

Abstract
Summary
A visualization suite for major forms of bulk and single-cell RNAseq data in R. dittoSeq is color blindness-friendly by default, robustly documented to power ease-of-use and allows highly customizable generation of both daily-use and publication-quality figures.
Availability and implementation
dittoSeq is an R package available through Bioconductor via an open source MIT license.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The tools available for analysis of sequencing-based transcriptomic data are quite diverse. Unfortunately, so are the data structures used by analysis tools for holding such data. Ideally, therefore, visualization tools would be structure agnostic. In the case of single-cell next generation sequencing (NGS) analysis, the demand for a universal and flexible visualization tool is especially high considering the popularity of the Seurat (Butler et al., 2018; Stuart et al., 2019) analysis package, as well as the plethora of analysis packages in Bioconductor (Amezquita et al., 2020) that generally utilize the SingleCellExperiment (SCE) structure. In order to optimally take advantage of all potential datasets and analysis tools, users often must work with data in both the Seurat and SCE formats, but preferably users would not also need to learn quirks of multiple, separate, structure-specific visualization tools. Although Seurat offers SCE compatibility via a conversion function, in addition to imposing jargon changes (such as ‘slot’ versus ‘assay’ or ‘pca’ rather than ‘PCA’), there are gaps in this compatibility stemming from how the function does not transfer features metadata (rowData) nor any more than two expression data matrices (assays). Any data not transferred is then inaccessible to plotting functions. Here, we present dittoSeq, a diverse and powerful visualization toolset that works natively with both structures, as well as the SummarizedExperiment structure (SE; the Bioconductor storage structure for bulk NGS data), to allow direct comparison across diverse analysis tools and out-of-the-box visualization of pre-processed data, no matter which structure. Further, dittoSeq is color blind-friendly by default, enables side-by-side visualization of single-cell and bulk RNAseq data and balances its powerful flexibility with intuitiveness and robust documentation.
2 Software description
2.1 Universal to the most common single-cell and bulk RNAseq data structures in R
dittoSeq was built with enabling side-by-side analysis of single-cell and bulk RNAseq data in mind. Thus, its visualizations rely on a set of helper functions (gene, isGene, getGenes, meta, metaLevels, isMeta, getMetas and getReductions) that properly navigate Seurat, SCE and SE object structures to retrieve necessary data. dittoSeq also allows import of DGEList bulk RNAseq data and raw matrices through conversion of such objects into an SCE via an importDittoBulk function. The most common tools used for differential gene expression of bulk RNAseq data are edgeR (Robinson et al., 2010), which uses the DGEList structure, and DESeq2 (Love et al., 2014), which uses a structure that extends the SE structure. Thus, by accepting Seurat, SCE and SE structures natively, and by providing a conversion tool for DGEList and raw data structures, dittoSeq becomes universally applicable to the most common RNAseq data structures in R.
2.2 Diverse visualizations that are powerfully customizable
Visualizations supported in dittoSeq have comparable breadth to Seurat’s but with improved handling of categorical data and enhanced customizability. Visualization functions include dimensionality reduction plots (dittoDimPlot and dittoDimHex), scatter plots (dittoScatterPlot and dittoScatterHex), heatmaps (dittoHeatmap), percent composition or expression across groups (dittoBarPlot and dittoPlot) and plotting of multiple features at once either as a single plot (dittoDotPlot and dittoPlotVarsAcrossGroups) or multiple plots (multi_dittoPlot, multi_dittoDimPlot and multi_dittoDimPlotVaryCells). All functions allow extensive customization via simple, discrete inputs that are robustly documented for ease-of-use. Examples of such customizations include: subsetting to certain cells or samples, changing sizes of data points and other representations, title adjustments, grouping data reordering and/or re-naming, automatic generation of annotations for heatmaps, overlay of trajectory analysis or density gradients onto scatter and dimensionality reduction plots, interactive plotting via plotly (Sievert, 2020) conversion and rasterization of plots with many points/cells for ease-of-use with vector-based figure editing software. Additionally, because most dittoSeq plot outputs are ggplot objects, extra layers and adjustments can be added manually via standard ggplot code. To make such manual alterations even easier, while also powering the publication-ready nature of dittoSeq plots, all functions also allow output of their underlying data via a ‘data.out’ input.
2.3 Color blindness-friendly by default
dittoSeq utilizes a modified version of the 8-color Okabe-Ito color panel—which is distinguishable by individuals with the most common forms of color blindness (Wong, 2011). By extending this panel to 40 colors, with lighter and darker repeats, we ensure that dittoSeq’s default color set is equally accessible to most users, yet also amenable to the many color requirement of complex scRNAseq data. Additional tweaks and options add to the color blindness compatibility of the package as well: default legend adjustments (enlarged keys); optional use of shapes, letter-overlay and/or faceting, in addition to coloring, when possible; optional labeling or circling of groups in scatter plots; and interactive plotting where data are displayed, in text, upon cursor hovering.
2.4 Example: visualizing expression of the human pancreas on the single-cell level
Figure 1 provides an example of how dittoSeq visualizations might be used to explore the cell type specific expression profiles of a human pancreas scRNAseq dataset (Baron et al., 2016) with visualizations that include: a UMAP plot with cell types labeled (Fig. 1a), a bar graph displaying cell type frequencies within each sample (Fig. 1b), a violin plot showing expression of a gene of interest across cell types (Fig. 1c) and a heatmap with metadata annotations (Fig. 1d). Code for producing these figures is available in Supplementary Material. dittoSeq figures like these, each obtained via a single line of code, allow viewing of expression data in multiple ways to power both initial, iterative, data interrogation as well as the production of precisely tuned, deliberately labeled, publication-quality figures.
Fig. 1.
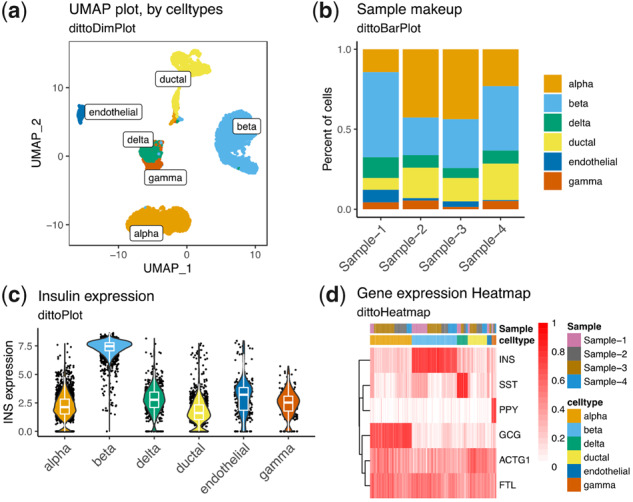
dittoSeq offers a plethora of highly customizable visualization options. Data for these figures come from Baron et al. (2016), subset to only some of the most common cell types for simplicity, then processed with a standard Seurat workflow. Plots were made with (a) dittoDimPlot, (b) dittoBarPlot, (c) dittoPlot and (d) dittoHeatmap. Data are available in the Gene Expression Omnibus at www.ncbi.nlm.nih.gov/geo and and can be accessed with GSE84133.
Supplementary Material
Acknowledgements
We would like to acknowledge Giuseppe D’Agostino, Rebecca Jaszczak and Aaron Lun for functionality and design discussions.
Funding
This work was supported by the National Institutes of Health [R21 AI120032], the Burroughs Wellcome Fund, and PREMIER, a National Institutes of Health / National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS) P30 Center for the Advancement of Precision Medicine in Rheumatology at UCSF [P30AR070155].
Conflict of Interest: none declared.
References
- Amezquita R.A. et al. (2020) Orchestrating single-cell analysis with Bioconductor. Nat. Methods, 17, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M. et al. (2016) A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cells, 3, 346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A. et al. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol., 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D. et al. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievert C. (2020) Interactive Web-Based Data Visualization with R, Plotly, and Shiny. Chapman and Hall/CRC, London. [Google Scholar]
- Stuart T. et al. (2019) Comprehensive integration of single-cell data. Cell, 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B. (2011) Points of view: color blindness. Nat. Methods, 8, 441–441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.