

Abstract
This research presents a reverse engineering approach to discover the patterns and evolution behavior of SARS-CoV-2 using AI and big data. Accordingly, we have studied five viral families (Orthomyxoviridae, Retroviridae, Filoviridae, Flaviviridae, and Coronaviridae) that happened in the era of the past one hundred years. To capture the similarities, common characteristics, and evolution behavior for prediction concerning SARS-CoV-2. And how reverse engineering using Artificial intelligence (AI) and big data is efficient and provides wide horizons. The results show that SARS-CoV-2 shares the same highest active amino acids (S, L, and T) with the mentioned viral families. As known, that affects the building function of the proteins. We have also devised a mathematical formula representing how we calculate the evolution difference percentage between each virus concerning its phylogenic tree. It shows that SARS-CoV-2 has fast mutation evolution concerning its time of arising. Artificial Intelligence (AI) is used to predict the next evolved instance of SARS-CoV-2 by utilizing the phylogenic tree data as a corpus using Long Short-term Memory (LSTM). This paper has shown the evolved viral instance prediction process on ORF7a protein from SARS-CoV-2 as the first stage to predict the complete mutant virus. Finally, in this research, we have focused on analyzing the virus to its primary factors by reverse engineering using AI and big data to understand the viral similarities, patterns, and evolution behavior to predict future viral mutations of the virus artificially in a systematic and logical way.
Keywords: Viral reverse engineering, COVID19, SARS-CoV-2, Long Short-term Memory (LSTM), Healthcare, Public health
1. Introduction
Viral reverse genetic engineering is a highly effective method for trace, analyze, visualize, and find clues about the current and future viral evolution using computing principles. Its success by dividing the virus into its primary factors will produce big data about viruses. It is used in the analysis, visualization, and artificial intelligence (AI) to predict the virus’s next evolved instance (Khan et al., 2021, Rehman et al., 2021b). Viral reverse genetic engineering is mainly used to help solve dilemmas such as SARS-CoV-2, which happened at the end of 2019 till nowadays. During the history of one hundred years, five viral families influenced human life. Also, most of these viruses are related or share some primary factors and characteristics. Genomic Characterization is a conventional laboratory method used to analyze genomes to know the generation tree (Phylogenetic) and genome sequencing. Viral reverse genetic engineering relies on genetic big data and AI with its phylogenetic tree. The entire data could be visualized to understand the patterns between viral families and behavioral evolution for each virus. Therefore, we would have an image of SARS-CoV-2 and how it differs from other viruses (Saba et al., 2020, Rehman et al., 2021a).
The main purpose of this research is to dig out whether SARS-CoV-2 follows a natural evolution behavior or not for other viruses through analyzing the big data of the virus to find key patterns. Then using the phylogenetic tree data that AI relays on in the next evolution instance. Because we believe that as much as data we can gather and clarify about the viruses, it will reflect on how we understand the viral behavior of SARS-CoV-2. Furthermore, interpreting the relation between the evolution process and natural selection. It is known that two important factors rule viral evolution. First, the natural selection factor is acquired under circumstances when two viruses with different strains meet and randomly share features to produce a new mutant viral copy. Second, the synthesis factor, where the viruses can be engineered for research and study. Finally, depending on the reverse viral engineering using AI and big data, we will detect whether the virus is evolved naturally or not.
In the past decades, many methodologies have been used in viral genetic analysis. It depends on the genetic characterization and phylogenic tree analysis—methods such as PCR and DNA sequencing (Yadav et al., 2020). The scientists used it to understand viral structure, statistics, and evolution (Khan et al., 2019). Furthermore, the use of AI is used to predict the protein structure of a given genome sequence. Therefore, these methodologies are used nowadays to help scientists increase their knowledge about viruses. Nowadays, there are valued efforts in AI for SARS-CoV-2 diagnoses to predict the infection results. Artificial intelligence (AI) may provide a method to augment the early detection of SARSCoV-2 infection. Moreover, it is a method that AI relies on the Chest Computed Tomography (CT) images from patients as a training dataset. The main aim was to build an AI model that could easily classify COVID-19 (+) patients in the early stages of SARS-CoV-2 infection using initial chest CT scans and related clinical details. It solves the time of determining the diagnosis result of the SARS-CoV-2 infection. A virus-specific reverse transcriptase-polymerase chain reaction (RT-PCR) is used in the laboratory to validate SARS-CoV-2, however, complete test period is of two days. (Mei et al., 2020).
Indeed, a new methodology is desired that could improve our understanding of the biological systems in general, especially in the 4.0 revolution industry era with AI and big data. It can make a scientific transition in technology and biology fields. Many scientists indicate the importance of AI and big data in the development of medical vaccines (Yousaf et al., 2019). It is difficult to overestimate the significance of predicting the emergence of new circulating influenza virus strains for subsequent annual vaccine production (Ayesha et al., 2021). The advent of high-throughput technology has resulted in a significant increase in biomedical data, such as genomic sequences, protein structures, and medical images, over the last few decades (Saba et al., 2020). As explained, our method is not diagnosing method. However, it aims to understand the SARS-CoV-2 data, behavior, and evolution to predict new possible viral instances of SARS-CoV-2. This will make our capabilities prepared for possible viral pandemics before it emerges.
To conclude, many scientists have indicated the importance of utilizing emerging technology to support the biological fields (Mughal et al., 2017, Mughal et al., 2018a, Mughal et al., 2018b). Nowadays, reverse engineering’s research idea using AI and big data can increase the efficiency of viral analysis to understand genome characterization and evolution behavior.
Further, this research is organized into few sections. For instance, Section 2 presents the background of the research. Section 3 explores the viral reverse process of COVID-19 virus; Section 4 exhibits the proposed methodology, experimental results and detailed discussion—finally, the conclusion and future work presented in Section 5.
2. Background
The main purpose is to know how SARS-CoV-2 differs from other viruses and whether it is evolved naturally or not. As well as what is the key data that AI can rely on in the prediction of the next possible viral evolution instance. Moreover, what is the best strategy to train this data and the prediction model that suits. For this purpose, we have decided to analyze and understand the pandemics caused by the five viral families that share some common features from 1918 till 2020 and compare it with our main use case SARS-CoV-2 presented in Table 1. Also, we developed a system to analyze each viral family with two matrices. First metrics, Amino Acids, Proteins and Genome Length to capture any patterns that are shared. The second metrics is the phylogenic tree to understand how each virus is mutating and evolving to compare it with the SARS-CoV-2 to understand the evolution process. To visualize each virus’s coherence in its family with Coronaviruses family to SARS-CoV-2 evolution behavior related to other viruses.
Table 1.
Viral families data identifications.
| Family | Viruses | Samples date | Samples size | Samples locations |
|---|---|---|---|---|
| Orthomyxoviridae | H1N1 (Spanish Flu) H2N2 (Asian Flu) H3N2 (Hong Kong Flu) H5N1 (Avian Flu) H1N1 (Swine Flu) |
1934–2013 | 5 viruses | USA, Puerto Rico Korea USA, New York China, Guangdong USA, California |
| Retroviridae | HIV-1 HIV-2 |
1981–2020 | 2 viruses 24 PEFD |
USA |
| Filoviridae | EBOV (Ebola Virus) | 1976–2020 | 1 virus 12 PEFD |
USA |
| Flaviviridae | HVC (Hepatitis-C) | 1989–2020 | 1 virus 12 PEFD |
USA |
| Coronaviridae | HCoV-OC43 HCoV-229E SARS-CoV-1 HCoV-NL63 HCoV-HKU1 MERS-CoV SARS-CoV-2 |
1960–2020 | 7 viruses 84 PEFD |
USA Canada, Toronto China, Wuhan |
Phylogenetic trees are an easy way to reflect millions or billions of years of evolution and the common history of many different species (Khan et al., 2021). Phylogenic trees are based on a hypothesis it is not necessarily definitive. However, it can give us an approximation about evolution. The Phylogenic Evolution Factor Difference (PEFD) is how fast mutations are in percentage concerning the root virus (Formula 1, 2). To find the key data that AI can rely on, we need to trace the ancestral viral roots. Moreover, to know how the mutation rate of SARS-CoV-2 is related to other viruses through the phylogenetic evolution factor difference.

We have built a Genetic Analysis (GA) program connected to the National Center for Biotechnology Information (NCBI) GenBank databases to retrieve and preprocess viral genomes, as presented in Fig. 1. Besides, after the preprocessing step from each genome, the following data will be extracted. First, Total Amino Acids, Total Proteins, Functional Proteins (length >= 25 Amino Acids), Amino Acids Occurrence and Genome Length. Second, Phylogenetic and Evolution Factor Difference (PEFD).
Fig. 1.

(a) Genetic Analysis (GA) for getting genome indicators. (b) Genetic Analysis (GA) for getting the Phylogenic tree and PEFD.
Finally, at the end of the result section, we have demonstrated the predicted result for the next evolved instance of SARS-CoV-2. To do this, we need to know the variables that we want to predict. First, we need to predict genome amino acids. Second, we need to predict the genome length. The prediction of the genome’s amino acids comes from understanding the structure of the genome. The genome is constructed of genes; every three genes represent one amino acid, start, or stop signs. It is called “Codons” to indicates the start and stop of amino acids as presented in Fig. 2. The group of amino acids constructs proteins. In bioinformatics, each amino acid assigned to three codes is called ‘Codons’ exhibited in Fig. 3. Therefore, we have proposed a strategy we called it three fit strategies by understanding the genetic structure. We make the machine learns by three genes: codons represent each amino acid presented in Fig. 4. In this paper, we predict the genome of ORF7a protein from SARS-CoV-2 as a first stage. We have used genome lengths from the phylogenetic tree to train the machine to understand its behavior through the evolution process. Our dataset is from the phylogenetic tree for the virus we want to predict. We have proposed an AI algorithm based on Long Short-Term Memory (LSTM) neural network since the dataset is based on linguistic representation.
Fig. 2.

Genome represented in three codons corresponding to one amino acid.
Fig. 3.
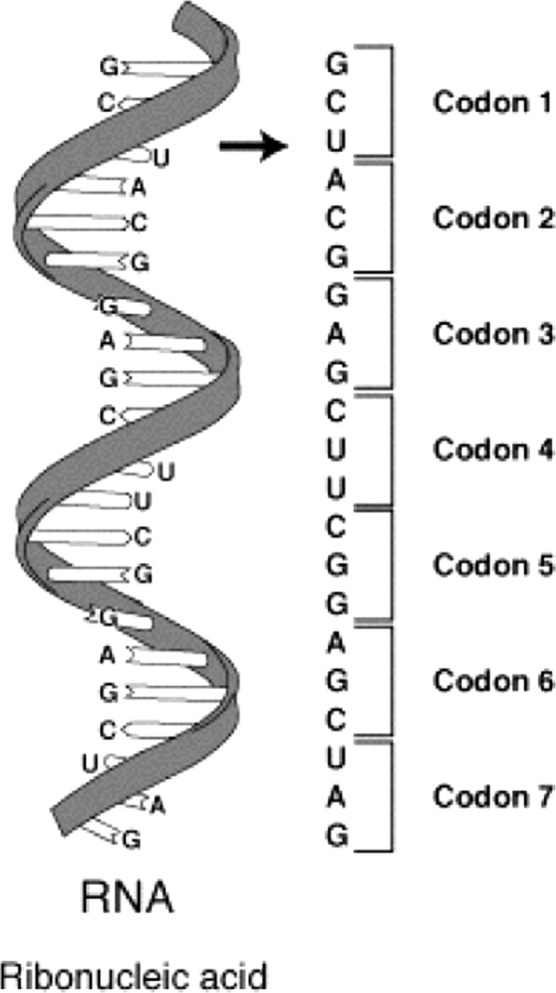
The mRNA structure with corresponding codons.
Fig. 4.

The training formation of the genomes in the Machine.
As known LSTM is mostly used in classification, prediction fault diagnosis based on time series data applications (Saba et al., 2021, Saba, 2021). RNN is a type of neural network that uses hidden units to analyze data streams. The performance of certain applications, such as text processing, speech recognition, and DNA sequences, is based on previous calculations. LSTM can keep large value dependencies for large sequences such as viral genomes, whether DNAs or RNAs. RNNs are usually fed input samples with a higher degree of interdependence. They also have a significant representation for storing data regarding previous time measures (Durbhaka et al., 2021, Khan et al., 2020). The genome codes “codons” are encoded into long string letters representing amino acids, start, and stop codons as in Fig. 4, Fig. 2. The model can be trained using genetic format as shown in Fig. 4, since it is based on predicting three letters of codons that form amino acids, start, and stop signs. Therefore, the machine will recognize the genome’s natural construction, as shown in Fig. 2, Fig. 3. This machine model is based on understanding how the viral genome is constructed as ribosomes act inside the cell in translating the genome codes into amino acids, start, and stop signs, therefore complete virus (Farabaugh, 2009). Finally, we want to have the precedence in identifying and characterizing future mutant viruses before it emerges in our existence such as SARS-CoV-2 and other pandemics that happened in the past one hundred years era. With more advanced and faster technologies comparing to other methods mentioned above.
3. Viral reverse engineering
Each viral family’s results are divided into five sub-sections with a brief history of each family and its analyzed viruses with each of the pandemic conditions. This process will let us understand the viral coherence between SARS-CoV-2 and all the viruses that happened for one hundred years from the smallest viral genes to viral behavior. At the end of the result section, we will compare it with the result related to SARS-CoV-2 as exhibited in Fig. 5.
Fig. 5.

Genetic reverse engineering process.
3.1. Orthomyxoviridae
Few viral diseases have played a more central role in the history of virology than influenza. As the First World War ended, the pandemic that swept the world in 1918 was the deadliest one ever, killing about 40–50,000 million people (Burrell et al., 2017). This section will dive into a family named Orthomyxoviridae that all Influenza viruses spread from it. As known, Influenza viruses and Coronaviruses share more identical shapes. However, Influenza viruses differ in some characteristics and the main one is the genome structure. Other viruses have one complete genome segment. On the other hand, the influenza virus’s genome is divided into seven segments. In fact, that difference does not affect the virus, but only in the preprocessing. Moreover, it played a major role in the past 100 years in the history of the pandemic. Table 2 presents a description of each influenza virus with its identification.
Table 2.
Influenza virus identification.
| Virus Name | Geo location | Collection date |
|---|---|---|
| H1N1 (Spanish Flu) | USA, Puerto Rico | 1934 |
| H2N2 (Asian Flu) | Korea | 1986 |
| H3N2 (Hong Kong Flu) | USA, New York | 2004 |
| H5N1 (Avian Flu) | China, Guangdong | 1996 |
| H1N1 (Swine Flu) | USA, California | 2009 |
We could not compare each genomic segment with other viruses with one complete genome segment due to the genomic structure’s differences. We have chosen H1N1 (Spanish, Swine) to analyze amino acids, proteins, and genome length, to capture similarities and patterns with respect to the SARS-CoV-2, because of its high impact on all Influenza viruses in 1918 and 2009. Without considering Phylogenic Evolution Factor Difference (PEFD) in the Influenza viruses’ section, because of the genome structure difference. The influenza A and B viruses both have eight negative-sense, single-stranded viral RNA (vRNA) segments in their genomes, while the influenza C virus only has seven (Bouvier and Palese, 2008). Finally, we have agreed that Coronaviruses and Influenza viruses’ main characteristics are considered the building blocks for the viral structure, such as amino acids, proteins, and genome length. Therefore, that gives us a clue about the virus functions between Coronaviruses and Influenza.
3.2. H1N1 (Spanish Flu)
Back at the be beginning of 1918, the H1N1 virus arises as a pandemic known as Spanish flu. It is considered the first influenza pandemic in the 20th century. Spanish flu does not originate in Spain. Actually, Spanish government was one of the first to recognize the existence of a new and strange disease in their country. During the first outbreak of influenza, which occurred in the summer of 1918, the virus spread rapidly. It affected a large proportion of the population and few to no people were immune to it, but only a few people died as a result of it (Mamelund, 2017). H1N1 spreads worldwide in the middle of World War I crisis. There is no consensus on the reason for H1N1 pandemic. Researchers claimed that, ”From unclear origin, it spread around the globe in three waves in 1918–19; nearly a third of the world population tallying 1.8 billion was infected, and an estimated 50–100 million died from the disease in less than a year” (Mamelund, 2017, The Spanish Influenza of 1918–20). However, World War I, was considered to be a fertile circumstance to produce viruses such H1N1. The genome data for H1N1 tabulated in Table 3.
Table 3.
H1N1 (Spanish flu)’s genome data.
| No. | Gnome/segment | Accessions | Total protein sequences | Functional proteins | Total amino acids | Highest amino acid occurrence (%) |
|---|---|---|---|---|---|---|
| 1 | Neuraminidase (NA) | NC_002018.1 | 38 | 5 | 430 | L: 12.3% |
| 2 | Hemagglutinin (HA) | NC_002017.1 | 42 | 8 | 548 | S: 8.4% |
| 3 | Matrix Protein (M1) Ion Channel (M2) |
NC_002016.1 | 27 | 3 | 316 | G: 7% |
| 4 | RNA Polymerase (PA) | NC_002022.1 | 3 | 1 | 742 | E: 10.5% |
| 5 | RNA Polymerase (PB1) | NC_002021.1 | 5 | 1 | 776 | L: 7.6% |
| 6 | RNA Polymerase (PB2) | NC_002023.1 | 3 | 1 | 778 | R: 8.2% |
| 7 | Nucleoprotein (NP) | NC_002019.1 | 2 | 1 | 520 | R: 9.9% |
| 8 | Non-Structural Protein (NS1) Non-Structural Protein (NS2) |
NC_002020.1 | 15 | 2 | 280 | L: 13.6% |
The accessions were taken from [29].
From analyzing H1N1 genome segments, we found that three segments have the highest number of protein sequences, which are HA, NA, and M1, M2. First, hemagglutinin (HA) is responsible for attaching and entering the cell. The main player is the viral surface glycoprotein, HA, which has two functions of binding to the receptor and fusion at the cell membrane (Mamelund, 2017). The hemagglutinin has the highest protein sequences among two with 42 sequences and 5 functional proteins with 430 amino acids. According to the collected data, hemagglutinin has a major function with a mutation rate higher than other segments. Therefore, it will lead the virus to develop itself to make new ways to attach and enter the cell body through evolution. Second, neuraminidase (NA) is the second-highest protein sequence among the two segments with 38 sequences and 8 functional proteins with length of 548 amino acids. The virus uses neuraminidase to exit the cell body by attaching the cell body from inside. Third, matrix protein (M1) and ion channel (M2) have the third-highest protein sequences with 27 sequences and 3 functional proteins with a length of 316 amino acids. Matrix protein is a mechanism that is responsible for making the genome segments stuck through tiny object-like strings inside the virus. Based on these findings, we evaluate that ”It has been postulated that the M1 protein forms a shell lining the inner surface of the viral envelope. This shell would act as a bridge between the internal components (RNPs and NEP) of the virion and the membrane proteins”. An ion channel is located on the viral body. When the virus enters the cell body ion channel, it will allow Acidic Interior particles to enter the viral body. Therefore, it will remove the matrix protein links and at that stage, the genome will be ready to go to the cell nucleus for the replication process. During viral entry, the proton-selective ion channel function of M2 protein promotes uncoating of the influenza virus ribonucleoprotein core after membrane fusion (Alvarado-Facundo et al., 2015).
Fig. 6 presents the amino acid variations of all the segments. Moreover, it indicates what the most shared amino acids with SARS-CoV-2 are. The most occurred amino acids are L in Neuraminidase with 12.3%, T in Hemagglutinin with 9% and G in (M1, M2) with 7% in all top three segments. As we discovered before, the main functions have happened in the top three proteins. Therefore, we can observe the most occurred amino acids as the main building blocks of these mutational changes due to it is acting like functional particles in the segment. This result helped us understand the important genetic patterns between SARS-CoV-2 and Influenza H1N1 in the last result section.
Fig. 6.

Amino Acids percentages per segment.
3.3. H1N1 (Swine Flu)
On April 15 and April 17, 2009, the Centers for Disease Control and Prevention (Saba et al., 2020) (CDC) identified two cases of human infection with a swine-origin influenza A (H1N1) virus (Dandagi and Byahatti, 2011). H1N1 refers to Swine flu that was reported in 2009. Through the transferring of the virus from pigs to humans. As Dandagi & Byahatti Claimed, “This strain can be transmitted from human to human (Yadav et al., 2020) and causes the normal symptoms of influenza”. (Dandagi & Byahatti, 2011, VIROLOGY). H1N1 (Swine) evolved in the past decades through many antigenic shifts between avian and swine virus strains.
From the data collected in Table 4 we can recognize the difference between the viral indications. And that is because of many antigenic shifts through time, according to the data. We have captured that the highest segments with total protein sequences are (M1, M2) and (NS1, NS2) with 6 sequences for each. Moreover, change can be recognized in the segment structure. This will affect the function of viral behavior. That clarifies how H1N1 can mutate faster concerning the circumstances since H1N1 1918. In the final result section, we have seen that SARS-CoV-2 has the same fast mutation behavior with time arisen.
Table 4.
H1N1 (Swine flu)’s genome data.
| No. | Gnome/segment | Accessions | Total protein sequences | Functional proteins |
Total amino acids |
Highest amino acids occurrence (%) |
|---|---|---|---|---|---|---|
| 1 | Neuraminidase (NA) | NC_026434.1 | 1 | 1 | 469 | G: 9.6%, I: 9.6% |
| 2 | Hemagglutinin (HA) | NC_026433.1 | 1 | 1 | 566 | S: 8.3% |
| 3 | Matrix Protein (M1) Ion Channel (M2) |
NC_026431.1 | 6 | 1 | 322 | A: 9.3% |
| 4 | RNA Polymerase (PA) | NC_026437.1 | 1 | 1 | 716 | E: 10.6% |
| 5 | RNA Polymerase (PB1) | NC_026435.1 | 1 | 1 | 757 | T: 7.8% |
| 6 | RNA Polymerase (PB2) | NC_026438.1 | 1 | 1 | 759 | V: 8.3% |
| 7 | Nucleoprotein (NP) | NC_026436.1 | 1 | 1 | 498 | R: 10% |
| 8 | Non-Structural Protein (NS1) Non-Structural Protein (NS2) |
NC_026432.1 | 6 | 1 | 282 | L: 10.3% |
The accessions were taken from [29].
In Fig. 7 we can find small differences in the variations of the amino acids. The highest amino acids in all segments are A (M1, M2) with 9.3% and L in (NS1, NS2) with 10.3%. This data indicates that changes in the amino acid structures require more than one antigenic shift through time. After the antigenic change between classical swine H1 and human seasonal H1 was detected, a significant antigenic difference still existed between classical swine H1 and human seasonal H1. (Garten et al., 2009). Hence, it is clear in the data between Spanish and Swine flu viruses.
Fig. 7.

Amino Acids percentages per segment.
3.4. Retroviridae
Retroviral viruses infect a vast number of animal species and are associated with many diseases in both humans and livestock (Retroviridae, 2017). It is a viral family that causes the viral infection Human immunodeficiency virus (HIV). HIV viruses spread in the middle of the 20 century and many humans were infected by HIV and known as HIV-1. And it is an early stage that will lead the virus to be in the perilous stage called AIDS. The first HIV was reported in 1959 from human sample blood in Kinshasa at Democratic Republic of Congo. The most ancestral available HIV-1 sequences were recovered from Kinshasa in 1959 (Rubio-Garrido et al., 2020). A human blood sample obtained in 1959 from west-central Africa holds earliest trace of the AIDS pandemic. The exact timing and circumstances of early events in the SIVcpz/HIV-1 zoonosis, however, are unclear (Gao et al., 1999b), because of two possible theories. First, chimpanzee as researchers claimed, “However, for many years, chimpanzees were not accepted as the source of HIV-1 because it remained unclear whether wild chimpanzees are naturally infected with this virus”. (Sharp & Hahn, 2010, THE ORGINES OF HIV-1 AND HIV-2). Second, according to the Medical Doctor and Minister Dr. Abdul Alim Muhammad, he convinced AIDS virus was a genocidal weapon used to spread it in Africa (Gardell, 1996). From the researcher’s variant claims and findings, we can sense that HIV existence, origins, and behavior are not usual. Before the end of the 20 century, HIV-1 evolved to HIV-2, a more aggressive HIV-1 virus. This section will dive and analyze HIV-1 and HIV-2 viruses to finally compare them with SARS-CoV-2. We have compared all the genomes with PEFD of HIV-1 and HIV-2 together because the genetic structure for both HIV-1 and HIV-2 has one complete genome segment instead of seven segments like Influenza viruses. Table 5 describes the HIV-1,2 that we have analyzed its data concerning SARS-CoV-2.
Table 5.
HIV identification.
| Virus name | Geo location | Release date |
|---|---|---|
| HIV-1 | USA | 2015 |
| HIV-2 | USA | 2015 |
Human immunodeficiency virus (HIV) is known as one of the spherical-shaped viruses common characteristic with Coronaviruses. Moreover, HIV has the same attaching mechanism and is detached from the cell body in Coronaviruses by the env-Glycoprotein Complex which are the proteins that cover the viral body of HIV. This section has done genetic analysis on the viral genomes and phylogenic evolution factor difference on the virus to capture the virus’s behavior through time and how fast HIV mutates, then compare it with our main use case SARS-CoV-2 at the final result section.
From Table 6, we can find the difference between viral indicators of HIV-1,2. As known, the first was HIV-1 with total and functional proteins higher and lowered length than HIV-2. The high variations between the genome data because the evolution process caused changes in the viral mutations in HIV-1 at the end of the 20th century is speedy. HIV-1, on the other hand, evolves rapidly. For this amount of diversity to have accumulated, the virus would have existed within human populations for several years before it was first identified (Sharp and Hahn, 2010).
Table 6.
HIVs genome data.
| Name | Total proteins | Accessions | Functional proteins | Total amino acids | Highest amino acids occurrence (%) | Total PEFD |
|---|---|---|---|---|---|---|
| HIV-1 | 243 | NC_001802.1 | 35 | 2818 | R: 9.5% | 147.26 |
| HIV-2 | 135 | NC_001722.1 | 25 | 3319 | R: 8.6% | 143.5 |
The accessions were taken from [29].
Fig. 8 indicates the change in the variations of amino acids. The highest three amino acids in both viruses are R, S, K, L and G. In HIV-1, the percentage of the amino acid is R(9.5%), S(9.4%) and K(8%). And in HIV-2, the percentage of the amino acid are R(8.6%), L(8.3%) and G(8%). This Amino acid changed through the evolutions process. However, it has a high occurrence in both viruses.
Fig. 8.

Amino Acids percentages per virus.
The diagram above shows us the behavior of the HIV-1 evolution. By applying the formula in PEFD (Formula 1 and 2). We can get the PEFD for HIV-1 concerning the other evolve instance of the virus and the total PEFD. At the beginning of other viral evolution, we can observe that it begins with 11.76% and continues to oscillate to 12.49%. Then the graph shows a smooth increase in evolution until it reaches 12.64%. On the other hand, At the beginning of other of the viral evolution of HIV-2, we have observed that it begins with 6.1% and increases to 18.5%. Then the graph shows a smooth decrease in the evolution process until it reaches 18.1%. The final total PEFD value for HIV-1 is 147.26 and HIV-2 is 143.5. The total PEFD results for both viruses are close to each, indicating that HIV-1 and HIV2 have much identical evolution speed. However, both viruses defer in the evolution behavior according to Fig. 9, Fig. 10. With keeping in mind that the duration of the viruses is between 1959 to 2020.
Fig. 9.

HIV-1 PEFD graph.
The accessions were taken from [29].
Fig. 10.

HIV-2 PEFD graph.
The accessions were taken from [29].
3.5. Filoviridae
The first discovery of Ebola virus was in Sudan, Africa as a result of outbreaks of hemorrhagic fever (Baron et al., 1983). It took around 20 years to spread again in 2014. By the evolution process through time caused mutational changes; therefore, Ebola spread again in 2014. We have analyzed the virus with PEFD to study the viral behavior of Ebola with respect to SARS-CoV-2. Ebola virus-like other viruses share similar attachment mechanisms using Glycoprotein (GP) (Gardell, 1996).
In Table 7, the identification of the Ebola virus from the NCBI databases. This Ebola virus sample is traced from states. We have done a genetic analysis with PEFD to capture the patterns and behavioral evolution of the Ebola virus and its evolution score.
Table 7.
Ebola virus identifications.
| Virus name | Geo location | Release date |
|---|---|---|
| EBOV | USA | 2015 |
Table 8 indicates to our understanding the variations in the total protein sequences, functional proteins, and length of the genome. Ebola virus has the longest length of 5849 amino acids between all viruses mentioned above. According to the analyzed data, the total and functional proteins are higher than Influenza and Retroviridae. Therefore, that is why Ebola virus symptoms are so deadly to Influenza and Retroviridae viruses. Lastly, we have compared Ebola result with SARS-CoV-2 to capture the difference in the behavior, especially as analyzed data shows that as the genome length increases and total proteins, the more aggressive the virus behavior is. In Fig. 11, we have observed that the top three amino acids used in Ebola virus are S(10.3%), L(9.7%) and R(7.9%). We have noted that the top three amino acids are captured in Influenza and Retroviridae viruses as high occurred amino acids.
Table 8.
Ebola virus genome data.
| Name | Total proteins | Accessions | Functional proteins | Total amino acids | Highest amino acids occurrence (%) | Total PEFD |
|---|---|---|---|---|---|---|
| EBOV | 465 | NC_014373.1 | 70 | 5849 | L: 9.7% | 193.1 |
The accessions were taken from [29].
Fig. 11.

Amino acid percentages.
Fig. 12 the PEFD of Ebola virus shows a smooth increase in the first three evolved viruses from 0% to 1.3%. Until we have captured a huge increase in the graph to 23.4% with a smooth increase, that is because of antigenic shifts in the viral genome. We have captured a smooth decrease in the evolution process in the last two evolved viruses to 21.1% and 22.2%. The total PEFD score for Ebola virus is (193.1) for the evolution time 1976 to 2020. Finally, the virus’s evolution speed is higher than the mentioned viruses with unstable behavior at the beginning, according to Fig. 13.
Fig. 12.

Ebola virus PEFD.
The accessions were taken from [29].
Fig. 13.

Amino acid percentages.
3.6. Flaviviridae
The hepatitis C virus is an RNA virus that belongs to the family Flaviviridae (Chen and Morgan, 2006, Lauer and Walker, 2001). Hepatitis-C infects and spread in the liver and causes a disease that the immune system cannot control to defends its spreads. Table 9 shows the identification of HVC virus from the NCBI GenBank databases. We have done genetic analysis with PEFD to capture the patterns with its viral behaviors in the evolution process to SARS-CoV-2. Table 10 clarifies that the Hepatitis-C virus (HCV) has more stable behavior related to other viruses. Hepatitis-C has total proteins 103 and 44 functional proteins with a length of 3097. As data showed before in Influenza, Retroviridae, and Filoviridae, we have captured that HCV has the highest functional protein sequences. The genome data was compared with SARS-CoV-2 in the final result section.
Table 9.
Hepatitis-C (HCV)’s identification.
| Virus name | Geo location | Release date |
|---|---|---|
| Hepatitis-C (HCV) | USA | 2018 |
Table 10.
Hepatitis-C virus (HCV)’s genome data.
| Virus name | Total proteins | Accessions | Functional proteins | Total amino acids | Highest amino acids occurrence (%) | Total PEFD |
|---|---|---|---|---|---|---|
| Hepatitis-C (HCV) | 103 | NC_038882.1 | 44 | 3097 | S: 12.7% | 31.9 |
The accessions were taken from [29].
From Fig. 13, we have observed that S(12.7%), P(11.7%) and R(10.1%) are the highest occurred amino acids in the Hepatitis-C virus. Moreover, we have noticed that amino acids S and R are high in Influenza, Retroviridae and Filoviridae. Therefore, the amino acid patterns indicate that the virus may share genes with those previous viruses.
In Fig. 14, we have captured the starting point of PEFD with 0.3% with smooth increasing. At the middle of the graph, at 4.1% after smooth increasing a small decreasing with 3%, the virus continues with the smooth increasing the evolution process. The hepatitis-C virus shows a more stable behavior in the evolution with 31.9 total PEFD scores for PEFD speed of Ebola with 193.1 and HIV-1 with 147.26 and HIV-2 with 143.5. With keeping in knowledge, the samples were taken for the discovery time till 2020.
Fig. 14.

Hepatitis-C (HCV)’s PEFD.
The accessions were taken from [29].
4. Proposed methodology
SARS-CoV-2 is the research’s main use case. This section evaluates the variations and differences of all Coronaviruses that happened in the past 100 years until we reach SARS-CoV-2 to the viruses that happened in the same era: Influenza Retroviruses, Filoviruses and Flaviviruses. In Table 11, identifications for each coronavirus that took from NCBI GenBank databases. We have done a study that includes genetic analysis, PEFD. Finally, an algorithm that predicts the next possible evolution instance of SARS-CoV-2 using Artificial Intelligence (AI) will be in the next section. Table 12 shows data of all seven coronaviruses. According to Table 12, Coronavirus family shows high genome length, total proteins, and functional proteins. The raising of Coronaviruses begun with HCoV-229E and HCoV-OC43 viruses. It shows a coherence in the total protein sequences with 32 sequences and syndromes since they are very close in terms of the time period. HCoV-2295 and HCoV-OC43 were the only HCoVs known before the SARS epidemic (Gaurav and Al-Nema, 2019a). Next, according to the total proteins and genome length of SARS-CoV is the closest to SARS-CoV-2 from genome structure for the PEFD result. Moreover, from Table 12 HCoV-NL63 shows low total protein sequences to other Coronaviruses. The next virus is HCoV-HKU1, discovered besides HCoV-NL63; however, they differ due to the evolution process. MERS-CoV virus has a high genome length with the lowest protein sequences among all Coronaviruses. SARS-CoV-2 shows the highest protein sequences and functional proteins among all viruses in the past 100 years. Lastly, in the final result, we have demonstrated the coherence between SARS-CoV-2 and other viruses. Fig. 15 shows the variations of all amino acid’s occurrences in all Coronaviruses. C, L, T, I, V and S are the most occurred amino acids. And some of the highest amino acids are in common with Influenza viruses, Retroviruses, Filoviruses and Flaviviruses. Influenza viruses share amino acid T and L, Retroviruses with S and L, Filoviruses with S and L, and Flaviviruses with S.
Table 11.
Coronaviruses identifications.
| Virus name | Geo location | Collection date |
|---|---|---|
| HCoV-229E | – | – |
| HCoV-OC43 | USA | – |
| SARS-CoV | Toronto, Canada | – |
| HCoV-NL63 | – | – |
| HCoV-HKU1 | USA | 2010 |
| MERS-CoV | – | 2012 |
| SARS-CoV-2 | China | 2019 |
Table 12.
Coronavirus’s genome data.
| Gnome | Accessions | Total Protein Sequences | Functional proteins | Total amino acids | Highest amino acids occurrence (%) | Total PEFD |
|---|---|---|---|---|---|---|
| HCoV-229E | NC_002645.1 | 762 | 28 | 8344 | C: 10%, L: 10% | 86.4 |
| HCoV-OC43 | NC_006213.1 | 794 | 92 | 9454 | L: 18.4% | 9.8 |
| SARS-CoV | NC_004718.3 | 804 | 43 | 8381 | L: 12.6% | 0.59 |
| HCoV-NL63 | NC_005831.2 | 370 | 60 | 9625 | L: 15.7% | 28 |
| HCoV-HKU1 | KF686346.1 | 775 | 41 | 9193 | L: 9.6% | 147.6 |
| MERS-CoV | NC_019843.3 | 273 | 68 | 9645 | L: 14.2% | 3.94 |
| SARS-CoV-2 | NC_045512 | 960 | 113 | 9350 | L: 18.3% | 0.134 |
The accessions were taken from [29].
Fig. 15.

Amino acid percentages per virus.
In Fig. 16 we have captured the evolution process of the HCoV-229E. It shows that the virus’s evolution process is stable concerning the started PEFD (0%). It continues by increasing in the evolution with sudden increasing in (7%) and (24.5%) points since it is the first Coronavirus captured in the 1960s era, its evolution behavior more stable with fast evolution speed concerning total PEFD score (86.4). Fig. 17 shows the evolution behavior of HCoV-OC43 virus discovered besides HCoV-299E. According to the total PEFD score (9.2), the virus has a slower evolution than HCoV-299E with no stable evolution behavior.
Fig. 16.

HCoV-229E PEFD.
The accessions were taken from [29].
Fig. 17.

HCoV-OC43 PEFD.
The accessions were taken from [29].
Fig. 18 describes the evolution process of the closest virus to SARS-CoV-2. SARS-CoV has a total PEFD score (0.59), the lowest evolution speed among mentioned viruses and its reasonable result concerning the time SARS-CoV emerges. Therefore, it indicates that the virus can keep evolving exponentially with little drops, sometimes with low evolution difference.
Fig. 18.

SARS-CoV PEFD.
The accessions were taken from [29].
Similarly, Fig. 19 describes the evolution behavior of HCoV-NL63. This virus spread after SARS-CoV. According to the total PEFD score (28) the evolution process is slow, with small evolution increasing. Then, we have captured a huge increasing behavior according to with PEFD score (18.2%). Ending with low drop-in in the evolution process. Fig. 20 presents the behavior of the HCoV-HKU1 virus it was discovered besides HCoV-NL63. According to the PEFD the virus has unstable behavior through the evolution, starting with (0%) ending with spike and smooth dropping by (21.2%). The total PEFD score (147.6) is fast evolution behavior concerning other Coronaviruses.
Fig. 19.

HCoV-NL63 PEFD.
The accessions were taken from [29].
Fig. 20.

HCoV-HKU1’s PEFD.
The accessions were taken from [29].
In Fig. 21, MERS-CoV shows more stable behavior according to the PEFD. The evolution process starts with one increasing spike from (0%) to (0.3%), followed by increasing, ending with smooth decreasing with (0.34%). The evolution speed of MERS-CoV is lower than other Coronaviruses, with a total PEFD score (3.94). Fig. 22 describes the behavioral evolution process of the SARS-CoV-2. According to the total PEFD score (0.134), the virus’s evolution speed is slower than other viruses. However, it is considered a fast evolution concerning the time that SARS-CoV-2 arises, one year. The PEFD describes the viral evolution concerning the time that virus spread in 2019 until 2020, almost one year. The evolution process starts with smooth increasing with (0.007%) then it continues with increasing, ending stable behavior with (0.02%).
Fig. 21.

MERS-CoV’s PEFD.
The accessions were taken from [29].
Fig. 22.

SARS-CoV-2’s PEFD.
The accessions were taken from [29].
4.1. SARS-CoV-2 genome prediction using AI
This section has utilized the reverse engineering technique using the big data gathered about the viral genomes. Furthermore, we got a clearer image of how viruses are differing in behavioral evolution. Therefore, we have designed an AI algorithm that utilizes the genetic code to predict the virus’s next evolution instance.
Since the genome data is represented in the linguistic letters, we have decided to predict the next viral evolution instance using long short-term memory (LSTM) neural network. Since it could be used in the sequence prediction. Because of the large size of one complete viral genome, we decided to predict one protein sequence of SARS-CoV-2. We have trained the machine from the phylogenic tree on the genetic sequences of SARS-CoV-2 “ORF7a protein” to predict the next evolved sequence as shown in Fig. 23. Once the machine produces a given sequence prediction, we have uploaded it to the “Robetta structure prediction tool” model the 3D structure of the protein sequence.
Fig. 23.

ORF7a protein prediction using LSTM neural network to predict the protein sequence. And “Robetta” structure prediction tool from Baker lab at University of Washington. (Main, 2020).
4.2. Experimental results and analysis
Influenza viruses HIVs share some of their structural characteristics with SARS-CoV-2. The hemagglutinin (HA) is the protein responsible for attaching to the cell body. Researchers claimed that it was found in the Influenza and SARS-CoV-2 viruses as a functional mechanism exhibited. From the data, we conclude that Influenza H1N1 has a common main feature with SARS-CoV-2, which is HA protein. Retroviruses shares some main behavioral characteristics with SARS-CoV-2. First, there is no consensus on the origin of both viruses. Second, SARS-CoV-2 and HIVs viruses have a similar attachment mechanism: “Spike protein and Glycoprotein” to the cell body. According to the amino acids percentage tables, we have noticed that the most active amino acids in both SARS-CoV-2 and HIV are S and L are used highly in three inserts. Lastly, SARS-Cov-2 and HIVs can both attacking white blood cells. Hence, it is possible that SARS-CoV-2 infects cells through a membrane fusion that involves the S protein Gaurav and Al-Nema (2019b).
Filovirus Ebola shares two main characteristics with SARS-CoV-2. First, the most occurred amino acids in SARS-CoV-2 and Ebola viruses are S and L. Moreover, SARS-CoV-2 and Ebola viruses have the same protein receptor, known as S protein or Glycoprotein Wang et al. (2020). Flavivirus Hepatitis-C shares two common characteristics with SARS-CoV-2. Which is the amino acid that used in both viruses is S. (Chan et al., 2020, Huang et al., 2020)
Finally, in Coronaviruses, we have captured that the closest virus to SARS-CoV-2 is SARS-CoV with high similarity in the genome and total PEFD score as depicted in Fig. 22.
4.3. Discussion
We have known how to utilize genetic data and AI to understand viral evolution, behavior, and patterns by applying reverse engineering techniques (Rehman et al., 2018, Mujeeb et al., 2019, Mittal et al., 2020). According to reverse engineering results, we have seen that viral families’ past 100 years share some significant characteristics with SARS-CoV-2. The highest occurred amino acids, genome length, genome structure, protein sequences and PEFD score. SARS-CoV-2 is from the highest viruses in genome lengths, total and functional protein sequences between all viral families. We have found that most of the analyzed viruses have common occurred amino acids percentage with SARS-CoV-2. These amino acids may reflect the SARS-CoV-2 functions such as attaching and detaching mechanisms responsible for attaching to the cell body. Furthermore, Influenza, HIV, and Ebola are sharing the same attaching mechanism in the viral surface. And as known, it is impossible that these viruses can be under antigenic reassortment process unless it is from the same viral families. Therefore, these facts support that the SARS-CoV-2 has developed with external interference factor.
We have reached a significant result by discovering the dataset that AI can rely on in the prediction step (Rehman et al., 2020). It has been achieved after diving into genetic analysis to understand viral big data. The machine model utilizes the viral genome in the virus’s phylogenic tree to understand the genetic code. LSTM sequence-to-sequence model shows an efficient result with 72% accuracy in the evolved instance prediction of ORF7a protein sequence. In the beginning, we have noticed that the accuracy is 4050%, and that is because the pattern consists of the dataset. However, if we have more data with the consistent change, this will increase the accuracy and this how we raised the accuracy from 40% to 72%. Finally, it is considered as a breaching step in the road to predict one complete viral genome.
5. Conclusion and future work
This research has presented how viruses emerged in last one hundred years to find clauses to understand SARS-CoV-2 patterns and its evolution behavior. In summary, reverse engineering techniques using AI and big data infection analysis can efficiently decompose the virus into its primary factors. These factors can be described as “virus big data”. It captured the viral patterns between all the viruses in the one-hundred-year era with the most common features with evolution behavior. This research shows how we can utilize the viral genome’s data from the virus’s phylogenetic tree to predict the same virus’s evolved instance. As the first stage in this research, we have taken one small protein sequence ORF7a of SARS-CoV-2 with a length of 29 amino acids then we have predicted the possible evolved instance of the protein. In the next paper we will start the last stage of analyzing and predicting viruses in two parts. Firstly, improve our Genetic Analysis (GA) system to automate the process of requesting, preprocessing, and analyzing the virus’s genome from the GenBank databases. Secondly, improving the AI algorithm to predict complete evolved SARS-CoV-2 virus as chains of predicted viruses as shown in the PEFD graphs through tarin the AI model on complete virus’s genome. Also, improving the accuracy since its at best result 72% by increasing the viral genome datasets’ variation with consistent patterns. This is how did improve the accuracy from 50% to 72%. This will improve the way of understanding SARS-CoV-2 evolution behavior and viral data.
CRediT authorship contribution statement
Ahmad M. Abu Haimed: Conceptualization, Methodology, Writing - original draft. Tanzila Saba: Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing - review & editing. Ayman Albasha: Software, Validation, Visualization. Amjad Rehman: Investigation, Methodology, Project Administration. Mahyar Kolivand: Formal analysis, Validation, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the research project [Genetic Virus Reverse Engineering] Prince Sultan University Saudi Arabia , [COVID19-CCIS-2020{60}]. The authors are thankful to Artificial Intelligence and Data Analytics (AIDA) Lab CCIS Prince Sultan University Saudi Arabia.
References
- Alvarado-Facundo E., Gao Y., Ribas-Aparicio R.M., Jiménez-Alberto A., Weiss C.D., Wang W. Influenza virus M2 protein ion channel activity helps to maintain pandemic 2009 H1N1 virus hemagglutinin fusion competence during transport to the cell surface. J. Virol. 2015;89(4):1975–1985. doi: 10.1128/JVI.03253-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayesha H., Iqbal S., Tariq M., Abrar M., Sanaullah M., Abbas I., et al. Automatic medical image interpretation: State of the art and future directions. Pattern Recognit. 2021;107856 [Google Scholar]
- Baron R.C., McCormick J.B., Zubeir O.A. Ebola virus disease in southern Sudan: hospital dissemination and intrafamilial spread. Bull. World Health Organ. 1983;61(6):997–1003. [PMC free article] [PubMed] [Google Scholar]
- Bouvier N.M., Palese P. The biology of influenza viruses. Vaccine. 2008;26(Suppl 4):D49–D53. doi: 10.1016/j.vaccine.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell C.J., Howard C.R., Murphy F.A. Orthomyxoviruses. Fenner and White’s Med. Virol. 2017;35:5–365. doi: 10.1016/b978-0-12-375156-0.00025-4. [DOI] [Google Scholar]
- Chan J.F.-W., Kok K.-H., Zhu Z., Chu H., To K.K.-W., Yuan S., Yuen K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting wuhan. Emerg. Microbes Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.L., Morgan T.R. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 2006;3(2):47–52. doi: 10.7150/ijms.3.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandagi G.L., Byahatti S.M. An insight into the swine-influenza A (H1N1) virus infection in humans. Lung India: Official Organ Indian Chest Soc. 2011;28(1):34–38. doi: 10.4103/0970-2113.76299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbhaka G.K., Selvaraj B., Mittal M., Saba T., Rehman A., Goyal L.M. Swarm-LSTM: Condition monitoring of gearbox fault diagnosis based on hybrid LSTM deep neural network optimized by swarm intelligence algorithms. CMC-Comput. Mater. Continua. 2021;66(2):2041–2059. [Google Scholar]
- Farabaugh P.J. Translational control and fidelity. Encycl. Microbiol. 2009;51:7–528. doi: 10.1016/b978-012373944-5.00106-1. [DOI] [Google Scholar]
- Gao F., Bailes E., Robertson D.L., Chen Y., Rodenburg C.M., Michael S.F., Cummins L.B., Arthur L.O., Peeters M., Shaw G.M., Sharp P.M., Hahn B.H. Origin of HIV-1 in the chimpanzee pan troglodytes troglodytes. Nature. 1999;397(6718):436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- Gardell M. In the Name of Elijah Muhammad: Louis Farrakhan and the Nation of Islam. first ed, first printing ed. Duke University Press Books; 1996. (The C. Eric Lincoln Series on the Black Experience). [DOI] [Google Scholar]
- Garten R.J., Davis C.T., Russell C.A., Shu B., Lindstrom S., Balish A., Sessions W.M., Xu X., Skepner E., Deyde V., Okomo-Adhiambo M., Gubareva L., Barnes J., Smith C.B., Emery S.L., Hillman M.J., Rivailler P., Smagala J., de Graaf M., et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325(5937):197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaurav A., Al-Nema M. Polymerases of coronaviruses. Viral Polym. 2019;27:1–300. doi: 10.1016/b978-0-12-815422-9.00010-3. [DOI] [Google Scholar]
- Gaurav A., Al-Nema M. Polymerases of coronaviruses. Viral Polym. 2019;27:1–300. doi: 10.1016/b978-0-12-815422-9.00010-3. [DOI] [Google Scholar]
- Huang Y., Yang C., Xu X., Xu W., Liu S. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sinica. 2020;41(9):1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M.A., Kadry S., Zhang Y.D., Akram T., Sharif M., Rehman A., Saba T. Prediction of COVID-19 - pneumonia based on selected deep features and one class kernel extreme learning machine. Comput. Electr. Eng. 2021;90 doi: 10.1016/j.compeleceng.2020.106960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M.A., Lali I.U., Rehman A., Ishaq M., Sharif M., Saba T., et al. Brain tumor detection and classification: A framework of marker-based watershed algorithm and multilevel priority features selection. Microsc. Res. Tech. 2019;82(6):909–922. doi: 10.1002/jemt.23238. [DOI] [PubMed] [Google Scholar]
- Khan M.A., Sharif T., Raza M., Saba T., Rehman A. Hand-crafted and deep convolutional neural network features fusion and selection strategy: an application to intelligent human action recognition. Appl. Soft Comput. 2020;87 [Google Scholar]
- Lauer G.M., Walker B.D. Hepatitis C virus infection. New England J. Med. 2001;345(1):41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- Main. (2020, December 16). Baker Lab at University of Washington. https://www.bakerlab.org/.
- Mamelund S.-E. Influenza, historical. Int. Encycl. Public Health. 2017;24:7–257. doi: 10.1016/b978-0-12-803678-5.00232-0. [DOI] [Google Scholar]
- Mei X., Lee H.C., Diao K., Huang M., Lin B., Liu C., Xie Z., Ma Y., Robson P.M., Chung M., Bernheim A., Mani V., Calcagno C., Li K., Li S., Shan H., Lv J., Zhao T., Xia J., Long Q., et al. 2020. Artificial intelligence-enabled rapid diagnosis of COVID-19 patients. medRxiv : the preprint server for health sciences, 2020.04.12.20062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A., Kumar D., Mittal M., Saba T., Abunadi I., Rehman A., Roy S. Detecting pneumonia using convolutions and dynamic capsule routing for chest X-ray images. Sensors. 2020;20(4):1068. doi: 10.3390/s20041068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal B., Muhammad N., Sharif M., Rehman A., Saba T. Removal of pectoral muscle based on topographic map and shape-shifting silhouette. BMC Cancer. 2018;18(1):778. doi: 10.1186/s12885-018-4638-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal B., Muhammad N., Sharif M., Saba T., Rehman A. Extraction of breast border and removal of pectoral muscle in wavelet, domain. Biomed. Res. 2017;28(11):5041–5043. [Google Scholar]
- Mughal B., Sharif M., Muhammad N., Saba T. A novel classification scheme to decline the mortality rate among women due to breast tumor. Microsc. Res. Tech. 2018;81(2):171–180. doi: 10.1002/jemt.22961. [DOI] [PubMed] [Google Scholar]
- Mujeeb S., Alghamdi T.A., Ullah S., Fatima A., Javaid N., Saba T. Exploiting deep learning for wind power forecasting based on big data analytics. Appl. Sci. 2019;9(20):4417. [Google Scholar]
- National Center for Biotechnology Information. (0000). https://www.ncbi.nlm.nih.gov/.
- Rehman A., Abbas N., Saba T., Mehmood Z., Mahmood T., Ahmed K.T. Microscopic malaria parasitemia diagnosis and grading on benchmark datasets. Microsc. Res. Tech. 2018;81(9):1042–1058. doi: 10.1002/jemt.23071. [DOI] [PubMed] [Google Scholar]
- Rehman A., Khan M.A., Mehmood Z., Saba T., Sardaraz M., Rashid M. Microscopic melanoma detection and classification: A framework of pixel-based fusion and multilevel features reduction. Microsc. Res. Tech. 2020;83(4):410–423. doi: 10.1002/jemt.23429. [DOI] [PubMed] [Google Scholar]
- Rehman A., Saba T., Ayesha N., Tariq U. Deep learning-based COVID-19 detection using CT and X-ray images: Current analytics and comparisons. IEEE IT Prof. 2021 doi: 10.1109/MITP.2020.3036820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A., Sadad T., Hussain A., Tariq U. Real-time diagnosis system of COVID-19 using X-ray images and deep learning. IEEE IT Prof. 2021 doi: 10.1109/MITP.2020.3042379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retroviridae A. 2017. Fenner’s veterinary virology; pp. 269–297. [DOI] [Google Scholar]
- Rubio-Garrido M., González-Alba J.M., Reina G., Ndarabu A., Barquín D., Carlos S., Galán J.C., Holguín Á. Current and historic HIV-1 molecular epidemiology in paediatric and adult population from Kinshasa in the democratic Republic of Congo. Sci. Rep. 2020;10(1):1–13. doi: 10.1038/s41598-020-74558-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba T. Computer vision for microscopic skin cancer diagnosis using handcrafted and non-handcrafted features. Microsc. Res. Tech. 2021 doi: 10.1002/jemt.23686. [DOI] [PubMed] [Google Scholar]
- Saba T., Abunadi I., Shahzad M.N., Khan A.R. Machine learning techniques to detect and forecast the daily total COVID-19 infected and deaths cases under different lockdown types. Microsc. Res. Tech. 2021 doi: 10.1002/jemt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba T., Mohamed A.S., El-Affendi M., Amin M. Brain tumor detection using fusion of hand crafted and deep learning features. Cogn. Syst. Res. 2020;59:221–230. [Google Scholar]
- Sharp P.M., Hahn B.H. The evolution of HIV-1 and the origin of AIDS. Philos. Trans. R. Soc. B. 2010;365(1552):2487–2494. doi: 10.1098/rstb.2010.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Xu W., Hu G., Xia S., Sun Z., Liu Z., Xie Y., Zhang R., Jiang S., Lu L. Acted article: SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell. Mol. Immunol. 2020:1–3. doi: 10.1038/s41423-020-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yadav A., Jha C.K., Sharan A. Optimizing LSTM for time series prediction in Indian stock market. Procedia Comput. Sci. 2020;167:2091–2100. doi: 10.1016/j.procs.2020.03.257. [DOI] [Google Scholar]
- Yousaf K., Mehmood Z., Awan I.A., Saba T., Alharbey R., Qadah T., Alrige M.A. A comprehensive study of mobile-health based assistive technology for the healthcare of dementia and Alzheimer’s disease (AD) Health Care Manag. Sci. 2019:1–23. doi: 10.1007/s10729-019-09486-0. [DOI] [PubMed] [Google Scholar]