

Significance Statement
Vesicoureteral reflux (VUR) is associated with progressive kidney disease. Familial aggregation supports a hereditary basis; however, its genetic architecture remains to be elucidated. The largest VUR copy number variant analysis and genome-wide association study to date accounts for multiple modes of inheritance and sex-specific effects in VUR, identifying three study-wide significant and five suggestive loci with large effects, containing canonical developmental genes including WDPCP and WNT5A. Results of experiments in mice support novel roles of Wnt5a in urogenital development. Altogether, 6% of patients carried high-risk genotypes. These findings have important implications for VUR screening.
Keywords: vesico-ureteral reflux, human genetics, genetics and development, pediatric nephrology
Visual Abstract

Abstract
Background
Vesicoureteral reflux (VUR) is a common, familial genitourinary disorder, and a major cause of pediatric urinary tract infection (UTI) and kidney failure. The genetic basis of VUR is not well understood.
Methods
A diagnostic analysis sought rare, pathogenic copy number variant (CNV) disorders among 1737 patients with VUR. A GWAS was performed in 1395 patients and 5366 controls, of European ancestry.
Results
Altogether, 3% of VUR patients harbored an undiagnosed rare CNV disorder, such as the 1q21.1, 16p11.2, 22q11.21, and triple X syndromes ((OR, 3.12; 95% CI, 2.10 to 4.54; P=6.35×10−8) The GWAS identified three study-wide significant and five suggestive loci with large effects (ORs, 1.41–6.9), containing canonical developmental genes expressed in the developing urinary tract (WDPCP, OTX1, BMP5, VANGL1, and WNT5A). In particular, 3.3% of VUR patients were homozygous for an intronic variant in WDPCP (rs13013890; OR, 3.65; 95% CI, 2.39 to 5.56; P=1.86×10–9). This locus was associated with multiple genitourinary phenotypes in the UK Biobank and eMERGE studies. Analysis of Wnt5a mutant mice confirmed the role of Wnt5a signaling in bladder and ureteric morphogenesis.
Conclusions
These data demonstrate the genetic heterogeneity of VUR. Altogether, 6% of patients with VUR harbored a rare CNV or a common variant genotype conferring an OR >3. Identification of these genetic risk factors has multiple implications for clinical care and for analysis of outcomes in VUR.
Vesicoureteral reflux (VUR), the retrograde flow of urine from the bladder toward the kidneys, caused by malfunction at the vesicoureteral junction, is a common cause of febrile urinary tract infection (UTI) and pediatric kidney failure.1,2 VUR occurs most often in isolation, but can arise in conjunction with more complex defects, such as hydronephrosis and renal dysplasia.3–5 VUR has a prevalence of 1%–2% in European populations6–9 and is much more prevalent in females (>70%), although males predominate in patients diagnosed between 0 and 2 years of age.10,11 VUR is highly familial, with a reported occurrence rate of 27%–51% among siblings, and 66% among offspring of affected individuals.9 Segregation analyses and linkage studies suggested the contribution of rare variants with large effects and multiple modes of inheritance.12–19
Urinary tract formation in the mouse begins at E9, when nephric ducts form and extend, joining the cloaca. Cloacal septation separates the hindgut from the urogenital sinus (UGS) between E11 and E14. Subsequently, the UGS differentiates into the bladder and urethra. The ureteric bud (UB) sprouts from nephric duct on E11, and its distal portion will form the ureter; however, it is initially joined to the nephric duct and not directly to the bladder. Mature ureter-bladder connections form between E10 and E13. Abnormalities in timing or morphogenesis at any stage can alter the site of ureter insertion, resulting in ectopic ureters or an abnormal connection at the vesicoureteral junction, causing VUR and obstruction.20–23
Relatively little is known about the genetics of VUR in humans.24 VUR is often present in syndromic disorders featuring congenital anomalies of the kidney and urinary tract (CAKUT).25 Mutations in ROBO2,26 SOX17,27 and TXNB28 have been implicated in rare families with isolated VUR, but most nonsyndromic patients are genetically unexplained. Genome-wide association studies (GWAS) of VUR have not been adequately powered.15,16,29 Recently, we reported that rare copy number variant (CNV) disorders also contribute to many urinary tract malformations, including VUR.30 Although CNV disorders were detected in 7.7% of patients with kidney malformations, they accounted for only 1%–2% of VUR patients.30 These findings motivated additional investigation of the genetics of VUR in larger cohorts, to confirm the contribution of CNV disorders, and explore alternative genetic models. Here, we combined genome-wide association and rare CNV analyses in the largest VUR cohort studied to date. We complemented these studies by analysis of expression pattern and developmental defects in Wnt5a mutant mice.
Methods
Subjects and Genotyping
All aspects of the study involving human research participants adhered to the principles of the Declaration of Helsinki, and the study protocol was approved by the Institutional Review Boards of Columbia University Medical Center and each participating recruitment site. Signed written informed consent by the participant and/or their parents or guardians was obtained according to the protocol of local Institutional Review Boards.
VUR patients and controls were obtained from the following study cohorts: VUR patients and healthy controls form Columbia University and US and international collaborating sites, patients from the Randomized Intervention for Children with VUR (RIVUR) study cohort,31 reflux nephropathy patients from the CKD in Children (CKiD) study,32,33 the Population Architecture using Genomics and Epidemiology consortium34 convenience controls and an Irish cohort comprising VUR-affected children, and unrelated controls.15 As we reported previously,30 21,498 convenience controls were obtained from collaborators and from dbGaP (see Data Availability and Accession Numbers), which comprised participants of genome-wide genotyping studies of complex traits that are not associated with VUR or other developmental defects. VUR patients from the RIVUR and Irish cohorts were diagnosed by voiding cystourethrogram (VCUG); CKiD VUR patients had a primary diagnosis of reflux nephropathy; patients from the Columbia cohort, including US and international collaborators, were ascertained by VCUG or other diagnostic imaging such as renal scintigraphy. All of the samples from the Columbia University and RIVUR cohorts were genotyped for this study on the Illumina MEGA array. For the Population Architecture using Genomics and Epidemiology cohort, genotyped on MEGA arrays, and the Irish cohort, genotyped on Affymetrix SNP 6.0 chips, we obtained access to intensity level raw genotyping data. Of these patients, 660 were included in our prior study.30
Initial Data Processing and Quality Control
Raw intensity probe level data were processed for each cohort using Illumina GenomeStudio v2011 (Illumina) or Affymetrix Power Tools software (Affymetrix) to generate genotyping calls. Using PLINK software,35 markers with call rate (CR) ≥95% and minor allele frequency (MAF) >1% were used to select subjects based on sample CR (≥95% or 99%, for association, or copy number analyses, respectively). We used KING software36 to estimate relatedness between samples within and across cohorts, and retained only unrelated samples (kinship coefficient <0.0884). After examining the possibility of sex chromosomal abnormalities that could be reportable in our CNV analysis (e.g., Klinefelter syndrome), we excluded samples with discrepancies between genotyped sex and self-reported sex. Principal component analyses (PCA) were carried out using Smartpca.37
CNV Analysis
PennCNV software38 was used to determine CNV calls and quality; CNVs with confidence scores <30 were excluded.30,38,39 CNV analyses were performed on hg18 coordinates, and subsequently mapped to hg19 using University of California Santa Cruz LiftOver software (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Illumina Genome Viewer 1.9.0 or Affymetrix ChAS were used to visualize CNVs and evaluate possible artifacts. CNVs were classified as pathogenic (known genomic disorders [GDs]) or likely pathogenic CNVs, based on previously described criteria30 adapted from recommendations from the American College of Medical Genetics guidelines40,41 and making use of annotations with RefGene, the Database of Genomic Variation and Phenotype in Humans using Ensembl Resources and International Standards for Cytogenomic Arrays (ISCA) databases, and curated lists of genes from the Online Mendelian Inheritance in Man and Mouse Genome Informatics databases and the literature. As previously described,30,42,43 a CNV was classified as pathogenic (known GD) when its genomic coordinates overlapped 70% or more of those of a well-characterized syndromic CNV, as defined in Database of Genomic Variation and Phenotype in Humans using Ensembl Resources44 and the literature39,45–47; whereas large, exon-intersecting CNVs with frequency in controls ≤0.02%, which did not overlap a benign or likely benign CNV in the ISCA database, were classified as likely pathogenic when they met one or more of the following conditions: overlapped 70% or more of the span of a pathogenic or likely pathogenic CNV in the ISCA database; intersected a gene associated with kidney disease or genitourinary tract defects or other developmental disorders in humans or mice; or overlapped 70% or more with the coordinates of a well-characterized syndromic CNV, with opposite copy number sign (loss or gain).
Our primary tests were to compare the burden of (1) the set of all large, rare CNVs and (2) a predefined set of 148 known GDs, between VUR patients and controls. The corrected P value threshold for testing whether VUR patients and controls significantly differ in the proportion of carriers of these two sets of variants is P=0.05/2=0.025. We next performed post-hoc tests for 18 individual GDs observed in VUR, with a corrected threshold of P=0.05/18=2.7×10−3. Rare CNV burden analyses were performed on autosomal, gene-intersecting CNVs of size ≥100 kb in patients and controls, with a frequency ≤0.1% in population subgroups based on PCA (Supplemental Figure 1), to avoid including CNVs that might be relatively common within ancestry groups represented in controls. Six controls, outliers in PCA without matching patients, were removed before burden analyses. For CNV burden analyses, the R v3.5.1 implementation of the two-sided Fisher’s exact test was used to compare proportions, and Kaplan–Meier survival curves representing the largest CNV per genome were compared between patients and controls using the nonparametric log-rank test implemented in the Python package lifelines (https://github.com/CamDavidsonPilon/lifelines/).
GWAS
For the purpose of GWAS, samples were first grouped by genotyping platform into four case-control datasets (Affymetrix SNP6.0; Illumina MEGA; Illumina Omni 1, 2.5, or OmniExpress; Illumina 660, 610, or 550 arrays).
After conducting PCA on each platform-specific dataset, only subjects of European ancestry were selected and the MEGA array dataset was further subdivided based on ancestry (Northwestern European, Polish, Italian, and Southeastern European), making a total of seven genetically matched case-control GWAS cohorts, referred to in the Results section. PCA was then performed separately on each of these seven GWAS cohorts. Tracy-Widom statistics48 from each of these PCAs were used to identify statistically significant PCs to be used as covariates in the association tests.
Imputation was performed separately for each cohort. After filtering out genotyped single nucleotide polymorphisms (SNPs) with a CR <95%, MAF <1%, or Hardy-Weinberg equilibrium exact test P value <0.0001 in controls and updating or removing SNPs with discrepancies in strand, alleles, position, and frequencies (1000 G-check-bim.pl perl script from the McCarthy Group; https://www.well.ox.ac.uk/∼wrayner/tools/), the Michigan Imputation Server (https://imputationserver.sph.umich.edu/) was used to carry out phasing (Eagle v2.3), and imputation (Minimac3) with the Haplotype Reference Consortium (r1.1 2016) reference panel. Imputation of chromosome X markers was run separately for males and females. DosageConvertor (v1.0.3 for autosomes, v1.0.4 for chromosome X) was used to convert dosage files from Minimac3 format to PLINK dose format. Only SNPs with high imputation quality (r2≥0.8) were retained.
Association analyses under additive, recessive, and dominant logistic regression models were then performed with Plink using logistic regression separately for each cohort with cohort-specific significant PCs included as covariates. For the additive model, dosage values for each SNP were used. For the recessive and dominant models, dosage values were first converted to “best guess” genotypes (i.e., genotypes with highest probability). All analyses were also repeated for males and females separately.
Association summary statistics from these GWAS, for 6,131,010 variants common to all seven cohorts, were meta-analyzed using the fixed-effects inverse variance weighted method implemented in METAL.49
We used a P value threshold of 7.58×10−9, (5×10−8 ÷ 2.2 ÷ 3) as a conservative threshold for genome-wide statistical significance of associations and 1.52×10−7 (20×5×10−8 ÷ 2.2 ÷ 3) for suggestive associations, to account for testing of multiple variants: the value of 5×10−8 is accepted as the standard threshold for genome-wide significant asscociations50; dividing by 2.2 accounts for the three genetic models tested, as shown by González et al.51; and dividing by three accounts for testing in males and females, both separately and combined.
Genomic inflation factor lambda was calculated using the estlambda function from the R package GenABEL. Quantile-quantile and Manhattan plots were generated in R using the qqman package. Regional plots for top were created with LocusZoom52 (http://csg.sph.umich.edu/locuszoom/), using LD information (r2) from the 1000 Genomes European population (November 2014).
Heritability and Polygenic Risk Scores
We used LDSC software53 (https://github.com/bulik/ldsc) to estimate VUR heritability and LDPred54 (https://github.com/bvilhjal/ldpred), to derive a polygenic risk score (PRS) for VUR, based on the summary statistics from our VUR GWAS under an additive model. Next, we used LDPred to generate the VUR-PRS for individuals of European ancestry in the CKiD study cohort (n=432). As opposed to genotypic risk scores, which are selectively based on top signals, or PRS estimated using linkage disequilibrium (LD)-based marker pruning and a P value threshold, PRS estimation from GWAS summary statistics with LDPred, uses a Bayesian approach with a priori on effect sizes and LD information from an external reference panel (1000 Genomes panel) to derive the posterior mean effect size of each marker.54 All common markers in our VUR GWAS summary statistics intersecting the reference panel, with a defined effect estimate value in the meta-analysis were used (n=5,061,103 markers).
Single Variant PheWAS in the Electronic Medical Records and Genomics and UK Biobank Datasets
Single variant PheWAS analysis under an additive model was performed for our GWAS eight top variants, using UK Biobank (UKBB; http://pheweb.sph.umich.edu/SAIGE-UKB/) and Electronic Medical Records and Genomics (eMERGE; https://emerge-network.org/) datasets. In the latter, we conducted the analyses in the whole cohorts and separately in the subgroup of participants under 21 years of age. International Classification of Disease (ICD) codes were converted ICD-9 codes (https://www.cms.gov/Medicare/Coding/ICD9ProviderDiagnosticCodes/codes) for PheWAS analysis,55 which mapped to totals of 1817 distinct phecodes. PheWAS was performed using the PheWAS R package.56 The package uses predefined “control” groups for each phecode “case” grouping. The case definition requires a minimum of two ICD-9 codes from the “case” grouping of each phecode. All phecodes were tested with logistic regression with each phecode case-control status as an outcome and genotype coded according to the GWAS model (additive, recessive, or dominant), adjusted for age, sex, site, and three PCs of ancestry as a predictor. We set the Bonferroni corrected statistical significance threshold to account for the number of phecodes used (0.05/1817=2.75×10−5). Additionally, we meta-analyzed PheWAS from the eMERGE (all age) and UKBB analyses, as implemented in the PheWAS R package.
Mice
All animal experiments followed protocols approved by the Institutional Animal Care and Use Committee at Columbia University. Hoxb7-Gfp: 129S.Cg-Tg (Hoxb7-EGFP)33Cos/J, Frank Costantini, Columbia University.57 Wnt5a Mutants: 004758 - B6; 129S7-Wnt5atm1Amc/J; Andrew P McMahon, University of Southern California.58 All work with mice was approved by and performed under the regulations of the Columbia University Institutional Animal Care and Use Committee. Animals were housed in the animal facility of Irving Cancer Research Center, Columbia University.
Mouse Genotyping
For Wnt5a ko genotyping was performed with the following primers: WT forward 5′-GAG GAG AAG CGC AGT CAA TC-3′; common 5′- CAT CTC AAC AAG GGC CTC AT-3′ and Mut forward 5′- GCC AGA GGC CAC TTG TGT AG-3′; wild type shows 484bp, mutant 400bp.
Histology
Histology was carried out using hematoxylin and eosin staining with the standard protocol. Paraffin sections were cleared in three washes with xylene for 2 minutes each. Samples were rehydrated by incubating successively in 100% ethanol for 5 minutes per change, two changes of 95% ethanol for 5 minutes per change, and two changes of 70% ethanol for 5 minutes per change. Slides were rinsed in tap water and samples were stained in hematoxylin solution for 3 minutes, rinsed, then incubated in 95% ethanol for 4 minutes. Slides were then stained with eosin Y solution for 2–3 minutes, then dehydrated with successive changes of ethanol and cleared with the slides xylene and mounted in Permount.
Immunostaining
Embryos were embedded in paraffin and serial sections were generated. For immunohistochemistry, paraffin sections were deparaffinized using HistoClear and rehydrated through a series of ethanol and 1XPBS washes. Antigen retrieval was performed by boiling slides for 15 minutes in pH 9 buffer or 30 minutes in pH 6 buffer. Primary antibodies in 1% horse serum were incubated overnight at 4°C. The next day, slides were washed with PBST three times for 10 minutes each and secondary antibodies were applied for 2 hours at room temperature (RT). 4′,6-diamidino-2-phenylindole (DAPI) was either applied as part of the secondary antibody cocktail or for 10 minutes, for nuclear staining, and then the slides were sealed with coverslips. E-cadherin (Ecad) staining: goat anti-mouse Ecad antibody (500 ng/ml, Paraffin or Cryo, #AF748; R&D Systems).
Whole-mount In Situ Hybridization
Embryos were fixed overnight at 4°C on a rocking table with 4% paraformaldehyde (PFA) in PBS. After fixation, embryos were washed three times for 10 minutes each in PBS with 0.1% Tween (PTW) at RT, incubated in 6% H2O2 for 15 minutes at RT, and then washed three times in PTW for 10 minutes each time. After the last wash, the PTW was replaced with 1 μg/ml of proteinase K for 15 minutes at RT. Proteinase K digestion was terminated by washing twice for 5 minutes each wash at RT with PTW, then tissue was postfixed for 10 minutes in 4% PFA/PTW/0.2% glutaraldehyde, washed three times for 5 minutes each in PTW, then placed in hybridization buffer (50% Formamide [Fluca,#47671], 1.3x SSC, pH 5.3; 5 m EDTA pH 8; Yeast RNA [50 ug/ml, #R6750; Sigma], 0.1% Tween 20; 0.4% Chaps; Heparin [100 µg/ml]) first, for 5 minutes at RT, and then for 1 hour at 68°C. Hybridization buffer was replaced with 0.4 ml hybridization buffer containing probe, and samples were incubated overnight at 68°C. Hybridization buffer was replaced with warmed hybridization buffer without probe at 68°C, washed three times for 30 minutes each, then washed in warmed hybridization buffer with Tris-buffered saline with 0.1% Tween 20 (TBST) three times for 30 minutes each at 68°C, and in TBST twice for 5 minutes each at RT. Embryos are preblocked in 0.5 ml 1× TBST +10% heat treated goat serum then placed in blocking solution with fresh 1× TBST +10% serum containing a 1:5000 dilution of alkaline phosphatase-conjugated anti-digoxigenin Fab fragments for o/n at 4°C. After the incubation with antibody samples were washed two times at RT with TBST, then placed in fresh TBST overnight at 4°C, and washed twice in alkaline phosphatase buffer (NTMT [100 mM Tris, pH 9.5, 50 mM MgCl2, 100 mM NaCl, 0.1% Tween 20 1%]). Finally, samples were incubated in substrate solution (NTMT, nitro blue tetrazolium chloride, 5-bromo-4-chloro-3-indolyl-phosphate; Roche) containing 0.5 mg/ml levamisole for 15 minutes to 2 hours at RT in the dark. Reactions were stopped by washing twice for 5 minutes each in TBST. Embryos were then postfixed in 4%PFA/PTW for o/n at 4°C.
RNAscope
Mouse bladders were fixed in 4% PFA for 24 hours at 4°C, then immersed in 30% sucrose in 1× PBS. Tissue was embedded in optimal cutting temperature compound at −80°C, then 14 μm sections were placed on Superfrost Plus Slides and airdried for 1–2 hours at −20°C. The slides were then washed with 200 ml 1× PBS for 5 minutes to remove the optimal cutting temperature compound, then baked for 30 minutes at 60°C. Tissue was postfixed in 10% neutral-buffered formalin for 90 minutes, then dehydrated through a series of ethanol washes followed by another baking step in 60°C for 10 minutes. Three to five drops of RNAscope Hydrogen Peroxide (ACD, 322335) were applied to cover the sections, incubated for 10 minutes then washed in distilled water twice. Target retrieval was performed by boiling slides in RNAscope1× Target Retrieval Reagent (ACD, 322000) that was heated to at least 99°C for 5 minutes. After rinsing in distilled water for 5 minutes, the slides were transferred to 100% ethanol for 5 minutes. Slides were placed on a humidified chamber and three to five drops of RNAscope Protease III (ACD, 322337) were applied to cover each section for 30 minutes at 40°C. Excess liquid was removed from the slides with distilled water washes, then three to five drops of the Wdpcp-C1 probe (ACD, 502151) was added to cover the sections for 2 hours at 40°C in the humidified chamber. Slides were kept overnight in 5× SSC and washed twice in 1× wash buffer (ACD, 310091) before continuing with the assay. Three to five drops of RNAscope Multiplex FL v2 Amp 1 (ACD, 323101) were applied to the sections then kept in humidified chamber for 30 minutes at 40°C. After two 2 minute washes in 1× wash buffer, three to five drops of RNAscope Multiplex FL horseradish peroxidase-C1 (ACD, 323104) were added onto the slides in the humidified chambers for 15 minutes at 40°C. After two 2-minute washes in 1× wash buffer, 150–200 µl of diluted tyramide signal amplification (ACD 322809) plus Cy3 (NEL744001KT, 1:1500; PerkinElmer) were added to the slides in the humidified chambers for 30 minutes at 40°C. After two 2-minute washes in 1× wash buffer, three to five drops of RNAscope Multiplex FL horseradish peroxidase Blocker (ACD, 323107) were added to the slides in the humidified chambers for 15 minutes at 40°C. DAPI was applied for 30 seconds at RT to each slide and incubated for 30 seconds at RT. DAPI was removed and one to two drops of Prolong Gold Antifade Mountant were applied on the slides to seal with coverslips.
Data Availability and Accession Numbers
Raw data that support the findings of this study are available from the corresponding authors on reasonable request. The data are also available on dbGaP (https://www.ncbi.nlm.nih.gov/gap) under the following accession numbers: phs001749.v1.p1, phs000650.v3.p1, phs000304.v1.p1, phs000169.v1.p1, phs000092.v1.p1, phs000199.v1.p1, phs000431.v1.p1, phs000356.v2.p1, phs001584.v1.p1 (and other, pending). Some restrictions may apply according to participants’ consent and privacy protection. All images generated from mouse experiments reported in this study will also be available from the corresponding authors.
Results
Rare CNV Analysis
We conducted a rare CNV analysis in a multiethnic cohort of 1737 VUR patients and 24,765 population controls. We found a significant excess burden of large, rare, gene-intersecting CNVs in VUR patients compared with controls (defined as size ≥100 kb, frequency in PCA-defined populations ≤0.1%; Figure 1, Supplemental Figures 2 and 3; P=3.54×10−7). This difference was principally attributable to enrichment for larger-sized CNVs (CNVs ≥500 kb; Supplemental Tables 1 and 2) in VUR. We estimated that 2.23% of VUR patients can be attributed to carrying a rare CNV of at least 500 kb (odds ratio [OR], 1.40; 95% confidence interval [95% CI], 1.16 to 1.67; P=3.96×10−4; Figure 1A, Supplemental Table 1). We next annotated CNVs for pathogenicity. Based on a survey of 148 known GDs, we found that VUR patients were significantly enriched in GDs, with 18 distinct, known GDs in 35 out of 1737 of patients and 162 out of 24,765 of controls (2.01% versus 0.65%, OR, 3.12; 95% CI, 2.10 to 4.54; P=6.35×10−8). The GD burden remained highly significant if we only considered the 1077 VUR patients newly reported in this study (OR, 4.05; 95% CI, 2.60 to 6.11, P=6.46×10−9). Recurrent GDs that achieved significance in post-hoc tests included 16p11.2 deletion, 22q11.21 duplication, and triple X syndromes (Table 1). GDs were detected among all subcohorts, including the CKiD and RiVUR cohorts, which excluded known chromosomal disorders in their entry criteria.
Figure 1.

Excess burden of large, rare CNVs in VUR. VUR patients are enriched in large (size ≥100 kb), rare (frequency in PCA-defined populations ≤0.1%), gene-intersecting CNVs. (A) Frequency of subjects with their largest CNV size at or above the indicated size thresholds (kb). (B) Proportion of CNV (size ≥100 kb) within the indicated size intervals (kb). The 100–500 kb interval is not shown. Blue bars represent VUR patients and black bars, controls.
Table 1.
VUR patients are enriched in known genomic disorders, compared with controls
| Genomic Disorder | Start (Mb) | End (Mb) | Size (Mb) | VUR | Controls | OR (95% CI) | P | ||
|---|---|---|---|---|---|---|---|---|---|
| Verbitsky et al. 2019 | This Report | Combined | |||||||
| n=660 | n=1077 | n=1737 | n=24,765 | ||||||
| All GDs | 7 | 28 | 35 | 162 | 3.12 (2.10 to 4.54) | 6.35×10−8 | |||
| Individual GDs | |||||||||
| 1q21.1 deletion | 145.37 | 147.83 | 2.21 | 0 | 5 | 5 | 14 | 5.10 (1.44 to 15.01) | 6.44×10−3 |
| 1q21.1 duplication | 146.47 | 147.83 | 1.36 | 1 | 0 | 1 | 15 | 0.95 (0.02 to 6.18) | 1 |
| 2q11.2 deletion | 96.74 | 98.02 | 1.28 | 0 | 1a | 1a | 0 | ∞ (0.37 to ∞) | 0.067 |
| 2q13 homozygous deletion | 110.86 | 111.98 | 0.12 | 0 | 1 | 1 | 0 | ∞ (0.37 to ∞) | 0.067 |
| 3pter-p25 deletion | 0.45 | 2.02 | 1.57 | 1 | 0 | 1 | 1 | 14.27 (0.18 to 1110) | 0.127 |
| 7q11.23 del (Williams-Beuren) | 72.72 | 74.34 | 1.62 | 0 | 1 | 1 | 0 | ∞ (0.37 to ∞) | 0.067 |
| 7q36 deletionb/critical deletion | 129.43§ | 159.12 | 29.60 | 1 | 1 | 2 | 0 | ∞ (2.68 to ∞) | 4.29×10−3 |
| 15q11.2 deletion (Prader-Willi/Angelman) | 23.65 | 28.54 | 5.01 | 1 | 1 | 2 | 0 | ∞ (2.68 to ∞) | 4.29×10−3 |
| 16p11.2 deletion | 29.59 | 30.20 | 0.61 | 1 | 3 | 4 | 5 | 11.43 (2.27 to 53.21) | 1.77×10−3 |
| 16p13.3 deletion (Rubinstein-Taybi) | 3.65 | 5.04 | 1.39 | 0 | 1 | 1 | 0 | ∞ (0.37 to ∞) | 0.067 |
| 16p13.11 deletion | 15.12 | 16.36 | 1.24 | 0 | 1 | 1 | 5 | 2.85 (0.06 to 25.50) | 0.33 |
| 16p13.11 duplication | 15.49 | 18.17 | 2.67 | 0 | 1 | 1 | 36 | 0.40 (0.01 to 2.35) | 0.51 |
| 17p12 deletion (HNPP) | 14.10 | 15.47 | 1.37 | 0 | 2 | 2 | 15 | 1.90 (0.21 to 8.19) | 0.31 |
| 17q12 duplication | 34.82 | 36.21 | 1.39 | 0 | 1 | 1 | 6 | 2.38 (0.05 to 19.61) | 0.38 |
| 22q11.21 deletion | 20.74 | 21.46 | 0.72 | 1 | 2a | 3a | 4 | 10.71 (1.57 to 63.37) | 8.05×10−3 |
| 22q11.21 duplication | 18.88 | 21.46 | 2.53 | 0 | 5 | 5 | 7 | 10.21 (2.55 to 37.39) | 6.45×10−4 |
| Xp21 hemizygous deletion | 6.46 | 8.14 | 1.67 | 0 | 1 | 1 | 3 | 4.75 (0.09 to 59.28) | 0.24 |
| Trisomy X | 0.06 | 155.26 | 154.91 | 1 | 2 | 3 | 1 | 42.81 (3.44 to 2.21×103) | 1.07×10−3 |
A screen for 148 known pathogenic deletions and duplications showed a significant excess of genomic disorders in VUR patients (P=6.35×10−8). Start, end positions, and size (Mb) are based on genome build hg19. Counts of carriers in VUR patients and population controls, ORs with respect to controls, their 95% CIs, and Fisher’s exact test P values are shown. VUR counts include patients previously reported, newly reported patients in this study, and the combined counts, shown in three separate columns. Fisher’s exact tests are based on the combined VUR counts, compared with controls. In controls, counts of all genomic disorder carriers are larger than the sum of those for individual ones shown in the table, which are only the ones present in VUR patients. HNPP, hereditary neuropathy with liability to pressure palsies.
One VUR case carries deletions at both 2q11.2 and 22q11.2; this case is counted only once in the sum of all GD carriers.
Coordinates for the 7q36 deletion.
An additional 16 (0.92%) patients carried a likely pathogenic CNV (Supplemental Table 3). Among them are heterozygous deletions in chr 2q31.1, encompassing BBS5 and ITGA6; 10q24.2, including HPSE2; 10q26.11–10q26.3, which includes FGFR2; 12p13.33, including CACNA1C; 12q21.31–12q21.33, encompassing NPHP6; and two instances of heterozygous deletions of COL4A5 in chr X, in females and duplications in 6q14.3, including TBX18; 16q24.1, encompassing FOXC2 and FOXF1; and 17q25.1–17q25.3, including ITGB4. Altogether, this analysis of a significantly larger cohort confirms the contribution of rare CNV GDs to the development of VUR and highlights its genetic heterogeneity.
Genome-wide Association Study
We investigated the contribution of common variants to VUR by performing a GWAS in seven cohorts of European ancestry (see Methods and Supplemental Table 4). After genetic matching and imputation, we performed meta-analysis in 1395 unrelated VUR patients and 5366 matched population controls, using 6,131,010 imputed variants common to all cohorts. Given that VUR is significantly more prevalent in females and its segregation is consistent with multiple modes of inheritance,18,19,59–61 we performed global and sex-specific association analyses under additive, recessive, and dominant models62 (Supplemental Tables 5–13, Supplemental Figure 3). Genome-wide significance thresholds were adjusted based on the number of models tested.
Significant and Suggestive GWAS Loci
Altogether we identified three significant and five suggestive loci (Figure 2, Table 2 and Supplemental Figure 4). Remarkably, the three significant loci conferred large effects (OR, 1.41–3.65). The most significant association was in Chr 2p15, under a recessive model (top SNP: rs13013890; OR, 3.65; 95% CI, 2.39 to 5.56; P=1.86×10−9), and mapped to an intron of WDPCP. Altogether, 3.3% of patients were homozygous for the risk allele, compared with 1.2% of controls. Coding mutations in WDPCP cause Bardet-Biedl syndrome (BBS15; MIM 615992), which features VUR as a known manifestation. Studies of Wdpcp mutant mice also revealed CAKUT-like phenotypes, including failure of the ureters to join the UGS, the primordium of the bladder and urethra.63 This locus also includes OTX1, and heterozygous microdeletions encompassing WDPCP and OTX1 have been reported in patients with genitourinary development defects.64 We did not detect such deletions in our CNV analysis of patients with VUR, indicating that this recessive signal is not attributable to hemizygosity at the WDPCP locus. Analysis of expression quantitative trait loci (eQTL) studies (Supplemental Tables 14–15) demonstrated that rs13013890 encodes a human blood eQTL for OTX1. We examined expression of Wdpcp and Otx1 in developing mouse embryos, focusing on the UB, which gives rises to the ureter, the bladder, and UGS. Analysis of Wdpcp at E11.5 revealed robust expression localized to the UB trunk, which will differentiate into the ureter (Supplemental Figure 5A shows expression at E11). Expression was also observed in the epithelium throughout the developing bladder and urethra at later stages (not shown). Analysis of Otx1 expression revealed highest levels in the UGS, detectable from E12 through late stages of development (Supplemental Figure 5B shows expression at E12).
Figure 2.
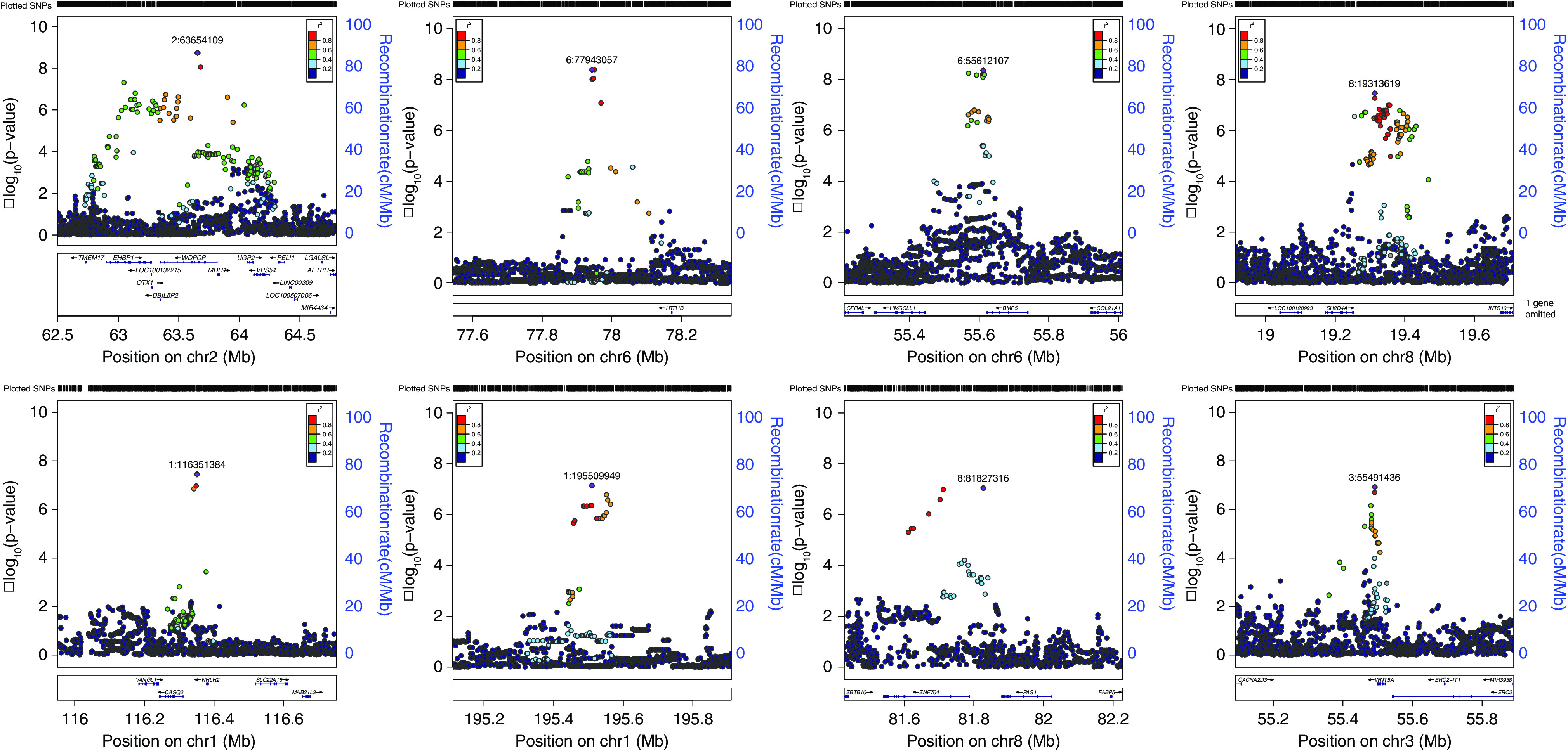
Regional association plots for loci associated with VUR. The calculated −log10 P values are shown according to genomic position (hg19). The most significant SNP is colored purple. Other colors indicate LD (r2; see inset) to top SNP, based on the 1000 Genomes European sample set.
Table 2.
VUR associations
| Top SNP | Chr. | Position | Nearest Genes | A1/A2 | Effect Allele (A1) freq. | OR (95% CI) | P | Model |
|---|---|---|---|---|---|---|---|---|
| rs13013890 | 2 | 63,654,109 | WDPCP, MDH1, OTX1, EHBP1, UGP2, VPS54 | A/G | 0.12 | 3.65 (2.39 to 5.56) | 1.86×10−9 | Recessive |
| rs10806089 | 6 | 77,943,057 | HTR1B | C/T | 0.34 | 2.75 (1.96 to 3.84) | 4.10×10−9 | Recessive (males) |
| rs1154855 | 6 | 55,612,107 | BMP5 | G/T | 0.48 | 1.41 (1.26 to 1.59) | 4.33×10−9 | Additive (females) |
| rs76292820 | 8 | 19,313,619 | CSGALNACT1 | T/C | 0.16 | 4.71 (2.72 to 8.17) | 3.46×10−8 | Recessive (males) |
| rs12759898 | 1 | 116,351,384 | NHLH2, VANGL1, CASQ2 | A/T | 0.08 | 1.61 (1.36 to 1.9) | 3.56×10−8 | Dominant |
| rs16838525 | 1 | 195,509,949 | AL357932.1 | C/T | 0.15 | 6.94 (3.43 to 14.06) | 7.21×10−8 | Recessive (males) |
| rs79060316 | 8 | 81,827,316 | ZNF704, PAG1 | T/C | 0.02 | 2.50 (1.79 to 3.50) | 9.07×10−8 | Dominant |
| rs503022 | 3 | 55,491,436 | WNT5A | A/C | 0.13 | 1.81 (1.45 to 2.25) | 1.19×10−7 | Additive (males) |
Top SNP each locus is shown (dbSNP 151). Positions are in University of California Santa Cruz hg19 coordinates. P values, ORs, and their 95% CIs are derived from meta-analysis of seven European ancestry cohorts (1395 VUR patients and 5366 controls; detailed counts per cohort are shown in Supplemental Table 4), under the indicated models. Chr., chromosome; freq., frequency; WDPCP, WD Repeat Containing Planar Cell Polarity Effector; MDH1, Malate Dehydrogenase 1; OTX1, Orthodenticle Homeobox 1; EHBP1, EH Domain Binding Protein 1; UGP2, UDP-Glucose Pyrophosphorylase 2; VPS54, Vacuolar Protein Sorting-Associated Protein 54; HTR1B, 5-Hydroxytryptamine Receptor 1B; BMP5, Bone Morphogenetic Protein 5; CSGALNACT1, Chondroitin Sulfate N-Acetylgalactosaminyltransferase 1; NHLH2, Nescient Helix-Loop-Helix 2; VANGL1, Vang Gogh-like Planar Cell Polarity Protein 1; CASQ2, Calsequestrin 2; ZNF704, Zinc Finger Protein 704; PAG1, Phosphoprotein Membrane Anchor With Glycosphingolipid Microdomains 1; WNT5A, Wnt Family Member 5A.
Among females (937 patients with VUR and 3129 controls), we found another genome-wide significant association on Chr 6p12.1 under an additive model (rs1154855; OR, 1.41; 95% CI, 1.26 to 1.59; P=4.33×10−9). The top SNP is located within the 3′ untranslated region of BMP5. We did not detect any eQTL associations for this SNP. The BMP family plays important roles in early embryonic development and urinary tract formation.65 Bmp5 is expressed in stroma abutting the urothelium of the bladder, and in the ureteral mesenchyme. Expression, which was strong at E11, continued through later stages of development (Supplemental Figure 5C shows expression at E14).
Among males (458 VUR patients and 2235 controls), we found a third significant association on Chr 6q14.1 under a recessive model (OR, 2.75; 95% CI, 1.96 to 3.84; P=4.10×10−9). Altogether, 15.7% of male patients were homozygous for the risk allele, compared with 9.2% of controls. The top SNP, rs10806089, is located within a low-abundance noncoding RNA (LOC101928570) and 203 kb of HTR1B. We did not detect any eQTL associations for this SNP. However, serotonergic pathways have been implicated in neural control of the lower urinary tract.66 Consistent with these data, Htr1b is expressed in cells scattered throughout the bladder mesenchyme and in the urethra, in a thin layer adjacent to the peripheral muscle (white arrowhead Supplemental Figure 5D).
In addition to these significant loci, for which we had adequate power (Supplemental Table 16), we found five suggestive associations (P value range 3.5×10−8–1.2×10−7, Table 2). Two of these loci, on Chr. 1p31.1 and Chr. 3p14.13, include genes that have been previously implicated in genitourinary developmental defects67,68: the Chr 1p13.1 locus includes VANGL1, a planar cell polarity protein implicated in neural tube defects67 and caudal dysgenesis syndrome, which can include urinary tract defects (rs12759898; OR, 1.61; 95% CI, 1.36 to 1.9; P=3.56×10−8; dominant model). The top Chr 3p14.13 signal is downstream of WNT5A, which has been implicated in kidney and ureteral development in mice (rs503022; OR, 1.81; 95% CI, 1.45 to 2.25; P=1.19×10−7; in males, additive model). WNT5A is highly intolerant to mutations and (probability of being loss of function intolerant, pLI, 0.99; missense Z-score, 1.98), and WNT5A mutations can cause Robinow syndrome (Online Mendelian Inheritance in Man # 180700). We previously reported a WNT5A loss of function variant in a patient with renal agenesis, nominating WNT5A as a candidate gene for isolated kidney malformations in humans.69
Wnt5a Signaling during Cloacal Morphogenesis is Critical for Nephric Duct Insertion and Ureter Maturation
The association with the WNT5A locus prompted us to further investigate the role of Wnt5a in development of the cloaca, which gives rise to the urethra and bladder.58,68,70,71 Wtn5a is expressed in the tail and cloacal mesenchyme at E9 (Figure 3, A and B; www.gudmap.org),72 and expression was localized in the mesenchyme surrounding primitive bladder and hindgut at E11 (Figure 3C). At later stages, Wnt5a was present in mesenchyme from the bladder neck to the distal urethra (Figure 3, D and E). Whole-mount analysis of Wnt5a knockouts at E15 revealed abnormal patterning along the anterior-posterior axis and kidney malformations as previously reported (fused kidneys and renal hypoplasia or renal agenesis, Figure 3, F and N and not shown). The cloaca normally septates between E11 and E13, separating the hindgut from the UGS by E15 (Figure 3G) and failure in this process is associated with anorectal malformations. In Wnt5a mutants at this stage, the cloaca persisted and remained connected to the hindgut (Figure 3O). UGS defects included rudimentary bladder and urethra (13 out of 18 embryos), absence of the bladder (5 out of 18 embryos) or defective urethral differentiation (Figure 3, N and O).
Figure 3.
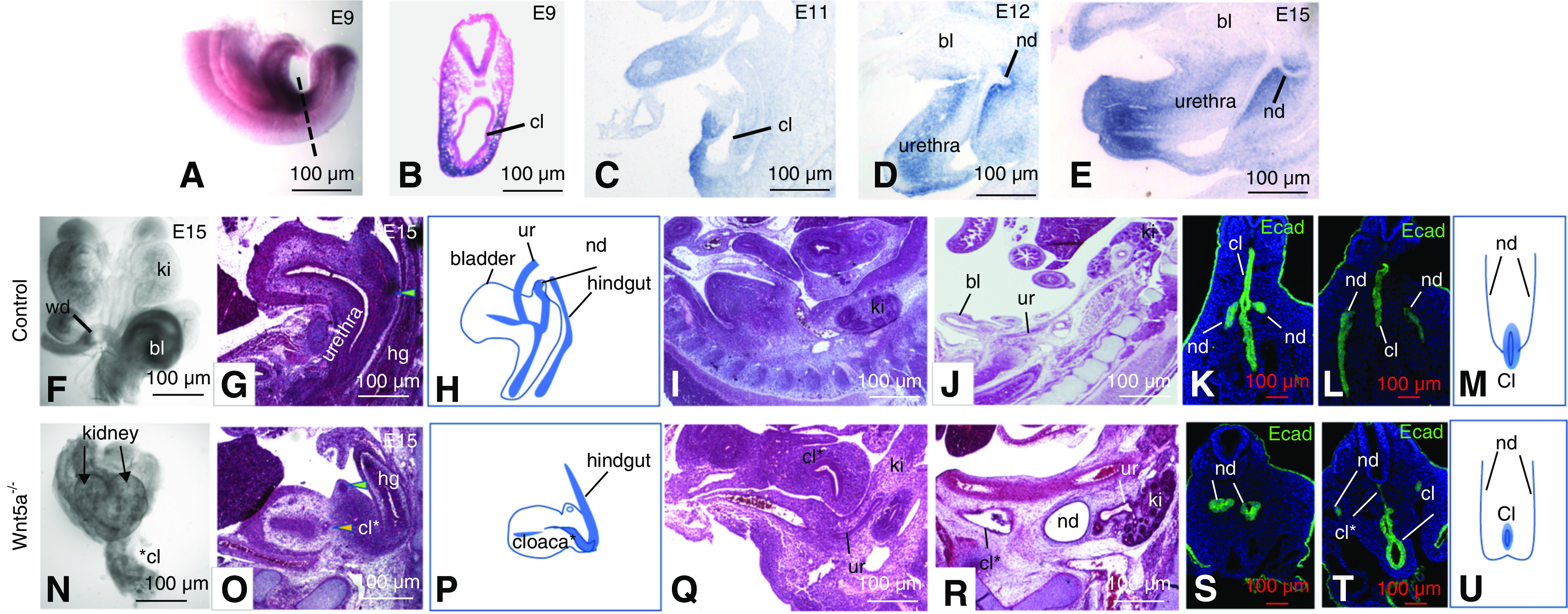
Wnt5a expression and its role in lower urinary tract morphogenesis. (A–E) In situ hybridization analysis. (A) Wnt5a expression at E9 in a whole mount embryo. (B) Section through a whole mount embryo showing Wnt5a expression surrounding the cloaca. (C–E) Wnt5a expression in sagittal sections from embryos at E11 (C), E12 (D), and E15 (E). (F and N) Whole-mount urogenital tracts from a control (F) and Wnt5a a mutant (N). (G and O) Hematoxylin and eosin–stained sections through the bladder/urethra and hindgut of controls (G) and Wnt5a mutants (O). Green arrowheads point to the nephric ducts. Yellow arrowhead in (O) points to the distal ureter. (H and P) Schematic representation of histology shown in (G and O). (I and Q) Sagittal sections through Wnt5a mutants at E12. (J and R) Sagittal sections through a control (J) and Wnt5a mutant embryo (R). (K, L, S, and T) Ecad staining (green) reveals nephric duct and cloacal epithelia at E9, in controls (K and L) and Wnt5a mutants (L and T). (M and U) Schematic showing nephric duct insertion into the cloaca in a wild-type embryo (M) and in a Wnt5a mutant embryo (U). bl, bladder; cl, cloaca; cl* truncated cloaca in Wnt5a mutant; ki, kidney; nd, nephric duct; ur, ureter; wd, Wolffian duct.
We next investigated the fate of nephric ducts and distal ureters. In mutants, the distal ureters, which normally join the bladder neck at E12 and E13, extended partway toward the cloacal region and ended blindly (Figure 3, I and Q). In embryos with rudimentary bladders, ureters generally terminated in the bladder wall outside the lumen, whereas in mutants with no sign of bladder development, ureters generally terminated at an abnormally anterior position and failed to reach the cloacal region (Figure 3, J and R). To investigate the origin of these abnormalities, we analyzed embryos at E9, when nephric ducts normally make primary connections with the cloaca Ecad staining of control embryos revealed nephric ducts that extended caudally, turned toward the midline, and joined the cloaca at the midpoint between the future hindgut and UGS (Figure 3, K–M). In mutants, cloacae were misplaced to an anterior position and the dorsal cloaca, which differentiates into the UGS, was truncated (Figure 3, S–U; cl* denotes the truncated portion of the cloaca). In addition to cloacal defects, nephric ducts in Wnt5a mutants either joined one another or ended blindly, and did not insert into the cloaca (Figure 3, S–U). The nephric duct termini in mutants were generally larger than normal, but we did not observe any sign of bifurcation as reported in other studies.68,70
PRS, Heritability, and Clinical Associations
Using LD score regression,53 we estimated the SNP-based heritability (h2) to be 15.24% under the additive model. We computed a VUR-PRS using LDPred54 under an additive model. To determine whether the VUR-PRS captures different CAKUT or kidney disease subtypes, we applied the VUR-PRS to participants of European ancestry in the CKiD study, a cohort composed of pediatric patients with different causes of kidney failure. However, the VUR-PRS was not different between non-VUR nephropathy causes of kidney disease (Supplemental Figure 6). We also did not detect any significant associations of the VUR-PRS with clinical parameters such as the severity of reflux or recurrence of UTI among patients with VUR.
We next performed a phenome-wide association study (PheWAS) of the top GWAS signals using two large cohorts: UKBB and eMERGE, followed by meta-analysis. We also conducted PheWAS on eMERGE participants under 21 years of age (pediatric eMERGE). Homozygosity for the WDPCP risk allele (rs13013890-A) was nominally associated with cystitis (Supplemental Tables 17–20; OR, 2.11; 95% CI, 1.22 to 3.65; P=7.78×10−3) in males, and with an increased risk of bladder neck obstruction (OR, 5.10; 95% CI, 2.23 to 11.69; P=1.17×10−4) and dystrophy of the genital tract (OR, 2.38; 95% CI, 1.39 to 4.07; P=1.48×10−3) in females. This locus was associated with 22 genitourinary traits at P<0.05. We also observed 46 nominal associations of other VUR risk loci with genitourinary traits (Supplemental Tables 17–20).
Discussion
In the largest genetic study of VUR conducted to date, we detected rare and common variants with large effects on this trait. Most studies of developmental disorders have focused on rare variants, but our study now demonstrates that common risk variants also contribute to congenital and developmental disorders. We also found that some risk loci conferred sex-specific effects. Moreover, we searched for loci under nonadditive models and detected two significant loci under a recessive inheritance. These data suggest that more systematic exploration of nonadditive inheritance and sex-specific analyses may increase the yield for GWAS and clarify the architecture of developmental disorders.
We found that 3% of patients with VUR have an unsuspected genomic disorder that is not clinically recognized, including those identified in major national cohorts such as RIVUR and CKiD, where a clinically detectable chromosomal disorder was an exclusion criterion. These findings extend our previous studies30 and clarify the spectrum of CNV disorders that contribute to VUR. In particular, we detected an enrichment of known GD in three loci (Chr. 16p11.2, 22q11.2, and Chr. X). These GD have been implicated in autism, schizophrenia, developmental delay, congenital cardiac, or vertebral defects,45,73–77 with variable penetrance. About one third of patients with the 22q11.2 microdeletion syndrome display an urinary tract defect.78,79 We previously showed that disruption of CRKL and TBX6 likely account for the kidney and urinary tract defects in the 22q11.2 and 16p11.2 syndromes, respectively.30,80,81 Recent studies have also implicated MAZ, another gene within the CNV 16p11.2 interval, in genitourinary defects.82 The 3% prevalence of CNV disorders has significant clinical implications, given the relatively high prevalence of VUR in the pediatric population. The detection of pathogenic CNVs can assist with diagnosis and prediction of complications, particularly for identification of patients at risk for poor neurocognitive outcomes.83 Because our dataset was sourced from multiple studies with different ascertainment criteria and phenotyping depth, we lacked access to complete data for all patients on kidney function and renal failure; presence of other genitourinary tract malformations; VUR staging and laterality and other clinical variables. However, we note that the high CNV burden was detected in cohorts with both mild (RIVUR) and severe (CKiD) outcomes, motivating further studies to clarify variability of renal function outcomes.
The GWAS for VUR identified loci with large effect, consistent with prior studies of developmental traits, such as hypospadias84,85 and bicuspid aortic valves.86 The top associations encompass genes that participate in embryonic development (WDPCP, BMP5, WNT5A, and VANGL1). The strongest association was an intronic variant in WDPCP, which encodes a planar polarity protein and has been implicated in Bardet Biedl-1587 and Oro-Digital-Facial syndromes,88 which can feature urinary tract malformations, including VUR.89,90 Wdpcp is expressed in the ureter during mouse embryonic development and its inactivation results in developmental abnormalities, including cloacal septation defects.63 We did not detect any coding mutations in LD with the top WDPCP signal, suggesting the noncoding variant(s) may influence the timing or location of expression of WDPCP or other genes in the interval. The large effect imparted by this locus warrants additional studies to identify the causal allele(s) and its utility in clinical prediction among families with the risk variants. WDPCP is also expressed in the lower urogenital tract, motivating further investigation of the potential association with bladder neck obstruction and female genitourinary disorders, as observed in the UKBB and eMERGE PheWAS. In addition, the GWAS findings should be followed up in independent cohorts for replication.
We performed detailed studies of Wnt5a mutant mice because heterozygous WNT5A mutations cause dominant Robinow syndrome, which can feature urinary tract defects, including VUR.91,92 We previously reported a WNT5A loss of function mutation in a patient with nonsyndromic kidney malformation.69 WNT5A signaling with its presumptive receptor ROR2, controls cell migration, convergent extension, planar cell polarity, and numerous aspects of organ development.58 WNT5A mediates noncanonical Wnt signaling via the calcium-signaling or planar cell polarity pathways.93 Wnt5a mutants display urinary tract phenotypes, including duplicated bifid/ureters and kidneys, renal hypoplasia, defective medullary patterning, defective ureteral insertion, and abnormal extension of the intermediate mesenchyme and defective genital tubercle morphogenesis.68,70,71 Here, we describe a novel role for WNT5A during early stages of bladder and urethral formation. We find that Wnt5a expression in the mesenchyme surrounding the cloaca is essential for cloacal morphogenesis and for insertion of nephric ducts, which are precursors of the UBs and the ureters. Defects in the nephric duct insertion process can impair connection between the upper and lower urinary tract and result in CAKUT-like phenotypes. These observations suggest WNT5A or a WNT5A-dependent signal from the cloaca may be important for directing nephric duct insertion, cloacal septation, and UGS differentiation. Abnormal position or differentiation of the UGS may be sufficient to impair nephric duct and ureter insertion, and may be the cause of the spectrum of phenotypes observed in Wnt5a mutants.
Important questions in the clinical management of VUR include risk prediction for complications such as renal failure and UTI, or screening criteria for asymptomatic siblings. In our study, about 6% of VUR patients carried high-risk genotypes, defined as major CNV disorders (3%) or homozygosity for the WDPCP risk allele (3%). Assessment of these loci may be useful for selecting siblings for screening. Recent studies have also shown the value of PRS for prediction of complex phenotypes.94,95 We had adequate power to detect common risk variants; doubling sample size could yield additional risk loci for VUR at lower MAF and/or OR (Supplemental Table 16) and more robust PRS. There are no long-term follow-up studies of VUR into adulthood, and the PheWAS approach may reveal long-term outcomes associated with genetic risk factors that cause VUR in childhood.
Disclosures
A. Gharavi reports having consultancy agreements with AstraZeneca Center for Genomics Research and Goldfinch Bio; reports receiving research funding from the Renal Research Institute; reports receiving honoraria from Sanofi; and being a scientific advisor or member of the bditorial boards of JASN, Kidney International, and Journal of Nephrology. Because A. Gharavi is an editor of the JASN, he was not involved in the peer-review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript. D. Crosslin reports consultancy agreements with UnitedHealth Group; research funding from UnitedHealth Group; and being a scientific advisor or membership with UnitedHealth Group. L. Gesualdo reports consultancy agreements with Medtronic, Retrophin; research funding from Philanthropic sources, Sanofi Genzyme; honoraria from Sanofi Genzyme, Mundipharma, Estor, AstraZeneca, Retrophin, and GSK; being a scientific advisor or membership with NDT Journal, Journal of Nephrology, Board of Directors (SIN, RPS, ERA-EDTA); and other interests/relationships: President of the Italian Renal Federation. E. Kenny reports honoraria from Illumina, Inc. and Regeneron Pharmaceuticals; being a scientific advisor or membership as Editorial board member of Frontiers in Ecology and Evolution and American Journal of Human Genetics; and being an advisory board member of American Society of Human Genetics. B. Levy reports having consultancy agreements with Igenomix, Myriad, Myome, and Natera; reports having an ownership interest in Natera; reports being a scientific advisor or member of Wiley—Prenatal Diagnosis, American College of Medical Genetics Foundation, CAP Cytogenetics committee, Cancer Genomics Consortium, and International Society for Prenatal Diagnosis. M. Miklaszewska reports honoraria from Medycyna Praktyczna – Pediatria. B. Warady reports having consultancy agreements with Akebia, Amgen, Baxter, Bayer, GlaxoSmithKline, Reata, and Relypsa; reports receiving research funding from Baxter Healthcare; reports receiving honoraria from Akebia, GlaxoSmithKline, Reata, Relypsa, and the University of Missouri; reports being a scientific advisor or member of the North American Pediatric Renal Trials and Collaborative Studies, the National Kidney Foundation, and Nephrologists Transforming Dialysis Safety Board of Directors. C. Willer reports their spouse works at Regeneron Pharmaceuticals. C. Wong reports receiving honoraria from Uptodate; being a scientific advisor or member of the Steering Committee for the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) funded CKD in Children study; reports participation in the past year on the ad hoc advisory committee to the National Kidney Foundation; reports being on the board of the New Mexico Pediatric Society in Albuquerque, NM; and reports other interests/relationships via a contract with NephCure International as part of the CurGN study (participating site). D. Barton reports being a scientific advisor or member of European Molecular Genetics Quality Network; and other interests/relationships with Rare Disease Ireland. D. Santoro reports receiving honoraria from Alexion, Astellas, AstraZeneca, and Mundipharma. E. Fiaccadori reports being a scientific advisor or member of the Editorial Board of the Journal of Nephrology, Editorial Board of Blood Purification; and has other interests/relationships as a member of the Italian Society of Nephrology and the European Society of Parenteral and Enteral Nutrition. F. Hildebrandt reports having consultancy agreements as cofounder of Goldfinch-Bio; reports having an ownership interest in Goldfinch-Bio; reports receiving honoraria from Sanofi; reports being a scientific advisor or member of Goldfinch-Bio; and has other interests/relationships as Cofounder and SAB member of Goldfinch-Bio. F. Lin reports receiving honoraria from lectures and seminars in academic institutions; and being a scientific advisor or member of the JASN editorial board. I. Ionita-Laza reports being a scientific advisor or member as the Associate Editor for Biometrics and Associate Editor of Statistics in Biosciences. I. Stanaway reports sole ownership of an LLC, Byrell Systems, registered in Washington State; and reports other interests/relationships as a member of the community service organization, the Odd Fellows, Ballard Chapter 170. J. Barasch reports having consultancy agreements with Abbvie (one meeting on AKI in 2014); reports receiving research funding from Abbott, and Alere supported one study in the clinical use of Neutrophil gelatinase–associated lipocalin 2009–2010; receiving honoraria from Harvard Medical School (reviewer and lecturer), March of Dimes (as a grant reviewer 2008–2018), and National Institutes of Health (NIH; as a grant reviewer 2020); reports patents and inventions with Abbott and Bioporto, and had licensed patents from Columbia University. K. Kiryluk reports being a scientific advisor or member of the Scientific Advisory Board of Gilead Sciences and Goldfinch-Bio; and has other interests/relationships through scientific partnerships with Aevi Genomics and AstraZeneca. M. Mizerska-Wasiak reports other interests/relationships as a member of the European Society Pediatric Nephrology, and the European Renal Association-European Dialysis Transplantation Association. M. Sampson reports having consultancy agreements with Kohlberg Kravis Roberts & Co. and Janssen Pharmaceutical; and being a scientific advisor or member of Natera. M. Szczepanska reports receiving research funding from Faculty of Medical Sciences in Zabrze, Medical University of Silesia in Katowice; reports receiving honoraria from Baxter, Roche, and Swixx; and reports having other interests/relationships with ESPN, ERA-EDTA, the Polish Society for Pediatrics, and the Polish Society for Pediatric Nephrology. M. Tkaczyk reports having consultancy agreements with Gerson Lehrman Group; and being President of the Polish Society for Pediatric Nephrology. P. Puri reports being Editor-in-Chief of Pediatric Surgery International. S. Sanna-Cherchi reports being a scientific advisor or member of Editorial Boards, with no royalties received, of Acta Biomedica, Journal of Nephrology, Kidney International, and Negative Results. All remaining authors have nothing to disclose.
Funding
The project is supported by NIDDK grants U54 DK104309 06 (to A.G. Gharavi, C.L. Mendelshon, and J.Barasch), R01 DK080099 and U01 HG008680 (to A.G. Gharavi), U01-DK 094530 (to C.L. Mendelsohn), R01-DK105124, RC2-DK116690, UH3-DK114926, and U01-HG008680 (to K. Kiryluk), 1R01DK103184, 1R21DK098531, and UL1 TR000040 (to S. Sanna-Cherchi), and R21DK119802-01A (to M. Verbitsky). Genotyping of Irish patients with VUR and support of J.M. Darlow and M.G. Dobson were funded by a grant from The Children’s Medical and Research Foundation/National Children’s Research Centre, and J.M. Darlow was subsequently supported by the (Irish) Health Research Board grant HRA-POR-2014–693. The eMERGE Phase III Network was funded by the National Human Genome Research Institute through grants U01HG8680 (Columbia University), U01HG8672 (Vanderbilt University Medical Center), U01HG8657 (Kaiser Permanente Washington Health Research Institute/University of Washington), U01HG8685 (Brigham and Women’s Hospital), U01HG8666 (Cincinnati Children’s Hospital Medical Center), U01HG6379 (Mayo Clinic), U01HG8679 (Geisinger Clinic), U01HG8684 (Children’s Hospital of Philadelphia), U01HG8673 (Northwestern University), MD007593 (Meharry Medical College), U01HG8676 (Partners Healthcare/Broad Institute), and U01HG8664 (Baylor College of Medicine).
Supplementary Material
Acknowledgments
The authors thank the patients who participated in this study. The POLYGENES network (http://www.polygenes.org), a collaborative network of Columbia University in New York and the Polish Registry of Congenital Malformations at Poznań University of Medical Sciences, Poznań, Poland recruited Polish patients with VUR for this study. Genotypes of the Irish controls were kindly provided to David E. Barton by Derek Morris and Prof. Aiden Corvin of Department of Psychiatry, Trinity College Dublin, and the International Schizophrenia Consortium. Parts of this study have also been conducted using the UKBB Resource under UKBB project ID number 41849.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020050681/-/DCSupplemental.
Supplemental Figure 1. PCA for CNV frequency filtering.
Supplemental Figure 2. Excess burden of large, rare CNVs in VUR compared with controls.
Supplemental Figure 3. VUR patients are enriched in larger rare CNVs as compared with controls.
Supplemental Figure 4. GWAS meta-analyses.
Supplemental Figure 5. Expression of Wdpcp, Otx1, Bmp5, and Htr1b in the mouse lower urinary tract.
Supplemental Figure 6. Distribution of VUR-PRS values across phenotypes in CKiD participants of European ancestry.
Supplemental Table 1. Excess burden of large, rare CNVs in VUR compared with controls.
Supplemental Table 2. VUR patients are enriched in large, rare CNVs, compared with controls.
Supplemental Table 3. Likely Pathogenic CNV.
Supplemental Table 4. GWAS cohorts.
Supplemental Table 5. GWAS meta-analysis additive model.
Supplemental Table 6. GWAS meta-analysis additive model in males.
Supplemental Table 7. GWAS meta-analysis additive model in females.
Supplemental Table 8. GWAS meta-analysis recessive model.
Supplemental Table 9. GWAS meta-analysis recessive model in males.
Supplemental Table 10. GWAS meta-analysis recessive model in females.
Supplemental Table 11. GWAS meta-analysis dominant model.
Supplemental Table 12. GWAS meta-analysis dominant model in males.
Supplemental Table 13. GWAS meta-analysis dominant model in females.
Supplemental Table 14. Blood eQTL.
Supplemental Table 15. GTEx eQTL.
Supplemental Table 16. Power calculations.
Supplemental Table 17. PheWAS on UKBB and eMERGE.
Supplemental Table 18. PheWAS on eMERGE.
Supplemental Table 19. PheWAS on eMERGE (pediatric only).
Supplemental Table 20. PheWAS on UKBB.
References
- 1.Stephens FD, Lenaghan D: The anatomical basis and dynamics of vesicoureteral reflux. J Urol 87: 669–680, 1962 [DOI] [PubMed] [Google Scholar]
- 2.United States Renal Data System : USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States, Bethesda, MD, National Institutes of Health, 2019. Available at https://www.usrds.org/annual-data-report. Accessed January 2, 2020
- 3.Farhat W, McLorie G, Geary D, Capolicchio G, Bägli D, Merguerian P, et al.: The natural history of neonatal vesicoureteral reflux associated with antenatal hydronephrosis. J Urol 164: 1057–1060, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Ichikawa I, Kuwayama F, Pope JC 4th, Stephens FD, Miyazaki Y: Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int 61: 889–898, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Ismaili K, Hall M, Piepsz A, Wissing KM, Collier F, Schulman C, et al.: Primary vesicoureteral reflux detected in neonates with a history of fetal renal pelvis dilatation: A prospective clinical and imaging study. J Pediatr 148: 222–227, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Arant BS Jr.: Vesicoureteric reflux and renal injury. Am J Kidney Dis 17: 491–511, 1991 [DOI] [PubMed] [Google Scholar]
- 7.Dillon MJ, Goonasekera CD: Reflux nephropathy. J Am Soc Nephrol 9: 2377–2383, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Peters PC, Johnson DE, Jackson JH Jr.: The incidence of vesicoureteral reflux in the premature child. J Urol 97: 259–260, 1967 [DOI] [PubMed] [Google Scholar]
- 9.Puri P, Gosemann JH, Darlow J, Barton DE: Genetics of vesicoureteral reflux. Nat Rev Urol 8: 539–552, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Steele BT, De Maria J: A new perspective on the natural history of vesicoureteric reflux. Pediatrics 90: 30–32, 1992 [PubMed] [Google Scholar]
- 11.Yeung CK, Godley ML, Dhillon HK, Gordon I, Duffy PG, Ransley PG: The characteristics of primary vesico-ureteric reflux in male and female infants with pre-natal hydronephrosis. Br J Urol 80: 319–327, 1997 [DOI] [PubMed] [Google Scholar]
- 12.Ashraf S, Hoskins BE, Chaib H, Hoefele J, Pasch A, Saisawat P, et al.: Mapping of a new locus for congenital anomalies of the kidney and urinary tract on chromosome 8q24. Nephrol Dial Transplant 25: 1496–1501, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger RH: A theory on the nature of transmission of congenital vesicoureteral reflux. J Urol 108: 249–254, 1972 [DOI] [PubMed] [Google Scholar]
- 14.Chapman CJ, Bailey RR, Janus ED, Abbott GD, Lynn KL: Vesicoureteric reflux: Segregation analysis. Am J Med Genet 20: 577–584, 1985 [DOI] [PubMed] [Google Scholar]
- 15.Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al.: Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux [published correction appears in Sci Rep 8: 459, 2018 10.1038/s41598-017-17992-w]. Sci Rep 7: 14595, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darlow JM, Dobson MG, Darlay R, Molony CM, Hunziker M, Green AJ, et al.: A new genome scan for primary nonsyndromic vesicoureteric reflux emphasizes high genetic heterogeneity and shows linkage and association with various genes already implicated in urinary tract development. Mol Genet Genomic Med 2: 7–29, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feather SA, Malcolm S, Woolf AS, Wright V, Blaydon D, Reid CJ, et al.: Primary, nonsyndromic vesicoureteric reflux and its nephropathy is genetically heterogeneous, with a locus on chromosome 1. Am J Hum Genet 66: 1420–1425, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanna-Cherchi S, Caridi G, Weng PL, Dagnino M, Seri M, Konka A, et al.: Localization of a gene for nonsyndromic renal hypodysplasia to chromosome 1p32-33. Am J Hum Genet 80: 539–549, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weng PL, Sanna-Cherchi S, Hensle T, Shapiro E, Werzberger A, Caridi G, et al.: A recessive gene for primary vesicoureteral reflux maps to chromosome 12p11-q13. J Am Soc Nephrol 20: 1633–1640, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Georgas KM, Armstrong J, Keast JR, Larkins CE, McHugh KM, Southard-Smith EM, et al.: An illustrated anatomical ontology of the developing mouse lower urogenital tract. Development 142: 1893–1908, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liaw A, Cunha GR, Shen J, Cao M, Liu G, Sinclair A, et al.: Development of the human bladder and ureterovesical junction. Differentiation 103: 66–73, 2018 [DOI] [PubMed] [Google Scholar]
- 22.Rasouly HM, Lu W: Lower urinary tract development and disease. Wiley Interdiscip Rev Syst Biol Med 5: 307–342, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams G, Fletcher JT, Alexander SI, Craig JC: Vesicoureteral reflux. J Am Soc Nephrol 19: 847–862, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG: Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 128: 4–15, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, et al.: Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 9: 358–364, 1995 [DOI] [PubMed] [Google Scholar]
- 26.Lu W, van Eerde AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, et al.: Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet 80: 616–632, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gimelli S, Caridi G, Beri S, McCracken K, Bocciardi R, Zordan P, et al.: Mutations in SOX17 are associated with congenital anomalies of the kidney and the urinary tract. Hum Mutat 31: 1352–1359, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gbadegesin RA, Brophy PD, Adeyemo A, Hall G, Gupta IR, Hains D, et al.: TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol 24: 1313–1322, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Eerde AM, Duran K, van Riel E, de Kovel CG, Koeleman BP, Knoers NV, et al.: Genes in the ureteric budding pathway: Association study on vesico-ureteral reflux patients. PLoS One 7: e31327, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, et al.: The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet 51: 117–127, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keren R, Carpenter MA, Hoberman A, Shaikh N, Matoo TK, Chesney RW, et al.: Rationale and design issues of the Randomized Intervention for Children With Vesicoureteral Reflux (RIVUR) study. Pediatrics 122[Suppl 5]: S240–S250, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furth SL, Cole SR, Moxey-Mims M, Kaskel F, Mak R, Schwartz G, et al.: Design and methods of the Chronic Kidney Disease in Children (CKiD) prospective cohort study. Clin J Am Soc Nephrol 1: 1006–1015, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong CJ, Moxey-Mims M, Jerry-Fluker J, Warady BA, Furth SL: CKiD (CKD in children) prospective cohort study: A review of current findings. Am J Kidney Dis 60: 1002–1011, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matise TC, Ambite JL, Buyske S, Carlson CS, Cole SA, Crawford DC, et al.; PAGE Study: The Next PAGE in understanding complex traits: Design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. Am J Epidemiol 174: 849–859, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al.: PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM: Robust relationship inference in genome-wide association studies. Bioinformatics 26: 2867–2873, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al.: PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17: 1665–1674, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, et al.: Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91: 987–997, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee: American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13: 680–685, 2011 [DOI] [PubMed] [Google Scholar]
- 41.South ST, Lee C, Lamb AN, Higgins AW, Kearney HM; Working Group for the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee: ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet Med 15: 901–909, 2013 [DOI] [PubMed] [Google Scholar]
- 42.Verbitsky M, Sanna-Cherchi S, Fasel DA, Levy B, Kiryluk K, Wuttke M, et al.: Genomic imbalances in pediatric patients with chronic kidney disease. J Clin Invest 125: 2171–2178, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westland R, Verbitsky M, Vukojevic K, Perry BJ, Fasel DA, Zwijnenburg PJG, et al.: Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int 88: 1402–1410, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al.: DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84: 524–533, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al.: A copy number variation morbidity map of developmental delay. Nat Genet 43: 838–846, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy UM, Page GP, Saade GR, Silver RM, Thorsten VR, Parker CB, et al.; NICHD Stillbirth Collaborative Research Network: Karyotype versus microarray testing for genetic abnormalities after stillbirth. N Engl J Med 367: 2185–2193, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al.: Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 367: 2175–2184, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patterson N, Price AL, Reich D: Population structure and eigenanalysis. PLoS Genet 2: e190, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willer CJ, Li Y, Abecasis GR: METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26: 2190–2191, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fadista J, Manning AK, Florez JC, Groop L: The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J Hum Genet 24: 1202–1205, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.González JR, Carrasco JL, Dudbridge F, Armengol L, Estivill X, Moreno V: Maximizing association statistics over genetic models. Genet Epidemiol 32: 246–254, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al.: LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 26: 2336–2337, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al.; ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; RACI Consortium: Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47: 1228–1235, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, et al.; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) study: Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet 97: 576–592, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.ICD-9-CM diagnosis and procedure codes: Abbreviated and full code titles. Baltimore, MD, U.S. Centers of Medicare & Medicaid Services, 2014. Available at www.cms.gov/Medicare/Coding/ICD9ProviderDiagnosticCodes/codes. Accessed May 14, 2017
- 56.Carroll RJ, Bastarache L, Denny JC: R PheWAS: Data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30: 2375–2376, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srinivas S, Goldberg MR, Watanabe T, D’Agati V, al-Awqati Q, Costantini F: Expression of green fluorescent protein in the ureteric bud of transgenic mice: A new tool for the analysis of ureteric bud morphogenesis. Dev Genet 24: 241–251, 1999 [DOI] [PubMed] [Google Scholar]
- 58.Yamaguchi TP, Bradley A, McMahon AP, Jones S: A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126: 1211–1223, 1999 [DOI] [PubMed] [Google Scholar]
- 59.Kitzler TM, Schneider R, Kohl S, Kolvenbach CM, Connaughton DM, Dai R, et al.: COL4A1 mutations as a potential novel cause of autosomal dominant CAKUT in humans. Hum Genet 138: 1105–1115, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pasch A, Hoefele J, Grimminger H, Hacker HW, Hildebrandt F: Multiple urinary tract malformations with likely recessive inheritance in a large Somalian kindred. Nephrol Dial Transplant 19: 3172–3175, 2004 [DOI] [PubMed] [Google Scholar]
- 61.Vivante A, Kleppa MJ, Schulz J, Kohl S, Sharma A, Chen J, et al.: Mutations in TBX18 cause dominant urinary tract malformations via transcriptional dysregulation of ureter development. Am J Hum Genet 97: 291–301, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Capozza N, Gulia C, Heidari Bateni Z, Zangari A, Gigli S, Briganti V, et al.: Vesicoureteral reflux in infants: What do we know about the gender prevalence by age? Eur Rev Med Pharmacol Sci 21: 5321–5329, 2017 [DOI] [PubMed] [Google Scholar]
- 63.Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, et al.: Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton. PLoS Biol 11: e1001720, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jorgez CJ, Rosenfeld JA, Wilken NR, Vangapandu HV, Sahin A, Pham D, et al.: Genitourinary defects associated with genomic deletions in 2p15 encompassing OTX1. PLoS One 9: e107028, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.King JA, Marker PC, Seung KJ, Kingsley DM: BMP5 and the molecular, skeletal, and soft-tissue alterations in short ear mice. Dev Biol 166: 112–122, 1994 [DOI] [PubMed] [Google Scholar]
- 66.Fan WJ, Chen SC, Hsieh TH, Lai CH, Lin YS, Peng CW, et al.: Influence of serotonergic mechanisms on the urine flow rate in male rats. Am J Physiol Regul Integr Comp Physiol 307: R1239–R1250, 2014 [DOI] [PubMed] [Google Scholar]
- 67.Iliescu A, Gravel M, Horth C, Gros P: Independent mutations at Arg181 and Arg274 of Vangl proteins that are associated with neural tube defects in humans decrease protein stability and impair membrane targeting. Biochemistry 53: 5356–5364, 2014 [DOI] [PubMed] [Google Scholar]
- 68.Pietilä I, Prunskaite-Hyyryläinen R, Kaisto S, Tika E, van Eerde AM, Salo AM, et al.: Wnt5a deficiency leads to anomalies in ureteric tree development, tubular epithelial cell organization and basement membrane integrity pointing to a role in kidney collecting duct patterning. PLoS One 11: e0147171, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanna-Cherchi S, Khan K, Westland R, Krithivasan P, Fievet L, Rasouly HM, et al.: Exome-wide association study identifies GREB1L mutations in congenital kidney malformations [published correction appears in Am J Hum Genet 101: 789–802, 2017 10.1016/j.ajhg.2017.09.018]. Am J Hum Genet 101: 1034, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yun K, Ajima R, Sharma N, Costantini F, Mackem S, Lewandoski M, et al.: Non-canonical Wnt5a/Ror2 signaling regulates kidney morphogenesis by controlling intermediate mesoderm extension. Hum Mol Genet 23: 6807–6814, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yun K, Perantoni AO: Hydronephrosis in the Wnt5a-ablated kidney is caused by an abnormal ureter-bladder connection. Differentiation 94: 1–7, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harding SD, Armit C, Armstrong J, Brennan J, Cheng Y, Haggarty B, et al.: The GUDMAP database--an online resource for genitourinary research. Development 138: 2845–2853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Kateb H, Khanna G, Filges I, Hauser N, Grange DK, Shen J, et al.: Scoliosis and vertebral anomalies: Additional abnormal phenotypes associated with chromosome 16p11.2 rearrangement. Am J Med Genet A 164A: 1118–1126, 2014 [DOI] [PubMed] [Google Scholar]
- 74.Glessner JT, Bick AG, Ito K, Homsy J, Rodriguez-Murillo L, Fromer M, et al.: Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res 115: 884–896, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, et al.: De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 41: 931–935, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, et al.: Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry 66: 947–956, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pescosolido MF, Gamsiz ED, Nagpal S, Morrow EM: Distribution of disease-associated copy number variants across distinct disorders of cognitive development. J Am Acad Child Adolesc Psychiatry 52: 414–430.e14, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kujat A, Schulz MD, Strenge S, Froster UG: Renal malformations in deletion 22q11.2 patients. Am J Med Genet A 140: 1601–1602, 2006 [DOI] [PubMed] [Google Scholar]
- 79.Wu HY, Rusnack SL, Bellah RD, Plachter N, McDonald-McGinn DM, Zackai EH, et al.: Genitourinary malformations in chromosome 22q11.2 deletion. J Urol 168: 2564–2565, 2002 [DOI] [PubMed] [Google Scholar]
- 80.Lopez-Rivera E, Liu YP, Verbitsky M, Anderson BR, Capone VP, Otto EA, et al.: Genetic drivers of kidney defects in the DiGeorge syndrome. N Engl J Med 376: 742–754, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanna-Cherchi S, Sampogna RV, Papeta N, Burgess KE, Nees SN, Perry BJ, et al.: Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med 369: 621–629, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haller M, Au J, O’Neill M, Lamb DJ: 16p11.2 transcription factor MAZ is a dosage-sensitive regulator of genitourinary development. Proc Natl Acad Sci U S A 115: E1849–E1858, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Verbitsky M, Kogon AJ, Matheson M, Hooper SR, Wong CS, Warady BA, et al.: Genomic disorders and neurocognitive impairment in pediatric CKD. J Am Soc Nephrol 28: 2303–2309, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geller F, Feenstra B, Carstensen L, Pers TH, van Rooij IA, Körberg IB, et al.: Genome-wide association analyses identify variants in developmental genes associated with hypospadias. Nat Genet 46: 957–963, 2014 [DOI] [PubMed] [Google Scholar]
- 85.van der Zanden LF, van Rooij IA, Feitz WF, Knight J, Donders AR, Renkema KY, et al.: Common variants in DGKK are strongly associated with risk of hypospadias. Nat Genet 43: 48–50, 2011 [DOI] [PubMed] [Google Scholar]
- 86.Yang B, Zhou W, Jiao J, Nielsen JB, Mathis MR, Heydarpour M, et al.: Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat Commun 8: 15481, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al.: Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 329: 1337–1340, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saari J, Lovell MA, Yu HC, Bellus GA: Compound heterozygosity for a frame shift mutation and a likely pathogenic sequence variant in the planar cell polarity—ciliogenesis gene WDPCP in a girl with polysyndactyly, coarctation of the aorta, and tongue hamartomas. Am J Med Genet A 167A: 421–427, 2015 [DOI] [PubMed] [Google Scholar]
- 89.Atmış B, Karabay-Bayazıt A, Melek E, Bişgin A, Anarat A: Renal features of Bardet Biedl syndrome: A single center experience. Turk J Pediatr 61: 186–192, 2019 [DOI] [PubMed] [Google Scholar]
- 90.Sharifian M, Dadkhah-Chimeh M, Einollahi B, Nafar M, Simforoush N, Basiri A, et al.: Renal transplantation in patients with Bardet-Biedl syndrome. Arch Iran Med 10: 339–342, 2007 [PubMed] [Google Scholar]
- 91.Bain MD, Winter RM, Burn J: Robinow syndrome without mesomelic ‘brachymelia’: A report of five cases. J Med Genet 23: 350–354, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roifman M, Marcelis CL, Paton T, Marshall C, Silver R, Lohr JL, et al.; FORGE Canada Consortium: De novo WNT5A-associated autosomal dominant Robinow syndrome suggests specificity of genotype and phenotype. Clin Genet 87: 34–41, 2015 [DOI] [PubMed] [Google Scholar]
- 93.Kohn AD, Moon RT: Wnt and calcium signaling: Beta-catenin-independent pathways. Cell Calcium 38: 439–446, 2005 [DOI] [PubMed] [Google Scholar]
- 94.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al.: Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50: 1219–1224, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, et al.: Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177: 587–596.e9, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data that support the findings of this study are available from the corresponding authors on reasonable request. The data are also available on dbGaP (https://www.ncbi.nlm.nih.gov/gap) under the following accession numbers: phs001749.v1.p1, phs000650.v3.p1, phs000304.v1.p1, phs000169.v1.p1, phs000092.v1.p1, phs000199.v1.p1, phs000431.v1.p1, phs000356.v2.p1, phs001584.v1.p1 (and other, pending). Some restrictions may apply according to participants’ consent and privacy protection. All images generated from mouse experiments reported in this study will also be available from the corresponding authors.