

Abstract
Metamorphic proteins switch between different folds, defying the protein folding paradigm. It is unclear how fold-switching arises during evolution. Using ancestral reconstruction and NMR, we studied the evolution of the metamorphic protein XCL1, which has two distinct folds with different functions, making it an unusual member of the chemokine family that generally adopts one conserved fold. XCL1 evolved from an ancestor with the chemokine fold. Evolution of a novel dimer interface, changes in structural constraints and molecular strain, and alteration of intramolecular protein contacts drove the evolution of metamorphosis. Then, XCL1 likely evolved to preferentially populate the non-canonical fold before reaching its modern-day near-equal population of folds. These discoveries illuminate how one sequence evolves to encode multiple structures, revealing principles for protein design and engineering.
One Sentence Summary:
The human protein XCL1 evolved to switch between two completely different folds with different functions.
Metamorphic proteins defy the protein folding paradigm, in which each amino acid sequence adopts one defined fold (monomorphic). They switch reversibly between entirely distinct folds, often with different functions, on the timescale of seconds (1). Metamorphic proteins undergo large-scale structural changes (e.g. alterations in secondary structure and hydrogen bonding networks), whereas allosteric proteins exhibit smaller-scale conformational dynamics (1, 2). While only ~6 metamorphic proteins have been well characterized (2–4), estimates suggest that metamorphic proteins may comprise up to 4% of proteins in the Protein Data Bank (PDB) (5). The emergence of new protein folds on evolutionary timescales has been examined (6–9), but it remains unclear how metamorphic folding evolves in a single protein.
Among metamorphic proteins, the human chemokine XCL1 (lymphotactin) undergoes one of the most significant structural switches, involving complete rearrangement of hydrogen bonding networks (Fig.1A–B) (10). XCL1 belongs to a family of 46 human chemokines: small, secreted proteins that direct immune cell migration. While non-XCL1 chemokines adopt a monomorphic, conserved α+β fold (chemokine fold) (11), XCL1 reversibly interconverts between the chemokine fold and a dimeric, all-β fold (alternate fold) with no structural similarity to other known proteins (Fig.1A) (10). Non-XCL1 chemokines execute two essential functions using one structure, but XCL1 divides these two functions between its two folds: the chemokine structure activates XCL1’s cognate G-protein coupled receptor (GPCR), while the alternate structure binds glycosaminoglycans (GAGs) (10). Additionally, the alternate structure is directly antimicrobial, like a subset of chemokines (12). In a family of proteins that adopt a single fold, how did XCL1 evolve to become metamorphic?
Fig. 1. Evolutionary history of XCL1.
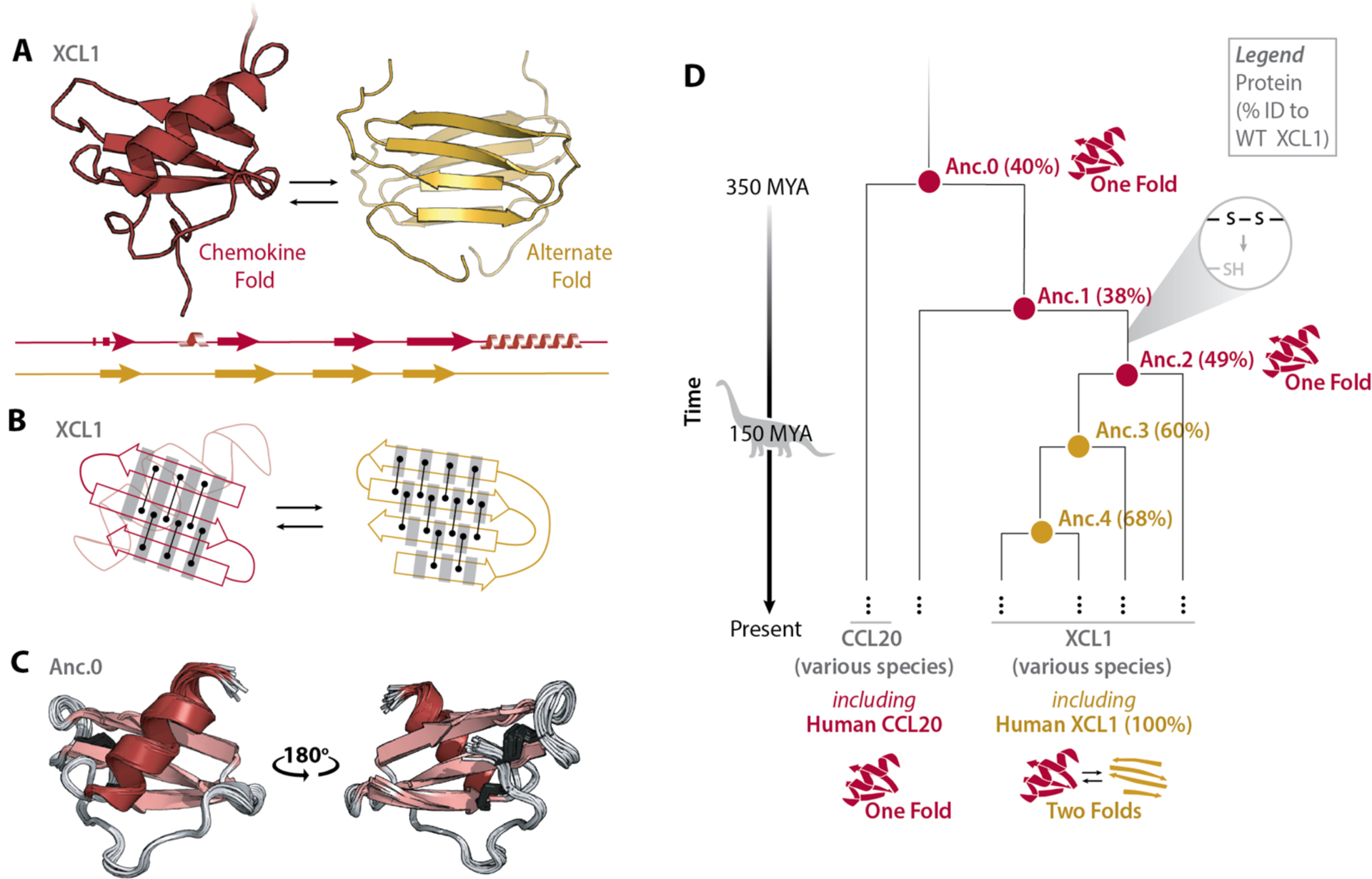
(A) Structures and secondary structure diagrams for XCL1’s chemokine fold (red) and alternate fold (gold). (B) Cartoon comparing hydrogen bonding networks in each fold’s β-strands. Black lines between dots represent pairs of hydrogen bonds between amino acids. Grey shading highlights the same set of residues. Interconversion between XCL1’s chemokine fold and alternate fold requires the β2 strand to rotate 180° and shift by one residue relative to the β1 and β3 strands, establishing an entirely new hydrogen bonding pattern. (C) Ensemble of the top 20 NMR structures for Anc.0 (PDB ID 7JH1), colored by secondary structure. Disulfide bonds are shown as dark grey sticks. (D) Simplified phylogenetic tree showing XCL1’s evolutionary history, beginning with the last shared ancestor (Anc.0) of XCL1 and another chemokine (CCL20). MYA, Million Years Ago. Nodes represent reconstructed ancestral sequences. For each ancestral sequence, % identity to extant human XCL1 is shown in parentheses.
To better understand the evolution of metamorphosis in XCL1, we inferred its phylogenetic tree using ancestral reconstruction (Fig.1D, Fig.S1). This technique utilizes multiple sequence alignments of modern-day proteins from different species to infer amino acid sequences of shared ancestors (13) and has been used to investigate various evolutionary questions (14–19). We created a multiple alignment for 457 chemokine sequences from 30 vertebrate species incorporating 14 chemokine structures (20), from which we inferred the phylogeny of XCL1 and related chemokines using maximum likelihood methods (i.e. the inferred ancestral sequences have the highest probability of producing the modern-day sequences (13, 21, 22)). Phylogenetic analysis indicates that XCL1 is most closely related to another, monomorphic chemokine, CCL20 (Fig.1D, Fig.S1) (12). We resurrected (i.e. expressed and purified) the last shared ancestor of XCL1 and CCL20 (Anc.0). Unlike the temperature- and salt-dependent structural equilibrium of XCL1 (23), the Anc.0 HSQC spectrum is unchanged from 10–50°C +/− NaCl (Fig. S2). We solved the Anc.0 NMR structure: it adopts the canonical chemokine fold (Fig.1C, Table S1). Despite sharing only 40% sequence identity with extant, human XCL1, Anc.0’s structure is highly similar to human XCL1’s chemokine fold (RMSD = 1.45Å) (Fig. 1A, C). Together, these data suggest that a metamorphic protein evolved from a monomorphic ancestor.
Non-XCL1 chemokines have two conserved disulfide bonds, one of which is incompatible with the alternate fold of XCL1. Restoring the missing disulfide bond to XCL1 makes it monomorphic, locked into the chemokine fold (24). Anc.0 has both chemokine disulfide bonds, so we sought to identify the interval where one disulfide was likely lost, because this may have imparted metamorphism. Maximum likelihood methods (13, 21, 22) identified several nodes along the evolutionary trajectory from Anc.0 to extant XCL1. By locating the oldest node whose sequence lacks the cysteines needed for one of the disulfides, we identified the interval where one disulfide was lost (Fig.1D), culminating in an ancestor called Anc.2 (Fig.1D). Anc.2 adopts a single fold at all temperature and salt conditions tested by NMR (10–50°C, +/− NaCl), indicating that disulfide loss alone did not enable metamorphism (Fig.1D, Fig.S3).
Given that loss of a conserved disulfide was necessary but insufficient to impart metamorphism to XCL1, what evolutionary changes resulted in the emergence of metamorphosis? We resurrected the remaining inferred ancestral sequences (Anc.3, Anc.4) and found by NMR that they both interconvert reversibly between two distinct folds (Fig.2A–C, Fig. S4–7) despite sharing only 60 and 68% sequence identity with human XCL1 respectively. This suggests that a wide range of sequences can encode metamorphism, because here metamorphism is encoded by sequences that differ at up to 40% of positions. ZZ-exchange experiments reveal that Anc.3 (kex=0.97 s−1) and Anc.4 (kex=2.6 s−1) exchange on the timescale of seconds, similar to human XCL1 (kex=0.90 s−1 (25)), consistent with XCL1 evolving to remain metamorphic over hundreds of millions of years, balancing occupancy of its two structures by keeping exchange rates in a narrow range.
Fig. 2. Evolutionary progression of metamorphic folding in XCL1.
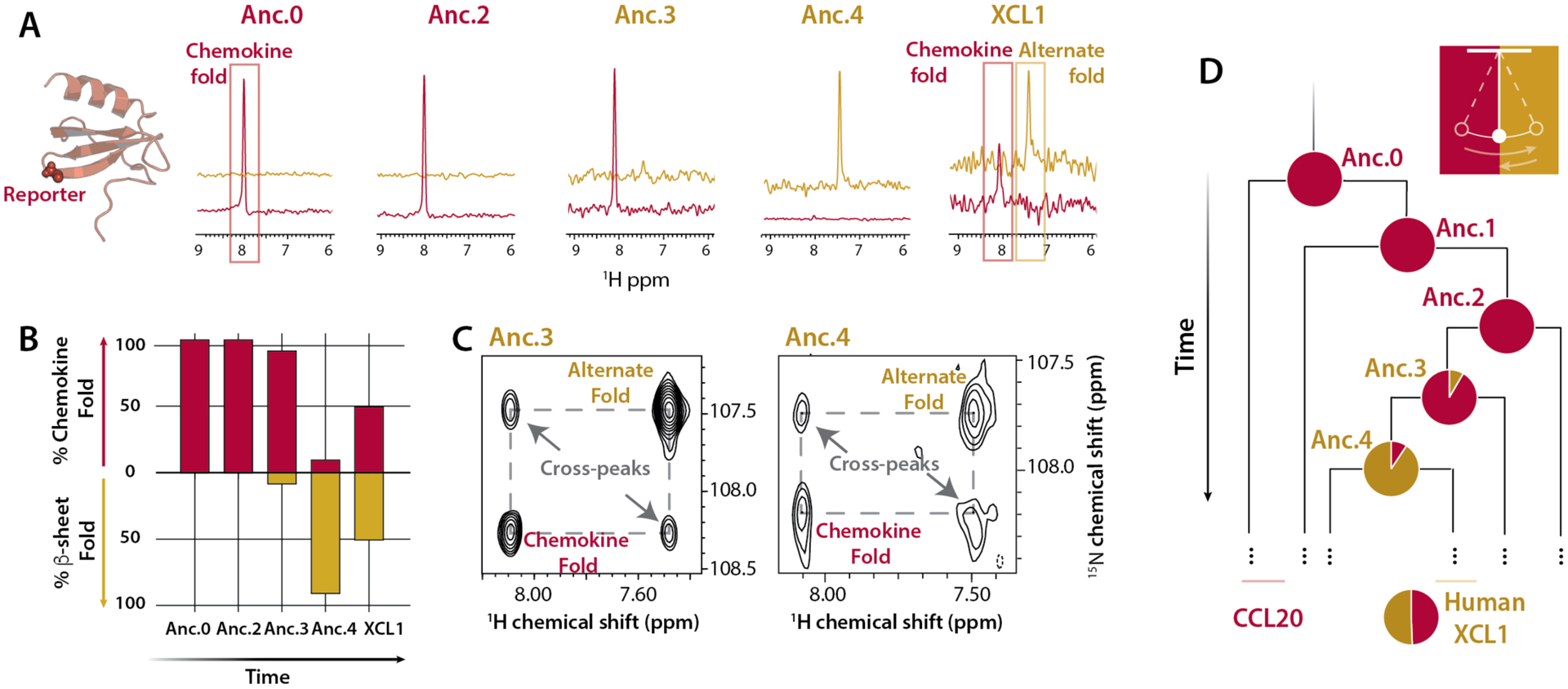
(A) 1D traces (from HSQC experiments performed at 37°C, 150 mM NaCl, 350 μM protein concentration) for a glycine peak (Gly44 in XCL1) that is diagnostic of XCL1’s metamorphic conformation, shown as spheres on Anc.0’s structure at left. Because Gly44 occupies distinct chemical environments in XCL1’s different native structures, there are two Gly44 peaks with chemical shifts of ~8.0 1H ppm (chemokine fold, red) and ~7.4 1H ppm (alternate fold, gold). (B) Fractional abundance of the chemokine and alternate folds were calculated as an average of relative HSQC peak volumes for two reporter residues (Gly44 and the indole NH of Trp55 in XCL1). All spectra were collected at 37°C with 150 mM NaCl and 350 μM protein concentration. (C) ZZ-exchange dynamic analysis (27) of Anc.3 at 40 °C and Anc.4 at 25 °C, both with 0 mM NaCl, using the glycine reporter. Cross peaks indicate the presence of structural interconversion. Conditions for each ancestor were chosen to maximize the presence of both folds in order to enhance detection of cross peaks. (D) Simplified phylogenetic tree indicating the back-and-forth evolutionary trajectory of XCL1 metamorphosis. Pie charts represent fractional occupancy of the chemokine fold (red) and the alternate fold (gold).
Because extant human XCL1 occupies its two folds in equal proportion, we sought to determine if Anc.3 and Anc.4 do also. HSQC peak intensities can quantify the fractional population of the different folds of XCL1, which is determined by the concentration-independent equilibrium constant for fold-switching. HSQC experiments show that under near-physiologic conditions (37°C, 150 mM NaCl) at identical protein concentrations, 92±3.7% of the Anc.3 population occupies the chemokine fold (Fig. 2A–B, D, Table S2). However, only 9.3±2.0% of the Anc.4 population occupies the chemokine fold (Fig. 2A–B, D, Table S2). To confirm the robustness of these results to statistical uncertainty in ancestrally reconstructed sequences, we expressed and purified “Alt.” ancestral proteins, where we replaced every residue with the next most likely residue if the next most likely residue was predicted with >20% probability, creating a “worst case” alternate at all positions ancestor, as is the standard in the field (26) (Fig. S8). Consistent with our results for the maximum likelihood ancestors, Alt.Anc.0 and Alt.Anc.2 are monomorphic, whereas Alt.Anc.3 and Alt.Anc.4 are metamorphic (Fig. S8), and Alt.Anc.4 populates the chemokine fold 11±1.4%. This suggests that XCL1 likely evolved from a single-fold ancestor (Anc.2), to a metamorphic ancestor that prefers the chemokine fold (Anc.3), to a metamorphic ancestor that prefers the alternate fold (Anc.4), to an extant metamorph that equally populates two different folds (Fig. 2B, D).
The aligned sequences of Anc.2 and Anc.3 differ in 26 positions distributed throughout sequence and structure (Fig. S1C). To uncover how these sequence changes introduced metamorphosis, we examined these positions based on three criteria: sequence comparison of positions at the alternate structure’s dimer interface, sequence comparison of positions likely to impact overall structural flexibility, and analysis of residue-residue contacts in the chemokine structure (Fig.3A). Another sequence-based criterion that has been used to predict protein metamorphosis is inaccurate secondary structure prediction (5, 28); however, each ancestral sequence, and WT XCL1, are predicted to adopt the secondary structure profile of the chemokine fold (Fig. S9).
Fig. 3. Metamorphic folding is enabled by the combined presence of changes in dimer interface, structural flexibility, and residue contact networks.
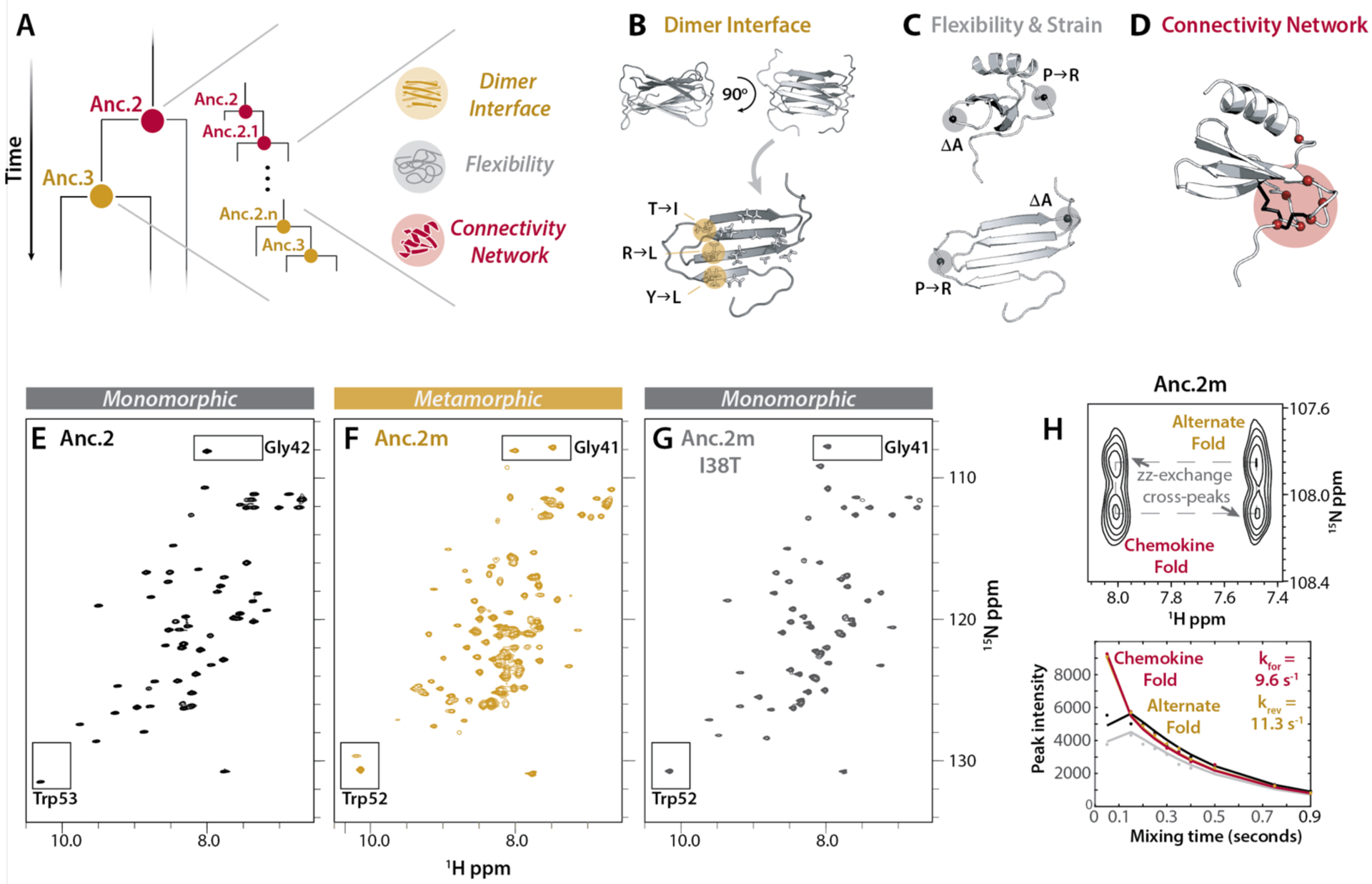
(A) Schematic of the hypothesis that metamorphosis evolved over time via a subset of key sequence changes from Anc.2 towards Anc.3. (B) Dimer interface positions that are polar in Anc.2 and hydrophobic in Anc.3. (C) Positions likely to be important for structural flexibility, and creation of strain in the chemokine fold. (D) Positions selected for replacement in Anc.2 using contact network analysis (Cα, red spheres). Anc.0 disulfides shown in black. (E-G) HSQC spectra for Anc.2 (E), Anc.2m (F) and Anc.2m I38T (G) (50°C, 20 mM NaPO4 (pH 6.0)). Boxes: conformational reporter residues. (H) ZZ-exchange analysis for Anc.2m (50°C, 20 mM NaPO4 (pH 6.0)) using reporter residue Gly41. Top: cross peaks indicate interconversion. Bottom: curves fit to peak intensities vs. time to calculate kinetic parameters. kforward (kfor), interconversion from the alternate fold towards the chemokine fold; kreverse (krev), interconversion in the reverse direction.
Several metamorphic proteins form protein-protein interfaces likely to stabilize one of their two structures, including RfaH (29), Selecase (30), and XCL1 (23). For XCL1, the alternate structure forms a stable dimer interface that is likely important for metamorphic folding, so we performed sequence comparison of Anc.2 and Anc.3 at XCL1 dimer interface positions. We found that three interface positions switch from polar or charged in Anc.2 to aliphatic side chains in Anc.3 (Y11I, T38I, and R43L) (Fig. 3B), making contacts with the rest of the apolar dimer interface more favorable. Sequence comparison also identified two changes between Anc.2 and Anc.3 that likely increase structural flexibility (P15R) or create strain (a deletion in the β1–β 2 loop at position 29) in the chemokine fold (Fig. 3C).
Networks of noncovalent residue-residue contacts are key indicators of protein conformation and stability (31, 32) that mediate essential biological phenomena (33–35). Because metamorphosis likely relies on a nuanced balance of conformational stability across multiple protein folds, we suspected that changes in noncovalent contact networks might contribute to the evolution of protein metamorphosis. We compared contact networks in human XCL1 versus Anc.0 and identified seven positions that make > 3 contacts in Anc.0 and < 3 contacts in XCL1, and whose identities differ between Anc.2 and Anc.3. We focused on these sequence substitutions because they decreased connectivity in Anc.0 (potentially permissive for interconversion) but minimized changes in connectivity in XCL1 (preserving the chemokine fold) (31) (Fig. 3D, S10), suggesting that mutations that permit rewiring of these contacts could enable metamorphic folding. These positions largely cluster around Anc.0’s second disulfide bond (Fig. 3D, S10C), “gluing” its structure in place. After the disulfide was lost, this remaining structural “glue” “dissolved” as metamorphosis evolved. Position 27, which points into the “glue” region, is a glutamine in Anc.0, Anc.2, and Anc.3. But in Anc.4, it is a bulky lysine, which is likely to sterically destabilize the chemokine fold, perhaps partly accounting for the fact that Anc.4 prefers the alternate fold. In human XCL1, this position is a threonine, perhaps facilitating XCL1’s shift to a 50/50 structural equilibrium.
To identify sequence changes sufficient to introduce metamorphic folding, we mutated positions in Anc.2 (monomorphic) to match Anc.3 (metamorphic) based on the findings described above (Table S3). We evaluated the resulting variants for fold-switching via HSQC experiments. We found that combining the three dimer interface, two flexibility-altering, and seven connectivity-altering substitutions (11/26 possible sequence changes; Anc.2m; Fig. 3A–D, Table S3) conferred the ability to populate the alternate fold (Fig. S11, Fig. 3E–H). Anc.2 variants incorporating all three dimer interface substitutions alone, or the two flexibility-altering changes alone, or the seven connectivity-altering changes alone are not metamorphic and only occupy the chemokine fold (Table S3), suggesting that these three sets of changes had to occur in combination to introduce metamorphic folding. Reverting the least disruptive mutation at the dimer interface (I38T) in Anc.2m disables metamorphosis, causing Anc.2m I38T to adopt exclusively the chemokine fold, indicating that the T38I mutation is necessary but insufficient for metamorphic folding (Fig. 3G, Table S3). This highlights how mutations that stabilize an alternate fold through formation of a protein-protein interface can critically impact metamorphic folding, as is seen in other metamorphic proteins (29). Together, these findings show that concurrent changes in interaction interfaces, structural flexibility, and noncovalent residue-residue contact networks act together to drive the evolution of protein metamorphosis.
Lattman and Rose proposed that a protein’s specific folded structure can be encoded either by amino acids distributed throughout its sequence (distributed control), or discrete sites (centralized control) (36). While both distributed (37) and centralized (38) control of folding have been described, the requirement for numerous, structurally distant mutations of different kinds to enable metamorphosis suggests that distributed control governs XCL1’s structure.
Despite growing interest in metamorphic proteins (2–5), the mechanism by which one sequence encodes two structures remains unclear. Here, ancestral reconstruction illuminates the molecular evolution of protein metamorphosis in XCL1. Metamorphic proteins have been hypothesized to represent evolutionary bridges, or “snapshots” of proteins “caught in the act” of evolving a new fold (8, 39, 40). Were this the case for XCL1, its ancestors would likely have progressed from populating only the chemokine fold to adopting exclusively the alternate fold. Rather, our data suggest that human XCL1 likely evolved from a metamorphic ancestor that may preferentially adopt the chemokine fold to one that may preferentially adopt the alternate fold, before evolving to occupy both structures in approximately equal proportion. This suggests that XCL1 is not evolving from one fold to a new one, but is evolving to remain metamorphic, indicating that metamorphosis may be a molecular phenotype that was selected for in XCL1 rather than a transient feature of an evolutionary intermediate.
Why would metamorphosis be favored? All chemokines activate GPCRs and bind GAGs, and some chemokines, including XCL1, are directly antimicrobial (12, 41–44). Non-XCL1 chemokines carry out these three functions using one fold, whereas XCL1’s chemokine fold activates its cognate GPCR and its alternate fold binds GAGs and is directly antimicrobial (10, 12, 41). As such, metamorphic folding could confer dynamic control over the fractional population, and therefore the activity, of each structure (Fig. 4A), avoiding the need to transcribe and translate multiple genes and degrade or inhibit multiple proteins in order to turn multiple functions on and off, thus allowing for fast switching between functional states mediated by distinct folds. It could also enhance a specific function (e.g. GAG binding), or permit acquisition of a new function (e.g. antimicrobial activity). Specifically, metamorphic folding may enable XCL1 to kill pathogens directly at the site of infection (alternate fold) and stimulate antigen cross-presentation to effector cells (chemokine fold), coordinating humoral and cell-mediated immune responses and allowing for spatiotemporal regulation of multiple functions (Fig. 4A). If fold-switching thus enhances the ability of other metamorphic proteins to carry out their biologic roles, then metamorphic proteins may be more common than previously thought. Why metamorphic folding evolved in XCL1 and not other chemokines, however, remains mysterious.
Fig. 4. Functional advantages and design insights provided by protein metamorphosis.
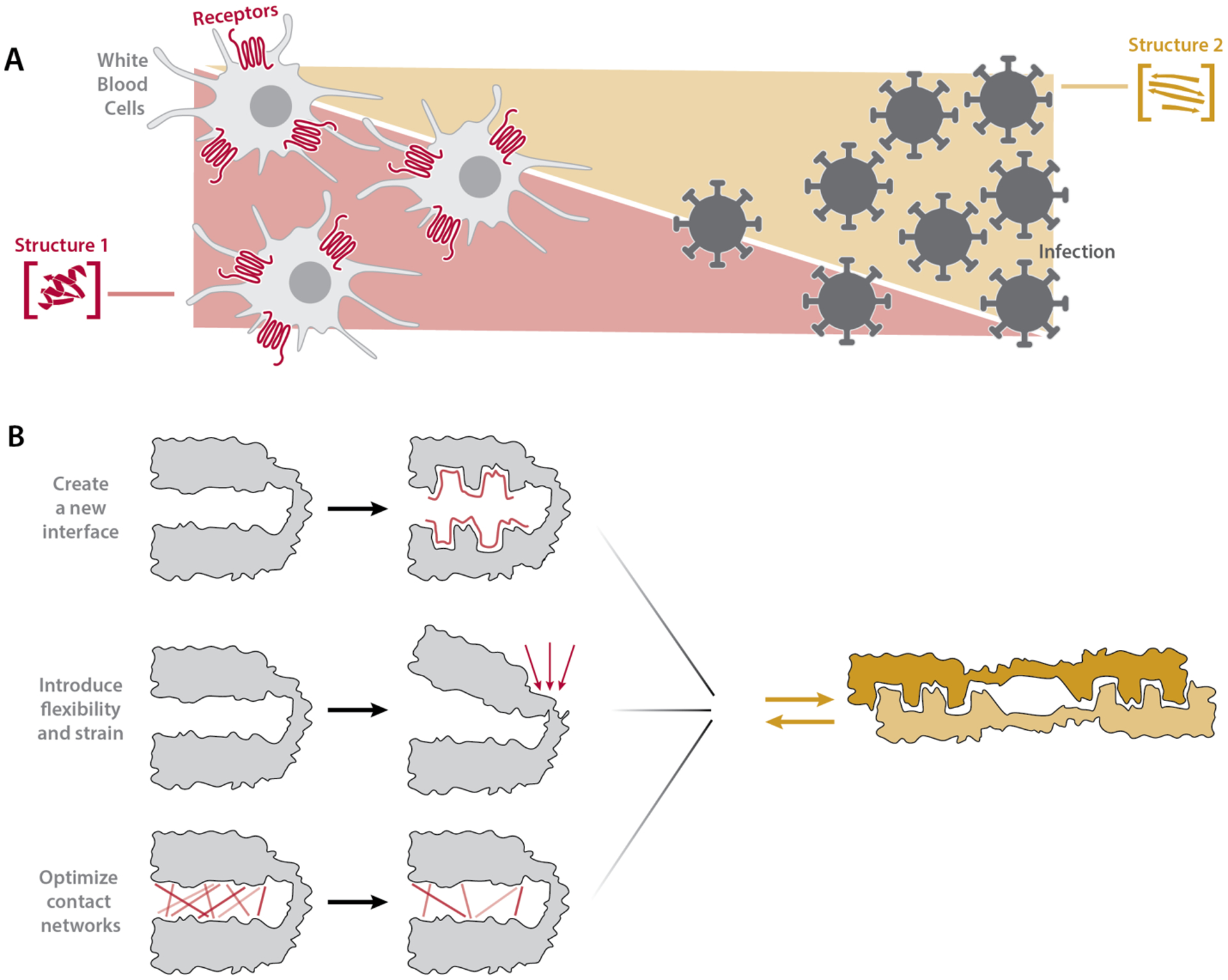
(A) Metamorphosis enables spatiotemporal control of protein function through modulation of the relative population of two different folded states in a concentration-dependent manner. For example, XCL1 can populate the antimicrobial fold close to a site of infection and the GPCR binding fold further away. (B) The emergence in nature or de novo design of fold-switching proteins may require (top) creation of a protein-protein interface that stabilizes the alternate fold, (middle) incorporation of molecular flexibility and strain, and (bottom) optimization of residue-residue contact networks to avoid thermodynamic or kinetic trapping in a single fold.
Analysis of proteins like XCL1 that reversibly switch between two well-defined folds can inform the de novo design of fold-switching proteins, which remains a challenge. Our work suggests that designed fold-switching proteins may need to incorporate protein-protein interfaces that stabilize one fold, structural constraints and strain to enable interconversion, and residue-residue contact networks that allow stable formation of both folds without trapping the protein in either fold (Fig. 4B). LOCKR, a designed protein switch system currently being developed for therapeutic use, has already been constructed by stabilizing a protein-protein interface, demonstrating the utility of this approach (45, 46). Moreover, the broad span of XCL1 sequence space compatible with metamorphic interconversion implies that design of fold-switching proteins could be within reach, especially with the help of principles uncovered in this study.
Supplementary Material
Acknowledgments:
We thank Cole Peterson for his helpful suggestions regarding the manuscript figures and text.
Funding: This work was supported in part by a grant from the State of Wisconsin Tax Check-Off Program for Cancer Research and the Medical College of Wisconsin Cancer Center, and NIH grants R56 AI103225, R37 AI058072, and S10 OD020000 (to B.F.V.), F30 CA236182 (to A.F.D), and F30 CA196040 to A.B.K.. A.F.D. and A.B.K are members of the NIH supported (T32 GM080202) Medical Scientist Training Program at the Medical College of Wisconsin. M.M.B. acknowledges the MRC (MC_U105185859) and ALSAC for funding.
Footnotes
Supplementary Materials:
Materials and Methods
Supplementary Text
Figures S1–S11
Table S1–S3
Competing interests: FCP and BFV have ownership interests in Protein Foundry, LLC.
Data and materials availability: The NMR structure of Anc.0 is available in the PDB (PDB ID 7JH1) and the BMRB (BMRB code 30777). Materials may be requested from the corresponding author.
References and Notes
- 1.Murzin AG, Biochemistry. Metamorphic proteins. Science 320, 1725–1726 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Dishman AF, Volkman BF, Unfolding the Mysteries of Protein Metamorphosis. ACS Chem Biol 13, 1438–1446 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lella M, Mahalakshmi R, Metamorphic Proteins: Emergence of Dual Protein Folds from One Primary Sequence. Biochemistry 56, 2971–2984 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Zamora-Carreras H, Maestro B, Sanz JM, Jimenez MA, Turncoat Polypeptides: We Adapt to Our Environment. Chembiochem 21, 432–441 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Porter LL, Looger LL, Extant fold-switching proteins are widespread. Proc Natl Acad Sci U S A 115, 5968–5973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peisajovich SG, Rockah L, Tawfik DS, Evolution of new protein topologies through multistep gene rearrangements. Nat Genet 38, 168–174 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Tokuriki N, Tawfik DS, Stability effects of mutations and protein evolvability. Curr Opin Struct Biol 19, 596–604 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Yadid I, Kirshenbaum N, Sharon M, Dym O, Tawfik DS, Metamorphic proteins mediate evolutionary transitions of structure. Proc Natl Acad Sci U S A 107, 7287–7292 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tokuriki N, Tawfik DS, Protein dynamism and evolvability. Science 324, 203–207 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Tuinstra RL et al. , Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci U S A 105, 5057–5062 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuloglu ES et al. , Monomeric solution structure of the prototypical ‘C’ chemokine lymphotactin. Biochemistry 40, 12486–12496 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nevins AM et al. , A Requirement for Metamorphic Interconversion in the Antimicrobial Activity of Chemokine XCL1. Biochemistry 55, 3784–3793 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Kumar S, Nei M, A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141, 1641–1650 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang BS, Kazmi MA, Sakmar TP, Synthetic gene technology: applications to ancestral gene reconstruction and structure-function studies of receptors. Methods Enzymol 343, 274–294 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Thornton JW, Need E, Crews D, Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science 301, 1714–1717 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Finnigan GC, Hanson-Smith V, Stevens TH, Thornton JW, Evolution of increased complexity in a molecular machine. Nature 481, 360–364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang BS, Jonsson K, Kazmi MA, Donoghue MJ, Sakmar TP, Recreating a functional ancestral archosaur visual pigment. Mol Biol Evol 19, 1483–1489 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Anderson DP et al. , Evolution of an ancient protein function involved in organized multicellularity in animals. Elife 5, e10147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitney DS, Volkman BF, Prehoda KE, Evolution of a Protein Interaction Domain Family by Tuning Conformational Flexibility. J Am Chem Soc 138, 15150–15156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pei J, Kim BH, Grishin NV, PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36, 2295–2300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guindon S, Delsuc F, Dufayard JF, Gascuel O, Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537, 113–137 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Guindon S, Gascuel O, A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Kuloglu ES, McCaslin DR, Markley JL, Volkman BF, Structural rearrangement of human lymphotactin, a C chemokine, under physiological solution conditions. J Biol Chem 277, 17863–17870 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tuinstra RL, Peterson FC, Elgin ES, Pelzek AJ, Volkman BF, An engineered second disulfide bond restricts lymphotactin/XCL1 to a chemokine-like conformation with XCR1 agonist activity. Biochemistry 46, 2564–2573 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyler RC, Wieting JC, Peterson FC, Volkman BF, Electrostatic optimization of the conformational energy landscape in a metamorphic protein. Biochemistry 51, 9067–9075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eick GN, Bridgham JT, Anderson DP, Harms MJ, Thornton JW, Robustness of Reconstructed Ancestral Protein Functions to Statistical Uncertainty. Mol Biol Evol 34, 247–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrow NA, Zhang O, Forman-Kay JD, Kay LE, A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J Biomol NMR 4, 727–734 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Mishra S, Looger LL, Porter LL, Inaccurate secondary structure predictions often indicate protein fold switching. Protein Sci 28, 1487–1493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burmann BM et al. , An alpha helix to beta barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell 150, 291–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.N. C-C Lopez-Pelegrin Mar, Cintas-Pedrola Anna, Herranz-Trillo Fatima, Bernado Pau, Peinado Juan R., Arolas Joan L., and Gomis-Ruth F. Xavier, Multiple Stable Conformations Account for Reversible Concentration-Dependent Oligomerization and Autoinhibition of a Metamorphic Metallopeptidase. Angewandte Chemie 126, 10800–10806 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Kayikci M et al. , Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat Struct Mol Biol 25, 185–194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greene LH, Protein structure networks. Brief Funct Genomics 11, 469–478 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Sente A et al. , Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat Struct Mol Biol 25, 538–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flock T et al. , Selectivity determinants of GPCR-G-protein binding. Nature 545, 317–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flock T et al. , Universal allosteric mechanism for Galpha activation by GPCRs. Nature 524, 173–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lattman EE, Rose GD, Protein folding--what’s the question? Proc Natl Acad Sci U S A 90, 439–441 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eaton KV et al. , Studying protein fold evolution with hybrids of differently folded homologs. Protein Eng Des Sel 28, 241–250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stewart KL, Dodds ED, Wysocki VH, Cordes MH, A polymetamorphic protein. Protein Sci 22, 641–649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumirov VK et al. , Multistep mutational transformation of a protein fold through structural intermediates. Protein Sci 27, 1767–1779 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.B. RE Cordes Matthew H.J., Walsh Nathan P., McKnight C. James and Sauer Robert T., An evolutionary bridge to a new protein fold. Nature Structural Biology 7, 1129–1132 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Dishman AF et al. , Switchable Membrane Remodeling and Antifungal Defense by Metamorphic Chemokine XCL1. ACS Infect Dis, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen LT, Vogel HJ, Structural perspectives on antimicrobial chemokines. Front Immunol 3, 384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hieshima K et al. , CCL28 has dual roles in mucosal immunity as a chemokine with broad-spectrum antimicrobial activity. J Immunol 170, 1452–1461 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Yang D et al. , Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol 74, 448–455 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Langan RA et al. , De novo design of bioactive protein switches. Nature 572, 205–210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng AH et al. , Modular and tunable biological feedback control using a de novo protein switch. Nature 572, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Peterson FC et al. , Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J Biol Chem 279, 12598–12604 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Fox JC et al. , Structural and agonist properties of XCL2, the other member of the C-chemokine subfamily. Cytokine 71, 302–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cornilescu G, Delaglio F, Bax A, Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13, 289–302 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Guntert P, Automated NMR structure calculation with CYANA. Methods Mol Biol 278, 353–378 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Linge JP, Habeck M, Rieping W, Nilges M, ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics 19, 315–316 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM, The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160, 65–73 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Delaglio F et al. , NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 54.Tollinger M, Skrynnikov NR, Mulder FA, Forman-Kay JD, Kay LE, Slow dynamics in folded and unfolded states of an SH3 domain. J Am Chem Soc 123, 11341–11352 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Kleist AB et al. , Solution NMR spectroscopy of GPCRs: Residue-specific labeling strategies with a focus on (13)C-methyl methionine labeling of the atypical chemokine receptor ACKR3. Methods Cell Biol 149, 259–288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsai J, Taylor R, Chothia C, Gerstein M, The packing density in proteins: standard radii and volumes. J Mol Biol 290, 253–266 (1999). [DOI] [PubMed] [Google Scholar]
- 57.L. LL Kim Allen K., Porter Lauren L., A method for predicting evolved fold switchers exclusively from their sequences. bioRxiv, (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.