrika G, Wisnivesky JP, Alcaide AB, Campo A, Seijo LM, Pueyo JC, Villanueva A, Lozano MD, Montes U et al. (2007) Assessing the relationship between lung cancer risk and emphysema detected on low‐dose CT of the chest. Chest 132, 1932–1938. [DOI] [PubMed] [Google Scholar]
- 11. Husebo GR, Nielsen R, Hardie J, Bakke PS, Lerner L, D'Alessandro‐Gabazza C, Gyuris J, Gabazza E, Aukrust P and Eagan T (2019) Risk factors for lung cancer in COPD ‐ results from the Bergen COPD cohort study. Respir Med 152, 81–88. [DOI] [PubMed] [Google Scholar]
- 12. Shah S, Blanchette CM, Coyle JC, Kowalkowski M, Arthur ST and Howden R (2019) Survival associated with chronic obstructive pulmonary disease among SEER‐Medicare beneficiaries with non‐small‐cell lung cancer. Int J Chron Obstruct Pulmon Dis 14, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sekine Y, Yamada Y, Chiyo M, Iwata T, Nakajima T, Yasufuku K, Suzuki M and Fujisawa T (2007) Association of chronic obstructive pulmonary disease and tumor recurrence in patients with stage IA lung cancer after complete resection. Ann Thorac Surg 84, 946–950. [DOI] [PubMed] [Google Scholar]
- 14. Zhai R, Yu X, Shafer A, Wain JC and Christiani DC (2014) The impact of coexisting COPD on survival of patients with early‐stage non‐small cell lung cancer undergoing surgical resection. Chest 145, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yi YS, Ban WH and Sohng KY (2018) Effect of COPD on symptoms, quality of life and prognosis in patients with advanced non‐small cell lung cancer. BMC Cancer 18, 1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caramori G, Ruggeri P, Mumby S, Ieni A, Lo Bello F, Chimankar V, Donovan C, Andò F, Nucera F, Coppolino I et al. (2019) Molecular links between COPD and lung cancer: new targets for drug discovery? Expert Opinion Ther Targets 23, 539–553. [DOI] [PubMed] [Google Scholar]
- 17. Yi G, Liang M, Li M, Fang X, Liu J, Lai Y, Chen J, Yao W, Feng X, Hu L et al. (2018) A large lung gene expression study identifying IL1B as a novel player in airway inflammation in COPD airway epithelial cells. Inflamm Res 67, 539–551. [DOI] [PubMed] [Google Scholar]
- 18. Zhang MY, Liu XX, Li H, Li R, Liu X and Qu YQ (2018) Elevated mRNA Levels of AURKA, CDC20 and TPX2 are associated with poor prognosis of smoking related lung adenocarcinoma using bioinformatics analysis. Int J Med Sci 15, 1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW et al. (2008) Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One 3, e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohan M, Bader R, Puxeddu I, Levi‐Schaffer F, Breuer R and Berkman N (2007) Enhanced osteopontin expression in a murine model of allergen‐induced airway remodelling. Clin Exp Allergy 37, 1444–1454. [DOI] [PubMed] [Google Scholar]
- 22. Donati V, Boldrini L, Dell'Omodarme M, Prati MC, Faviana P, Camacci T, Lucchi M, Mussi A, Santoro M, Basolo F et al. (2005) Osteopontin expression and prognostic significance in non‐small cell lung cancer. Clin Cancer Res 11, 6459–6465. [DOI] [PubMed] [Google Scholar]
- 23. Hu Z, Lin D, Yuan J, Xiao T, Zhang H, Sun W, Han N, Ma Y, Di X, Gao M et al. (2005) Overexpression of osteopontin is associated with more aggressive phenotypes in human non‐small cell lung cancer. Clin Cancer Res 11, 4646–4652. [DOI] [PubMed] [Google Scholar]
- 24. Schneider S, Yochim J, Brabender J, Uchida K, Danenberg KD, Metzger R, Schneider PM, Salonga D, Hölscher AH and Danenberg PV (2004) Osteopontin but not osteonectin messenger RNA expression is a prognostic marker in curatively resected non‐small cell lung cancer. Clin Cancer Res 10, 1588–1596. [DOI] [PubMed] [Google Scholar]
- 25. Shijubo N, Uede T, Kon S, Maeda M, Segawa T, Imada A, Hirasawa M and Abe S (1999) Vascular endothelial growth factor and osteopontin in stage I lung adenocarcinoma. Am J Respir Crit Care Med 160, 1269–1273. [DOI] [PubMed] [Google Scholar]
- 26. Boldrini L, Donati V, Dell'Omodarme M, Prati MC, Faviana P, Camacci T, Lucchi M, Mussi A, Santoro M, Basolo F et al. (2005) Prognostic significance of osteopontin expression in early‐stage non‐small‐cell lung cancer. Br J Cancer 93, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Du W, Chen Z, Xiang C (2017) Upregulation of PD‐L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp Cell Res 359, 449–457. [DOI] [PubMed] [Google Scholar]
- 28. Giopanou I, Lilis I, Papaleonidopoulos V, Agalioti T, Kanellakis NI, Spiropoulou N, Spella M and Stathopoulos G (2017) Tumor‐derived osteopontin isoforms cooperate with TRP53 and CCL2 to promote lung metastasis. Oncoimmunology 6, e1256528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang C, Wu M, Zhang L, Shang LR, Fang JH and Zhuang SM (2016) Fibrotic microenvironment promotes the metastatic seeding of tumor cells via activating the fibronectin 1/secreted phosphoprotein 1‐integrin signaling. Oncotarget 7, 45702–45714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shan M, Yuan X, Song LZ, Roberts L, Zarinkamar N, Seryshev A, Zhang Y, Hilsenbeck S, Chang SH, Dong C et al. (2012) Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci Transl Med 4, 117ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Papaporfyriou A, Loukides S, Kostikas K, Simoes DCM, Papatheodorou G, Konstantellou E, Hillas G, Papiris S, Koulouris N and Bakakos P (2014) Increased levels of osteopontin in sputum supernatant in patients with COPD. Chest 146, 951–958. [DOI] [PubMed] [Google Scholar]
- 32. Ali MN, Mori M, Mertens TCJ, Siddhuraj P, Erjefält JS, Önnerfjord P, Hiemstra PS and Egesten A (2019) Osteopontin expression in small airway epithelium in Copd is dependent on differentiation and confined to subsets of cells. Sci Rep 9, 15566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petta V, Loukides S, Kostikas K, Papaioannou AI, Papatheodorou G, Cholidou K, Tomos I, Papiris S, Koulouris NG and Bakakos P (2018) Serum osteopontin in patients with lung cancer and chronic obstructive pulmonary disease: does the co‐existence make the difference? J Thorac Dis 10, 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inoue M and Shinohara ML (2011) Intracellular osteopontin (iOPN) and immunity. Immunol Res 49, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang G, Chu P, Chen M, Zhao C, Chen S, Li X, Yang G and Chang C (2019) Osteopontin promotes rat hepatocyte proliferation both in vitro and in vivo . Artifi Cells Nanomed Biotechnol 47, 3745–3757. [DOI] [PubMed] [Google Scholar]
- 36. Ayoub C, Nozza A, Denault A, Deschamps A and Dupuis J (2014) Pulmonary production of osteopontin in humans: effects of left ventricular systolic dysfunction and cardiopulmonary bypass. J Card Fail 20, 377.e9–13. [PubMed] [Google Scholar]
- 37. Lee SJ, Kim SH, Kim W, Lim S, Lee SH, Kim YE, Cho YJ, Jeong YY, Kim HC, Lee JD et al. (2014) Increased plasma osteopontin in frequent exacerbator and acute exacerbation of COPD. Clin Respir J 8, 305–311. [DOI] [PubMed] [Google Scholar]
- 38. Chang YS, Kim HJ, Chang J, Ahn CM, Kim SK and Kim SK (2007) Elevated circulating level of osteopontin is associated with advanced disease state of non‐small cell lung cancer. Lung Cancer 57, 373–380. [DOI] [PubMed] [Google Scholar]
- 39. Rud AK, Boye K, Oijordsbakken M, Lund‐Iversen M, Halvorsen AR, Solberg SK, Berge G, Helland A, Brustugun OT and Mælandsmo GM (2013) Osteopontin is a prognostic biomarker in non‐small cell lung cancer. BMC Cancer 13, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malumbres M and Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- 41. Li W, Zheng G, Xia J, Yang G, Sun J, Wang X, Wen M, Sun Y, Zhang Z and Jin F (2018) Cell cycle‐related and expression‐elevated protein in tumor overexpression is associated with proliferation behaviors and poor prognosis in non‐small‐cell lung cancer. Cancer Sci 109, 1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Basu AK (2018). DNA damage, mutagenesis and cancer. Int J Mol Sci 19, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reck M, Heigener DF, Mok T, Soria JC and Rabe KF (2013) Management of non‐small‐cell lung cancer: recent developments. Lancet 382, 709–719. [DOI] [PubMed] [Google Scholar]
- 44. Chen Z, Fillmore CM, Hammerman PS, Kim CF and Wong KK (2014) Non‐small‐cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 14, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- 47. Scagliotti GV and Novello S (2012) The role of the insulin‐like growth factor signaling pathway in non‐small cell lung cancer and other solid tumors. Cancer Treat Rev 38, 292–302. [DOI] [PubMed] [Google Scholar]
- 48. Ho GYF, Zheng SL, Cushman M, Perez‐Soler R, Kim M, Xue X, Wang T, Schlecht NF, Tinker L, Rohan TE et al. (2016) Associations of insulin and IGFBP‐3 with lung cancer susceptibility in current smokers. J Natl Cancer Inst 108, djw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Min HY, Boo HJ, Lee HJ, Jang HJ, Yun HJ, Hwang SJ, Smith JK, Lee HJ and Lee HY (2016) Smoking‐associated lung cancer prevention by blockade of the beta‐adrenergic receptor‐mediated insulin‐like growth factor receptor activation. Oncotarget 7, 70936–70947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vincent EE, Elder DJ, Curwen J, Kilgour E, Hers I and Tavaré JM (2013) Targeting non‐small cell lung cancer cells by dual inhibition of the insulin receptor and the insulin‐like growth factor‐1 receptor. PLoS One 8, e66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duffy MJ, Synnott NC, McGowan PM, Crown J, O'Connor D and Gallagher WM (2014) p53 as a target for the treatment of cancer. Cancer Treat Rev 40, 1153–1160. [DOI] [PubMed] [Google Scholar]
- 52. Yue X, Zhao Y, Xu Y, Zheng M, Feng Z and Hu W (2017) Mutant p53 in cancer: accumulation, gain‐of‐function, and therapy. J Mol Biol 429, 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morissette MC, Vachon‐Beaudoin G, Parent J, Chakir J and Milot J (2008) Increased p53 level, Bax/Bcl‐x(L) ratio, and TRAIL receptor expression in human emphysema. Am J Respir Crit Care Med 178, 240–247. [DOI] [PubMed] [Google Scholar]
- 54. Goldkorn T and Filosto S (2010) Lung injury and cancer: Mechanistic insights into ceramide and EGFR signaling under cigarette smoke. Am J Respir Cell Mol Biol 43, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographic of the patients included for analysis.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.