

Abstract
Much public research suggests that autoimmune diseases such as rheumatoid arthritis (RA) are induced by aberrant “self” immune responses attacking autologous tissues and organ components. However, recent studies have reported that autoimmune diseases may be triggered by dysbiotic composition changes of the intestinal bacteria and an imbalance between these bacteria and intestinal immune systems. However, there are a few solid concepts or methods to study the putative involvement and relationship of these inner environmental factors in RA pathogenesis. Fortunately, Collagen-Induced Arthritis (CIA) and Collagen Antibody-Induced Arthritis (CAIA) models have been widely used as animal models for studying the pathogenesis of RA. In addition to RA, these models can be extensively used as animal models for studying complicated hypotheses in many diseases. In this review, we introduce some basic information about the CIA and CAIA models as well as how to apply these models effectively to investigate relationships between the pathogenesis of autoimmune diseases, especially RA, and the dysbiosis of intestinal bacterial flora.
1. Introduction
Autoimmune diseases such as rheumatoid arthritis (RA) have been believed to be caused by an overactivated immune system recognizing and attacking “self” tissues as “foreign” tissues. Unfortunately, triggers of this immune activation have not yet been elucidated.
RA susceptibility is linked to the major histocompatibility complex (MHC) class II genes [1–3], and the severity of RA in patients who have the HLA-DR4 gene tends to be high. However, RA is not a disease specifically induced in only HLA-DR4 positive individuals; rather, it may depend on conditions in hosts and environmental factors including lifestyle habits (smoking) [4], diet [5], periodontal diseases [6–8], gastrointestinal diseases [9], mental stress [10], dysbiosis of intestinal bacteria [11, 12], and immunological dysfunctions correlated with aging [13].
Recent advances in 16S ribosomal RNA analysis of fecal bacteria show dysbiosis, an abnormal balance of intestinal bacteria, is a common factor in many autoimmune diseases including RA [14–18]. Other common factors may include changes in substances produced by intestinal bacteria and destruction of mucosal barriers and gastrointestinal defense mechanisms. In addition, it is also important to assess the contribution of antibody reactions, which play important roles in immune defense [19, 20].
Collagen-Induced Arthritis (CIA) and Collagen Antibody-Induced Arthritis (CAIA) are useful animal models of RA and have been used for the functional analysis of pathological genes and for evaluating the anti-inflammatory and anti-RA drugs. Based on significant accumulated information, both models can be useful to investigate the contribution of conditions in hosts and environmental factors in autoimmune diseases. In this review, we introduce some basic information about CIA and CAIA, and new approaches as well as strategies for using these models to understand better the contribution of bacteria in the pathogenesis of RA.
2. CIA and CAIA as Animal Models of RA
2.1. Development of CIA
2.1.1. Induction of CIA
Arthritis animal models such as adjuvant arthritis, CIA, and CAIA are convenient for RA research because the severity of inflammation is easily evaluated and quantitatively analyzed. The widely used CIA model was developed by Trentham et al. and consists of immunizing Wistar rats with type II collagen, who subsequently develop polyarthritis in the paws [21]. After their discovery, chronic and progressive arthritis models were induced in rats [22, 23] and mice [24] as well as in some primates (squirrel monkeys [25], rhesus [26], cynomolgus [27], and marmosets [28]) by immunization with type II collagen emulsified in Complete Freund's Adjuvant (CFA). These models are now widely used and considered standard RA models in preclinical studies.
2.1.2. Specificity of Serum Antibody: Nonimmunogenicity of Autologous Type II
The CIA models have similar immunological and pathological characteristics to RA. CIA can be induced in several mouse strains expressing susceptible major histocompatibility complex (MHC) genes. CIA susceptibilities in mice depend on the MHC H-2 haplotype [29], similar to that in humans where RA severities depend on the MHC class II molecule, HLA-DR [1–3]. Antibodies in the serum from RA patients react to their autoantigen, human type II collagen (HII), and heterologous chicken and bovine type II collagen (CII and BII) as well, because they recognize a common epitope in type II collagen (Figure 1). These cross-reactivities were observed in CIA models in which serum antibodies cross-reacted to immunized CII or BII and their own rat or mouse type II collagen (MsII) [30, 31].
Figure 1.

Specificity of anti-type II collagen antibodies in RA patients. Cross-reactivity of autoantibodies (Ab), purified from RA patient serum using an HII-affinity column: bovine type II collagen (BII), chick type II collagen (CII), and human type II collagen (HII). (a) HII versus BII. (b) HII versus CII. (c) HII versus CII.
This evidence suggests that RA patients may be sensitized to heterologous type II collagen similar to that in CIA animals. Of note, serum from RA patients does not contain HII-specific antibodies [31], and the immunization of mice with MsII did not induce arthritis because the MsII is not recognized as an antigen by their immune systems [32]. This suggests that autologous type II collagen is not an antigen that induces antibody production.
CIA is not a direct RA animal model because of its different disease mechanisms. For example, the concentration of serum anti-type II collagen antibodies in arthritic CIA mice is significantly higher than in RA patients. DBA/1 mice develop CIA within 6–8 weeks after immunization, and the serum anti-type II collagen antibodyconcentration can reach mg/ml levels. In contrast, the serum anti-type II antibody concentration in RA patient is in μg/ml levels. RA patients harboring the HLA-DR4 gene with severe arthritic inflammation have low levels of anti-type II collagen autoantibodies, in contrast to CIA-susceptible DBA/1 mice. Therefore, RA can be characterized into two types, both of which are related to environmental factors, but differ regarding how antibodies induce pathology [19, 20].
2.1.3. Induction of Arthritis by Serum Antibody Transfer
CIA can be induced by the transfer of serum from arthritic CIA mice into naïve mice [33]. Furthermore, the injection of a serum IgG fraction containing anti-type II collagen antibodies from RA patients also induced arthritis in naïve mice [34]. This evidence suggests autoantibodies against autologous type II collagen are essential for the induction of CIA.
2.1.4. Contribution of T Cells in the Induction of CIA
Type II collagen is a T cell dependent antigen and researchers have suggested that CIA and RA are induced by T cell activation. A study reported that the transfer of T cell clones established by repeated type II collagen stimulation induced arthritis in mice [35]. However, the T cell clones were actually specific to pepsin which was used in the extraction of collagen, and this result misled the researchers into believing that T cells were reactive to autologous MsII [36]. Finally, a study, which used pepsin-free CB-peptides of MsII obtained by cyanogen bromide (CB) digestion, confirmed that mouse T cells only recognized species-specific epitopes of immunized type II collagen and did not react to common epitopes of heterogeneous type II collagen and MsII (unpublished data). However, these findings cannot conclude that T cells independently involve progression and aggravation of arthritis. In arthritis, T cells differentiate into various subtypes and release inflammatory and anti-inflammatory cytokines and chemokines, all of which contribute to the augmentation of inflammation and degradation of connective tissues. We found that irradiation of 2 Gy X-rays in CIA mice induced more severe inflammation in the areas of the tails where the adjuvant was injected, indicating the importance of T cells in inflammatory conditions (unpublished data).
2.2. Development of CAIA
2.2.1. Arthritogenic Epitopes on Type II Collagen in CIA and RA
The restricted location of arthritogenic epitopes recognized by autoantibodies in the sera from CIA mice and RA patients was studied using CB-peptides of type II collagen. When DBA/1 mice were immunized with CII or BII, arthritogenic epitopes were found only in CB11 (Figure 2), the eleventh fragment by CB cleavage [37], and when B10.RIII mice were immunized with BII, arthritogenic epitopes were found in CB8 [38]. In CIA-susceptible Wistar rats, the autoantibodies recognized CB11. In contrast, in monkeys [39] and humans [40], arthritogenic epitopes were widely distributed in CB7 through CB11 depending on the individual MHC backgrounds.
Figure 2.

Localization of arthritogenic epitopes on cyanogen bromide (CB) digested type II collagen fragments (CB 6, 8, 9.7, 10, 11, and 12). The circles represent the epitopes of the arthritogenic monoclonal antibodies on endoproteinase (LysC)-digested CB11 fragments (LysC1, LysC2, and LysC3).
2.2.2. Induction of Mouse Arthritis with a Cocktail of Monoclonal Antibodies
The distribution of CB11 arthritogenic epitopes in DBA/1 mice was demonstrated by the immunization of mice with the renatured CB11 fragment of type II collagen [37]. Based on this discovery, we succeeded in inducing arthritis, called Collagen Antibody-Induced Arthritis (CAIA), in normal mice by the intravenous (IV) administration of an antibody cocktail containing four monoclonal antibodies that recognized the CB11 arthritogenic epitopes (Figure 3) [41].
Figure 3.

Arthritis induced by the IV injection of an anti-type II collagen monoclonal antibody cocktail. Arthritis in DBA/1 mice on day 8 after the IV injection of the four-clone monoclonal antibody cocktail (10 mg), which recognizes the multispecies common epitope on the CB11 fragment of type II collagen. Mice developed polyarthritis in the paws.
Moreover, by adding another monoclonal antibody into the four-clone antibody cocktail, we established a more effective arthritis model [42]. Three of the clones, A2-10 (Ig G2a), D1-2G (IgG2b), and D2-112 (IgG2b), recognize the LysC1 (124–129) fragment consisting of 167 amino acids obtained by digesting CB11 with LysC, an endoproteinase, and the two other clones, F10-21 (IgG2a) and D8-6 (IgG2a) recognize the LysC2 (291–374) fragment (Figure 2). Due to the ability of the monoclonal antibody injection to bypass antibody production steps, the CAIA model has limitations for studies on antigen recognition by antigen-presenting cells and T cells. This was confirmed in CAIA induction using scid/scid mice who lack T cells [43]. However, CAIA can be used as a negative control with CIA studies to study the mechanism of these antigen recognition and antibody production steps.
2.2.3. Synergistic Effects of Type II Collagen Antibodies and Bacterial Toxins
CIA severity was enhanced by oral (PO) administration of mycoplasma arthritidis mitogen (MAM) [44], a T cell mitogen, or Staphylococcal enterotoxin B from Staphylococcus aureus (SEB) [45] before and after arthritis onset. In CAIA, we studied the arthritogenic effect of lipopolysaccharide derived from Escherichia coli O111:B4 (E. coli-LPS), a bacterial toxin produced from intestinal Gram-negative bacteria. Although the IV administration of a low dose (1 mg) of the four-clone monoclonal antibody cocktail failed to induce arthritis, a subsequent intraperitoneal (IP) administration of E. coli-LPS (50 μg) on day 3 induced severe arthritis within 24–48 hours, which reached a plateau after 7-8 days (Figure 4(a)).
Figure 4.
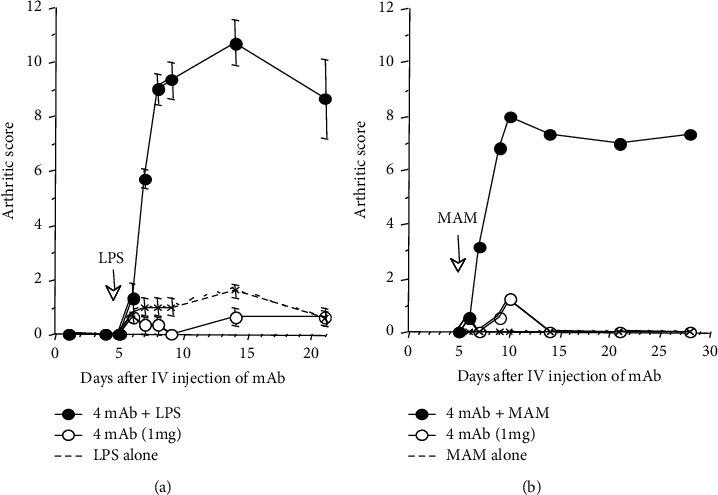
Synergistic effects of bacterial toxins to trigger and exacerbate arthritis in CAIA mice. CAIA in two mouse strains (DBA/1 and B10.RIII) was established by IV injection of the arthritogenic four-clone monoclonal antibody cocktail (1 mg) on day 0 and subsequent IP injection of 50 μg of lipopolysaccharide (LPS) from E. coli (a), or mycoplasma arthritidis mitogen (MAM) (b) on day 3. Mice developed arthritis within 24–48 hours after receiving the toxins and the arthritic score plateaus 7-8 days later and can last up to 30 days. (a) mAb plus LPS in DBA/1 mice. (b) mAb plus MAM in B10.RIII mice.
While neither the low-dose antibody cocktail or LPS alone was able to induce arthritis, the combination induces arthritis synergistically. Similar synergistic effects with the antibody cocktail were observed with MAM) (Figure 4(b)) and SEB (data not shown). This indicated that bacterial toxins nonspecifically play critical pathogenic roles with autoantibodies and contribute to the induction and progression of arthritis.
2.3. Attentions in CIA and CAIA as Models of RA
2.3.1. Differences between CIA and CAIA
The susceptibility of CIA and CAIA depends on the MHC H-2 haplotypes and species of heterogeneous type II collagen used for immunization. In mouse CIA, H-2q (DBA/1), H-2r (B10.RIII) and H-2s (SJL) haplotypes show high CIA susceptibility [24, 46, 47], while H-2b (C57BL/6) shows low CIA susceptibility, as observed in their respective levels of serum anti-mouse type II collagen autoantibodies. To induce CIA in these low responder strains, changing the immunological methods, such as an extra booster injection of heterologous type II collagen emulsified 1 : 1 with CFA containing a high concentration (5 mg/ml) of Mycobacterium tuberculosis (M. tuberculosis) on day 21 [48, 49], or the inhibition of cytokines such as IFN-γ [50], induce more frequent arthritis, and are useful for a specific experimental purpose. For gene function analysis, establishing gene modified mice requires 1-2 years or more. As C57BL/6 mice, which are a common transgenic mouse strain, are low responders to CIA induction, they may need to be backcrossed with highly susceptible DBA/1 mice [48]. In some cases, reproducing the arthritic phenotype in neonate mice is unreliable and inconsistent.
On the other hand, because CAIA bypasses anti-type II collagen autoantibody production, CAIA can be induced at 100% incidence, in a shorter period, and in many mouse strains who carry H-2q (DBA/1), H-2r (B10.RIII), H-2b (C57BL/6), H-2d (BALB/c) [42], and H-2s (Scid/scid C.B-17) [43]. An IP injection combining the monoclonal antibody cocktail and LPS reproducibly induces especially severe arthritis in widely available strains such as BALB/c, as well as C57BL/6 mice which is available for genetic modifications. Therefore, CAIA is a more useful model for the functional analysis of arthritis-related genes using various congenic, transgenic, and knockout mice [51].
Similar to human RA, CIA mice show increased rheumatoid factor (RF) and anti-citrullinated protein/peptide antibodies (ACPA) [52, 53]. Although humans tend to show a sex-bias in RA development [54, 55], mice did not seem to show any sex-bias in CIA [55] and CAIA (unpublished data). Interestingly, the sex-bias could be reproduced in human gene transgenic mice. With inducing human HLA-DR4 genes in mice, the female mice developed higher incidences of severe arthritis than male mice [56]. Thus, the transgenic mice can be useful for analyzing human genes related to RA development in CIA.
Table 1 shows the similarities and differences between CIA and CAIA models and compares these with the features of human RA.
Table 1.
Similarities and differences between CAIA and CIA animal models. CFA: Complete Freund's adjuvant, RA: rheumatoid arthritis, CIA: Collagen-Induced Arthritis, and CAIA: Collagen Antibody-Induced Arthritis.
| CIA model | CAIA model | |
|---|---|---|
| Induction method | Type II collagen/CFA | Monoclonal antibodies against type II collagen |
| Feature of arthritis | Polyarthritis | Polyarthritis |
| Similarities to human RA | (i) Activates adaptive immune system. (ii) Produces antibodies against specific epitopes on type II collagen in joints. (iii) Synovial inflammation due to infiltrating immune cells. (iv) Cartilage and bone destruction. (v) Increases RF and ACPA |
(i) Antibodies against specific epitopes on type II collagen in joints. (ii) Synovial inflammation due to infiltrating immune cells. (iii) Cartilage and bone destruction. |
| Advantages | (i) Study CIA development in relation to the adaptive immune systems. (ii) Study immunity specific to cartilage. (iii) No sex-bias |
(i) Induces arthritis in many mouse strains. (ii) Develops consistent arthritis severity and with almost 100% incidence. (iii) Effectively studies test articles in a short study period (1-2 weeks). (iv) RF and ACPA negative. (v) No sex-bias |
| Disadvantages | (i) Restricted to select mouse strains. (ii) Animal housing conditions can affect arthritis severities and incidences. (iii) A long study period due to the slow onset of arthritis (1-2 months). (iv) Strongly affected by CFA presence. |
(i) Does not involve the full spectrum of immune activation. (ii) Animal housing conditions can affect arthritis severities and incidences. |
2.3.2. Incidence and Severity of CIA: Study Variations by Emulsion and Animal Housing
Although CIA is a useful model to study the mechanism of arthritis induction, some important points should be considered to reduce variability in CIA. It is very difficult to obtain consistent reproducibility among long-term animal experiments. CIA would be considered a long-term experiment as it requires four weeks for developing arthritis and another three weeks to reach maximum severities of arthritis. For example, the incidence and severity of arthritis can range from 0% to 100%, just depending on the cage.
Housing conditions can affect the phenotype of CIA. Animals must be housed in specific pathogen-free (SPF) conditions without stress. Due to the aggressive nature of select mouse strains (DBA/1), fighting among cage-mates may be inevitable, leading to injuries and stress which may affect CIA development.
Qualities of the emulsion of CFA and type II collagen used to immunize mice are also important. When inducing CIA in many mice, a higher volume of emulsion is required and may need to be prepared with several emulsion batches. The qualities and stabilities of the emulsions in between the preparations can affect the consistency of developing CIA.
To obtain better results with these variations, the same group of mice should not be placed in a single cage; instead, the group of mice should be housed randomly in several cages. However, CAIA can avoid these problems because it yields approximately 100% incidence with reproducible arthritis severity in short periods (within 2 weeks).
2.3.3. Importance of a Negative Control Group: Immunological Effects of CFA and LPS in Arthritis Induction
In immunological function research, the most critical consideration in CIA is the effect of CFA which affects immune cells and the intestinal barrier [57, 58]. CIA requires subcutaneous (SC) injection of an emulsion of CFA with type II collagen into mouse tails. Therefore, a negative control group consisting of mice injected with an emulsion of CFA with PBS should be included to assess the effects of CFA in host immune systems. T cells in CIA mice are predominantly activated by the CFA in the emulsion, not by type II collagen. Dynamics of inflammatory or anti-inflammatory cytokines and chemokines in published reports might, in reality, be related to T cells affected by an adjuvant, such as CFA [59, 60]. With the same concept in mind for CAIA experiments, a negative control group consisting of an LPS injection alone should be used.
3. Risk Factors in Pathogenesis of RA
3.1. Internal Factors for RA
RA is a complex inflammatory disease associated with many inflammatory factors, such as prostaglandins, cyclooxygenase I and II, complement system, inflammatory and anti-inflammatory cytokines, chemokines, and signal transduction of immune cells, which are closely related to the enhancement and repression of inflammatory reactions. Therefore, these factors have been used as target molecules for reducing inflammation by anti-RA drugs including inhibitors, antagonists, and antibodies. On the other hand, the contribution of specific genes in arthritis induction has been also determined using genetic modification technologies such as congenic, transgenic, and knockout mice. Many researchers have been studying the role of T cells in the production of various cytokines and chemokines in inflammation, and the roles of transcriptional factors associated with the production of the internal factors [61].
However, these inflammatory factors in RA might be affected not only by the internal immune responses but also by external pathological factors. Identifying external environmental factors which affect the inflammatory responses must be the next essential step to understand pathogenesis in RA.
3.2. Dysbiosis as an External Factor in RA: A Balance between Intestinal Bacteria, Mucosal Barrier, and Immune Function of the Digestive Tract
Generally, in the digestive tract, physiological inflammation (homeostasis) is maintained by balancing the mucosal immune function and barrier function, and intestinal bacterial composition. However, this balance may be disrupted by increased intestinal mucosa permeabilities [19, 62] which are affected by drugs, such as nonsteroidal anti-inflammatory drugs(NSAIDs), intestinal disorders caused by diarrhea and constipation [63], and mental stress [64–66]. As a result, excessive environmental factors absorbed from the intestinal mucosa shift physiological inflammation to pathological inflammation, leading to chronic inflammatory diseases. Indeed, sera and synovial tissues from RA patients contained bacterial DNA and peptidoglycan [67], suggesting the dysfunction of their mucosal barriers.
Recent reports suggested that vegetarian diets can change the fecal bacterial population in RA patients, subsequently causing remission of clinical symptoms [68, 69]. These results may indicate a relationship between intestinal bacteria and RA pathology [14–18]. More specifically, 16S ribosomal RNA analysis, which can analyze fecal bacteria, demonstrated existing an imbalance of intestinal bacteria in RA patients. These results suggest that the dysbiosis of intestinal commensal bacteria, rather than specific individual bacteria, is related to the pathogenesis of RA. Moreover, as dysbiosis was also observed in inflammatory bowel disease (IBD) [16, 17] and, in spondyloarthritis [70], it might be a common condition in many autoimmune diseases.
For example, it was reported that Bifidobacteria, Bacteroides-Porphyromonas-Prevotella group, Bacteroides fragilis subgroup, and Eubacterium rectale-Clostridium coccoides group microbes were significantly decreased in RA patients compared with fibromyalgia patients [14]. Moreover, it was also reported that Prevotella copri levels increased and Bacteroides levels decreased in the feces of new-onset untreated RA patients [71]. An analysis of bacteria in fecal, dental, and saliva samples by metagenomic shotgun sequencing and a metagenome-wide association study compared RA patients with healthy volunteers. The study revealed that no Haemophilus but increased Lactobacillus salivarius in RA patients [72].
4. Examples of RA Risk Factor Studies Using CIA and CAIA
As described above, CIA and CAIA are very useful models to identify RA-related factors and to study the roles of these factors in the pathogenesis of arthritis. Here, we introduce some suggested experiments using modified protocols based on CIA and CAIA as follows.
4.1. Oral Collagen-Induced Arthritis (Oral CIA): A Model to Mimic Intestinal Absorption of Antigens
DBA/1 mice received oral administration of heterologous type II collagen for 15 weeks (5 times/week) and developed low severity arthritis as shown in Figures 5(a) and 5(b) (Oral CIA), accompanied by increased anti-MsII antibodies. This study indicated that ingesting dietary heterologous type II collagen for many weeks developed low levels of antibodies against MsII, which can induce arthritis [73].
Figure 5.

Oral Collagen-Induced Arthritis (oral CIA) and nonantibody mediated LPS-induced arthritis. Oral administration of heterologous type II collagen for a prolonged period can elicit very mild arthritis (oral CIA) in mice, (a) and (b), compared with typical severe arthritis in general CIA without LPS injection (c).
4.2. Nonantibody Mediated LPS-Induced Arthritis: Induction by LPS Alone in Immune-Deficient Mice
In continuation of the previous section above, oral coadministration of LPS (10 μg) in the Oral CIA mice led to more severe arthritis associated with higher antibody levels against MsII. To confirm the role of intestinal permeability in this synergistic effect of LPS, the mice orally received a normal dose of indomethacin (40 μg/mouse) and ovoinhibitor (2 mg/mouse), a protease inhibitor in egg white, known to enhance the permeability of the digestive tract. These mice showed suppressed immune function of the digestive tract, along with low levels of antibodies against the orally administered type II collagen. On the other hand, these mice showed higher susceptibility against LPS, resulting in 50% mortality of mice after the IP injection of 50 μg LPS. In addition, the surviving mice developed very severe arthritis without antibodies against MsII. This kind of severe arthritis had not been observed in general CIA and CAIA (Figure 5(c)) [73]. This mouse arthritis model may mimic pathologically similar arthritis in RA patients without HLA-DR4 gene and autoantibodies. These results suggest the importance of mucosal barrier permeability and absorption of bacterial toxins, such as LPS, in the digestive tract in disease induction.
4.3. Studies of the Effect of Bacterial Toxins in CAIA
As described before, an IP injection of bacterial toxins such as LPS, MAM, and SEB after an initial IV injection of a low dose of anti-typeII collagen antibody cocktail (1 mg) induced severe arthritis (Figure 4), indicating the model can be used for studying the effects of environmental factors in arthritis induction. Therefore, pathological effects of LPS from Porphyromonas gingivalis (P. gingivalis) (Pg-LPS), which cause periodontitis and contribute to progressive RA [74–76], were studied using this protocol. An IP injection of Pg-LPS (50 μg) failed to develop arthritis in DBA/1 mice who initially received an IV injection of the type II collagen antibody cocktail (1 mg) (data not shown) although E. coli-LPS (50 μg) induced severe arthritis under the same conditions. These results suggest the pathological role of P. gingivalis in inducing arthritis may be unrelated to Pg-LPS, but rather associated with changing the intestinal bacteria composition and mucosal barrier function, as reported by Nakajima et al. [77]. Inducing arthritis using a combination of the anti-type II collagen antibody cocktail and bacterial toxins might be useful to screen environmental factors and study their pathological roles in arthritis induction.
4.4. Studies of the Pathological Roles of Intestinal Bacteria Toxins in CAIA: Influence of Aging
Yoshino et al. reported that the oral administration of LPS on day 50 after type II collagen immunization rebooted severe arthritis in DBA/1 mice in the CIA model [78]. This LPS administration was tested in the CAIA model using mice aged 8 weeks and 8 months. In the 8-month-old mice, 1 mg of LPS orally given on day 3 after the type II collagen antibody cocktail injection (1.5 mg) induced arthritis within 24–48 hours and reached a plateau after 7-8 days with high serum IL-6 levels. The severity of arthritis correlated with serum IL-6 levels. However, in the 8-week-old mice,this same procedure failed to develop arthritis and also to increase serum IL-6 levels. This result indicated that increased animal age is related to lowered intestinal barrier function and increased susceptibility against LPS toxicity due to a decreased mucosal defense function [79]. These protocols might be useful to evaluate increased permeability and decreased barrier function and their correlation with sensitivity against bacterial toxins related to aging and intestinal diseases.
4.5. Studies of Intestinal Bacterial Effects on Arthritis
Bacterial species in the oral cavity and intestine may be linked to MHC molecules in humans and mice [80]. Totaro et al. showed that DNA from P. gingivalis, which is a pathogenic bacteria strain, was found at a higher rate in the synovial membranes of RA patients who have the HLA-DRβ1 allele than in other patients [81]. In addition, Gomez et al. showed that Clostridium-like bacteria are dominant in the intestine of CIA-sensitive DRβ1 0401 allele transgenic mice, while Porphyromonadaceae and Bifidobacteria are dominant in CIA-resistant DRβ1 0402 allele transgenic mice. The DRβ1 0402 mice also showed sex and age-influenced gut microbiome composition, but not the DRβ1 0401 mice who even had altered gut permeability [82]. Recently, several reports suggested a link between gut bacteria and CIA development using oral administration of several bacteria strains in mice. For example, among several beneficial bacteria strains altering disease outcomes [83], Lactobacillus helveticus reduced the severity of arthritis significantly [84]; Lactobacillus casei administration during induction of CIA suppressed anti-type II collagen antibody levels and delayed onset and reduced severity of CIA [85]. Interestingly, not only live bacteria but also heat-killed Lactobacillus reuteri administration inhibited CIA and CAIA induction [86]. Moreover, Liu et al. compared fecal bacteria species between arthritic and nonarthritic DBA/1 mice in CIA and demonstrated Lactobacillus was dominant in arthritic mice but,Bacteroidaceae and Lachnospitaceae proliferated in nonarthritic mice. They also showed that germ-free mice can become CIA-susceptible when they receive intestinal bacteria from CIA arthritic mice [57].
Alternatively, Jubair et al. reported effects of mixed broad-spectrum antibiotics (ampicillin, metronidazole, neomycin, and vancomycin), which change intestinal bacterial composition, in CIA [58]. Eliminating intestinal bacteria with the antibiotic treatments suppressed the severity of arthritis by approximately 40% and decreased anti-type II collagen antibody levels. Interestingly, dysbiosis and inflammation of the intestinal mucosa had already occurred before the onset of arthritis in DBA/1 mice who were immunized with an emulsion of CFA and type II collagen. These mice also have higher intestinal mucosa permeability confirmed by oral administration of fluorescein isothiocyanate (FITC) conjugated dextran. Another study showed that the administration of mixed broad-spectrum antibiotics reduced severity of arthritis, correlating with decreased Th17 cells and their IL-17 production in the intestinal lamina propria of the mice [87, 88].
These findings suggested CFA may affect bacteria composition, host immune systems, and barrier functions of the intestinal mucosa in CIA. To confirm this hypothesis, 8-week-old DBA/1 mice receiveda single immunization with an emulsion of type II collagen and CFA containing low concentration of M. tuberculosis (1 mg/ml). This immunization generally fails to induce CIA. Six weeks after the initial immunization, an oral administration of LPS (3 mg) induced arthritis in nonarthritic mice (Figure 6). These results indicate that the 14-week-old mice, who are usually resistant to LPS by oral administration, absorbed LPS from the intestine. This result indicated that CFA significantly affects mucosal barrier functions and increases the absorption of bacterial toxins, such as LPS, which are generally not absorbed in intestine.
Figure 6.

Exacerbating arthritis by oral administration of LPS in nonarthritic CIA mice. Mice were immunized with an emulsion of type II collagen and 1 mg/ml CFA. Nonarthritic mice failed to develop significant arthritis; however, the oral administration of LPS on day 42 induced severe arthritis in the nonarthritic mice (square), similar to the arthritic mice (circle).
These results clearly showed that dysbiosis, the bacterial composition change in the intestine, is closely related to the induction, progression, and severity of arthritis. These results might adapt to studies using gnotobiotic mice with which intestinal bacteria are removed or transferred. Furthermore, these studies suggested that CFA affects host immune systems and intestinal bacteria, resulting in inflammation of the intestinal mucosa. Thus, CFA plays important role in the development of arthritis, which is useful knowledge for future studies.
4.6. Studies on the Effects of Dysbiosis and Oral Bacteria on Arthritis
Dental clinical research reported that RA patients with periodontitis, especially associated with P. gingivalis [89] and Aggregatibacter actinomycetemcomitans [90], suffer from more severe arthritis [75–77]. It was demonstrated that P. gingivalis infections, which led to periodontitis, aggravated arthritis in CIA mice [91–95]. Regarding pathological roles of P. gingivalis, Nakajima et al. recently published that oral administration of P. gingivalis in C57BL/6 mice changed bacterial flora composition. The change suppressed mRNA expression of tight junction proteins in mucosal membranes and induced high serum endotoxin levels by increasing the permeability of the intestinal mucosal barrier [77]. The same group reported that the repeated oral administration of P. gingivalis exacerbated CIA, with increased numbers of intestinal bacteria, such as Bacteroides and Prevotella,leading to induce the proliferation of Th17 cells and increase blood IL-17 levels [96]. However, the oral administration of P. gingivalis did not change the anti-type II collagen antibody levels and failed to activateP. gingivalis proliferation in the intestine. Dysbiosis can also be developed by oral antibiotics. Depleting the microbiota with oral feeding of an antibiotic cocktail prior to the induction of CIA reduced severity of arthritis and the levels of serum inflammatory cytokines and anti-CII antibodies [58]. However, enrofloxacin, an antibiotic, aggravated CIA and increased the levels of serum IFN-g, IL-17A, and IL-6 [97].
Furthermore, it is important to note the intestinal bacteria composition changes during the development of CIA [57, 87]; Firmicutes and Proteobacteria were increased and Bacteroides and lactobacillus were decreased. Even progressing arthritis development shifts bacteria composition in CIA mice [98]. The composition change may be associated with the stress and pain in the developing disease as well as housing conditions [57, 58, 97]. The housing condition factor requires more attention because viral or bacterial contamination in mice can reduce the severity of arthritis in CIA [99] and CAIA (unpublished observed data). Therefore, specific pathogen-free conditions are highly recommended in the arthritis studies, at least.
Further studies are required to analyze pathogenesis of dysbiosis in diseases. CIA and CAIA are useful models because oral inoculation of a target bacterium in mice is an easy procedure and then observe the dysbiosis and the resulting impact on the severity of arthritis.
4.7. Studies of the Adverse Effects of Antiautoimmune Drugs that Influence the Intestinal Bacteria
The administration of immunosuppressants, steroids, and NSAIDs, which are used to treat autoimmune diseases, may cause intestinal dysbiosis. Among these drugs, methotrexate (MTX) has been used as an effective positive control drug in both CIA and CAIA [100, 101]. However, it was reported that MTX treatment decreased the numbers of Prevotella [102], Enterococcus faecium [103], and Bacteroides group [104] in the intestines of RA patients. Bacteria number changes in the intestinal flora including these bacteria might be closely related to the effectiveness of MTX and remission of RA. Our report indicated that the IgA and IgG levels against P. gingivalis significantly increased in sera from RA patients treated with MTX for a long period, suggesting MTX may change the intestinal bacterial balance including P. gingivalis [20, 100, 105]. As previously described, immune suppressed mice become highly susceptible against LPS toxicity and develop severe and destructive arthritis with a single IP injection of LPS. Therefore, a long duration of treatment with immunosuppressive RA drugs might further reduce the immune function in RA patients, who have already a low immune response and worsen their RA prognosis.
Recently, it has been reported that immune checkpoint inhibitors, such as programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) activate T cells and can induce autoimmune-disease-like symptoms [106–108]. However, in CIA using DBA/1 mice, the administration of anti-PD-L1 antibodies decreased the severity of arthritis and serum IL-17 and IL-23 levels [109]. On the other hand, administration of soluble PD-1, which was detected at high levels in the sera or synovial fluid in RA patients, aggravates CIA by activating Th1 and Th17 cells [110]. In RA patients, the functional balance between Th17 cells and Tregs was changed [111].Moreover, in CIA, the administration of IL-27, which suppresses Th17 differentiation, decreased the severity of CIA and increased Th17 positive Tregs in the spleen [112].
Activating T cells by suppressing the PD-1/PD-L1 pathway with immune checkpoint inhibitors induces autoimmune-disease-like-symptoms in cancer patients, but it is still under investigation as to how the activation of pathways of Th17 or Tregs affects the phenotypes in CIA and CAIA. In addition, the relationship between the immune checkpoint inhibitors and intestinal bacteria should be closely analyzed. CIA and CAIA might be useful models to investigate the relationships between cancer immunity and the induction of autoimmune-disease-like symptoms.
5. Conclusion
Recently, many studies have been reported on the relationship between autoimmune diseases and intestinal bacteria. Many autoimmune diseases including RA,which are considered chronic diseases, have common pathogenesis in decreasing intestinal barrier function and mucosal immune function. CIA and CAIA, animal models of arthritis, have been used for many inflammation related studies, but not sufficiently used for studies investigating bacterial flora in the pathogenesis of arthritis.
As described in this review, CIA and CAIA can be used for studying the contribution of gene background, host immune ability, bacterial flora, and intestinal mucosal barriers to the pathogenesis of RA, using many parameters beyond just arthritic scores. We believe that the CIA and CAIA models and their published protocols will be useful to investigate the many factors involved in autoimmune diseases, and we hope this review proves to be a useful guide for studying new concepts in many diseases.
Conflicts of Interest
The authors declare that they have received financial support from Chondrex, Inc.
References
- 1.Gregersen P. K. HLA class II polymorphism: implications for genetic susceptibility to autoimmune disease. Laboratory Investigation. 1989;61:5–19. [PubMed] [Google Scholar]
- 2.Newton J., Harney S., Wordsworth B., Brown M. A review of the MHC genetics of rheumatoid arthritis. Genes & Immunity. 2004;5(3):151–157. doi: 10.1038/sj.gene.6364045. [DOI] [PubMed] [Google Scholar]
- 3.Stastny P., Ball E. J., Khan M. A., Olsen N. J., Pincus T., Gao X. HLA-DR4 and other genetic markers in rheumatoid arthritis. Rheumatology. 1988;27(2):132–138. doi: 10.1093/rheumatology/xxvii.suppl_2.132. [DOI] [PubMed] [Google Scholar]
- 4.Chang K., Yang S., Kim S., Han K., Park S., Shin J. Smoking and rheumatoid arthritis. International Journal of Molecular Sciences. 2014;15(12):22279–22295. doi: 10.3390/ijms151222279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stamp L. K., James M. J., Cleland L. G. Diet and rheumatoid arthritis: a review of the literature. Seminars in Arthritis and Rheumatism. 2005;35(2):77–94. doi: 10.1016/j.semarthrit.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Jajoo N. S., Shelke A. U., Bajaj R. S. Periodontitis and rheumatoid arthritis: the common thread. Clinical Reviews in Bone and Mineral Metabolism. 2020;18(1–3):18–30. doi: 10.1007/s12018-020-09271-6. [DOI] [Google Scholar]
- 7.Bartold P. M., Marshall R. I., Haynes D. R. Periodontitis and rheumatoid arthritis: a review. Journal of Periodontology. 2005;76(11):2066–2074. doi: 10.1902/jop.2005.76.11-s.2066. [DOI] [PubMed] [Google Scholar]
- 8.McHugh J. Rheumatoid arthritis: new model linking periodontitis and RA. Nature Reviews Rheumatology. 2017;13(2):p. 66. doi: 10.1038/nrrheum.2016.221. [DOI] [PubMed] [Google Scholar]
- 9.Craig E., Cappelli L. C. Gastrointestinal and hepatic disease in rheumatoid arthritis. Rheumatic Disease Clinics of North America. 2018;44:89–111. doi: 10.1016/j.rdc.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cutolo M., Straub R. H. Stress as a risk factor in the pathogenesis of rheumatoid arthritis. Neuroimmunomodulation. 2006;13(5-6):277–282. doi: 10.1159/000104855. [DOI] [PubMed] [Google Scholar]
- 11.Kishikawa T., Maeda Y., Nii T., et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Annals of the Rheumatic Diseases. 2020;79(1):103–111. doi: 10.1136/annrheumdis-2019-215743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeoh N., Burton J. P., Suppiah P., Reid G., Stebbings S. The role of the microbiome in rheumatic diseases. Current Rheumatology Reports. 2013;15:p. 314. doi: 10.1007/s11926-012-0314-y. [DOI] [PubMed] [Google Scholar]
- 13.Weyand C. M., Goronzy J. J. Aging of the immune system. Mechanisms and therapeutic targets. Annals of the American Thoracic Society. 2016;13(5):S422–S428. doi: 10.1513/annalsats.201602-095aw. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaahtovuo J., Munukka E., Korkeamäki M., Luukkainen R., Toivanen P. Fecal microbiota in early rheumatoid arthritis. The Journal of Rheumatology. 2008;35(8):1500–1505. [PubMed] [Google Scholar]
- 15.Catrina A. I., Deane K. D., Scher J. U. Gene, environment, microbiome and mucosal immune tolerance in rheumatoid arthritis. Rheumatology. 2016;55(3):391–402. doi: 10.1093/rheumatology/keu469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tlaskalova-Hogenova H., Tuckova L., Mestecky J., et al. Interaction of mucosal microbiota with the innate immune system. Scandinavian Journal of Immunology. 2005;62(s1):106–113. doi: 10.1111/j.1365-3083.2005.01618.x. [DOI] [PubMed] [Google Scholar]
- 17.Sartor R. B., Muehlbauer M. Microbial host interactions in IBD: implications for pathogenesis and therapy. Current Gastroenterology Reports. 2007;9(6):497–507. doi: 10.1007/s11894-007-0066-4. [DOI] [PubMed] [Google Scholar]
- 18.Hara N., Alkanani A. K., Ir D., et al. The role of the intestinal microbiota in type 1 diabetes. Clinical Immunology. 2013;146(2):112–119. doi: 10.1016/j.clim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Terato K., Do C. T., Shionoya H. Slipping through the cracks: linking low immune function and intestinal bacterial imbalance to the etiology of rheumatoid arthritis. Autoimmune Diseases. 2015;2015:12. doi: 10.1155/2015/636207.636207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terato K., Waritani T., Fukai R., Shionoya H., Itoh H., Katayama K. Contribution of bacterial pathogens to evoking serological disease markers and aggravating disease activity in rheumatoid arthritis. PLoS One. 2018;13(2) doi: 10.1371/journal.pone.0190588.e0190588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trentham D. E., Townes A. S., Kang A. H. Autoimmunity to type II collagen an experimental model of arthritis. Journal of Experimental Medicine. 1977;146(3):857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffiths M. M., DeWitt C. W. Immunogenetic control of experimental collagen-induced arthritis in rats. II. ECIA susceptibility and immune response to type II collagen (calf) are linked to RT1. European Journal of Immunogenetics. 1981;8(6):463–470. doi: 10.1111/j.1744-313x.1981.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths M. M., Eichwald E. J., Martin J. H., Smith C. B., Dewitt C. W. Immunogenetic control of experimental type II collagen-induced arthritis. Arthritis & Rheumatism. 1981;24(6):781–789. doi: 10.1002/art.1780240605. [DOI] [PubMed] [Google Scholar]
- 24.Courtenay J. S., Dallman M. J., Dayan A. D., Martin A., Mosedale B. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283(5748):666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 25.Cathcart E. S., Hayes K. C., Gonnerman W. A., Lazzari A. A., Franzblau C. Experimental arthritis in a nonhuman primate. I. Induction by bovine type II collagen. Laboratory Investigation; a Journal of Technical Methods and Pathology. 1986;54(1):26–31. [PubMed] [Google Scholar]
- 26.Yoo T. J., Kim S. Y., Stuart J. M., et al. Induction of arthritis in monkeys by immunization with type II collagen. Journal of Experimental Medicine. 1988;168(2):777–782. doi: 10.1084/jem.168.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terato K., Arai H., Shimozuru Y., et al. Sex-linked differences in susceptibility of cynomolgus monkeys to type II collagen-induced arthritis. Evidence that epitope-specific immune suppression is involved in the regulation of type II collagen autoantibody formation. Arthritis & Rheumatism. 1989;32(6):748–758. doi: 10.1002/anr.1780320613. [DOI] [PubMed] [Google Scholar]
- 28.Vierboom M. P., Breedveld E., Kondova I., Hart B. A. Collagen-induced arthritis in common marmosets: a new nonhuman primate model for chronic arthritis. Arthritis Research & Therapy. 2010;12(5):p. R200. doi: 10.1186/ar3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Protocol for the successful induction of collagen-induced arthritis (CIA) in mice. 2017. https://www.chondrex.com/documents/Mouse%20CIA.pdf.
- 30.Terato K., Shimozuru Y., Katayama K., et al. Specificity of antibodies to type II collagen in rheumatoid arthritis. Arthritis & Rheumatism. 1990;33(10):1493–1500. doi: 10.1002/art.1780331006. [DOI] [PubMed] [Google Scholar]
- 31.Terato K., DeArmey D. A., Ye X. J., Griffiths M. M., Cremer M. A. The mechanism of autoantibody formation to cartilage in rheumatoid arthritis: possible cross-reaction of antibodies to dietary collagens with autologous type II collagen. Clinical Immunology and Immunopathology. 1996;79(2):142–154. doi: 10.1006/clin.1996.0061. [DOI] [PubMed] [Google Scholar]
- 32.Holmdahl R., Jansson L., Larsson E., Rubin K., Klareskog L. Homologous type II collagen induces chronic and progressive arthritis in mice. Arthritis & Rheumatism. 1986;29(1):106–113. doi: 10.1002/art.1780290114. [DOI] [PubMed] [Google Scholar]
- 33.Stuart J. M., Dixon F. J. Serum transfer of collagen-induced arthritis in mice. Journal of Experimental Medicine. 1983;158(2):378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wooley P. H., Luthra H. S., Singh S. K., Huse A. R., Stuart J. M., David C. S. Passive transfer of arthritis to mice by injection of human anti-type II collagen antibody. Mayo Clinic Proceedings. 1984;59(11-12):737–743. doi: 10.1016/s0025-6196(12)65583-9. [DOI] [PubMed] [Google Scholar]
- 35.Seki N., Sudo Y., Yoshioka T., et al. Type II collagen-induced murine arthritis. I. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. Journal of Immunology. 1988;140(5):1477–1484. [PubMed] [Google Scholar]
- 36.Andersson M., Holmdahl R. Analysis of type II collagen-reactive T cells in the mouse I. Different regulation of autoreactivevs. non-autoreactive anti-type II collagen T cells in the DBA/1 mouse. European Journal of Immunology. 1990;20(5):1061–1066. doi: 10.1002/eji.1830200517. [DOI] [PubMed] [Google Scholar]
- 37.Terato K., Hasty K. A., Cremer M. A., Stuart J. M., Townes A. S., Kang A. H. Collagen-induced arthritis in mice. Localization of an arthritogenic determinant to a fragment of the type II collagen molecule. Journal of Experimental Medicine. 1985;162(2):637–646. doi: 10.1084/jem.162.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myers L. K., Miyahara H., Terato K., Seyer J. M., Stuart J. M., Kang A. H. Collagen-induced arthritis in B10.RIII mice (H-2r): identification of an arthritogenic T-cell determinant. Immunology. 1995;84(4):509–513. [PMC free article] [PubMed] [Google Scholar]
- 39.Shimozuru Y., Yamane S., Fujimoto K., et al. Collagen-induced arthritis in nonhuman primates: multiple epitopes of type I collagen can induce autoimmune-mediated arthritis in outbred cynomolgus monkeys. Arthritis & Rheumatism. 1998;41(3):507–514. doi: 10.1002/1529-0131(199803)41:3<507::aid-art17>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 40.Goto M., Yoshinoya S., Miyamoto T., et al. Stimulation of interleukin-1α and interleukin-1β release from human monocytes by cyanogen bromide peptides of type II collagen. Arthritis & Rheumatism. 1988;31(12):1508–1514. doi: 10.1002/art.1780311207. [DOI] [PubMed] [Google Scholar]
- 41.Terato K., Hasty K. A., Reife R. A., Cremer M. A., Kang A. H., Stuart J. M. Induction of arthritis with monoclonal antibodies to collagen. Journal of Immunology. 1992;148(7):2103–2108. [PubMed] [Google Scholar]
- 42.Hutamekalin P., Saito T., Yamaki K., et al. Collagen antibody-induced arthritis in mice: development of a new arthritogenic 5-clone cocktail of monoclonal anti-type II collagen antibodies. Journal of Immunological Methods. 2009;343(1):49–55. doi: 10.1016/j.jim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Kagari T., Doi H., Shimozato T. The importance of IL-1β and TNF-α, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. The Journal of Immunology. 2002;169(3):1459–1466. doi: 10.4049/jimmunol.169.3.1459. [DOI] [PubMed] [Google Scholar]
- 44.Cole B. C., Griffiths M. M. Triggering and exacerbation of autoimmune arthritis by the mycoplasma arthritidis superantigen mam. Arthritis & Rheumatism. 1993;36(7):994–1002. doi: 10.1002/art.1780360717. [DOI] [PubMed] [Google Scholar]
- 45.Wooley P. H., Cingel B. Staphylococcal enterotoxin B increases the severity of type II collagen induced arthritis in mice. Annals of the Rheumatic Diseases. 1995;54(4):298–304. doi: 10.1136/ard.54.4.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wooley P. H. Collagen-induced arthritis in the mouse. Methods in Enzymology. 1988;162:361–373. doi: 10.1016/0076-6879(88)62091-x. [DOI] [PubMed] [Google Scholar]
- 47.Wooley P. H., Luthra H. S., Griffiths M. M., Stuart J. M., Huse A., David C. S. Type II collagen-induced arthritis in mice. IV. Variations in immunogenetic regulation provide evidence for multiple arthritogenic epitopes on the collagen molecule. Journal of Immunology. 1985;135(4):2443–2451. [PubMed] [Google Scholar]
- 48.Campbell I. K., Hamilton J. A., Wicks I. P. Collagen-induced arthritis in C57BL/6 (H-2b) mice: new insights into an important disease model of rheumatoid arthritis. European Journal of Immunology. 2000;30(6):1568–1575. doi: 10.1002/1521-4141(200006)30:6<1568::aid-immu1568>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 49.Kai H., Shibuya K., Wang Y., et al. Critical role of M. tuberculosis for dendritic cell maturation to induce collagen-induced arthritis in H-2b background of C57BL/6 mice. Immunology. 2006;118(2):233–239. doi: 10.1111/j.1365-2567.2006.02361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu C.-Q., Song Z., Mayton L., Wu B., Wooley P. H. IFNγ deficient C57BL/6 (H-2b) mice develop collagen induced arthritis with predominant usage of T cell receptor Vβ6 and Vβ8 in arthritic joints. Annals of the Rheumatic Diseases. 2003;62(10):983–990. doi: 10.1136/ard.62.10.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chondrex Inc. Collagen antibody induced arthritis in mice. 2020. https://www.chondrex.com/documents/Mouse-CAIA.pdf.
- 52.Förster M., Raposo B., Ekman D., et al. Genetic control of antibody production during collagen-induced arthritis development in heterogeneous stock mice. Arthritis & Rheumatism. 2012;64(11):3594–3603. doi: 10.1002/art.34658. [DOI] [PubMed] [Google Scholar]
- 53.Kidd B. A., Ho P. P., Sharpe O., et al. Epitope spreading to citrullinated antigens in mouse models of autoimmune arthritis and demyelination. Arthritis Research & Therapy. 2008;10(5):p. R119. doi: 10.1186/ar2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ortona E., Pierdominici M., Maselli A., Veroni C., Aloisi F., Shoenfeld Y. Sex-based differences in autoimmune diseases. Annali dell’Istituto Superiore di Sanità. 2016;52:205–212. doi: 10.4415/ANN_16_02_12. [DOI] [PubMed] [Google Scholar]
- 55.Cutolo M. Sex and rheumatoid arthritis: mouse model versus human disease. Arthritis & Rheumatism. 2007;56(1):1–3. doi: 10.1002/art.22322. [DOI] [PubMed] [Google Scholar]
- 56.Taneja V., Behrens M., Mangalam A., Griffiths M. M., Luthra H. S., David C. S. New humanized HLA-DR4-transgenic mice that mimic the sex bias of rheumatoid arthritis. Arthritis & Rheumatism. 2007;56(1):69–78. doi: 10.1002/art.22213. [DOI] [PubMed] [Google Scholar]
- 57.Liu X., Zeng B., Zhang J., et al. Role of the gut microbiome in modulating arthritis progression in mice. Scientific Reports. 2016;6:p. 30594. doi: 10.1038/srep30594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jubair W. K., Hendrickson J. D., Severs E. L., et al. Modulation of inflammatory arthritis in mice by gut microbiota through mucosal inflammation and autoantibody generation. Arthritis & Rheumatology. 2018;70(8):1220–1233. doi: 10.1002/art.40490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ratkay L. G., Zhang L., Tonzetich J., Waterfield J. D. Complete Freund’s adjuvant induces an earlier and more severe arthritis in MRL-lpr mice. Journal of Immunology. 1993;151(9):5081–5087. [PubMed] [Google Scholar]
- 60.Geboes L., Klerck B. D., Balen M. V., et al. Freund’s complete adjuvant induces arthritis in mice lacking a functional interferon-γ receptor by triggering tumor necrosis factor α-driven osteoclastogenesis. Arthritis & Rheumatism. 2007;56(8):2595–2607. doi: 10.1002/art.22791. [DOI] [PubMed] [Google Scholar]
- 61.Kondo Y., Yokosawa M., Kaneko S., et al. Review: transcriptional regulation of CD4+ T cell differentiation in experimentally induced arthritis and rheumatoid arthritis. Arthritis & Rheumatology. 2018;70(5):653–661. doi: 10.1002/art.40398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Basset C., Holton J., O’Mahony R., Roitt I. Innate immunity and pathogen-host interaction. Vaccine. 2003;21:p. S12. doi: 10.1016/s0264-410x(03)00195-6. [DOI] [PubMed] [Google Scholar]
- 63.Khalif I. L., Shapina M. V. Inflammatory bowel disease treatment in eastern Europe: current status, challenges and needs. Current Opinion in Gastroenterology. 2017;33(4):230–233. doi: 10.1097/mog.0000000000000370. [DOI] [PubMed] [Google Scholar]
- 64.Anderlik P., Szeri I., Bános Z., Barna Z. Bacterial translocation after cold stress in young and old mice. Acta Microbiologica Hungarica. 1990;37(3):289–294. [PubMed] [Google Scholar]
- 65.Velin A. K., Ericson A. C., Braaf Y., Wallon C., Soderholm J. D. Increased antigen and bacterial uptake in follicle associated epithelium induced by chronic psychological stress in rats. Gut. 2004;53(4):494–500. doi: 10.1136/gut.2003.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Selkirk G. A., McLellan T. M., Wright H. E., Rhind S. G. Mild endotoxemia, NF-κB translocation, and cytokine increase during exertional heat stress in trained and untrained individuals. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2008;295(2):R611–R623. doi: 10.1152/ajpregu.00917.2007. [DOI] [PubMed] [Google Scholar]
- 67.Van Der Heijden I. M., Wilbrink B., Tchetverikov I., et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis & Rheumatism. 2000;43(3):593–598. doi: 10.1002/1529-0131(200003)43:3<593::aid-anr16>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 68.Peltonen R., Nenonen M., Helve T., Hänninen O., Toivanen P., Eerola E. Faecal microbial flora and disease activity in rheumatoid arthritis during a vegan diet. Rheumatology. 1997;36(1):64–68. doi: 10.1093/rheumatology/36.1.64. [DOI] [PubMed] [Google Scholar]
- 69.Kjeldsen-Kragh J. Rheumatoid arthritis treated with vegetarian diets. The American Journal of Clinical Nutrition. 1999;70(3):594S–600S. doi: 10.1093/ajcn/70.3.594s. [DOI] [PubMed] [Google Scholar]
- 70.Breban M., Tap J., Leboime A., et al. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Annals of the Rheumatic Diseases. 2017;76(9):1614–1622. doi: 10.1136/annrheumdis-2016-211064. [DOI] [PubMed] [Google Scholar]
- 71.Scher J. U., Sczesnak A., Longman R. S., et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2 doi: 10.7554/elife.01202.e01202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang X., Zhang D., Jia H., et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature Medicine. 2015;21(8):895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 73.Terato K., Ye X. J., Miyahara H., Cremer M. A., Griffiths M. M. Induction of chronic autoimmune arthritis in DBA/1 mice by oral administration of type II collagen and Escherichia coll lipopolysaccharide. Rheumatology. 1996;35(9):828–838. doi: 10.1093/rheumatology/35.9.828. [DOI] [PubMed] [Google Scholar]
- 74.de Pablo P., Dietrich T., McAlindon T. E. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. Journal of Rheumatology. 2008;35:70–76. [PubMed] [Google Scholar]
- 75.Liao F., Li Z., Wang Y., Shi B., Gong Z., Cheng X. Porphyromonas gingivalis may play an important role in the pathogenesis of periodontitis-associated rheumatoid arthritis. Medical Hypotheses. 2009;72(6):732–735. doi: 10.1016/j.mehy.2008.12.040. [DOI] [PubMed] [Google Scholar]
- 76.Chou Y.-Y., Lai K.-L., Chen D.-Y., Lin C.-H., Chen H.-H. Rheumatoid arthritis risk associated with periodontitis exposure: a nationwide, population-based cohort study. PLoS One. 2015;10(10) doi: 10.1371/journal.pone.0139693.e0139693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakajima M., Arimatsu K., Kato T., et al. Oral administration of P. Gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0134234.e0134234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshino S., Sasatomi E., Mori Y., Sagai M. Oral administration of lipopolysaccharide exacerbates collagen-induced arthritis in mice. Journal of Immunology. 1999;163(6):3417–3422. [PubMed] [Google Scholar]
- 79.Terato K., Harper D. S., Griffiths M. M., et al. Collagen-induced arthritis in mice: synergistic effect of E. coli lipopolysaccharide bypasses epitope specificity in the induction of arthritis with monoclonal antibodies to type II collagen. Autoimmunity. 1995;22(3):137–147. doi: 10.3109/08916939508995311. [DOI] [PubMed] [Google Scholar]
- 80.Toivanen P., Vaahtovuo J., Eerola E. Influence of major histocompatibility complex on bacterial composition of fecal flora. Infection and Immunity. 2001;69(4):2372–2377. doi: 10.1128/iai.69.4.2372-2377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Totaro M., Cattani P., Ria F., et al. Porphyromonas gingivalis and the pathogenesis of rheumatoid arthritis: analysis of various compartments including the synovial tissue. Arthritis Research & Therapy. 2013;15(3):p. R66. doi: 10.1186/ar4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gomez A., Luckey D., Yeoman C. J., et al. Loss of sex and age driven differences in the gut microbiome characterize arthritis-susceptible 0401 mice but not arthritis-resistant 0402 mice. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0036095.e36095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Azad M. A. K., Sarker M., Li T., Yin J. Probiotic species in the modulation of gut microbiota: an overview. Biomed Research International. 2018;2018:8. doi: 10.1155/2018/9478630.9478630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamashita M., Matsumoto K., Endo T., et al. Preventive effect of lactobacillus helveticus SBT2171 on collagen-induced arthritis in mice. Frontiers in Microbiology. 2017;8:p. 1159. doi: 10.3389/fmicb.2017.01159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kato I., Endo-Tanaka K., Yokokura T. Suppressive effects of the oral administration of Lactobacillus case I on type II collagen-induced arthritis in DBA/1 mice. Life Sciences. 1998;63(8):635–644. doi: 10.1016/s0024-3205(98)00315-4. [DOI] [PubMed] [Google Scholar]
- 86.Jia H., Ren S., Wang X. Heat-killed probiotic regulates the body’s regulatory immunity to attenuate subsequent experimental autoimmune arthritis. Immunology Letters. 2019;216:89–96. doi: 10.1016/j.imlet.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 87.Rogier R., Evans-Marin H., Manasson J., et al. Alteration of the intestinal microbiome characterizes preclinical inflammatory arthritis in mice and its modulation attenuates established arthritis. Scientific Reports. 2017;7:p. 15613. doi: 10.1038/s41598-017-15802-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Evans-Marin H., Rogier R., Koralov S. B., et al. Microbiota-dependent involvement of Th17 cells in murine models of inflammatory arthritis. Arthritis & Rheumatology. 2018;70:1971–1983. doi: 10.1002/art.40657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mikuls T. R., Thiele G. M., Deane K. D., et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis & Rheumatism. 2012;64(11):3522–3530. doi: 10.1002/art.34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoshida A., Nakano Y., Yamashita Y., et al. Immunodominant region of Actinobacillus actinomycetemcomitans 40-kilodalton heat shock protein in patients with rheumatoid arthritis. Journal of Dental Research. 2001;80(1):346–350. doi: 10.1177/00220345010800010901. [DOI] [PubMed] [Google Scholar]
- 91.Marchesan J. T., Gerow E. A., Schaff R., et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis Research & Therapy. 2013;15(6):p. R186. doi: 10.1186/ar4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maresz K. J., Hellvard A., Sroka A., et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD) PLoS Pathogens. 2013;9 doi: 10.1371/journal.ppat.1003627.e1003627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gully N., Bright R., Marino V., et al. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PLoS One. 2014;9(6) doi: 10.1371/journal.pone.0100838.e100838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chukkapalli S., Rivera-Kweh M., Gehlot P., et al. Periodontal bacterial colonization in synovial tissues exacerbates collagen-induced arthritis in B10.RIII mice. Arthritis Research & Therapy. 2016;18:p. 161. doi: 10.1186/s13075-016-1056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sandal I., Karydis A., Luo J., et al. Bone loss and aggravated autoimmune arthritis in HLA-DRβ1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Research & Therapy. 2016;18:p. 249. doi: 10.1186/s13075-016-1143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato K., Takahashi N., Kato T., et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Scientific Reports. 2017;7:p. 6955. doi: 10.1038/s41598-017-07196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dorożyńska I., Majewska-Szczepanik M., Marcińska K., Szczepanik M. Partial depletion of natural gut flora by antibiotic aggravates collagen induced arthritis (CIA) in mice. Pharmacological Reports. 2014;66(2):250–255. doi: 10.1016/j.pharep.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 98.Nemoto N., Takeda Y., Nara H., et al. Analysis of intestinal immunity and flora in a collagen-induced mouse arthritis model: differences during arthritis progression. International Immunology. 2019;32(1):p. 49. doi: 10.1093/intimm/dxz058. [DOI] [PubMed] [Google Scholar]
- 99.Cho Y.-G., Cho M.-L., Min S.-Y., Kim H.-Y. Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis. Autoimmunity Reviews. 2007;7(1):65–70. doi: 10.1016/j.autrev.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 100.Lange F., Bajtner E., Rintisch C., Nandakumar K. S., Sack U., Holmdahl R. Methotrexate ameliorates T cell dependent autoimmune arthritis and encephalomyelitis but not antibody induced or fibroblast induced arthritis. Annals of the Rheumatic Diseases. 2005;64(4):599–605. doi: 10.1136/ard.2004.026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Neurath M. F., Hildner K., Becker C., et al. Methotrexate specifically modulates cytokine production by T cells and macrophages in murine collagen-induced arthritis (CIA): a mechanism for methotrexate-mediated immunosuppression. Clinical & Experimental Immunology. 1999;115(1):42–55. doi: 10.1046/j.1365-2249.1999.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou X., Nardini C. A method for automated pathogenic content estimation with application to rheumatoid arthritis. BMC Systems Biology. 2016;10:p. 107. doi: 10.1186/s12918-016-0344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sayers E., MacGregor A., Carding S. R. Drug-microbiota interactions and treatment response: relevance to rheumatoid arthritis. AIMS Microbiology. 2018;4(4):642–654. doi: 10.3934/microbiol.2018.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nayak R. R., Stapleton-Gray K., O’Loughlin C., Fischbach M., Turnbaugh P. J. Arthritis & Rheumatology. Vol. 69. Hoboken, NJ, USA: Wiley; 2017. [Google Scholar]
- 105.Tsuji F., Yoshimi M., Katsuta O., Takai M., Ishihara K., Aono H. Point mutation of tyrosine 759 of the IL-6 family cytokine receptor, gp130, augments collagen-induced arthritis in DBA/1J mice. BMC Musculoskeletal Disorders. 2009;10:p. 23. doi: 10.1186/1471-2474-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Postow M. A., Callahan M. K., Wolchok J. D. Immune checkpoint blockade in cancer therapy. Journal of Clinical Oncology. 2015;33(17):p. 1974. doi: 10.1200/jco.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Puri A., Homsi J. The safety of pembrolizumab in metastatic melanoma and rheumatoid arthritis. Melanoma Research. 2017;27(5):519–523. doi: 10.1097/cmr.0000000000000387. [DOI] [PubMed] [Google Scholar]
- 108.Kumar P., Saini S., Prabhakar B. S. Cancer immunotherapy with check point inhibitor can cause autoimmune adverse events due to loss of Treg homeostasis. Seminars in Cancer Biology. 2020;64:29–35. doi: 10.1016/j.semcancer.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 109.Wang G., Hu P., Yang J., Shen G., Wu X. The effects of PDL-Ig on collagen-induced arthritis. Rheumatology International. 2011;31(4):513–519. doi: 10.1007/s00296-009-1249-0. [DOI] [PubMed] [Google Scholar]
- 110.Liu C., Jiang J., Gao L., et al. Soluble PD-1 aggravates progression of collagen-induced arthritis through Th1 and Th17 pathways. Arthritis Research & Therapy. 2015;17:p. 340. doi: 10.1186/s13075-015-0859-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee G. The balance of Th17 versus treg cells in autoimmunity. International Journal of Molecular Sciences. 2018;19(3):p. 730. doi: 10.3390/ijms19030730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moon S.-J., Park J.-S., Heo Y.-J., et al. In vivo action of IL-27: reciprocal regulation of Th17 and Treg cells in collagen-induced arthritis. Experimental & Molecular Medicine. 2013;45(10):p. e46. doi: 10.1038/emm.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]