

Significance Statement
There are at least 14 different renal tubule segments, each with characteristic cell types with distinct functions. Although the advent of RNA sequencing (RNA-seq) has greatly improved our understanding of gene expression in these renal epithelial cell types, detailed mapping of transcripts has been limited by methods that tend to be biased toward transcript ends. Coupling full-length RNA-seq analysis with renal tubule microdissection characterized gene expression along the mouse renal tubule, including mapping of transcript abundance and alternative exon usage. The data provide a comprehensive view of gene expression along the nephron and collecting duct, made available to scientists via a user-friendly web resource.
Keywords: RNA-seq, alternative splicing, renal tubule, microdissection
Abstract
Background
The repertoire of protein expression along the renal tubule depends both on regulation of transcription and regulation of alternative splicing that can generate multiple proteins from a single gene.
Methods
A full-length, small-sample RNA-seq protocol profiled transcriptomes for all 14 renal tubule segments microdissected from mouse kidneys.
Results
This study identified >34,000 transcripts, including 3709 that were expressed in a segment-specific manner. All data are provided as an online resource (https://esbl.nhlbi.nih.gov/MRECA/Nephron/). Many of the genes expressed in unique patterns along the renal tubule were solute carriers, transcription factors, or G protein–coupled receptors that account for segment-specific function. Mapping the distribution of transcripts associated with Wnk-SPAK-PKA signaling, renin-angiotensin-aldosterone signaling, and cystic diseases of the kidney illustrated the applications of the online resource. The method allowed full-length mapping of RNA-seq reads, which facilitated comprehensive, unbiased characterization of alternative exon usage along the renal tubule, including known isoforms of Cldn10, Kcnj1 (ROMK), Slc12a1 (NKCC2), Wnk1, Stk39 (SPAK), and Slc14a2 (UT-A urea transporter). It also identified many novel isoforms with segment-specific distribution. These included variants associated with altered protein structure (Slc9a8, Khk, Tsc22d1, and Scoc), and variants that may affect untranslated, regulatory regions of transcripts (Pth1r, Pkar1a, and Dab2).
Conclusions
Full-length, unbiased sequencing of transcripts identified gene-expression patterns along the mouse renal tubule. The data, provided as an online resource, include both quantitative and qualitative differences in transcripts. Identification of alternative splicing along the renal tubule may prove critical to understanding renal physiology and pathophysiology.
Human and rodent genomes contain around 20,000 protein-coding genes.1 The identity of each of the individual cell types throughout the body are determined by what mix of these 20,000 genes are expressed. However, the repertoire of gene expression in each cell type is expanded even more greatly by alternative exon usage, allowing a single gene to produce more than one mRNA species and more than one protein. These alternatively spliced isoforms may result in differences in protein structure and function, or differences in mRNA properties (e.g., mRNA stability, localization, and translation efficiency).2 Previously, we mapped gene expression along the rat renal tubule by microdissecting each of 14 renal tubule segments through the use of RNA sequencing (RNA-seq) in microdissected tubule segments,3 producing a widely used gene-expression resource. Although successfully mapping >9000 transcripts, the methodology used did not allow a comprehensive accounting of alternative exon usage along the renal tubule. Furthermore, although the rat is used extensively in studies of renal physiology, most contemporary studies of renal physiology and pathophysiology use the mouse. Popular single-cell RNA-seq (scRNA-seq) methods have achieved profiling of cell type–specific gene expression, but have limited sequencing depth and preclude capture of full-length transcripts.4 Renal tubule microdissection coupled with small-sample RNA-seq has the potential to circumvent these limitations by generating adequate read coverage of full-length mRNAs, while identifying cell type–specific alternative splicing.
Here, we use a full-length, small-sample RNA-seq protocol to characterize transcriptomes for all 14 renal tubule segments microdissected from mouse kidneys. The resulting sensitivity and precision allowed us to use the data to comprehensively characterize alternative-splicing differences among the different renal epithelial cell types. We also provide a web-based resource that allows users to explore and download data for future studies.
Methods
Mouse Renal Tubule Microdissection
Male, 6- to 8-week-old, C57BL/6 mice (Taconic) were used for renal tubule microdissection. All animal work in this study was conducted in accordance with National Institutes of Health (NIH) animal protocol H-0047R4. Mice were euthanized via cervical dislocation. The kidneys were perfused via the left ventricle with ice-cold Dulbecco's PBS (DPBS; Thermo Scientific) to remove blood cells, followed by reperfusion with the dissection buffer (5 mM HEPES, 120 mM sodium chloride [NaCl], 5 mM potassium chloride [KCl], 2 mM calcium chloride [CaCl2], 2.5 mM disodium phosphate, 1.2 mM magnesium sulfate, 5.5 mM glucose, 5 mM Na acetate, pH 7.4) with 1 mg/ml collagenase B (Roche). We harvested the kidneys, obtained thin tissue slices along the cortical-medullary axis, and proceeded to digestion. The dissociation was carried out in dissection buffer containing collagenase B (1–2 mg/ml) and hyaluronidase (1.2–2 mg/ml; Worthington Biochemical) at 37°C with frequent agitation for 30 minutes. We monitored the digestion until the optimal microdissectable condition was reached. Most renal tubules were readily microdissectable under this condition, except the inner medullary collecting ducts (IMCDs), which required a higher enzyme concentration (2 mg/ml) and a longer digestion time (up to 90 minutes at 37°C). The microdissection was carried out under a Wild M8 dissection stereomicroscope equipped with on-stage cooling. The renal tubules were washed in dishes containing ice-cold DPBS by pipetting to remove contaminants before RNA extraction using a Direct-zol RNA MicroPrep kit (Zymo Research, Irvine, CA). Four to eight tubules were collected for each sample.
Small-Sample RNA-Seq
These steps were performed as previously reported.5 We eluted RNA in 10 µl water and immediately proceeded to generation of cDNA using the SMART-Seq V4 Ultra Low RNA Kit (Takara Bio, Mountain View, CA). Of the resulting cDNA from 14 PCR cycles, 1 ng was “tagmented” and bar coded by using a Nextera XT DNA Sample Preparation Kit (Illumina). The final libraries were purified by AmPure XP magnetic beads (Beckman Coulter, Indianapolis, IN) and quantified using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA). An equal amount of index libraries were pooled and sequenced (paired-end 50 bp) on an Illumina Hiseq 3000 platform.
RNA-seq reads were aligned by STAR (2.5.4a)6 to the mouse Ensembl genome (release 94) with the corresponding annotation file (release 94). We processed the unique genomic alignment for alignment visualization on the University of California, Santa Cruz (UCSC) Genome Browser, or Jbrowse.7 Transcripts per million (TPM) and expected counts were obtained using RSEM (1.3.0).8 We excluded samples with <75% uniquely mapped reads and <10 million uniquely mapped reads for downstream analysis. Unless otherwise specified, the computational analyses were performed on the NIH Biowulf High-Performance Computing platform.
Differential Expression Analysis and Hierarchic Clustering
Differentially expressed genes (DEGs) were identified using R package edgeR.9 Raw expected counts (RSEM output) were used as input for this analysis. Each renal tubule segment was compared against the rest of the segments. Genes from these comparisons with a maximum TPM of ≥25, absolute log fold change ≥2, and false discovery rate <0.05 were merged. A total of 3709 nonredundant genes were included in the following analysis. Hierarchic clustering analysis was done in R using the Pearson correlation. TPM values were used in the log2(TPM+1) scale. Distance between samples or between genes is defined as as.dist(1−correlation). Heatmaps were generated using the heatmaply package.
Isoform Analysis
Alternative-splicing analysis was performed with MAJIQ 2.1.10 To facilitate the identification process of local splicing variations (LSVs), RNA-seq data were remapped using STAR in a two-pass mode6 before MAJIQ analysis. Exon’s percent spliced in (PSI or Ψ) was estimated for each renal segment with replicates. Similar to the gene differential analysis described above, the individual renal segment was compared with other segments to estimate the change in each splicing junction’s relative inclusion level, termed ΔPSI (ΔΨ). High-confidence differential splicing variants were selected on the basis of P(ΔΨ >0.2)>0.95. All nonredundant, differentially spliced LSVs along the renal tubule were included in the final list. Splicing variants and splice graphs were visualized using Voila.10 Representative LSVs from each renal segment were further analyzed with MAJIQ-SPEL10 on the Galaxy server (https://galaxy.biociphers.org/galaxy/root?tool_id=majiq_spel). The alignments were visualized on the UCSC Genome Browser, and tracks were downloaded for figure preparation. Exon-level or user-defined region read counts were obtained using featureCounts (subread/2.0.1). The counts were normalized to exon length before being normalizing to the reference exon.
Comparison of Small-Sample RNA-Seq and Published Single-Cell RNA-Seq Data
The single-cell RNA-seq data published by Ransick et al.11 were downloaded from the Gene Expression Omnibus (GEO; GSE129798) and processed with Seurat using the parameters mentioned in the original paper. Cell clusters were identified and projected on UMAP (Supplemental Figure 2A). Epithelial cell clusters were further annotated using the cell type–specific markers proposed in the original paper (Supplemental Figure 2, B–D). The average gene expression for each cell cluster is provided in Supplemental Table 2. The top ten DEGs of each epithelial cell cluster were mapped to the small-sample RNA-seq data (Supplemental Figure 2E, Supplemental Table 2).
Quantitative PCR
cDNAs from the cortical thick ascending limb of the loop of Henle (CTAL), distal convoluted tubule (DCT), connecting tubule (CNT), cortical collecting duct (CCD), and outer medullary collecting duct (OMCD) (n=3 mice for each segment) were used for quantitative PCR (qPCR). Primers targeting Stk39 full-length (forward primer A and reverse primer) and exon 5a–containing isoform (forward primer B and reverse primer) were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Supplemental Figure 4B). qPCR assays were performed using FastStart Universal SYBR Green Master mix (Sigma) according to the manufacturer’s protocol with minor modifications. In the 384-well plate (Applied Biosystems), 15-μl reactions (10 ng cDNA) were performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems). Each reaction was performed twice, and qPCR data were analyzed using the 2ΔΔCt method. Final values were log2 transformed (Supplemental Figure 4C).
Immunohistochemistry and RNAscope In Situ Hybridization
The mouse kidney tissue was prepared as described previously.5 Mice underwent cervical dislocation and were perfused with ice-cold DPBS followed by 4% paraformaldehyde in DPBS. Kidneys were then fixed for 2 hours in 4% paraformaldehyde before transferring to 20% sucrose at 4°C overnight. The blocks were further processed in the National Heart, Lung, and Blood Institute (NHLBI) Pathology Facility. Sections (6 µm thick) were cut. Frozen sections were thawed at room temperature for 10–20 minutes and rehydrated in PBS for 10 minutes. After blocking for 30 minutes with 1% BSA and 0.2% gelatin, primary antibodies were applied overnight at 4°C. Sections were washed three times for 5 minutes in PBS. The secondary antibody incubation was carried out for 1 hour at room temperature.
RNAscope was performed on fixed frozen tissue sections according to RNAscope Multiplex Fluorescent Reagent Kit version 2 user manual (document #323100-USM). TSA Plus fluorophores at the concentration of 1:1500 were used to develop signals. Once the signals were developed for each channel, a standard immunohistochemistry protocol was followed for antibody staining. The confocal images were taken in the NHLBI Light Microscopy Core.
Immunohistochemistry Antibodies and RNAscope Probes
The antibody used in this study is chicken anti-Slc12a1 (C20; Knepper laboratory). The probe used in this study is Ptger3 (catalog number 501831-C3; Advanced Cell Diagnostics).
Website
The website was built around the Shiny package in R. Data tables were displayed using the DT package. The plotly, ggplot2, and heatmaply packages were used to create the interactive heatmaps or feature plots. The RSQLite, dbplyr, and pool packages were used to write and fetch data from the Genotype-Tissue Expression (GTEx) V8 RNA-seq database.
Results
Transcriptomic Landscape of Microdissected Mouse Renal Tubule Segments
To provide a comprehensive gene-expression landscape and alternative-splicing landscape of renal epithelial cells, we carried out full-length RNA-seq in all 14 renal tubule segments microdissected from mouse kidneys, including proximal tubules (PTs; initial segment of the PT [PTS1], proximal straight tubule in cortical-medullary rays [PTS2], and last segment of the proximal straight tubule in the outer stripe of outer medulla [PTS3]); thin limbs of Henle’s loop (the short descending limb of the loop of Henle [DTL1], long descending limb of the loop of Henle in the outer medulla [DTL2], long descending limb of the loop of Henle in the inner medulla [DTL3], and thin ascending limb of the loop of Henle [ATL]); thick limbs (medullary thick ascending limb of the loop of Henle [MTAL] and cortical thick ascending limb of the loop of Henle [CTAL]); distal convoluted tubule (DCT); and all collecting duct segments (connecting tubule [CNT], cortical collecting duct [CCD], outer medullary collecting duct [OMCD], and inner medullary collecting duct [IMCD]) (Figure 1).3,12–14 The resulting data are viewable at https://esbl.nhlbi.nih.gov/MRECA/Nephron/ or Supplemental Table 1. For the DCT, it was impossible to see well-defined boundaries between DCT1 and DCT2 (Figure 1B), so we did not separately dissect these. In total, we collected 93 samples from 53 mice (87 renal tubules plus six glomeruli; https://esbl.nhlbi.nih.gov/MRECA/G/), with an average of 85% uniquely mapped reads (Supplemental Figure 1A), an average of 3.7% mitochondrial fraction (Supplemental Figure 1B), and no systematic batch effects (principal component analysis, Supplemental Figure 1C), indicating high data quality. Using a threshold of TPM greater than one, a median of 13,444 transcripts (including noncoding genes) were detected in each segment (Supplemental Table 1).
Figure 1.
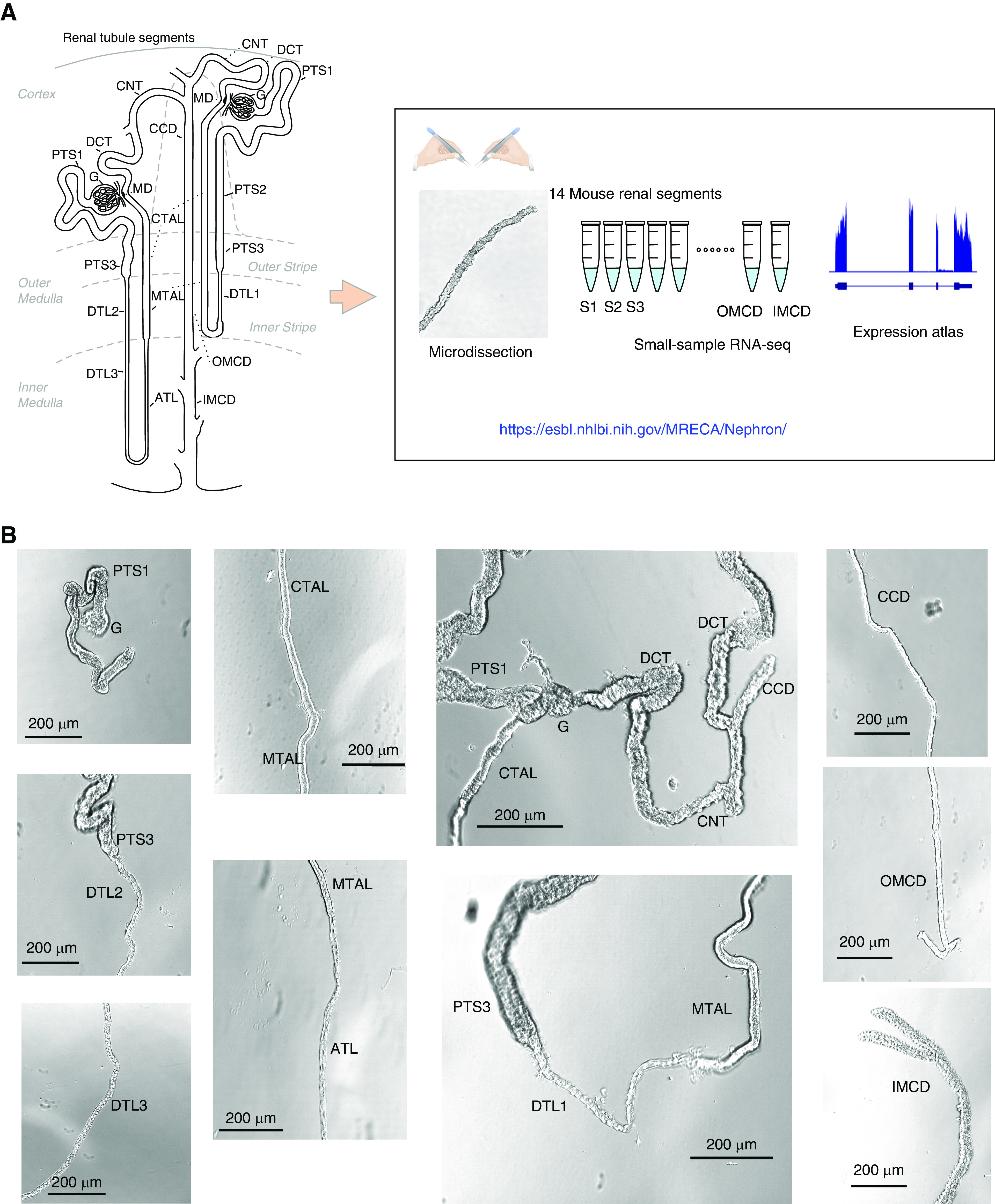
Overview of renal tubule cell nomenclature and experimental design. (A) The scheme12,13 shows the connection of both a short-looped and a long-looped nephron to the CD system. Small-sample RNA-seq coupled with microdissection was used to quantify gene expression in all 14 mouse renal tubule segments. The data are provided in a user-friendly website. (B) Representative images of microdissected mouse renal tubule segments. The images were captured by the Invitrogen EVOS XL Core Cell Imaging System. Scale bars, 200 µm. G, glomerulus; PTS1, the initial segment of the proximal tubule; PTS2, proximal straight tubule in cortical medullary rays; PTS3, last segment of the proximal straight tubule in the outer stripe of outer medulla; DTL1, the short descending limb of the loop of Henle; DTL2, long descending limb of the loop of Henle in the outer medulla; DTL3, long descending limb of the loop of Henle in the inner medulla; ATL, thin ascending limb of the loop of Henle; MTAL, medullary thick ascending limb of the loop of Henle; CTAL, cortical thick ascending limb of the loop of Henle; MD, mecula densa; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct.
As shown in Figure 2A, the distribution of recognized kidney cell type–specific markers along the renal tubule matched well with prior knowledge.15 For example, megalin (Lrp2) was found chiefly in PTs, the bumetanide-sensitive Na-K-2Cl cotransporter 2 (Slc12a1) was restricted to MTAL and CTAL, the thiazide-sensitive Na+-Cl− cotransporter (Slc12a3) and the Ca2+-binding protein parvalbumin (Pvalb) were confined to DCT, whereas aquaporin-2 (Aqp2) was limited to CD segments, including the CNT. Overall, these data illustrate a high degree of concordance among replicates, demonstrating the reproducibility and accuracy of the small-sample RNA-seq approach applied to microdissected tubules. This fidelity of the data was further revealed by comparing the small-sample RNA-seq data with recently published single-cell data (see Methods) (Supplemental Figure 2, Supplemental Table 2).11
Figure 2.
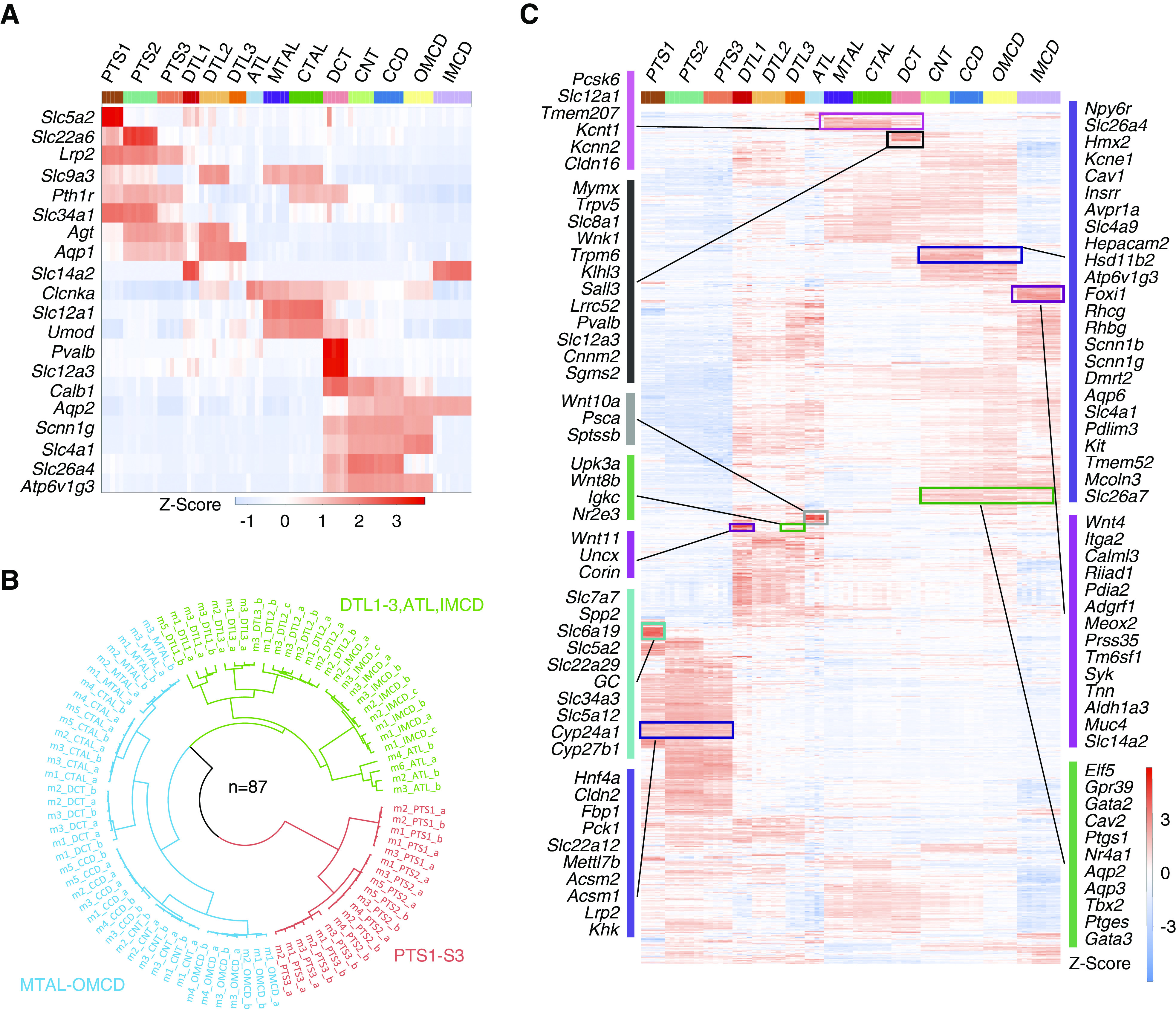
Gene expression along renal tubule segments. (A) Heatmap showing the expression pattern of classic renal tubule markers. Columns are individual renal tubule replicates and rows are marker genes. Color bar (top) indicates different renal tubule segments. Red color indicates high expression, and blue color indicates low expression. Z-score was calculated from log2(TPM+1) using the scale function. (B) Three major renal tubule clusters were revealed in which the PTs were clustered together (red), segments from the MTAL through the OMCD formed a second group (blue), and thin limbs of the loop of Henle and IMCDs formed a third group (green). The polar dendrogram was built by hierarchic clustering analysis. Sample identifiers refer to both biologic and technical information. (C) Heatmap displaying the scaled expression pattern of 3709 DEGs along the renal tubule segment. The full gene list is provided in Supplemental Table 4.
Next, we performed differential gene-expression analysis on each renal tubule segment separately and identified 3709 DEGs (Supplemental Table 3). On the basis of the degree of similarity of their DEG expression patterns, three major renal tubule clusters were revealed (Figure 2B). The transcripts accounting for these patterns are presented in Figure 2C and Supplemental Table 4, many of which are products of novel segment-specific genes.
The three PT segments (PTS1, PTS2, and PTS3) exhibited comparable expression patterns, which differed markedly from those of other tubule segments (Figure 2C). Some transcripts are exclusively localized to the PTS1 (turquoise cluster, Figure 2C), and are linked to glucose reabsorption (Slc5a2 [SGLT2]), lactate reabsorption (Slc5a12 [Na-lactate cotransporter]), vitamin D metabolism (Gc [vitamin D–binding protein], Cyp24a1 [the 24-hydroxylase], and Cyp27b1 [the 1-hydroxylase]), and amino acid reabsorption (Slc7a7 [L-type amino acid transporter], Slc6a19 [neutral amino acid transporter AT1]). Other transcripts are selectively expressed in all three PTs (violet cluster, Figure 2C), such as the transcription factor (TF) Hnf4a, the tight-junction protein claudin-2 (Cldn2), phosphoenolpyruvate carboxykinase (Pck1), and megalin (Lrp2).
The thin limb cell types have not been studied extensively. Although there are strong similarities among the thin limb segments (Figure 2C), they can be distinguished by certain segment-specific genes: DTL1 selectively expresses the serine protease Corin, the homeobox TF Uncx, and the urea channel Slc14a2. The long descending limb of the loop of Henle in the outer medulla selectively expresses the secreted activin-antagonist protein follistatin (Fst), the GPI-linked adhesion protein Cdh13, and the protein kinase Stk32a. The long descending limb of the loop of Henle in the inner medulla selectively expresses the nuclear receptor Nr2e3, the Ig κ chain Igkc, and the secreted protein dermokine (Dmkn). ATL selectively expresses IGF-binding protein 2 (Igfbp2); the GPI-linked, prostate-specific cell antigen (Psca); serine palmitoyltransferase small subunit B (Sptssb); and the secreted TNF-α inhibitor protein Ccdc3.
In the TAL-DCT-CNT region, some transcripts with less well-characterized functions were selectively enriched. For example, TALs selectively expressed Tmem207 and Pcsk6 (proprotein convertase subtilisin/kexin-6). Tmem207, which codes for a single-pass type I membrane protein, showed restricted expression in kidney versus all other tissues (GTEx, https://www.gtexportal.org/home/gene/TMEM207), suggesting a specialized role in the kidney. Pcsk6 acts as a secreted enzyme that activates atrial peptide-converting enzyme (i.e., Corin, a serine peptidase) in the heart, thereby regulating BP.16 Interestingly, very high levels of Corin were found exclusively in DTL1, where it can potentially be released by Pcsk6 derived from the neighboring MTAL.16 In DCT, some transcripts with no prior known functional roles in the DCT were identified. For example, LRRC52 is a K+ channel auxiliary γ subunit that modulates the function of Slowpoke-homolog K channels, including Maxi-K (Kcnma1)17 and Kcnu1.18
In the CNT-CD region of the renal tubule, we found genes already known to be selectively expressed in principal cells (PCs; Aqp2, Aqp3, Gata3, and Scnn1g) and intercalated cells (ICs; Slc4a1, Slc26a4, Foxi1, Atp6v1g3, and Aqp6). The IMCD, present in the region of the kidney with the highest osmolality, however, exhibited a unique gene-expression pattern that included IMCD-enriched genes linked to epithelial cell differentiation (such as Pax2, Wnt7b, Wnt4, and Gata3).19,20
To further identify unique patterns of gene distribution from Figure 2C, we mapped solute carriers (Figure 3A, Supplemental Table 5), TFs (Figure 3B), G protein–coupled receptors (Figure 3C), aquaporins (Supplemental Figure 1D), and ion channels (Supplemental Figure 1E) along the renal tubule. In addition, segment-specific, ligand-receptor pairs are listed in Supplemental Table 6. To support the fidelity of the data, immunofluorescence, coupled with in situ hybridization with RNAscope, confirmed the expression of Ptger321 in some, but not all, cells of the TAL (Figure 3D).
Figure 3.

Gene-expression patterns. (A) Dot plot displaying major non-PT solute carriers along the mouse nephron. See Supplemental Table 5 for PT-selective solute carriers. (B) Dot plot displaying important TFs. Several TFs were selectively expressed in DTLs. Examples include atonal TF (Atoh8) and UNC homeobox (Uncx) in DTL1; and nuclear receptor (Nr2e3) and paired-like homeodomain 2 (Pitx2) in DTL2–3. Several TFs were found in DCT, including Sall3 (a zinc-finger TF) and Emx1 (a homeobox TF). The mesenchyme homeobox 2 (Meox2) and T-Box TF 3 (Tbx3) were found to be highly expressed in IMCD. (C) G protein–coupled receptors (GPCRs) along the mouse nephron segments. The analysis also identified some GPCRs without well-characterized kidney functions, including the cholecystokinin A receptor (Cckar) in PT. In addition, serotonin receptor (Htr4) and thyroid-stimulating hormone receptor (Tshr) were selectively found in DTL1, PGE receptor 2 (Ptger2) was found in DTL3, and GPCR110 (Adgrf1) was found in IMCD. Dot size and color indicate expression (log2[TPM+1]). (D) Immunostaining coupled with RNAscope showing Ptger3 expression (green dots) in Slc12a1 cells (red) in mouse kidney sections. Scale bar, 20 µm.
The Landscape of Alternative Splicing in Mouse Renal Tubules
Our full-length RNA-seq protocol enables us to systematically map alternative exon usage along the renal tubule. We examined alternative-splicing events using MAJIQ,10 which quantifies reads that span splice sites (see Methods). The relative inclusion level of each junction (PSI or Ψ) in each renal tubule segment was estimated using all replicates. In total, we detected 931 confident alternative-splicing sites across 683 genes (Supplemental Table 7). These types of alternative splicing include exon skipping (64.7%), alternative 5′ splicing (2.0%), alternative 3′ splicing (0.75%), and complex splicing (the combination of aforementioned, 32.5%).10
Our data revealed distinct, segment-specific isoforms of many genes, including known isoforms of Cldn10, Kcnj1 (ROMK), Slc12a1 (NKCC2), Wnk1, Stk39 (SPAK), and Slc14a2 (urea transporter A) (Supplemental Figures 3, A–F, and 4 for Stk39). Some novel isoforms were also identified, including those that have effects on protein structures (changes of amino acid sequence) (Figure 4, Supplemental Figure 5) or mRNA properties (changes of noncoding sequence) (Figure 5, Supplemental Figure 6). Alternative-splicing events are regulated by RNA-binding proteins and, more specifically, by splicing factors. The differentially expressed splicing factors are listed in Supplemental Table 8.
Figure 4.

Alternative splicing occurs within protein-coding regions. Distribution of (A) Slc9a8, (B) Khk, (C) Tsc22d1, and (D) Scoc isoforms along renal segments. Mapping is visualized in the UCSC Genome Browser. The structure of the gene is shown at the top of the track, with exons connected by blue rectangles, and introns indicated by thin lines with arrows. The splicing graph at the top of the panel was generated using MAJIQ-SPEL. The paths on the splicing graph indicate the splicing junctions. The same color codes were used for exons in the splice graph and UCSC Genes Track. Gene expression is shown on the right. Quantifications of relative abundance (PSI) of indicated exons are shown in violin plot at the bottom (sum of PSIs=1). PSI was calculated by MAJIQ and visualized by MAJIQ-SPEL. Track height scale, auto. The bottom protein alignment track is annotated on the basis of the UniProt/SwissProt database.
Figure 5.
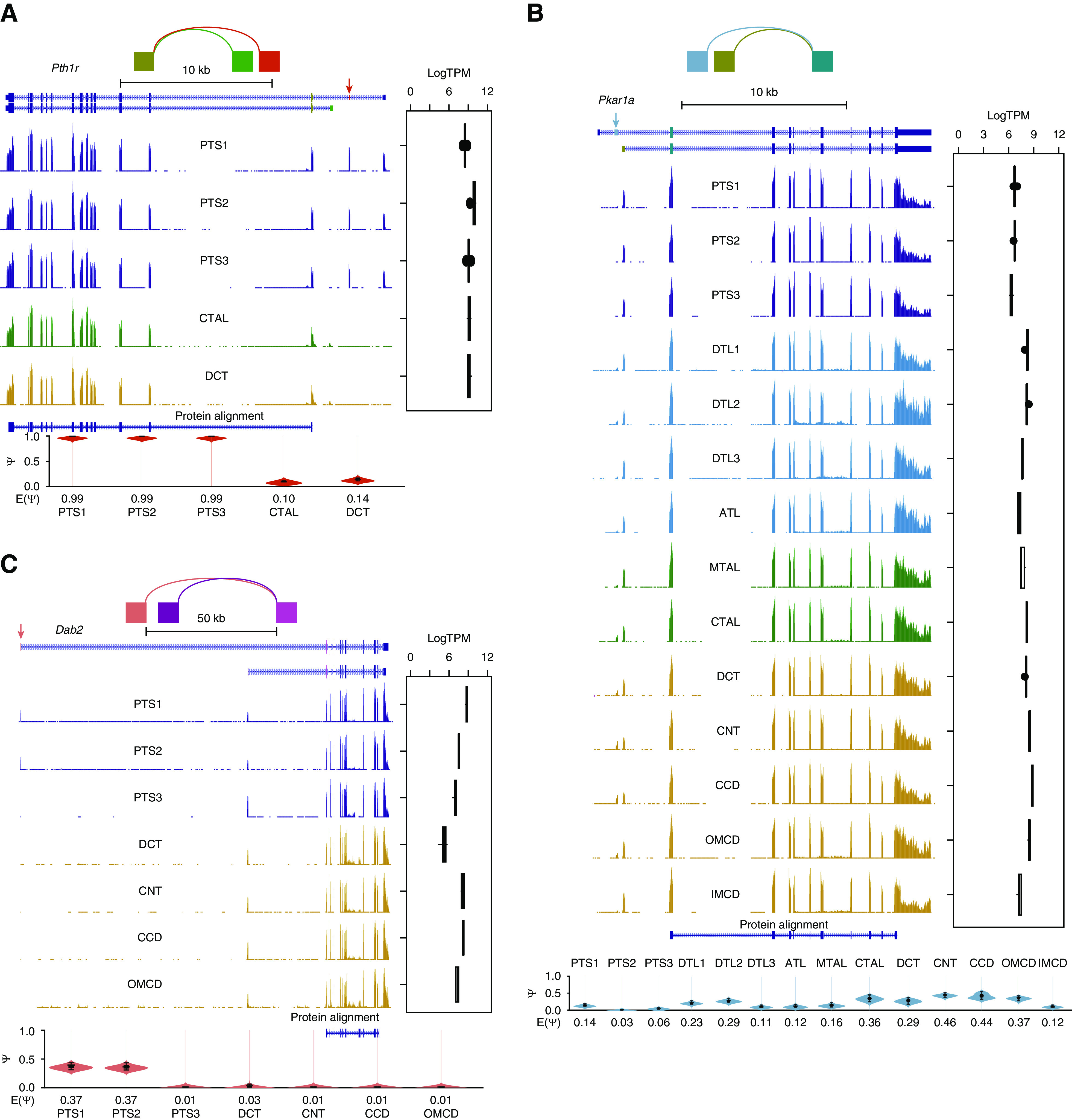
Alternative splicing occurs within nonprotein-coding regions. Distribution of (A) Pth1r, (B) Pkar1a, and (C) Dab2 isoforms along renal segments. Mapping is visualized in the UCSC Genome Browser. The structure of the gene is shown at the top of the track, with exons connected by blue rectangles and introns indicated by thin lines with arrows. The splicing graph at the top of the panel was generated using MAJIQ-SPEL. The paths on the splicing graph indicate the splicing junctions. The same color codes were used for exons in the splice graph and UCSC Genes Track. Gene expression is shown on the right. Quantifications of relative abundance (PSI) of indicated exons are shown in violin plot at the bottom (sum of PSIs=1). PSI was calculated by MAJIQ and visualized by MAJIQ-SPEL. Track height scale, auto. The bottom protein alignment track is annotated on the basis of the UniProt/SwissProt database.
Figure 4 shows alternative events that have effects on the protein sequence. Examples include Na/hydrogen exchanger 8 (Slc9a8), which is abundantly expressed in PTS2 and PTS3 (Figure 4A).22 The analysis showed a dominant isoform in PTS2 and PTS3 that differs from that expressed in other segments. The predicted protein lacks four membrane-spanning helices. In addition, an extra, unannotated, noncoding exon (exon in red) was found to be present in this isoform (Figure 4A). The functional difference between these protein variants remains to be determined. The analysis also revealed alternative splicing of ketohexokinase (Khk), which is abundantly expressed in PTs and is critical for fructose metabolism. It was seen to have a dominant alternative isoform in PTs (Figure 4B). Specifically, the ATP binding domain–containing isoform needed for kinase activity was dominant in PTS1, PTS2, and PTS3, consistent with the known metabolic role of Khk in PTs. The isoform present in nonproximal segments presumably has some other role. We also identified a regulatory protein, Tsc22d1, showing a DCT-specific isoform with an extra 5′ coding exon (Figure 4C). This DCT-specific isoform (ENSMUST00000048371, 1077 amino acids) is much longer (by 934 amino acids) than the isoform seen in the other tubule segments (ENSMUST00000022587, 143 amino acids). This short isoform is believed to function as a leucine zipper TF. The extended amino-terminal region in the DCT-specific isoform shows many low-complexity regions (typically associated with RNA binding), and contains a Pat1 domain implicated in regulation of RNA stability (E9QLZ1). This observation suggests specialized roles of Tsc22d1 in DCT related to post-transcriptional regulation of gene expression. Another example is the Golgi short coiled-coil protein (Scoc) (Figure 4D), which is associated with autophagy induced by amino acid starvation.23 It is expressed in all renal segments, but shows distinct isoforms in PTs versus all of the other segments. The PTs express the canonic isoform (Q78YZ6-1), whereas the other segments express an isoform that lacks amino acids 1–43 (Q78YZ6-2). Other similar examples of alternative splicing are provided in Supplemental Figure 5, including proton-coupled peptide transporter (Slc15a2), microtubule affinity regulating kinase 3 (Mark3), myosin light chain 6 (Myl6), and mitochondrial ketone body metabolism enzyme (Hmgcl).
Figure 5 shows examples of alternative splicing that exhibit changes in noncoding sequences but produce the same protein sequence. Differences in noncoding sequences may mediate differences in mRNA stability, translocation, or translation.1 One example is the parathyroid hormone 1 receptor (Pth1r), which shows comparable expression in both PTs and CTAL/DCT (Figure 5A). The isoform in PTs differs from that in the CTAL/DCT in the 5′ noncoding exons. Examples also include the protein kinase cAMP-dependent type I regulatory subunit α (Pkar1a), which shows isoforms that differ at the 5′ exons (Figure 5B). The mixture of both “long” and “short” isoforms is most prominent in CNT-CCD-OMCD, where Prkar1a plays a crucial role in aquaporin-2 regulation. It remains to be determined whether the alternative splicing of Pkar1a could contribute to its kinase activity through the effect on Pkar1a mRNA’s properties. Similarly, alternative 5′ exons were found in disabled homolog 2 (Dab2) (Figure 5C), a gene that has been shown to link to CKD development.24 Specifically, in PTS1 and PTS2, Dab2 expressed both the long and short isoform, whereas in PTS3/DCT-OMCD only the short one was dominant. Additional similar alternative-splicing examples are provided in Supplemental Figure 6, including Ca-sensing receptor (Casr), γ-glutamyltransferase 1 (Ggt1), fibroblast growth factor 1 (Fgf1), and mitochondrial ketone body metabolism enzyme (Bdh).
Resource Web Page
To allow users facile access to the curated data, we have set up a publicly accessible web resource at https://esbl.nhlbi.nih.gov/MRECA/Nephron/. This resource has several tabs allowing interrogation in different ways: (1) “Transcriptomic landscape” provides the transcript expression values for all 14 renal tubule segments. From this page, visualization of isoform distributions are possible via Jbrowse,7 or through the UCSC Genome Browser. (2) “Gene groups” integrates the HUGO Gene Nomenclature Committee database, providing additional gene category analyses. (3) “Differential gene expression” supports the interactive visualization of renal segment-selective genes. (4) “Comparative transcriptomics” compares the gene expression between human tissues (GTEx RNA-seq) and mouse renal tubule data described in this study. (5) “Download” provides the download links for data. The web resource allowed the identification of a cell-surface protein, Embigin (Emb), which is present in all cortical renal tubules, except the PTs. Thus, a FACS-based approach using anti-Embigin could be used to enrich DCT cells for scRNA-seq analysis.25
In the following, we provide examples of use of the web resource to identify the distributions of transcripts associated with three physiologic/pathophysiologic areas of investigation: (1) Wnk-SPAK-PKA signaling along the renal tubule (Figure 6); (2) renin-angiotensin-aldosterone signaling along the renal tubule (Figure 7); and (3) distributions of transcripts associated with genetically caused cystic kidney diseases (Figure 8). These distribution patterns are also consistent across published single-cell data (tab 11 in Supplemental Table 2).11
Figure 6.
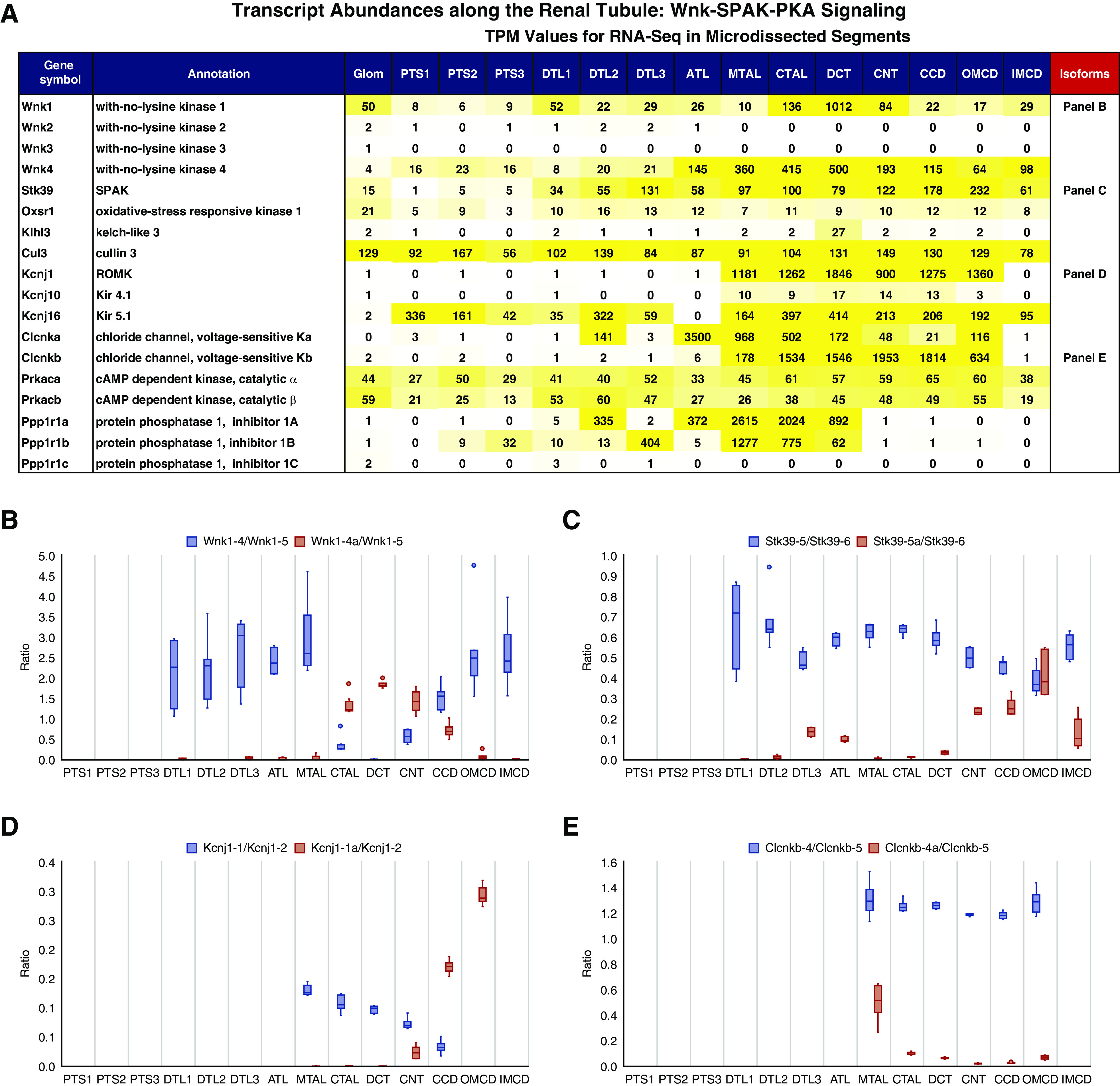
Distributions of transcripts associated with Wnk-SPAK-PKA signaling. (A) Transcript abundance (TPM) of major Wnk signaling components along the renal tubule. The yellow color indicates expression levels. Quantification of exon-inclusion level (ratio) for (B) Wnk1, (C) SPAK, (D) Kcnj1, and (E) Clcnkb. Gene symbols are followed by exon numbers. The ratio was calculated on the basis of the relative abundance of spliced exons (blue) over reference exons (orange). Read counts were calculated using featureCounts (see Method).
Figure 7.

Distributions of transcripts associated with renin-angiotensin-aldosterone signaling. Transcript abundance (TPM) of major renin-angiotensin-aldosterone signaling components along the renal segments. The yellow color indicates expression levels. The scRNA-seq data are from Chen et al.5
Figure 8.
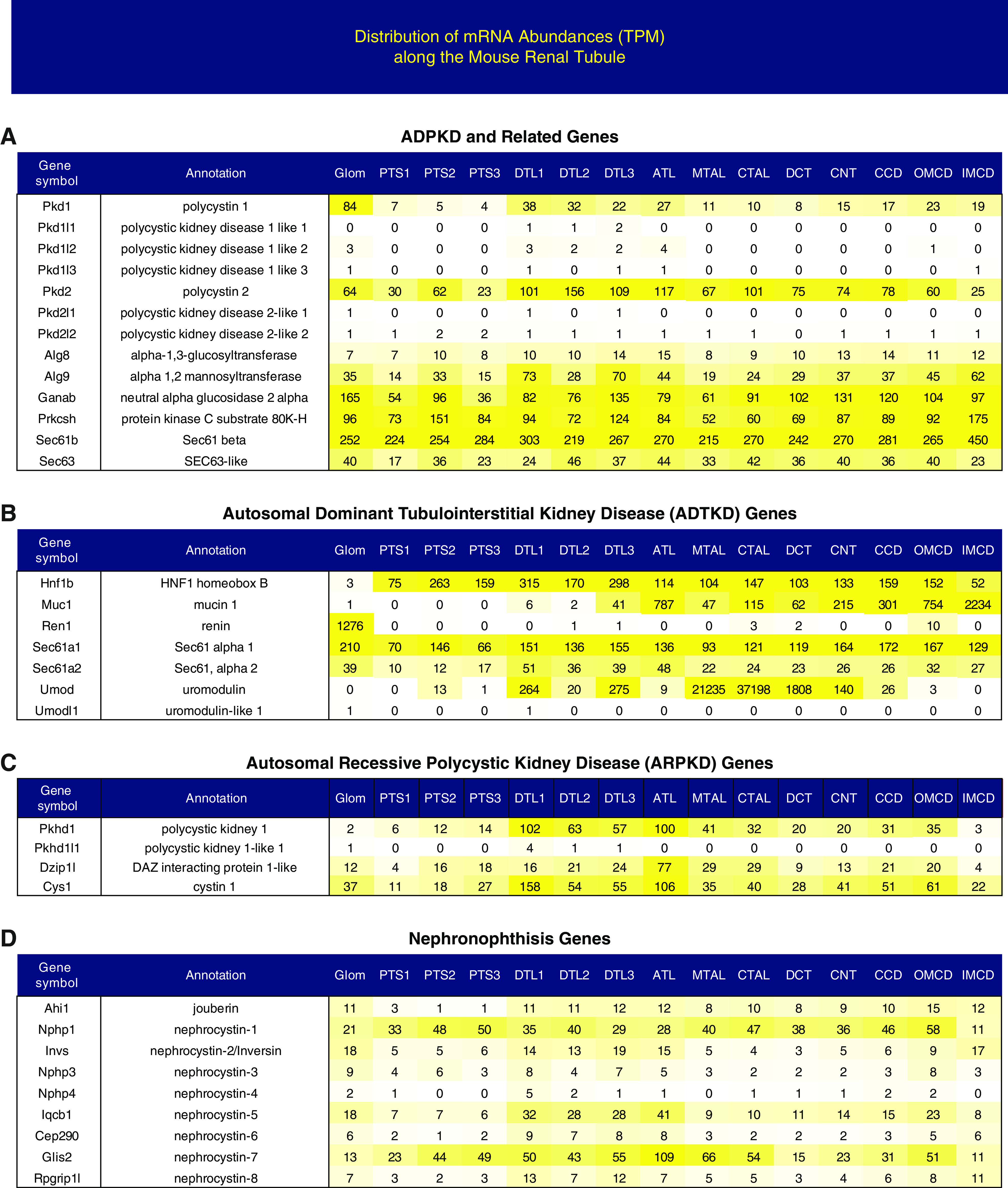
Distributions of transcripts associated with genetic cystic kidney diseases. Transcript abundance (TPM) of genetically caused cystic kidney disease transcripts along the renal segments. The yellow color indicates expression levels. ADPKD, autosomal dominant polycystic kidney disease.
Wnk-SPAK-PKA Signaling
The Wnk-SPAK and PKA protein kinase signaling pathways regulate NaCl absorption by the DCT, through regulation of the Na-Cl cotransporter (Slc12a3), and TAL, through regulation of the Na-K-2Cl cotransporter (Slc12a1).26 Figure 6A shows the distributions of transcripts corresponding to major components of Wnk-SPAK and PKA signaling.27,28 Of the four WNK family members, Wnk1 and Wnk4, but not Wnk2 and Wnk3, transcripts were found in renal tubule segments. In contrast to the relatively uniform expression of Wnk4 (ATL-IMCD), Wnk1 is enriched in DCT, expressed at a level at least ten-fold greater than in other segments. However, Wnk1 in the DCT consists virtually exclusively of an isoform missing the initial exons 1–4 and, therefore, missing the kinase domain (Figure 6B, Supplemental Figure 3D). This isoform is termed KS-WNK1.29 The Wnks are regulated, in part, by a RING-type E3-ubiquitin ligase complex comprising the adaptor protein Klhl3, which is selectively expressed in the DCT, and the ubiquitin ligase (Cul3), which is more widely expressed. Downstream from Wnk4 are two kinases (SPAK [Stk39] and Oxsr1) that are widely expressed in the distal part of the nephron and CD. However, the CD (CNT-IMCD), rather than the TAL or DCT (see Discussion),30 express a SPAK variant that uses an alternative promoter (exon 5a, Figure 6C, Supplemental Figure 3E, Supplemental Figure 4). Subsequent qPCR analysis confirmed our observations (Supplemental Figure 4, B and C). Further analysis indicated that the exon 5a isoform is present in PCs (Supplemental Figure 4D). The apical K channel Kcnj1 (ROMK) mediates K secretion and has two well-characterized isoforms, one in distal nephron segments and one in CDs (Figure 6D, Supplemental Figure 3B). Basolateral K and Cl channels play important roles, in part, by setting the membrane voltage.28 The K channels Kcnj10 and Kcnj16 typically form a heterodimer. As shown in Figure 6A, the distribution of Kcnj16 is broader than Kcnj10, suggesting it has a distinct role in some segments, perhaps forming heteromers with other K channels.31 Intracellular Cl has been implicated in regulation of Wnk432 and is determined, in part, by basolateral Cl channels. The kidney-type Cl channels, Clcnka and Clcnkb, are found in the distal nephron and CD segments. Although Clcnka has been characterized in the literature as being specific to the ATL,33 it appears to be expressed throughout the ATL-OMCD region. Clcnkb is expressed as different splice variant isoforms (not previously reported) with an alternative exon seen chiefly in the MTAL (Figure 6E). Recently, there has been increased recognition of the role of particular protein phosphatase regulators of the Ppp1r1 family in the DCT.34 As seen in Figure 6A, these phosphatase inhibitor proteins are selectively expressed in distal nephron segments, but not in the CDs.
Renin-Angiotensin-Aldosterone Signaling
The renin-angiotensin-aldosterone system plays a central role in regulation of renal NaCl reabsorption and, therefore, in the regulation of BP and extracellular fluid volume. Figure 7 shows the distribution of transcripts associated with renin-angiotensin-aldosterone signaling along the renal tubule. scRNA-seq data from our prior publication5 indicates expression in cortical ICs (A-IC and B-IC) and PCs. The distributions are largely in accord with the current literature, with a couple of exceptions. First, the glucocorticoid-metabolizing enzyme Hsd11b2 and the mineralocorticoid receptor appear to be expressed predominantly in CD segments, but show expression extending upstream to the DCT, compatible with the original proposal that aldosterone could directly regulate transport in the DCT.35–37 The DCT1 and DCT2 segments were too short individually to be dissected independently for this study, but proved resolvable by scRNA-seq.25 Interestingly, the mineralocorticoid receptor was strongly expressed in the B-IC, compatible with current literature,38,39 although Hsd11b2 showed little or no expression in these cells. Second, the angiotensin II receptor Agtr1a was found to be expressed predominantly in the PT, with little or no expression distally at an mRNA level.11,25
Genetically Caused Cystic Kidney Diseases
In accord with recent classification systems,40–43 we divide these transcripts into four groups as labeled in Figure 8. Autosomal dominant polycystic kidney disease is the most common genetic cause of CKD.40 The two genes most often harboring mutations, Pkd1 and Pkd2, appear to be expressed throughout the nephron and CDs (Figure 8A). Transcripts in the other categories, autosomal dominant tubulointerstitial kidney disease (Figure 8B), autosomal recessive polycystic kidney disease (Figure 8C), and nephronophthisis (Figure 8D), all tend to be widely expressed, with a couple of exceptions. Mucin-1 (Muc1) is selectively expressed in CD segments and uromodulin (Umod), classically viewed as TAL specific, appears to be expressed also in the descending thin limb segments and the DCT.44
Discussion
Here, we used full-length, small-sample RNA-seq coupled with renal tubule microdissection to investigate the transcriptome landscape and alternative-splicing landscape of all 14 mouse renal tubule segments. The resulting data are viewable at https://esbl.nhlbi.nih.gov/MRECA/Nephron/. Specific segments were identified on the basis of morphology, topology, and location in the kidney, and (unlike scRNA-seq) small-sample RNA-seq does not rely on particular marker genes for identification.
These data provide a comprehensive list of segment-specific genes, with the potential to reveal important, but unknown, functions of genes in kidney. Given the scope of this manuscript, we only focused on certain aspects of these gene-expression profiles (e.g., solute carriers) and on particular physiologic and pathophysiologic processes (e.g., Wnk-SPAK-PKA signaling). Consequently, the data are likely to be useful for a wide number of additional applications (e.g., different biotypes of transcripts). Furthermore, the methodology described here can be extended in important additional directions, e.g., comparisons between male and female, and between strains or species.11,45–47 An important aspect of these data is that they add information on alternative-splicing differences along the renal tubule, adding to other recent evidence.25,48 The Smart-seq–based protocol, which provides uniform coverage of the entire transcript, allowed us to comprehensively map alternative exon usage.
In a previous study, we used RNA-seq to profile microdissected tubules from a different rodent species, the rat.3 Not surprisingly, most of the gene-expression data are consistent between mouse and rat (see tab 10 in Supplemental Table 2). However, the rat data were generated >5 years ago when the technology was still evolving, resulting in some apparent differences compared with the current mouse data (e.g., for Corin and Psca). Such differences are likely due to limitations in the sensitivity and exon-coverage uniformity of the prior method, and to a less-complete annotation of the genome of the rat versus the mouse. We believe, however, that both rat and mouse datasets will continue to be useful. Perhaps the chief benefit of the new dataset is that it allows detailed, comprehensive mapping of transcript splicing.
Many of the identified splice-variant isoforms show segment specificity, adding a new, additional layer of gene-expression variation, markedly extending prior knowledge. One example is Stk39 (SPAK). A prior study49 showed that exon 5a of Stk39 was present in the renal medulla rather than the cortex, and the authors suggested this result may be due to the presence of exon 5a in the MTAL. Our RNA-seq data, confirmed by qPCR, indicate that the exon 5a isoform is instead expressed in the CD system (including the OMCD), and not in MTAL, providing an alternative interpretation of the prior data showing exon 5a in the medulla.
The biologic significance of these isoforms has not been thoroughly investigated individually. In addition, alternative splicing is a tightly regulated process that may be mediated by both cis- and trans-regulatory elements.50 We found a number of differentially expressed splicing factors among renal segments. These factors may play a role in the regulation of alternative splicing. Overall, alternative splicing increases the complexity of the transcriptome and proteome. Much work remains, both experimentally and computationally, to go from alternative splicing, to protein function, and ultimately to renal function.
Disclosures
Because M.A. Knepper is an editor of JASN, he was not involved in the peer-review process for this manuscript. A guest editor oversaw the peer-review and decision-making process for this manuscript. All remaining authors have nothing to disclose.
Funding
This work was primarily funded by NHLBI Division of Intramural Research projects ZIA-HL001285 and ZIA-HL006129 (to M. Knepper). This work was also supported by an NIH Clinical Center 2020 Bench-to-Bedside Award.
Supplementary Material
Acknowledgments
The authors thank Dr. Stefan Somlo, Dr. Michael Caplan, Dr. Robert Kleta, and Dr. Olivier Devuyst for advice regarding genes involved in cystic diseases of the kidney.
Next-generation sequencing was done in the NHLBI DNA Sequencing Core Facility (Dr. Yuesheng Li, Director). Confocal images were taken in the NHLBI Confocal Microscopy Core Facility (Dr. Christian Combs, Director). Microdissected tubules were imaged in the NHLBI Induced Pluripotent Stem Cells Core (Dr. Jizhong Zhou, Director). Tissue sections were prepared in the NHBLI Pathology Facility (Dr. Zu-Xi Yu, Director).
Dr. Lihe Chen and Dr. Mark A. Knepper designed research, analyzed data, and made figures; Dr. Lihe Chen performed experiments and designed websites; Dr. Lihe Chen, Dr. Chun-Lin Chou, and Dr. Mark A. Knepper contributed reagents, discussed the results, and drafted the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Data Sharing Statement
Sequencing data have been deposited in the GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150338).
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020101406/-/DCSupplemental.
Supplemental Figure 1. RNA-seq quality control and gene expression patterns along the renal tubules.
Supplemental Figure 2. Comparison of small-sample RNA-seq and published single-cell RNA-seq data.
Supplemental Figure 3. Known alternative splicing examples in kidney.
Supplemental Figure 4. Distribution of Stk39 isoforms along renal tubule segments.
Supplemental Figure 5. Additional alternative splicing examples that change protein structure.
Supplemental Figure 6. Additional alternative splicing examples.
Supplemental Table 1. Mouse renal tubule gene expression.
Supplemental Table 2. Analysis of published single-cell RNA-seq data.
Supplemental Table 3. Mouse renal tubule differentially expressed genes.
Supplemental Table 4. Differentially expressed genes in Figure 2B.
Supplemental Table 5. Proximal tubule selective solute carriers.
Supplemental Table 6. Ligand-receptor pairs.
Supplemental Table 7. Alternative splicing.
Supplemental Table 8. Splicing factors.
Supplemental Table 9. Exon counts.
References
- 1.ENCODE Project Consortium: An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al.: Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JW, Chou CL, Knepper MA: Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Li T, Liu F, Chen Y, Yao J, Li Z, et al.: Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol Cell 73: 130–142.e5, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, et al.: Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A 114: E9989–E9998, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al.: STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buels R, Yao E, Diesh CM, Hayes RD, Munoz-Torres M, Helt G, et al.: JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol 17: 66, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, Dewey CN: RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson MD, McCarthy DJ, Smyth GK: edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, et al.: A new view of transcriptome complexity and regulation through the lens of local splicing variations. eLife 5: e11752, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ransick A, Lindström NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, et al.: Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev Cell 51: 399–413.e7, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kriz W, Bankir L: A standard nomenclature for structures of the kidney. The Renal Commission of the International Union of Physiological Sciences (IUPS). Kidney Int 33: 1–7, 1988 [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Clark JZ, Nelson JW, Kaissling B, Ellison DH, Knepper MA: Renal-tubule epithelial cell nomenclature for single-cell RNA-sequencing studies. J Am Soc Nephrol 30: 1358–1364, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Limbutara K, Chou CL, Knepper MA: Quantitative proteomics of all 14 renal tubule segments in rat. J Am Soc Nephrol 31: 1255–1266, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW, Knepper MA: Representation and relative abundance of cell-type selective markers in whole-kidney RNA-seq data. Kidney Int 95: 787–796, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S, Cao P, Dong N, Peng J, Zhang C, Wang H, et al.: PCSK6-mediated corin activation is essential for normal blood pressure. Nat Med 21: 1048–1053, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan J, Aldrich RW: BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci U S A 109: 7917–7922, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng XH, Yang C, Xia XM, Liu M, Lingle CJ: SLO3 auxiliary subunit LRRC52 controls gating of sperm KSPER currents and is critical for normal fertility. Proc Natl Acad Sci U S A 112: 2599–2604, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JS, Valerius MT, McMahon AP: Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134: 2533–2539, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Grote D, Souabni A, Busslinger M, Bouchard M: Pax 2/8-regulated Gata 3 expression is necessary for morphogenesis and guidance of the nephric duct in the developing kidney. Development 133: 53–61, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Breyer MD, Jacobson HR, Davis LS, Breyer RM: In situ hybridization and localization of mRNA for the rabbit prostaglandin EP3 receptor. Kidney Int 44: 1372–1378, 1993 [DOI] [PubMed] [Google Scholar]
- 22.Goyal S, Vanden Heuvel G, Aronson PS: Renal expression of novel Na+/H+ exchanger isoform NHE8. Am J Physiol Renal Physiol 284: F467–F473, 2003 [DOI] [PubMed] [Google Scholar]
- 23.McKnight NC, Jefferies HB, Alemu EA, Saunders RE, Howell M, Johansen T, et al.: Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J 31: 1931–1946, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, et al.: Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med 24: 1721–1731, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Chou C-L, Knepper MA: Targeted Single-Cell RNA-seq Identifies Minority Cell Types of Kidney Distal Nephron. J Am Soc Nephrol 32: 886–896, 2021. 10.1681/ASN.2020101407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH: The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadchouel J, Ellison DH, Gamba G: Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016 [DOI] [PubMed] [Google Scholar]
- 28.Welling PA: Roles and regulation of renal K channels. Annu Rev Physiol 78: 415–435, 2016 [DOI] [PubMed] [Google Scholar]
- 29.Delaloy C, Lu J, Houot AM, Disse-Nicodeme S, Gasc JM, Corvol P, et al.: Multiple promoters in the WNK1 gene: One controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol 23: 9208–9221, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, et al.: A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derst C, Karschin C, Wischmeyer E, Hirsch JR, Preisig-Müller R, Rajan S, et al.: Genetic and functional linkage of Kir5.1 and Kir2.1 channel subunits. FEBS Lett 491: 305–311, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ: Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi K, Uchida S, Mizutani S, Sasaki S, Marumo F: Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol 12: 1327–1334, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Picard N, Trompf K, Yang CL, Miller RL, Carrel M, Loffing-Cueni D, et al.: Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J Am Soc Nephrol 25: 511–522, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velázquez H, Bartiss A, Bernstein P, Ellison DH: Adrenal steroids stimulate thiazide-sensitive NaCl transport by rat renal distal tubules. Am J Physiol 270: F211–F219, 1996 [DOI] [PubMed] [Google Scholar]
- 36.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA: The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci U S A 95: 14552–14557, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng L, Poulsen SB, Wu Q, Esteva-Font C, Olesen ETB, Peng L, et al.: Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 30: 1454–1470, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pham TD, Verlander JW, Wang Y, Romero CA, Yue Q, Chen C, et al.: Aldosterone regulates pendrin and epithelial sodium channel activity through intercalated cell mineralocorticoid receptor-dependent and -independent mechanisms over a wide range in serum potassium. J Am Soc Nephrol 31: 483–499, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata S, Rinehart J, Zhang J, Moeckel G, Castañeda-Bueno M, Stiegler AL, et al.: Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lanktree MB, Haghighi A, di Bari I, Song X, Pei Y: Insights into autosomal dominant polycystic kidney disease from genetic studies [published online ahead of print July 20, 2020]. Clin J Am Soc Nephrol 10.2215/CJN.02320220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benzing T, Schermer B: Clinical spectrum and pathogenesis of nephronophthisis. Curr Opin Nephrol Hypertens 21: 272–278, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, et al.: Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers 5: 60, 2019 [DOI] [PubMed] [Google Scholar]
- 43.Bergmann C: Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front Pediatr 5: 221, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tokonami N, Takata T, Beyeler J, Ehrbar I, Yoshifuji A, Christensen EI, et al.: Uromodulin is expressed in the distal convoluted tubule, where it is critical for regulation of the sodium chloride cotransporter NCC. Kidney Int 94: 701–715, 2018 [DOI] [PubMed] [Google Scholar]
- 45.Li Q, McDonough AA, Layton HE, Layton AT: Functional implications of sexual dimorphism of transporter patterns along the rat proximal tubule: Modeling and analysis. Am J Physiol Renal Physiol 315: F692–F700, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, et al.: Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tahaei E, Coleman R, Saritas T, Ellison DH, Welling PA: Distal convoluted tubule sexual dimorphism revealed by advanced 3D imaging. Am J Physiol Renal Physiol 319: F754–F764, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wineberg Y, Bar-Lev TH, Futorian A, Ben-Haim N, Armon L, Ickowicz D, et al.: Single-cell RNA sequencing reveals mRNA splice isoform switching during kidney development. J Am Soc Nephrol 31: 2278–2291, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCormick JA, Ellison DH: Distal convoluted tubule. Compr Physiol 5: 45–98, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ: Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14: 153–165, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.