

Abstract
There is a vital need to develop in vitro models of the developing human brain to recapitulate the biological effects that toxic compounds have on the brain. To model perineural vascular plexus (PNVP) in vitro, which is a key stage in embryonic development, human embryonic stem cells (hESC)-derived endothelial cells (ECs), neural progenitor cells, and microglia (MG) with primary pericytes (PCs) in synthetic hydrogels in a custom-designed microfluidics device are cocultured. The formation of a vascular plexus that includes networks of ECs (CD31+, VE-cadherin+), MG (IBA1+), and PCs (PDGFRβ+), and an overlying neuronal layer that includes differentiated neuronal cells (βIII Tubulin+, GFAP+) and radial glia (Nestin+, Notch2NL+), are characterized. Increased brain-derived neurotrophic factor secretion and differential metabolite secretion by the vascular plexus and the neuronal cells over time are consistent with PNVP functionality. Multiple concentrations of developmental toxicants (teratogens, microglial disruptor, and vascular network disruptors) significantly reduce the migration of ECs and MG toward the neuronal layer, inhibit formation of the vascular network, and decrease vascular endothelial growth factor A (VEGFA) secretion. By quantifying 3D cell migration, metabolic activity, vascular network disruption, and cytotoxicity, the PNVP model may be a useful tool to make physiologically relevant predictions of developmental toxicity.
Keywords: developmental toxicity, engineered organoids, human microphysiological systems, microfluidics devices, synthetic hydrogels
1. Introduction
There is a growing need to measure toxicity to human tissues due to the increasing number of environmental contaminants.[1–3] Contaminants such as pharmaceuticals and industrial waste are being detected in air, food, and water.[2,4] The developing human brain is particularly vulnerable to developmental neurotoxicants through maternal exposure, and there has been a significant increase in neurological birth defects and brain disorders over the past three decades.[1,5–9] During early brain development, the human brain is highly vulnerable to developmental neurotoxicants, particularly prior to and during the formation of the blood–brain barrier, where the primary source of protection is the maternal placental barrier. Toxins which cross the maternal barrier may directly influence neurogenesis or angiogenesis processes that can result in neurodevelopmental abnormalities or lifelong vulnerabilities in the adult blood–brain barrier.[10] Capturing this key moment in human brain development is important to our understanding of chemical compounds that may affect brain disorders.
In humans, vascularization of the brain and the formation of the early blood–brain barrier occur during weeks 6–8 post conception. At gestational weeks 6–7, a vascular plexus termed the perineural vascular plexus (PNVP) forms on the outer pial layer of the developing neural tube and is separated from the adjacent neuronal layer by a cellular and extracellular matrix (ECM) membrane consisting of glial end feet and ECM proteins (laminin, reelin) termed the glial limiting membrane (Figure 1A).[11–13] During week 8, angiogenic sprouts infiltrate from the PNVP on the pial (lateral) surface and invade into the cortex toward the luminal (medial) surface.[12–14] Neural progenitor cells, which later form neurons, astrocytes, and oligodendrocytes of the mature cortex and blood–brain barrier, along with pyramidal neurons form a guiding scaffold for the infiltrating vasculature from the outer pial surface toward the inner ventricular surface.[12–14] Microglia and radial glial cells play a key role in mediating the invasion of vascular sprouts and later branching.[15,16] The PNVP invasion stage establishes a dynamic interface composed of microglia (MG), endothelial cells (ECs), pericytes (PCs), neural progenitor cells (NPCs), and the glial limiting membrane.[12,14,17] Ultimately, establishment of the early brain barrier from the PNVP results in the thickening and patterning of cerebral cortex [12,14] and later maturation of the blood–brain barrier along with astrogliogenesis and astrocyte maturation.[10]
Figure 1.
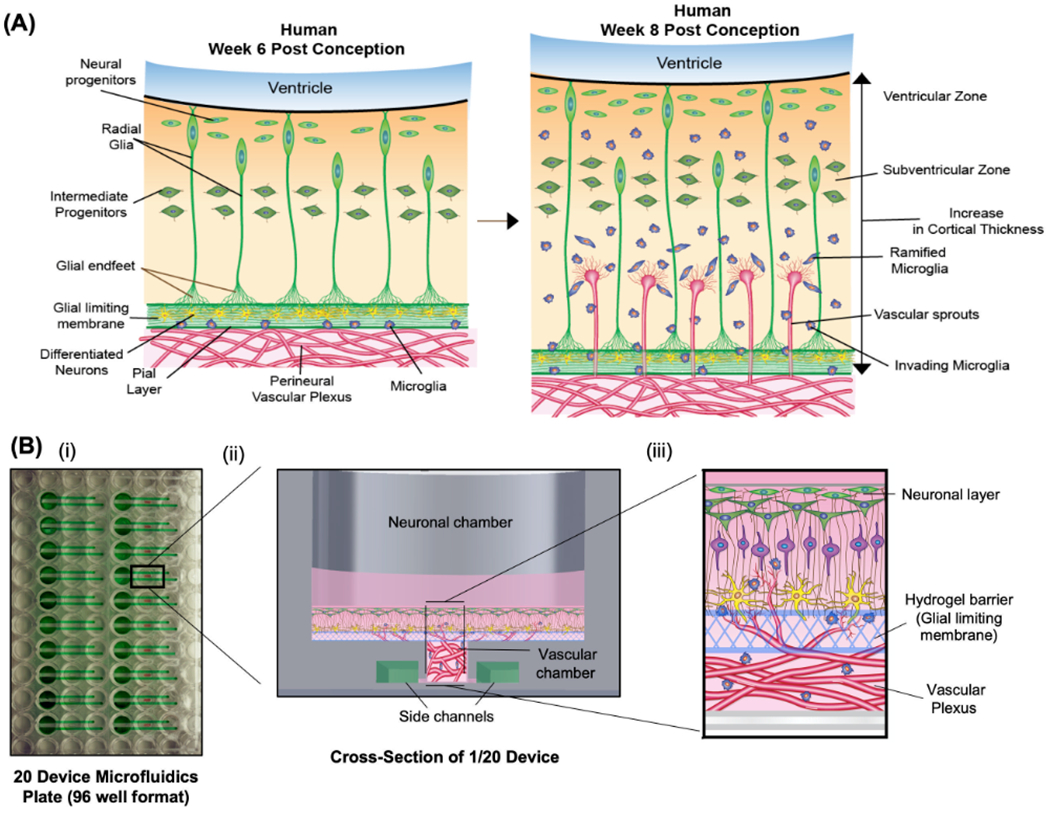
Overview of the establishment of PNVP model. A) Schematic diagram demonstrates early cellular and molecular processes that occur during the initial vascularization of the developing brain from the perineural vascular plexus in humans. B) (i) Overview of a custom designed microfluidics plate containing 20 devices per 96-well plate. (ii) Overview of one device describing different chambers and side channels. (iii) Schematic diagram demonstrates the cross-sectional view of the establishment of PNVP model in vitro.
Prior studies screening for developmental neurotoxicants have typically focused on either 2D or 3D angiogenesis and neurogenesis assays separately, and have not captured developmental timing or discrete multicellular interactions [1]. The advent of pluripotent stem cells and 3D cell culture technologies has attempted to create 3D brain cultures, often termed “organoids,” to recapitulate the early embryonic developmental stages.[18–20] However, these assays have not yet captured the essential PNVP point in brain development and are also not produced with the goal of chemical compound screening. Existing organoids are typically embryoid body-like aggregates cultured free-floating in media, or adherently grown on Matrigel,[21] or other extracellular matrix-coated dishes.[20] The limitations of current organoid models, often, include a lack of 3D vasculature,[18] the presence of animal-derived matrices (e.g., Matrigel)[1] and the absence of a layered structure that mimics specific features of human tissue development.
In this study, we have used synthetic hydrogels and human cells to model the PNVP developmental stages in vitro. We differentiated human embryonic stem cells into i) cortical NPCs (matured until week 6 to match developmental timing), ii) ECs, and iii) MG, and we used human primary PCs. We cocultured the stem cell-derived cell types and the human primary PCs in a 96-well, tubeless, passive flow microfluidics device (Figure 1B). We optimized coculture conditions and developed an experimental system to model the PNVP at various developmental stages and to allow for easy setup of multiple PNVP wells for screening of chemical compounds. Importantly, synthetic poly(ethylene glycol) (PEG) hydrogels functioned as a synthetic ECM for three purposes: i) to assemble a 3D PNVP;[22] ii) to serve as a defined mimic of the glial limiting membrane; and iii) to provide a substrate for cortical neuronal layer development (Figure 1). Our lab has previously established synthetic hydrogel formulations that are supportive of both angiogenic and neurogenic processes, as well as neurovascular integration.[1,22–31] The PEG formulations include pendant cell adhesion peptides for cell interaction, and a protease-labile linkage to allow for hydrogel remodeling. To study the extent to which xenobiotics (e.g., drugs, toxicants) induced pathological responses at the PNVP stage, we treated our in vitro model with putative neurotoxicants that are known to target specific cell populations, and we examined their impact on PNVP formation and integration. The PNVP model provided information on both primary and secondary effects of potential toxicants in the context of a key point of vulnerability in human brain development.
2. Results
2.1. Development of Human PNVP Model In Vitro
We established the early cellular processes of the perineural vascular plexus (Figure 1A) in a custom designed microfluidics plate. Each plate contained 20 microwells, and each microwell included discrete input and output ports (Figure 1B), as detailed in the Experimental Section. Each microwell consisted of i) an underlying vascular chamber loaded with ECs, PCs, and MG within a PEG hydrogel network designed to support the formation of 3D microvasculature, ii) an overlying neuronal chamber designed to support the formation of a neuronal layer, and iii) a PEG hydrogel mimic of the glial limiting membrane separating the vascular chamber and the neuronal chamber (Figure 1B). We showed that the neuronal and vascular chambers were distinct from each other by adding FITC-Dextran (70 kD, concentration 0.25 mg mL−1) in the neuronal chamber and detecting no FITC-Dextran in the collected medium in the vascular chamber during a two-day incubation (data not shown).
The formation of our PNVP model generally mimicked the stages of natural PNVP formation. After the assembly of a vascular network in the vascular chamber (Figure 2A, day 5), the vascular network along with microglia migrated through the glial limiting membrane (Figure 2B, day 14) and integrated with the neuronal layer (Figure 2C, day 21), consistent with the stages of PNVP development.[12–14] The distribution of endothelial cells and microglia within the model at day 5, 14, and 21 was quantified (Figure 3A). Increased brain-derived neurotrophic factor (BDNF) secretion by the vascular network over time likely supported the formation of new capillary sprouts from existing capillaries in the microvasculature in the vascular chamber, and survival of neuronal cells in the neuronal chamber (Figure 3B).[32–34] Whereas, BDNF secretion by neuronal cells in the neuronal chamber likely aided neurogenesis by supporting neuronal outgrowth and differentiation.[32–34] Since secreted metabolites demonstrate an active physiological state, we also characterized metabolite secretion by both the vascular network and the neuronal layer over time using proton-nuclear magnetic resonance (1H NMR) spectroscopy.[35,36] We observed distinct patterns of key biomarkers (glucose, aspartate, choline, alanine, lactate, 3-methyl-2-oxovale, 3-aminoisobutyrate, 2-oxoglutarate) in both chambers, similar to physiological systems in vivo (Figure 3C).[35,36] Different PEG formulations were created of varying crosslink percentages and examined for which formulation allowed vascular sprouting and the migration of endothelial cells and pericytes under the influence of VEGF gradients (data not shown). We found that a PEG formulation with similar peptides and crosslinking percentage to the vascular chamber (crosslink 50%) enabled vascular sprouting, so we used this formulation for the glial limiting membrane mimic.
Figure 2.
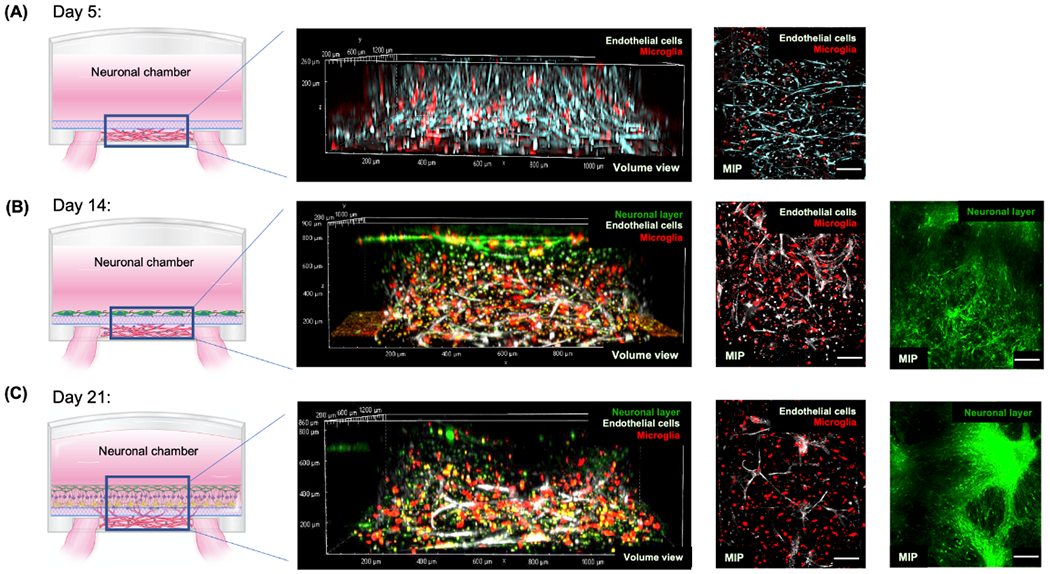
The time-course development of PNVP model in vitro. A) At day 5, vascular network formation in the vascular chamber was visualized using live Cy5-stained endothelial cells (ECs) and cell tracker red-stained microglia (MG). Three-dimensionality of the vascular network was shown by the volume view of the vascular network (second column). The overall vascular network was displayed as maximum intensity projections (MIPs). B) At day 14, the remodeling of the vascular network, the development of the neuronal layer, and their integration was visualized using live Cy5-stained ECs, cell tracker red-stained MG, and cell tracker green-stained differentiating neuronal progenitor cells (NPCs). The integration of the vascular network and neuronal layer was shown by the volume view. The overall vascular network and neuronal layer was displayed as maximum intensity projections (MIPs). C) At day 21, the further maturation of PNVP model was visualized using live Cy5-stained ECs, cell tracker red-stained MG, and cell tracker green-stained differentiating NPCs. Scale bars: 100 μm.
Figure 3.
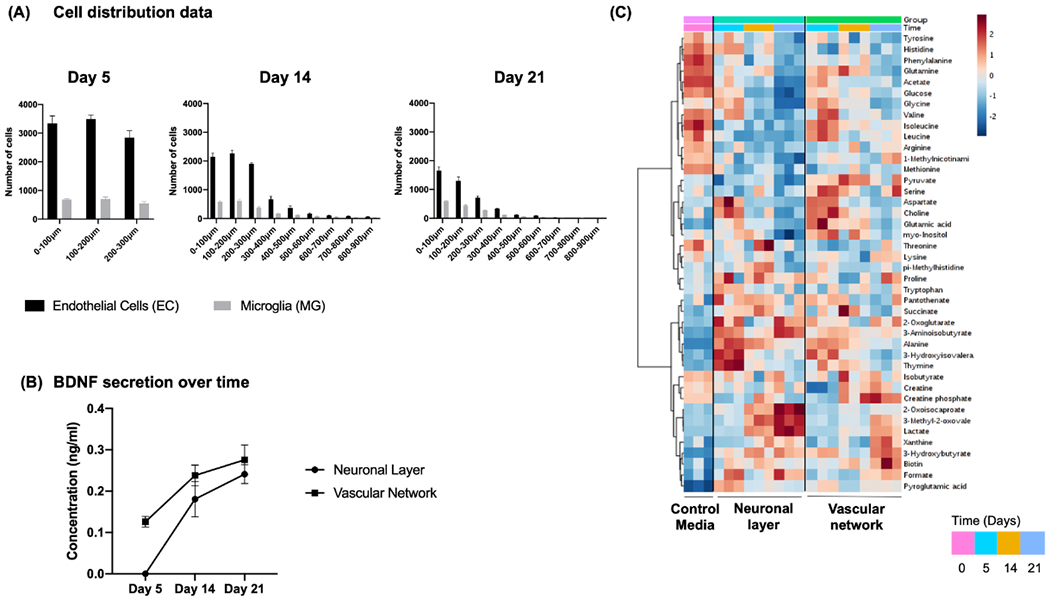
Characterization of PNVP model at day 5, 14, and 21. A) The distribution of endothelial cells and microglia at every 100 μm intervals within PNVP model at day 5, 14, and 21. B) BDNF secretion over time by neuronal layer and vascular network within the neuronal and vascular chamber, respectively. C) Heatmap showing the metabolic profiles of neuronal layer and vascular network analyzed at day 0, 5, 14, and 21. Heatmap was generated by hierarchical clustering using Pearson correlation as a distance measurement and Ward clustering algorithm. Legend shows auto scaled values from 2 to −2 assigned to the relative metabolite concentrations. Individual metabolites are shown in the rows and group averages are shown in the columns of the heatmap.
2.2. Vascular Network Assembly Was Inhibited by Vascular Disrupting Compounds
Vascular cells assembled into 3D networks in the vascular chamber and were negatively affected by known vascular disrupting compounds. At day 5, EC (CD31+), and PC (SM22α+) markers were apparent throughout the 3D vascular network in the vascular chamber (Figure 4A).[1,37] The vascular disruptors Sunitinib and Imatinib significantly decreased EC and MG migration at low (0.8 × 10−6 M), medium (8 × 10−6 M), and high (80 × 10−6 M) concentrations. Pyridaben also inhibited EC and MG migration, but only at high concentration (Figure 4B). Sunitinib, Imatinib, and Pyridaben added at high concentration (80 × 10−6 M) decreased vascular network branching, decreased vascular nodes, and decreased VEGFA secretion. Increased lactate dehydrogenase (LDH) release data showed that high concentrations of vascular disruptors induced cytotoxicity in both the neuronal chamber and the vascular chamber (Figure 4B).
Figure 4.
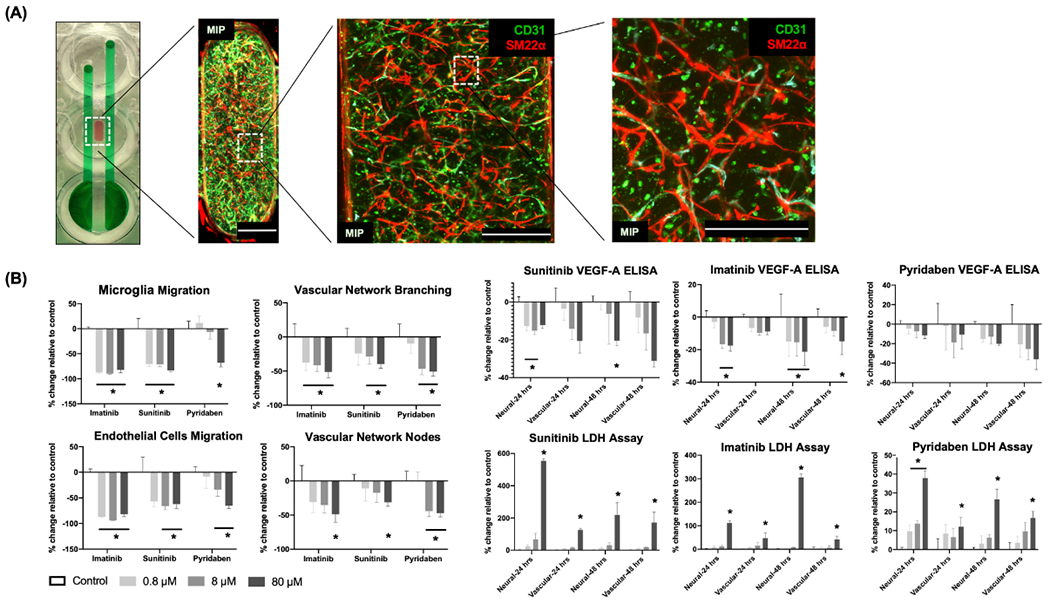
Vascular network characterization and effects induced by vascular disruptors. A) At day 5, standard fixation and immunostaining protocols characterized the formation of vascular network within the vascular chamber of a device in the microfluidics plate by labeling endothelial cells and pericytes with CD31 expression (green) and SM22α (red). Samples were imaged in 3D with confocal microscopy and displayed as maximum intensity projections (MIPs). Scale bars: 500 μm. B). At day 16, concentration-dependent toxicity profiles (endothelial cells migration, microglia migration, vascular network branching and nodes, VEGFA secretion, and LDH release in neural (or, neuronal) and vascular chambers) were measured as percent change, compared to vehicle controls. Vascular disruptors (Imatinib, Sunitinib, and Pyridaben) were added at low (0.8 × 10−6 M), medium (8 × 10−6 M), and high (80 × 10−6 M) concentrations for two days. Data are plotted as mean ± SEM and analyzed using a two-way ANOVA followed by Dunnett’s post hoc test. Statistical significance is indicated by * for p < 0.05.
2.3. The Migration of MG Incorporated into Vascular Plexus Was Inhibited by Microglial Disruptor
Microglia became incorporated with the vascular network and played a key role in endothelial cell migration and vascular network remodeling. At day 5, MG (IBA1+ and CD45+), and PC (SM22α+) markers were expressed throughout the 3D vascular network in the vascular chamber (Figure 5A).[1] Microglia migration was significantly reduced in the absence of the neuronal layer on top of the glial limiting membrane (Figure 5B), indicating that the neuronal layer was chemotactic for MG. Mancozeb treatment (CSF1R inhibitor) for 48 h at 0.03 × 10−6, 0.3 × 10−6, 2 × 10−6, and 6 × 10−6 M resulted in decreased EC and MG migration. However, Mancozeb did not decrease vascular network branching, vascular nodes, or VEGFA secretion, except at the highest concentration added (Figure 5C). We also observed that Mancozeb exhibited cytotoxicity at higher concentrations in the neuronal chamber (Figure 5C).
Figure 5.
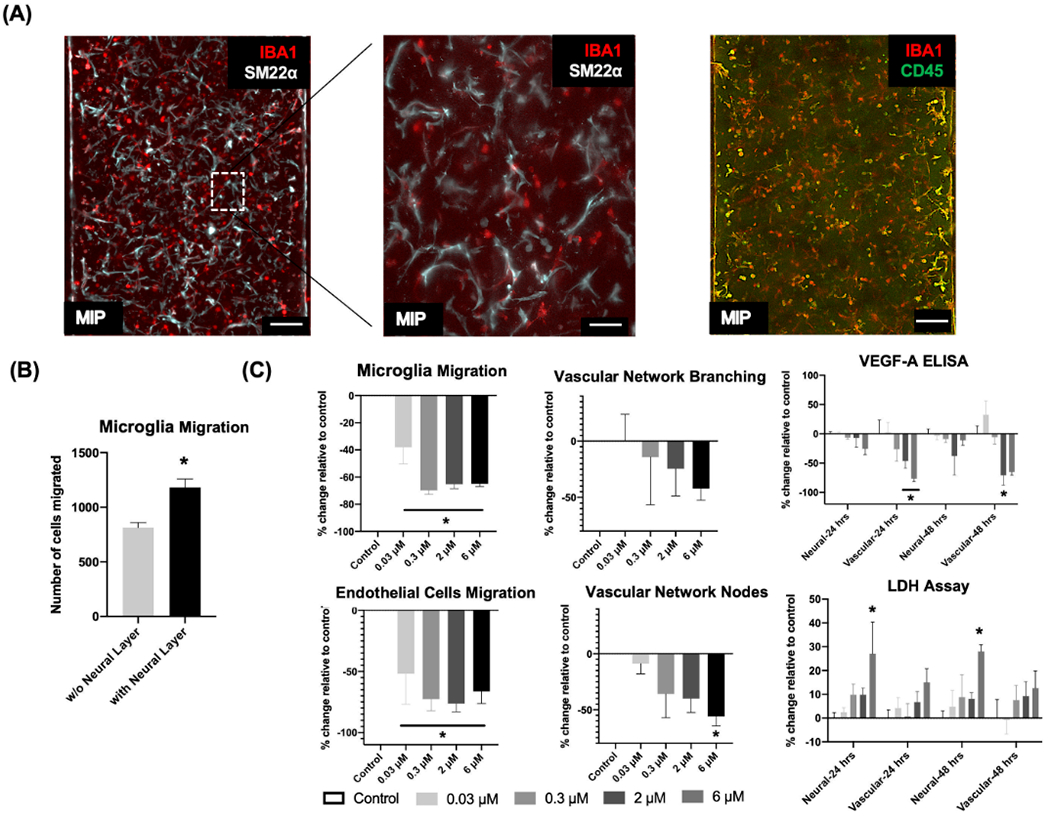
Characterization of microglia incorporated in the vascular network and effects induced by Mancozeb. A) At day 5, standard fixation and immunostaining protocols characterized the incorporation of microglia in the vascular network by labeling microglia with IBA1 expression (red) or CD45 (green), and labeling pericytes with SM22α (Cy5, white). The colocalized expression of CD45, or IBA1 and SM22α indicated the incorporation of microglia in the vascular network. Samples were imaged in 3D with confocal microscopy and displayed as maximum intensity projections (MIPs). Scale bars: 100 μm. B) The migration of microglia in the presence, or absence of the neuronal layer in the neural (or, neuronal) chamber was measured. Data are plotted as mean ± SEM and analyzed using a t-test. Statistical significance is indicated by * for p < 0.05. C) At day 16, concentration-dependent toxicity profiles (endothelial cell migration, microglia migration, vascular network branching and nodes, VEGFA secretion, and LDH release in neuronal and vascular chambers) were measured as percent change, compared to vehicle controls. Mancozeb was added at 0.03 × 10−6, 0.3 × 10−6, 2 × 10−6, and 6 × 10−6 M concentrations for two days. Data are plotted as mean ± SEM and analyzed using a one-way ANOVA followed by Dunnett’s post hoc test. Statistical significance is indicated by * for p < 0.05.
2.4. NPCs Differentiate into Radial Glial, Astrocyte, and Intermediate Neuronal Precursors and Are Affected by Teratogenic Compounds
The neuronal layer of the PNVP model differentiated into neuronal subpopulations, and the migration of cells toward the neuronal layer was affected by the addition of known teratogens. By day 14, the neuronal layer differentiated into neurons (γIII Tubulin+), astrocyte progenitors (GFAP+), and radial glial cells (Nestin+ and Notch2NL+) (Figure 6A).[1,38] The teratogens Thalidomide and Valproate (NMDAR inhibitors) significantly decreased EC and MG migration when added at low (0.8 × 10−6 M), medium (8 × 10−6 M), and high (80 × 10−6 M) concentrations for 48 h (Figure 6B). At high concentrations of both compounds we observed significantly decreased vascular network branching, decreased vascular nodes, and decreased release of VEGFA (Figure 6B). Interestingly, we observed that Thalidomide and Valproate exhibited cytotoxicity at higher concentrations in the neuronal chamber (Figure 6B).
Figure 6.
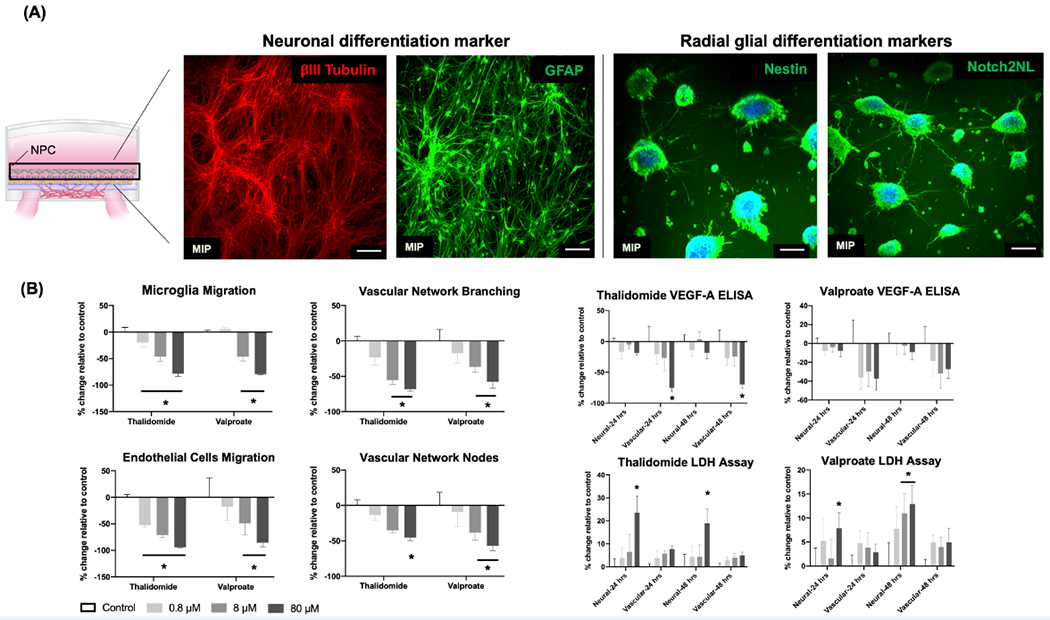
Characterization of neuronal differentiation into other cell types and effects induced by teratogens. A) At day 14, standard fixation and immunostaining protocols characterized the differentiation of neuronal progenitor cells (NPCs) into neurons with γIII Tubulin expression (red), astrocytes with GFAP expression (green), and radial glial cells with Nestin expression (green) and Notch2NL (green). Samples were imaged in 3D with confocal microscopy and displayed as maximum intensity projections (MIPs). Scale bars: 100 μm. B) At day 16, concentration-dependent toxicity profiles (endothelial cells migration, microglia migration, vascular network branching and nodes, VEGFA secretion, and LDH release in neural (or, neuronal) and vascular chambers) were measured as percent change, compared to vehicle controls. Teratogens (Thalidomide and Valproate) were added at low (0.8 × 10−6 M), medium (8 × 10−6 M), and high (80 × 10−6 M) concentrations for two days. Data are plotted as mean ± SEM and analyzed using a two-way ANOVA followed by Dunnett’s post hoc test. Statistical significance is indicated by * for p < 0.05.
3. Discussion and Conclusion
The human brain is at its most vulnerable during the early stages of development, and one key stage of vulnerability is the initial vascularization of the developing brain from the associated perineural vascular plexus (PNVP), where the blood–brain barrier has yet to fully form (Figure 1). Disruptions in PNVP formation may have detrimental effects throughout development and into adulthood, and such disruption is often overlooked when assessing potential developmental neurotoxicants, or developing new neural therapies.[10] Many researchers have attempted to establish brain organoid models, which are often complex, 3D models that simultaneously represent several early developmental processes. Here we sought to mimic discrete, key stages of PNVP assembly by achieving two specific objectives. i) Assembly of a 3D microvascular network, a glial limiting membrane, and a neuronal layer in a microfluidics device, and ii) assess the toxic effects of vascular disruptors and teratogens in the assembled PNVP model.
We confirmed the assembly of ECs, PCs, and MG into 3D networks (Figures 4A and 5A) and the differentiation of the neuronal layer into neuronal subpopulations (Figure 6A). We also used live staining dyes to visualize the remodeling of the vascular network and 3D migration of ECs and MG toward the neuronal layer on top of the glial limiting membrane over three weeks (Figure 2). Previous studies have shown that the BDNF derived from endothelial cells during brain development promoted the formation of new capillaries from existing capillaries and supported neuronal survival during neurogenesis in parallel.[32–34] In addition, BDNF derived from neuronal cells has been shown to support neuronal outgrowth and differentiation into other neuronal precursors.[32–34] We observed similar patterns in our PNVP model in vitro, as BDNF secretion was associated with formation of capillary sprouts and the presence of neurogenesis. This observation further suggested that our model functionally mimicked aspects of PNVP development (Figure 3B). We also used NMR spectroscopy to measure the levels of key metabolites in our culture system to examine the extent of physiological functionality of the neuronal layer and the vascular network (Figure 3C). We observed decreasing levels of glucose, aspartate, choline, alanine, and increasing levels of lactate, 3-methyl2-oxovale, 3 aminoisobutyrate, 2-oxoglutarate over time in both chambers. These changes were in correlation with the patterns observed in neurogenesis and vascular network development in previously published studies, which further suggested that the overall model was metabolically active over time.[35,36]
Our PEG hydrogel formulations were able to serve three distinct purposes. First, the PEG hydrogel in the vascular chamber supported assembly of a vascular network. The density of the cell adhesion peptide ligand Arg-Gly-Asp (RGD) and the matrix metalloproteinase (MMP) degradation peptide linkage (KCGPQGIWGQCK), as well as the mechanical stiffness of the PEG hydrogels was chosen to support 3D vascular network assembly, based on our prior studies.[22,24–31,37,39] Second, an artificial glial limiting membrane was created by overlaying the vascular chamber with a degradable PEG hydrogel formulation (crosslink 50%) that was chosen to support neurovascular interactions and mimic the combined vascular and cortical ECM of the natural glial limiting membrane seen in vivo.[1,23] Finally, the degradable PEG hydrogel barrier delayed vascular infiltration from the underlying vascular layer into the neuronal layer. This allowed NPCs placed on the surface of the synthetic hydrogel at day 5 to develop into a defined neuroepithelial layer, thus establishing a mimic of a neurovascular unit.[1,40] Therefore, the time required for PEG degradation in conjunction with neuronal maturation required to induce MG/EC migration serves as a natural barrier to this phenomenon, thereby causing this delay. Taken together, these observations indicate that synthetic PEG hydrogel formulations served as appropriate multifunctional ECMs for assembly of a PNVP mimic.
Interestingly, the absence of the neuronal layer on top of the glial limiting membrane significantly reduced the migration of microglia (Figure 5B). This result is consistent with the stages of corticogenesis and angiogenesis, which involve reciprocal signaling and occur in opposite directions.[12–14] An enzyme-linked immunosorbent assay (ELISA) showed that the neuronal layer secreted VEGFA, which is a likely mechanism by which the neuronal layer recruited endothelial cell migration. Microglia have also been shown to play a key role in the sprouting of vascular networks through the interplay of CSF1R and VEGF receptors,[15,16] perhaps explaining the effects of the neuronal layer on MG migration.
To validate biological signaling taking place within our model, individual cellular interactions were blocked through the addition of vascular, neuronal, and microglial targeted inhibitors. Vascular network disruptors (Sunitinib, Imatinib, and Pyridaben) at higher concentrations disrupted vascular network formation, and lower concentrations reduced vascular sprouting toward the overlying neuronal layer (Figure 4B). Sunitinib and Imatinib have been shown to block VEGF and PDGFR receptors, which likely disrupted vascular network assembly and cell migration. Other studies have used VEGFR2 inhibitors (e.g., SU5416, PTK787) to inhibit endothelial migration into the neuronal layer.[41,42] Mancozeb treatment (CSF1R inhibitor) inhibited microglial and endothelial cells’ migration toward the neuronal layer (Figure 5B). Previous studies have shown that microglia secrete VEGFC, which further interacts with CSF1R and VEGFA to induce vascular sprouting. Thus, Mancozeb might have blocked CSF1R receptors and reduced vascular sprouting.[15,16] Other CSF1R inhibitors (e.g., pexidartinib, PLX7486) have been shown to inhibit microglia attraction to invading ECs and NPCs.[43] Thalidomide and Valproate (NMDAR inhibitors) affected neuronal metabolism and induced cytotoxicity, resulting in decreased VEGFA output, which would further decrease endothelial migration into the neuronal layer (Figure 6B). Thalidomide and Valproate have been found to induce teratogenic effects by disrupting neuronal communication in early developmental stages.[44–46] In addition, NMDAR inhibitors (e.g., memantine, dextromethorphan) have been used to affect neuronal health and neurological signaling.[47,48]
Taken together, these observations suggest that our PNVP model produces cell responses consistent with what would be expected in response to well-known inhibitory compounds. Acute toxicity and metabolic profiles were monitored using the LDH cytotoxicity assay to capture the release of lactate dehydrogenase due to cellular apoptosis, and an ELISA to capture the release of VEGF-A as a functional output.[49,50] We evaluated the cytotoxicity and functional effect of each compound on the vascular chamber and the neuronal chamber separately—since our device had two separately addressable chambers—to examine whether each compound was affecting angiogenesis more than corticogenesis or vice versa. Our data showed that the neuronal layer and the vascular network exhibited different and independent cytotoxicity profiles under the effects of different compounds. Similarly, we found that Sunitinib, Imatinib, and Valproate exhibited higher cytotoxicity on the neuronal layer than on the vascular network, which resulted in lower VEGFA production, thus resulting in significant reduction in cellular migration toward the neuronal layer (Figures 4B and 6B).[50] These data further suggest that our model has the potential to predict cytotoxicity of compounds at early developmental stages, and the ability to probe individual the tissue-specific toxicity of chemical compounds.
A number of features of the PNVP model could be valuable in future studies of putative chemical toxicants. First, the model is translucent during its formation, which allows for optical imaging during the process of tissue development. Future studies can apply real time imaging to visualize the integration and disruption of the vascular network and the neuronal layer during or after treatment with putative toxicants. Second, the model is constructed using pluripotent stem cell-derived precursor cells, which may enable incorporation of cells with specific genetic backgrounds to be included to understand toxicant susceptibility or study disease establishment. Finally, the cell types included and the synthetic materials used as ECMs in the device can be readily modified to represent different developmental scenarios, so the approach could be more broadly applied to other multilayered tissue structures that form during human development.
4. Experimental section
Cell Culture and Maintenance:
Media formulations and differentiation protocols for differentiating human ESCs into NPCs and MG/macrophages precursor cells were adapted from Schwartz et al.[1] The media formulations and differentiation protocol for differentiating human ESCs into ECs and culturing human primary PC were adapted from Kaushik et al.[37] The composition of all mediums used for this study is described in Table S1 of the Supporting Information.
Microfluidics Device Design and Construction:
Micromilling was used to create devices.[51] Devices were constructed from 96-well microplates (20 devices per plate) using computer-aided design (SolidWorks, Dassault Systèmes, France). To generate toolpaths, SprutCAM software was used and devices were CNC milled (Tormach, Waunakee, WI) into clear 96-well nontissue culture-treated plates (Corning Inc.). Three wells of a 96-well plate were used to create one device through micromilling. Each micromilled device consisted of a vascular microwell (1 mm wide, 2 mm long) connected to two side channels (1 mm wide, 13–16 mm long) both sides by a 100 μm gap. Plates were cleaned by sonication in 100% isopropyl alcohol. Milled plates were washed with water, dried with compressed air, then heated to 70 °C and solvent bonded with acetonitrile. Bonded devices were cooled at room temperature. Excess polystyrene was trimmed from the outside of the device using a handheld razor blade to complete device construction. Devices were then treated with UV for 15 min to sterilize and then transferred to a biosafety hood for cell culture.
Setting Up the PNVP Model in the Microfluidics Plate for Toxicity Screening:
One day before setting up the PNVP models, cultured ECs were stained with SiR-Actin dye (Cytoskeleton, Denver, CO) at 1:10 000 dilution in E7V media. MG were stained with cell tracker red dye (ThermoFisher Scientific, Madison, WI) at 1:7500 dilution in the microglia medium and incubated for 15 min. Then the medium was replaced with fresh media; the stain-containing medium was centrifuged at 300 g for 5 min to remove the stain and recover any microglia floating in the media.
On the day of setting up the microfluidics plates (day 0), PEG hydrogel solution was prepared and endothelial cells, pericytes, and microglia cells were trypsinized, encapsulated, and polymerized according to methods published in the previous papers.[1,37] After trypsinizing EC, PC, and MG using their corresponding protocols, the cells were added in the ratio of EC:PC:MG = 10:5:2 in a 15 mL conical tube, centrifuged at 300 g for 5 min, and the cell pellet was mixed with the hydrogel solution. The hydrogel solution consisted of 40 mg mL−1 8-arm PEG-NB, 4.8 × 10−3 M MMP-degradable peptide (50% molar ratio of cysteines to norbornene groups), 2 × 10−3 M CRGDS, and 0.05% (wt/wt) photoinitiator (Irgacure 2959) in 0.01 × 10−3 M PBS. The cell-hydrogel solution was pipetted into center well of the microfluidics plate (3.1 μL per well) and polymerized for 4 min under a UV lamp (365 nm, 5–10 mW cm−2, UVP XX-15L, Fisher). Cells at the following concentrations in the center well ECs 3400 μL−1, PCs 1700 μL−1, and MG 680 μL−1. Then, 40 μL hydrogel solution was pipetted on top of the polymerized gel in the vascular chamber and polymerized for another 4 min under the UV lamp. The overlay layer was the glial limiting membrane as represented in Figure 1C. 200 μL E7V medium supplemented with 1× nonessential amino acids (ThermoFisher Scientific, Madison, WI) and 1× GlutaMAX (ThermoFisher Scientific, Madison, WI) was added in the center well (on top of the polymerized hydrogels) and 150 μL supplemented E7V medium was added to the feed well. Medium was replaced from the center well and output ports every other day. On day 5, differentiated NPCs stained with cell tracker green dye (ThermoFisher Scientific, Madison, WI) were trypsinized and 50 000 cells in N2 medium were added on top of the overlay gel; medium only was added in the feed well. The N2 medium was replaced every other day.
For toxicity treatments, 150 μL N2 medium containing toxicants (Sunitinib, Imatinib, Pyridaben, Mancozeb, Thalidomide, Valproate) at different concentrations were added to the center well and feed well on day 14. All compounds were purchased from Sigma-Aldrich, St. Louis. Stock solutions (10 × 10−3 M) of all toxicants were prepared in DMSO. Control wells were treated with 0.1% DMSO as a vehicle. Spent medium was collected and fresh medium containing compounds were added at 24 and 48 h of the treatment for the VEGFA ELISA and LDH cytotoxicity assay. Along with visualizing the vascular network, microglia, and neuronal layer during the establishment of PNVP model, migration of endothelial cells and microglia stained with live markers were captured by confocal microscopy after toxicity treatments. For VEGFA ELISA, collected medium samples from the vascular chamber and neuronal chamber were diluted 80 times for VEGFA and analyzed for VEGFA quantification using a Human VEGF Quantikine ELISA kit (R&D systems, Minneapolis, MN) as per manufacturer’s instructions. For BDNF ELISA, non-diluted medium samples were analyzed BDNF quantification using a Human Free BDNF Quantikine ELISA kit (R&D systems, Minneapolis, MN) as per manufacturer’s instructions. For LDH cytotoxicity estimation, LDH release was calculated in the collected medium samples from the vascular chamber and neuronal chamber using Pierce LDH cytotoxicity assay kit (ThermoFisher Scientific, Madison, WI) as per manufacturer’s instructions. For 1 H NMR spectroscopy, medium samples from the vascular chamber and neuronal chamber were processed and analyzed for different metabolites concentration using a procedure described in Ayuso et al.[52]
Immunocytochemistry and Confocal Fluorescence Microscopy:
Immunocytochemistry: After 5, 14, and 21 days of coculture, PNVP organoids in the microfluidics plate were fixed with 4% paraformaldehyde for 2 h. Fixed samples were then washed with 1× PBS for three times for 5 min, then blocked and permeabilized with 0.25% Triton X-100, 10% donkey serum (DS) in PBS for 1 h, and followed by incubation in primary antibody solution (1% DS, 0.25% Triton X-100, 1% BSA in 0.01 m PBS) at the manufacturer’s suggested dilutions for 72 h at 4 °C. Hydrogels were washed five times with PBS for 30 min, then incubated in secondary fluorescent antibody solution (1% DS, 0.25% Triton X-100, 1% BSA in PBS) overnight at 4 °C. Hydrogels were washed five times with PBS for 30 min and placed into PBS for imaging.
Antibodies:
Anti-CD31 (Agilent Technologies, Santa Clara, CA), Anti-VE-cadherin (R&D Systems, Minneapolis, MN), Anti-PDGFRγ (Cell Signaling Technologies, Danvers, MA), Anti-αSMA (Abcam, Cambridge, MA), Anti-IBA1 (Abcam, Cambridge, MA), Anti-CD45 (BD Biosciences, San Jose, CA), Anti-γIII Tubulin (R&D Systems, Minneapolis, MN), Anti-GFAP (Abcam, Cambridge, MA), Anti-Nestin (Abcam, Cambridge, MA), Anti-Notch2NL (Santa Cruz Biotechnology, Dallas, TX), Donkey antirabbit Alexa-Fluor 568 (ThermoFisher Scientific, Madison, WI), Donkey anti-mouse Alexa-Fluor 488 (ThermoFisher Scientific, Madison, WI), Donkey anti-goat Alexa-Fluor 647 (ThermoFisher Scientific, Madison, WI).
Imaging:
A Nikon A1 confocal microscope was used to image the PNVP organoid in the microfluidics plate after toxicity treatment (using live dyes) and immunostaining. All image volumes were captured using a 10× objective at 10 μm intervals, and a pinhole size of 1.2. 488, 568, and 647 nm laser lines were used.
Statistics:
Data are presented as the mean ± SEM. Sample sizes used for toxicity treatment included n = 3 replicates for all compound treatments. Two-way/one-way ANOVA followed by Dunnett’s multiple comparisons test and t-test between two groups were performed using GraphPad Prism version 8.00 for Mac (GraphPad Software, San Diego, CA). Statistical significance was defined as p < 0.05.
Supplementary Material
Acknowledgements
This research was supported by the Environmental Protection Agency STAR Center (Grant Nos. 835737 and K99ES028744) (B.P.J.) and the National Institutes of Health (U01TR002383 and R01NS109427).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Gaurav Kaushik, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA.
Kartik Gupta, Department of Surgery, University of Wisconsin-Madison, 1111 Highland Ave., Madison, WI 53705, USA.
Victoria Harms, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA.
Elizabeth Torr, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA.
Jonathan Evans, Department of Biomedical Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA.
Hunter J. Johnson, Department of Biomedical Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
Cheryl Soref, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA.
Suehelay Acevedo-Acevedo, Department of Biomedical Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA.
Jessica Antosiewicz-Bourget, Morgridge Institute for Research, 330 N Orchard St, Madison, WI 53715, USA.
Daniel Mamott, Morgridge Institute for Research, 330 N Orchard St, Madison, WI 53715, USA.
Peyton Uhl, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA.
Brian P. Johnson, Department of Biomedical Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
Sean P. Palecek, Department of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
David J. Beebe, Department of Pathology and Laboratory Medicine, University of Wisconsin-Madison, 1685 Highland Ave., Madison, WI 53705, USA, University of Wisconsin Carbone Center Research, 600 Highland Ave., Madison, WI 53792, USA, Department of Biomedical Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
James A. Thomson, Morgridge Institute for Research, 330 N Orchard St, Madison, WI 53715, USA
William T. Daly, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA.
William L. Murphy, Department of Orthopedics and Rehabilitation, University of Wisconsin-Madison, 1111 Highland Ave., WIMR 5418, Madison, WI 53705, USA, Department of Biomedical Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA.
References
- [1].Schwartz MP, Hou Z, Propson NE, Zhang J, Engstrom CJ, Costa VS, Jiang P, Nguyen BK, Bolin JM, Daly W, Wang Y, Stewart R, Page CD, Murphy WL, Thomson JA, Proc. Natl. Acad. Sci. USA 2015, 112, 12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Grandjean P, Landrigan PJ, Lancet Neurol. 2014, 13, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fabre KM, Livingston C, Tagle DA, Exp. Biol. Med. 2014, 239, 1073. [DOI] [PubMed] [Google Scholar]
- [4].Landrigan PJ, Curr. Opin. Pediatr. 2010, 22, 219. [DOI] [PubMed] [Google Scholar]
- [5].Crofton KM, Mundy WR, Lein PJ, Bal-Price A, Coecke S, Seiler AEM, Knaut H, Buzanska L, Goldberg A, ALTEX 2011, 28, 9. [PubMed] [Google Scholar]
- [6].van de Leemput J, Boles NC, Kiehl TR, Corneo B, Lederman P, Menon V, Lee C, Martinez RA, Levi BP, Thompson CL, Yao S, Kaykas A, Temple S, Fasano CA, Neuron 2014, 83, 51. [DOI] [PubMed] [Google Scholar]
- [7].Kaushik G, Huber DP, Aho K, Finney B, Bearden S, Zarbalis KS, Thomas MA, Biochem. Biophys. Res. Commun. 2016, 474, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaushik G, Xia Y, Yang L, Thomas MA, BMC Genomics 2016, Suppl 3, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kaushik G, Thomas MA, Sustainable Chem. Pharm. 2019, 13, 100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Saili KS, Zurlinden TJ, Schwab AJ, Silvin A, Baker NC, Hunter ES, Ginhoux F, Knudsen TB, Birth Defects Res. 2017, 109, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Celin AR, Rapacioli M, Gonzalez MA, Ballarin VL, De Plazas SF, López-Costa JJ, Flores V, PLoS One 2015, 10, e0116343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Eichmann A, Thomas JL, Cold Spring Harbor Perspect. Med. 2013, 3, a006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Paredes I, Himmels P, Ruiz de Almodóvar C, Dev. Cell 2018, 45, 10. [DOI] [PubMed] [Google Scholar]
- [14].Bautch VL, James JM, Cell Adhes. Migr. 2009, 3, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ginhoux F, Lim S, Hoeffel G, Low D, Huber T, Front. Cell. Neurosci. 2013, 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reemst K, Noctor SC, Lucassen PJ, Hol EM, Front. Hum. Neurosci. 2016, 10, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marín-Padilla M, Front. Neuroanat. 2012, 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kelava I, Lancaster MA, Dev. Biol. 2016, 420, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, Smith SJ, Huguenard JR, Geschwind DH, Barres BA, Pasca SP, Nat. Methods 2015, 12, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, Horvath TL, Vaccarino FM, Proc. Natl. Acad. Sci. USA 2012, 109, 12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, Nature 2013, 501, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zanotelli MR, Ardalani H, Zhang J, Hou Z, Nguyen EH, Swanson S, Nguyen BK, Bolin J, Elwell A, Bischel LL, Xie AW, Stewart R, Beebe DJ, Thomson JA, Schwartz MP, Murphy WL, Acta Biomater. 2016, 35, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barry C, Schmitz MT, Propson NE, Hou Z, Zhang J, Nguyen BK, Bolin JM, Jiang P, McIntosh BE, Probasco MD, Swanson S, Stewart R, Thomson JA, Schwartz MP, Murphy WL, Exp. Biol. Med. 2017, 242, 1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Toepke MW, Impellitteri NA, Theisen JM, Murphy WL, Macromol. Mater. Eng. 2013, 298, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jongpaiboonkit L, King WJ, Lyons GE, Paguirigan AL, Warrick JW, Beebe DJ, Murphy WL, Biomaterials 2008, 29, 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jongpaiboonkit L, King WJ, Murphy WL, Tissue Eng., Part A 2008, 15, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koepsel JT, Loveland SG, Schwartz MP, Zorn S, Belair DG, Le NN, Murphy WL, Integr. Biol. 2012, 4, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Koepsel JT, Nguyen EH, Murphy WL, Integr. Biol. 2012, 4, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Belair DG, Le NN, Murphy WL, Chem. Commun. 2014, 50, 15651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Murphy WL, McDevitt TC, Engler AJ, Nat. Mater. 2014, 13, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nguyen EH, Daly WT, Le NNT, Farnoodian M, Belair DG, Schwartz MP, Lebakken CS, Ananiev GE, Saghiri MA, Knudsen TB, Sheibani N, Murphy WL, Nat. Biomed. Eng. 2017, 1, 0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marie C, Pedard M, Quirié A, Tessier A, Garnier P, Totoson P, Demougeot C, Cereb J. Blood Flow Metab. 2018, 38, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rossi C, Angelucci A, Costantin L, Braschi C, Mazzantini M, Babbini F, Fabbri ME, Tessarollo L, Maffei L, Berardi N, Caleo M, Eur. J. Neurosci. 2006, 24, 1850. [DOI] [PubMed] [Google Scholar]
- [34].Kermani P, Hempstead B, Trends Cardiovasc. Med. 2007, 17, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ramm P, Couillard-Despres S, Plötz S, Rivera FJ, Krampert M, Lehner B, Kremer W, Bogdahn U, Kalbitzer HR, Aigner L, Stem Cells 2009, 27, 420. [DOI] [PubMed] [Google Scholar]
- [36].Choi WT, Tosun M, Jeong HH, Karakas C, Semerci F, Liu Z, M. Maletić-Savatić, BMC Syst. Biol. 2018, 12, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kaushik G, Gil DA, Torr E, Berge ES, Soref C, Uhl P, Fontana G, Antosiewicz-Bourget J, Edington C, Schwartz MP, Griffth LG, Thomson JA, Skala MC, Daly WT, Murphy WL, Adv. Healthcare Mater. 2019, 8, e1801186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, Mantalas GL, Novak AM, van den Bout A, Bishara A, Rosenkrantz JL, Lorig-Roach R, Field AR, Haeussler M, Russo L, Bhaduri A, Nowakowski TJ, Pollen AA, Dougherty ML, Nuttle X, Addor MC, Zwolinski S, Katzman S, Kriegstein A, Eichler EE, Salama SR, Jacobs FMJ, Haussler D, Cell 2018, 173, 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang J, Chu LF, Hou Z, Schwartz MP, Hacker T, Vickerman V, Swanson S, Leng N, Nguyen BK, Elwell A, Bolin J, Brown ME, Stewart R, Burlingham WJ, Murphy WL, Thomson JA, Proc. Natl. Acad. Sci. USA 2017, 114, E6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nguyen EH, Dombroe MJ, Fisk DL, Daly WT, Sorenson CM, Murphy WL, Sheibani N, Appl. In Vitro Toxicol. 2019, 5, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Latham AM, Bruns AF, Kankanala J, Johnson AP, Fishwick CWG, Homer-Vanniasinkam S, Ponnambalam S, Br. J. Pharmacol. 2012, 165, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O’Reilly T, Persohn E, Rösel J, Schnell C, Stover D, Theuer A, Towbin H, Wenger F, Woods-Cook K, Menrad A, Siemeister G, Schirner M, Thierauch KH, Schneider MR, Drevs J, Martiny-Baron G, Totzke F, Marmé D, Cancer Res. 2000, 60, 2178. [PubMed] [Google Scholar]
- [43].Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D, Immunother J. Cancer 2017, 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vargesson N, Birth Defects Res., Part C 2015, 105, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhao H, Wang Q, Yan T, Zhang Y, Xu H. j., Yu H. p, Tu Z, Guo X, Jiang Y. h., Li X. j, Zhou H, Zhang YQ, Transl. Psychiatry 2019, 9, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lloyd KA, Biosci. Horiz. 2013, 6, hzt003. [Google Scholar]
- [47].Hewitt DJ, Clin. J. Pain. 2000, 16, S73. [DOI] [PubMed] [Google Scholar]
- [48].Olivares D, Deshpande VK, Shi Y, Lahiri DK, Greig NH, Rogers JT, Huang X, Curr. Alzheimer Res. 2013, 9, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kaja S, Payne AJ, Naumchuk Y, Koulen P, Curr. Protoc. Toxicol. 2017, 72, 2.26.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barber M, Andrews WD, Memi F, Gardener P, Ciantar D, Tata M, Ruhrberg C, Parnavelas JG, Cereb. Cortex 2018, 28, 2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guckenberger DJ, De Groot TE, Wan AMD, Beebe DJ, Young EWK, Lab Chip 2015, 15, 2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ayuso JM, Gillette A, Lugo-Cintrón K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP, Skala MC, Beebe DJ, EBioMedicine 2018, 37, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.