

Abstract
Pulmonary fibrosis is a progressive lung disease characterized by myofibroblast accumulation and excessive extracellular matrix deposition. We sought to investigate the role of FKBP13 (13-kD FK506-binding protein), an endoplasmic reticulum–resident molecular chaperone, in various forms of pulmonary fibrosis. We first characterized the gene and protein expression of FKBP13 in lung biopsy specimens from 24 patients with idiopathic pulmonary fibrosis and 17 control subjects. FKBP13 expression was found to be elevated in the fibrotic regions of idiopathic pulmonary fibrosis lung tissues and correlated with declining forced vital capacity and dyspnea severity. FKBP13 expression was also increased in lung biopsy specimens of patients with hypersensitivity pneumonitis, rheumatoid arthritis, and sarcoidosis-associated interstitial lung disease. We next evaluated the role of this protein using FKBP13−/− mice in a bleomycin model of pulmonary fibrosis. Animals were assessed for lung function and histopathology at different stages of lung injury including the inflammatory (Day 7), fibrotic (Day 21), and resolution (Day 50) phases. FKBP13−/− mice showed increased infiltration of inflammatory cells and cytokines at Day 7, increased lung elastance and fibrosis at Day 21, and impaired resolution of fibrosis at Day 50. These changes were associated with an increased number of cells that stained positive for TUNEL and cleaved caspase 3 in the FKBP13−/− lungs, indicating a heightened cellular sensitivity to bleomycin. Our findings suggest that FKBP13 is a potential biomarker for severity of interstitial lung diseases and that it has a biologically relevant role in protecting mice against bleomycin-induced injury, inflammation, and fibrosis.
Keywords: interstitial lung disease, inflammation, endoplasmic reticulum stress, unfolded protein response, FKBP13
Clinical Relevance
The endoplasmic reticulum chaperone FKBP13 (13-kD FK506-binding protein) was previously shown to be associated with progressive fibrotic lung disease and hypothesized to be a potential therapeutic target to prevent progressive fibrotic lung disease. We demonstrate here that FKBP13 expression in lung tissue is positively correlated with the clinical severity of fibrotic lung disease but that it appears to play a protective biological role in the disease process.
Pulmonary fibrosis results in the progressive scarring and stiffening of the lungs, eventually leading to respiratory failure (1). Most cases are diagnosed as idiopathic pulmonary fibrosis (IPF), but secondary causes such as genetic susceptibility, environmental pollutants, medications, and radiation exposure have been identified. Although a complete understanding of the cellular mechanisms involved in the pathogenesis of fibrotic lung disease remains elusive, accumulating evidence demonstrates that injury to the lung epithelium stimulates the accumulation of myofibroblasts, which are responsible for the excessive deposition of extracellular-matrix components into the interstitium, causing decreased lung compliance and impaired gas exchange (1).
The endoplasmic reticulum (ER) is the site of protein biosynthesis and folding. Disruption of homeostasis in this organelle causes ER stress and activation of the unfolded protein response (UPR), which have been shown to contribute to the development of various fibrotic disorders, including IPF (2, 3). Activation of the UPR pathway in mammalian cells is believed to be triggered by the dissociation of GRP78 (glucose-regulated protein 78) from three ER transmembrane proteins: IRE1 (inositol-requiring enzyme 1), ATF6 (activating transcription factor 6), and PERK (protein kinase RNA–like ER kinase). Activation of these pathways results in the upregulation of transcription factors and chaperones that increase the protein-folding capacity of the ER and promote the degradation of misfolded or unfolded proteins (4). Failure to restore ER homeostasis can lead to the initiation of apoptotic pathways through the transcription factor CHOP (C/EBP-homologous protein).
A distinct family of immunophilins termed the FKBPs (FK506-binding proteins) has been noted to play an important role in both connective-tissue remodeling and the ER stress response (5, 6). The mechanism by which FKBPs facilitate remodeling is believed to occur through their PPIase (peptidyl–prolyl cis/trans–isomerase) activity, which alters the peptidyl–prolyl bonds in proteins. FKBP65 is one such member of the family that is upregulated in lung tissue of patients with IPF and that is necessary for collagen production by lung fibroblasts (5). FKBP65 associates with GRP78 and the collagen-specific chaperone HSP47 during collagen assembly, and its deficiency in mouse embryos results in altered collagen trafficking and aggregation of procollagen within the ER (7–9). Here, we characterize the role of another 1 of the 15 members of FKBP family, FKBP13 (13-kD FKBP), in patients with IPF and in an experimental mouse model of bleomycin-induced lung fibrosis.
FKBP13, which is encoded by the FKBP2 gene, is localized to the lumen of the ER and is induced during ER stress (10–12). It also shares 50% homology with other UPR chaperones, GRP78 and GRP94, suggesting that FKBP13 itself also functions as a chaperone (11). Boon and colleagues (2009) (13) observed that FKBP13 gene expression was upregulated fivefold in the lung tissue of patients with progressive IPF compared with those with stable disease. Similar observations have been made in a cohort of patients with progressive hypersensitivity pneumonitis (14). Unlike FKBP65, FKBP13 has lower activity toward post-translationally modified procollagen and does not appear to play a role in collagen folding (15). Recently, it was shown that FKBP13 protects plasma cells from ER stress–induced apoptosis by promoting the degradation of misfolded immunoglobulins (16). We postulated that the upregulation of FKBP13 in fibrotic lung tissue acts to protect cells against ER stress. However, as shown in our previous studies, its overall effect on the development of fibrosis depends on the specific cell types that are affected (17).
To further investigate the roles of FKBP13 in the pathogenesis of pulmonary fibrosis, we began by constructing a tissue microarray (TMA) comprising lung biopsy specimens from patients with IPF and control subjects. This was used to investigate the expression pattern of FKBP13 at the protein and mRNA levels and to assess the association between FKBP13 expression and lung function in the patients. We then generated FKBP13-knockout (FKBP13−/−) mice and subjected them to a model of bleomycin-induced pulmonary fibrosis. In these experiments, we observed that FKBP13−/− mice are more susceptible to injury by bleomycin, resulting in more severe pulmonary fibrosis at lower doses of the drug. These data suggest that FKBP13 plays a protective role in the pathogenesis of pulmonary fibrosis.
Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/2019.12.15.858340) and an abstract (https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2014.189.1_MeetingAbstracts.A6336).
Methods
Human Lung Resected Tissue
All procedures using human tissues were approved by the Hamilton Integrated Research Ethics Board (11-3559 and 13-523-C). Formalin-fixed, paraffin-embedded (FFPE) human lung tissues were obtained from the biobank for lung diseases at St. Joseph’s Hospital in Hamilton, Ontario. IPF (n = 30), hypersensitivity pneumonitis (n = 5), rheumatoid arthritis (n = 6), and sarcoidosis (n = 6) cases were selected on the basis of clinical history, radiographic appearance, and a pattern of usual interstitial pneumonia or fibrotic granulomas as determined by trained molecular pathologists and radiologists. Noninvolved tissue samples from lung-cancer cases (n = 17) were used as controls. Upon confirmation of a positive diagnosis, patient slides were scanned using the Olympus VS120 Slide Scanner (Olympus Corp.), and 0.6-mm diameter fibrotic and nonfibrotic cores were selected to be placed into a TMA block. Fibrotic regions were identified on the basis of features of usual interstitial pneumonia (UIP), including spatial heterogeneity in the parenchyma, architectural distortion, and fibroblastic foci. In total, 316 cores were selected, with at least 3 cores for each specified area. Forced vital capacity (FVC) and modified Medical Research Council dyspnea scores were obtained for the corresponding cases. Pulmonary function testing was conducted within a median of 48 days (range, 1–191 d) from the date of the biopsy.
Histology
Tissue slides were stained with hematoxylin and eosin for cellular analysis and tissue architectural analysis, Picrosirius Red and Masson’s Trichrome for collagen analysis, anti-αSMA (α-smooth muscle actin) (M0851; Agilent Dako) for the identification of myofibroblasts, GRP78 (N-20; Santa Cruz Biotechnology, Inc.) and XBP1 to assess UPR activation, and anti-FKBP13 to visualize FKBP13 (MAB4356; R&D Systems, Inc.). TUNEL staining was performed using the TACS 2 TdT Fluorescein Kit (4812-30-K; Trevigen). Stained slides were scanned with the Olympus VS120 Slide Scanner (Olympus Corp.) and analyzed with HALO image-analysis software (Indica Labs). The slide scanner utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. A molecular pathologist (A.N.) was consulted for histopathological analysis of FKBP13-expressing cell types.
Gene Expression Analysis
RNA was extracted from FFPE tissues using the RNeasy FFPE kit (QIAGEN). RNA concentrations were measured using a NanoDrop spectrophotometer and version 3.3.0 software (NanoDrop Technologies LLC). The NanoString nCounter platform (NanoString Technologies, Inc.) was used to quantify the expression of FKBP13 in human FFPE samples as previously described (17). Statistical analysis was performed using the nSolver Analysis Software version 4.0 (NanoString Technologies, Inc.). Raw gene expression data were normalized to five housekeeping genes (HPRT1, POLR2A, PPIA, TBP, TUBA1A) that were selected by the built-in geNorm algorithm (18).
Single-Cell RNA Sequencing
Preprocessed data using the Cell Ranger pipeline (10× Genomics) were obtained from the Gene Expression Omnibus using accessing number GSE122960. Out of 16 available samples, all 8 samples from donors and 4 samples from patients with IPF were downloaded and used for further analysis. Post-processing was performed following the description provided by Reyfman and colleagues (2018) (19) using the Seurat (20) package in R (R Foundation for Statistical Computing). The t-distributed stochastic neighbor embedding plot, expression plot, and violin plots were created using the Seurat package. Cell populations were defined by using the genes reported to be differentially expressed between the cell populations (19).
Animal Experiments
All animal work was approved by the Animal Research Ethics Board of McMaster University under protocol number 12.02.06. Male C57BL6/J mice aged 10–12 weeks were bred and housed at the McMaster University Central Animal Facility. Animals were kept on a 12-hour light/dark cycle at a controlled temperature of 20–25°C and ambient humidity of ∼50%. The animals were allowed access to food and water and were fed ad libitum.
Generation of FKBP13-Deficient Mice by Gene Targeting
FKBP13-deficient animals were initially generated in 129SvEvBrd embryonic stem cells (Taconic Biosciences, Inc.), and chimeric mice were made on a C57BL/6J (The Jackson Laboratory) background to produce F2 homozygous mutants by Dr. Barbara Hendrickson (University of Chicago). Jackson Laboratory rederived these mice in the C57BL/6J strain, and they were further backcrossed for seven generations onto a C57BL6/J background. Mice heterozygous (FKBP13+/−) and homozygous (FKBP13−/−) for the FKBP13 mutation were characterized by Southern blot analyses of tail genomic DNA. Northern blot analyses of RNA from wild-type (WT; FKBP13+/+) brain, kidney, liver, and spleen revealed easily detectable 0.6-kb FKBP13 mRNA. However, no FKBP13 mRNA was detected in FKBP13−/− mouse–derived tissues, despite loading similar amounts of RNA for all samples, as judged by actin probe hybridization. Immunostaining for FKBP13 using an anti-FKBP13 antiserum (a gift from Dr. S. Burakoff, Dana-Faber Cancer Institute) confirmed a lack of FKBP13 protein in the FKBP13−/− lung tissue.
Bleomycin Administration and Collection of Samples
Experimental pulmonary fibrosis was induced by intratracheal intubation of bleomycin (National Drug Code [NDC] 61703-332-18; Hospira) in isoflurane-anesthetized mice (Manufacturing Technology Centre, Pharmaceuticals) at 0.04 or 0.06 U per mouse. Bleomycin was solubilized in 50 μl of sterile saline. Lung tissues and BAL fluid (BALF) were isolated and processed as described previously (17). BALF differential cell counts were then performed by counting 300 leukocytes and using hemocytological procedures to classify the cells as neutrophils, macrophages, eosinophils, or lymphocytes and multiplying the relative abundance of each cell type by the BALF total cell count. Mouse lung tissues were fixed and embedded in paraffin, and a TMA containing 135 cores that were each 1.5 mm in diameter was generated using the TMA Master platform (3DHISTECH Ltd.). Three cores were obtained from each mouse lung, providing complete coverage of the lung area. Histological staining was performed on this TMA as described above. Semiquantitative histological analysis of pulmonary fibrosis was performed through Ashcroft scoring as described previously (17).
Immunofluorescence
Immunofluorescence staining of FKBP13 (MAB4356; R&D Systems, Inc.) and E-cadherin (ab1416; Abcam) was performed. After deparaffinization and saturation of nonspecific sites with BSA (5%, 30 min), cells were incubated with primary antibodies overnight in a humidified chamber at 4°C. Conjugated secondary antibodies were used at a dilution of 1:2,000. Slides were mounted in ProLong Gold with DAPI (ProLong Gold antifade regent with DAPI, P36931; Life Technologies). Images were taken using an epifluorescence microscope (Olympus IX81; Olympus Corp.) and analyzed with MetaMorph image-analysis software (Molecular Devices, LLC).
Assessment of Pulmonary Mechanics
Lung-function measurements, including pressure–volume loops and quasistatic elastance were performed as described previously (17).
Statistical Analysis
Results were expressed as the mean ± SEM for hypothesis testing or as the mean ± SD for descriptive statistics. Two groups were compared with a two-tailed unpaired Student’s t test. When more than two groups were compared, a one-way ANOVA followed by a Newman-Keuls multiple-comparison test was used. Pearson’s r was used to calculate the linear correlation coefficients. Statistical tests were employed using GraphPad Prism 7 (GraphPad Software, Inc.). A P value of <0.05 was considered to indicate statistical significance.
Results
FKBP13 Expression Is Increased in the Fibrotic Regions of Patients with IPF and Is Correlated with Clinical Severity
FKBP13 was previously shown to be upregulated at the mRNA level in patients with progressive IPF and other interstitial lung diseases (13). Here, we aimed to characterize the expression of FKBP13 by immunohistochemistry (IHC) in the lungs of patients with IPF. A TMA was designed containing fibrotic and nonfibrotic cores taken from lung biopsy specimens of 24 patients with IPF and 17 control subjects. Demographic information and clinical characteristics of these patients are presented in Table 1. The amount of FKBP13 was higher in the fibrotic regions of IPF tissues than in nonfibrotic regions and healthy control tissues (Figures 1A and 1B). Total RNA isolated from FFPE tissues was assessed for FKBP13 using NanoString gene expression analysis. Consistent with the IHC findings, the amount of FKBP13 was elevated in the fibrotic tissues (Figure 1C). To determine the specific cellular subpopulations that express FKBP13, the open-access single-cell RNA-sequencing data set by Reyfman and colleagues (2018) (19) was analyzed. Cells were clustered using t-distributed stochastic neighbor embedding, and clusters were assigned to cell populations on the basis of the genes reported as markers (see Figures E1A and E1B in the data supplement). With the pooled data from lung biopsy specimens of four patients with IPF and eight control subjects, FKBP13 was found to be ubiquitously expressed by all cellular subpopulations in the lung (Figure E1C) (19). FKBP13 was noted to be significantly upregulated in the IPF plasma-cell population.
Table 1.
Baseline and Functional Characteristics of the IPF Study Population
| Characteristic | IPF (N = 24) |
|---|---|
| Age, yr | 59.0 ± 10.1 |
| Male sex, n (%) | 17 (70.8) |
| Smoker (former and active), n (%) | 16 (66.6) |
| Time since diagnosis of IPF, yr | 2.64 ± 2.82 |
| FVC, percentage of predicted value | 62.9 ± 23.2 |
| FEV1, percentage of predicted value | 64.5 ± 20.3 |
| DlCO, percentage of predicted value | 42.8 ± 10.9 |
| mMRC dyspnea score* | 1.9 ± 1.2 |
| Use of supplemental oxygen, n (%) | 1 (4.2) |
Definition of abbreviations: FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; IPF = idiopathic pulmonary fibrosis; mMRC = modified Medical Research Council.
Data are presented as the mean ± SD, unless stated otherwise. Demographic information for the lung-cancer control group was unavailable. Lung biopsy specimens from the cohort described in this table form the tissue microarray assembled for Figure 1.
Dyspnea was evaluated using the mMRC scale. Scores range from 0 to 4, with higher scores indicating worse dyspnea.
Figure 1.
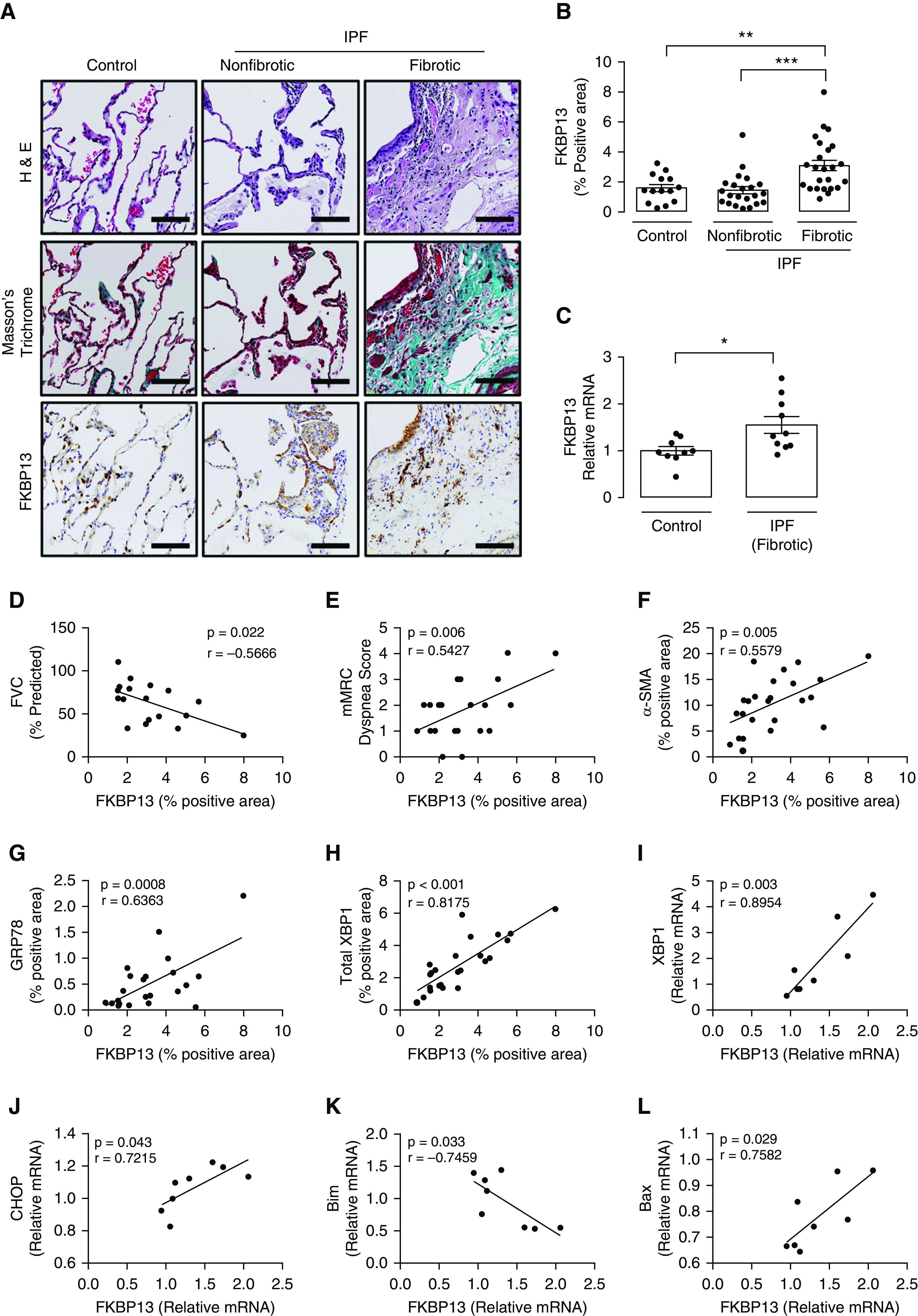
FKBP13 (13-kD FK506-binding protein) expression is elevated in fibrotic idiopathic pulmonary fibrosis (IPF) tissue and is correlated with disease severity, ER stress, and apoptosis markers. A tissue microarray containing fibrotic and nonfibrotic cores from 24 patients with IPF and 17 control subjects was stained for FKBP13 and other markers by using immunohistochemistry (IHC) and was quantified by using HALO image-analysis software. The NanoString nCounter platform was used to assess the gene expression of unfolded protein response (UPR) and apoptosis markers in the extracted cores. (A and B) Representative FKBP13 immunostaining images and quantification by HALO. Corresponding Masson’s Trichrome and H&E staining from serial sections are shown. Images were acquired using the automated Olympus VS120 slide scanner, which utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. (C) Comparison of FKBP13 mRNA expression in fibrotic IPF and control tissue cores by NanoString gene expression analysis. (D–H) Correlation of FKBP13-positive immunostained area with percent-predicted FVC; mMRC dyspnea score; and α-SMA (α-smooth muscle actin)-, GRP78 (glucose-regulated protein 78)-, and total XBP1–positive area. (I–L) Correlation of FKBP13 mRNA expression with XBP1, CHOP (C/EBP-homologous protein), Bim, and Bax in the fibrotic regions of human IPF lung tissues. Each point represents the average protein or mRNA expression value for an individual patient. Data is presented as mean ± SEM. Scale bars, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control. ER = endoplasmic reticulum; FVC = forced vital capacity; H&E = hematoxylin and eosin; mMRC = modified Medical Research Council.
Within the cohort of patients with IPF, the expression of FKBP13 in the fibrotic cores was correlated with clinical parameters of disease severity. Specifically, we found a negative correlation with FVC and a positive correlation with patient-reported dyspnea scores (Figures 1D and 1E). We further assessed the correlation of FKBP13 with other molecular markers of fibrosis, ER stress, and apoptosis that have been linked to the development of IPF (21). At the protein level, FKBP13 was positively correlated with the myofibroblast marker αSMA and the UPR markers GRP78 and total XBP1 (Figures 1G–1I). This was corroborated at the mRNA level, with FKBP13 showing positive correlations with GRP78, total XBP1, CHOP, and Bax and a negative correlation with Bim (Figures 1K–1M). As an ER stress–inducible gene, the upregulation of FKBP13 in fibrotic IPF tissues may therefore be a result of physiological UPR activation to increase protein-folding capacity.
We hypothesized that FKBP13 would also be upregulated in other forms of fibrotic lung disease. A second TMA was constructed to assess the expression of FKBP13 in hypersensitivity pneumonitis, rheumatoid arthritis, and sarcoidosis-associated interstitial lung disease, in addition to an independent cohort of patients with IPF. Baseline demographic and clinical data are shown in Table 2. Histopathological analysis of the FKBP13 staining by a molecular pathologist showed that FKBP13 appears primarily localized in the bronchial and alveolar epithelium and in alveolar macrophages (Figure 2A). FKBP13 expression was found to be significantly elevated in the fibrotic regions of these tissues compared with control tissues (Figure 2B). These findings provide further support to the potential protective role of FKBP13 in interstitial lung diseases.
Table 2.
Baseline and Functional Characteristics of Mixed Interstitial Lung Disease Study Population
| Characteristic | Idiopathic Pulmonary Fibrosis (N = 6) | Hypersensitivity Pneumonitis (N = 5) | Rheumatoid Arthritis (N = 6) | Sarcoidosis (N = 6) |
|---|---|---|---|---|
| Age, yr | 59.2 ± 8.4 | 55.8 ± 11.9 | 63.3 ± 6.2 | 54.2 ± 14.7 |
| Male sex, n (%) | 2 (33.3) | 1 (20.0) | 5 (83.3) | 2 (33.3) |
| Smoker (former and active), n (%) | 2 (33.3) | 3 (60.0) | 5 (83.3) | 3 (50.0) |
| Time since diagnosis of IPF, yr | 1.0 ± 0.8 | 1.7 ± 1.0 | 4.8 ± 2.9 | 0.6 ± 0.4 |
| FVC% predicted | 57.2 ± 17.0 | 66.8 ± 17.8 | 61.5 ± 6.1 | 65.3 ± 17.2 |
| DlCO% predicted | 64.0 ± 17.3 | 43.0 ± 13.0 | 32.2 ± 5.9 | 52.5 ± 11.0 |
| mMRC dyspnea score* | 1.8 ± 1.3 | 2.0 ± 1.0 | 2.6 ± 0.5 | 1.5 ± 1.0 |
Data are presented as the mean ± SD, unless stated otherwise. Demographic information for the lung-cancer control group was unavailable. Lung biopsy specimens from the cohort described in this table form the tissue microarray assembled for Figure 2.
Dyspnea was evaluated using the mMRC scale. Scores range from 0 to 4, with higher scores indicating worse dyspnea.
Figure 2.

FKBP13 expression is elevated in other forms of interstitial lung disease (ILD). A second tissue microarray was constructed using lung biopsy specimens from patients with ILD secondary to HP, RA, and sarcoidosis. A smaller, independent cohort of patients with IPF was also included. (A) Representative H&E and FKBP13 immunostaining. Histological hallmarks of each disease, as determined by a molecular pathologist, are shown (row 2). Specific FKBP13-expressing cell types, including macrophages (row 4), AEC (row 5), and BEC (row 6), were identified by the pathologist on the basis of morphology. Images were acquired using the automated Olympus VS120 slide scanner, which utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. (B) Quantification of FKBP13 staining by HALO. No bronchial epithelial cells were observed in control lung. Each open circle represents the average protein or mRNA expression value for an individual patient. The open squares denote the regions in the low-magnification images that are zoomed in on the subsequent rows. The coordinates of the corresponding high-magnification image are shown next to each square. Data is presented as mean ± SEM. Scale bars, 100 μm *P < 0.05 and **P < 0.01 versus control. AEC = alveolar epithelial cells; BEC = bronchial epithelial cells; HP = hypersensitivity pneumonitis; RA = rheumatoid arthritis.
FKBP13 Expression Is Increased in the Lung Tissue of Bleomycin-treated Mice
Because of the upregulation of FKBP13 in various fibrotic lung diseases and its association with declining lung function and ER stress, we further explored the role of this protein in the development of pulmonary fibrosis by generating FKBP13-knockout (FKBP13−/−) mice. We induced pulmonary fibrosis using a single intratracheal 0.06-U dose of bleomycin, and FKBP13 expression was assessed by IHC. In WT mice, FKBP13 was significantly upregulated at Day 7 and Day 21 after bleomycin administration (Figures 3A and 3B). On the basis of histopathological analysis by a molecular pathologist, it was noted that FKBP13 was primarily localized in bronchial and alveolar epithelial cells, which was similar to the localization in IPF tissues (Figure 3A). There was no FKBP13 staining present in FKBP13−/− mice, providing validation for the knockout. Dual immunofluorescence analysis of WT lung tissues further confirmed the colocalization of FKBP13 with the epithelial-cell marker E-cadherin (Figure 3C). Of note, the colocalization of FKBP13 and E-cadherin peaked at Day 7 (90.8 ± 2.1%) before returning to baseline at Day 21 (Figure 3D).
Figure 3.

FKBP13 expression is induced by bleomycin (Bleo) in mice. Wild-type (WT) C57BL/6 mice and FKBP13−/− mice (n = 4/group) were treated with Bleo (0.06 U/mouse) and assessed after 7 and 21 days. Lung tissue was stained for FKBP13 by IHC and IF. (A) Representative FKBP13 immunostaining images. Images were acquired using the automated Olympus VS120 slide scanner, which utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. Magnified images demonstrating FKBP13-positive cells are shown in the inset. (B) Quantification of FKBP13%-stained area in WT mice by HALO image-analysis software. No FKBP13 staining was detected in FKBP13−/− lungs. *P < 0.05 versus untreated mice. (C) Dual immunofluorescence analysis confirmed the colocalization of FKBP13 (red) with the epithelial-cell marker E-cadherin (E-Cad; green) in WT mice treated with Bleo. DAPI was used as a nuclear marker. (D) Percentage of FKBP13-positive area colocalizing with E-Cad. Scale bars, 100 μm. **P < 0.01 and ***P < 0.001 versus untreated mice. #P < 0.05 and ###P < 0.001 versus Day 7. IF = immunofluorescence.
FKBP13 Knockout Increases Susceptibility to Bleomycin-induced Pulmonary Fibrosis
To determine whether FKBP13−/− mice developed a fibrotic response to bleomycin administration that was different from that of control mice, two doses of bleomycin, 0.04 U or 0.06 U/mouse, were delivered intratracheally to WT and FKBP13−/− mice. Mice were then assessed for changes in lung function at Day 21 using a mechanical ventilator (flexiVent; Scientific Respiratory Equipment Inc.). At the lower dose of bleomycin (0.04 U), WT mice were unaffected with respect to lung function and histopathology, whereas FKBP13−/− mice displayed increased static lung elastance (Figures 4A and E2A). At the higher dose of bleomycin (0.06 U), both WT and FKBP13−/− mice experienced a similar elevation in lung elastance at Day 21 (Figures 4B and E2B).
Figure 4.

FKBP13−/− mice are more susceptible to changes in lung function induced by a low dose of Bleo. WT and FKBP13−/− mice (n = 6–8 per group) were treated with a low dose (0.04 U/mouse) and high dose (0.06 U/mouse) of Bleo, and lung function, as well as fibrosis, was assessed after 21 days (see illustration). (A and B) Elastance measurements were derived from the PV loops (Figure E2). FKBP13−/− mice are more susceptible to fibrosis induced by a low dose of Bleo. (C and D) Masson’s Trichrome staining to assess collagen deposition and quantification of the Trichrome-positive area by using HALO image-analysis software. (E and F) α-SMA immunostaining to assess myofibroblast accumulation and quantification by HALO. Images were acquired using the automated Olympus VS120 slide scanner, which utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. Scale bars, 100 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. untreated mice. #P < 0.05, and ###P < 0.001 between genotypes. PV = pressure–volume.
To determine whether the higher elastance measurement of the FKBP13−/− lungs at Day 21 correlated with an increase in fibrogenesis, histopathological analysis of the lung tissues was performed. Consistent with the lung-stiffness measurements, the lower dose of bleomycin led to increased collagen deposition in the lung parenchyma of the FKBP13−/− mice, whereas WT mice were unaffected (Figures 4C and 4D). The fibrotic lung parenchyma of FKBP13−/− mice also showed increased αSMA immunostaining compared with WT mice, indicating an accumulation of myofibroblasts (Figures 4E and 4F). At the higher bleomycin dose, the extent of fibrosis and myofibroblast accumulation was relatively similar in both strains at Day 21. Taken together, these results show that FKBP13−/− mice are more susceptible to bleomycin-induced lung injury, suggesting that FKBP13 is involved in processes that protect against either injury or fibrogenesis.
FKBP13 Deficiency Increases Lung Inflammation after Bleomycin Administration
To determine whether the exaggerated fibrotic processes in the FKBP13−/− mice are due in part to their susceptibility to bleomycin-induced pulmonary inflammation, we assessed the inflammatory profile of the BALF collected from the mice, which included total cell infiltrates, cell differentials, and TGF-β1 and IL-6 concentrations. This was done both at the peak of the inflammatory phase (Day 7) and at the peak of the fibrotic phase (Day 21) after bleomycin instillation. At Day 7, the lower dose of bleomycin (0.04 U) led to an influx of inflammatory cells in both WT and FKBP13−/− mice compared with their respective control animals; however, the FKBP13−/− mice showed a twofold greater influx over the WT mice (Figure 5A). FKBP13−/− mice had increased infiltration of macrophages, neutrophils, and lymphocytes at Day 7 compared with WT mice (Figures 5B–5D). There were no differences in the amount and phenotype of inflammatory cells at Day 21 after bleomycin administration. The concentration of the proinflammatory cytokine IL-6 were elevated in both strains at Day 7 but were elevated by a twofold greater extent in FKBP13−/− compared with WT lungs (Figure 5E). The concentration of the profibrotic cytokine TGF-β1 was also higher in FKBP13−/− mice than in WT mice at Day 7 (Figure 5F). At Day 21, the concentration of TGF-β1 was below detection. These data demonstrate that although both strains experience a proinflammatory response at Day 7, the response is intensified in the FKBP13−/− mice.
Figure 5.
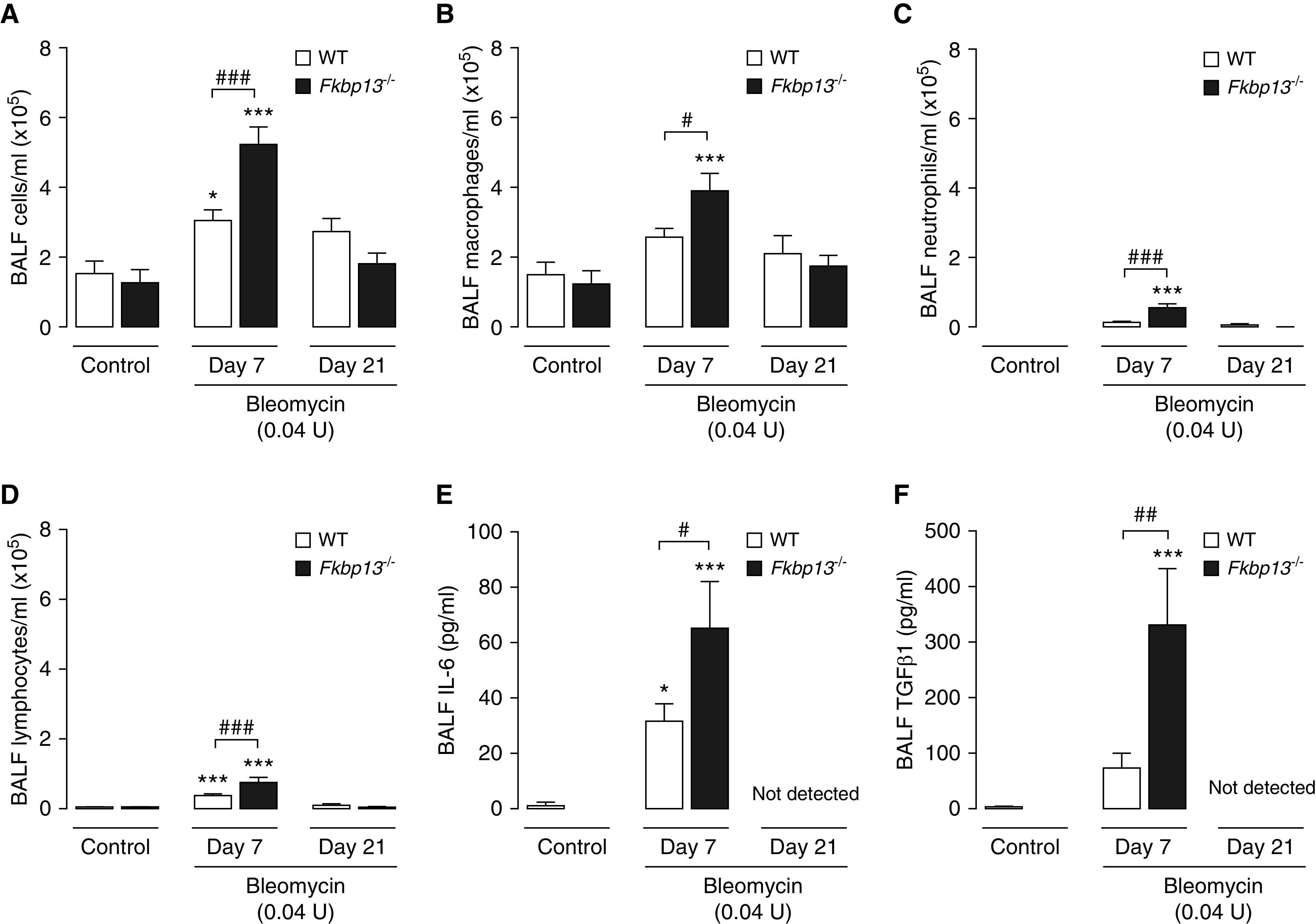
FKBP13−/− mice have enhanced cellular infiltration and profibrotic cytokine signaling in response to a low dose of Bleo. WT and FKBP13−/− mice (n = 6–8/group) were treated with the low dose (0.04 U/mouse) of Bleo, and BAL fluid (BALF) was collected at Day 7 to assess their inflammatory profiles. BALF cells were stained with Wright-Giemsa and the (A) total cells, (B) macrophages, (C) neutrophils, and (D) lymphocytes were counted. (E and F) IL-6 and TGF-β1 concentrations in the BALF were assessed by using ELISA. *P < 0.05 and ***P < 0.001 vs. untreated mice. #P < 0.05, ##P < 0.01, and ###P < 0.001 between genotypes.
FKBP13−/− Mice Are More Susceptible to Bleomycin-induced Apoptosis
Epithelial injury and apoptosis are recognized as precipitating events in the development of pulmonary fibrosis (22). To assess apoptosis in the bleomycin-treated mice, lung tissues were stained for TUNEL and cleaved caspase 3. At Day 7 after administration of the low dose of bleomycin, FKBP13−/− mice demonstrated a significant increase in the number of TUNEL-positive cells (Figures 6A and 6B). Higher numbers of apoptotic cells were also seen in the knockout lung tissues at Day 21, as determined by cleaved caspase 3 immunostaining (Figures 6C and 6D). Histopathological analysis suggested that type 2 alveolar epithelial cells were the predominant cell type affected.
Figure 6.

FKBP13−/− lungs have increased amount of UPR activation and apoptotic cells. WT and FKBP13−/− mice (n = 6–8/group) were treated with the low dose (0.04 U/mouse) of Bleo, and lung tissues were assessed for UPR and apoptosis markers by using IHC, in situ hybridization, and Western blot. (A and B) TUNEL staining and quantification of positive cells (arrow) at Day 7 by using HALO image-analysis software. Scale bars, 100 μm. (C and D) Cleaved caspase 3 immunostaining and quantification of positive cells (arrow) at Day 21 by using HALO image-analysis software. Scale bars, 100 μm. Images were acquired using the automated Olympus VS120 slide scanner, which utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. Magnified regions shown in inset image. *P < 0.05 versus untreated mice. #P < 0.05 between genotypes.
Resolution of Bleomycin-induced Pulmonary Fibrosis is Impaired in FKBP13−/− Mice
A characteristic of the bleomycin model is the reversibility of the fibrosis in the afflicted animals (23). To determine whether FKBP13 deficiency affects this process, lung stiffness and fibrosis measurements were made 50 days after the administration of high-dose (0.06 U) bleomycin. As reported earlier, this dose causes both strains to develop a similar severity of fibrosis at Day 21 (Figure 4). At Day 50, lung stiffness in the WT mice returned to baseline, whereas it remained elevated in the FKBP13−/− mice (Figures 7A and 7B). The fibrotic tissue area also persisted in the FKBP13−/− mice, whereas it normalized in the WT mice (Figures 7C and 7D), suggesting that FKBP13 deficiency impairs fibrosis resolution.
Figure 7.

FKBP13−/− mice have impaired resolution of Bleo-induced pulmonary fibrosis. WT and FKBP13−/− mice (n = 4–6/group) were treated with high-dose Bleo and assessed for changes in lung function and fibrosis at Day 50. (A and B) PV loops as assessed by using flexiVent and derivation of elastance. (C and D) Masson’s Trichrome staining of lung tissues for assessment of fibrosis and quantification of Trichrome-positive area by using HALO. Images were acquired using the automated Olympus VS120 slide scanner, which utilizes a stitching algorithm to reconstruct the whole specimen from overlapping image tiles. Scale bars, 100 μm. **P < 0.01 versus untreated mice. #P < 0.05 and ##P < 0.01 between genotypes.
Discussion
In this study, we investigated the role of the ER-resident molecular chaperone, FKBP13, in patients with diagnosed IPF and in an in vivo system that models the pathogenesis of pulmonary fibrosis. Previously, FKBP13 has been identified as a gene that is upregulated in the lung tissue of patients with progressive IPF, as defined by a ≥10% decline in FVC and ≥15% decline in DlCO over the 12 months after the biopsy (13). Furthermore, FKBP13 expression was found to be elevated in a cohort of patients with rapidly progressive chronic hypersensitivity pneumonitis, a disease that is also characterized by inflammation and progressive fibrosis (14). In our present study, we corroborate these findings at the protein level, with higher expression of FKBP13 detected in the fibrotic lung tissue of patients with IPF, hypersensitivity pneumonitis, rheumatoid arthritis, and sarcoidosis than that detected in nonfibrotic regions and control tissues. We also report that FKBP13 expression in IPF fibrotic lung tissue correlates with parameters of disease severity such as FVC and patient-reported dyspnea scores, suggesting that it could be useful as a biomarker for disease severity and progression. Our studies in transgenic mice confirmed a strong pathophysiological relevance of this molecule in all stages of lung injury, including the inflammatory, fibrotic, and resolution phases. We demonstrate that complete knockout of FKBP13 in mice results in increased susceptibility to bleomycin-induced pulmonary fibrosis and impaired resolution of fibrosis.
Bleomycin is known to induce widespread ER stress in the mouse lung (17, 24, 25). Because of the heterogeneous nature of this organ, an important task that remains is determining the specific cellular subpopulations that experience this ER stress and their resulting contribution to fibrotic remodeling. Activation of the UPR in response to ER stress initially leads to an adaptive adjustment of protein-folding capacity through the upregulation of ER resident molecular chaperones, attenuation of protein synthesis, degradation of misfolded or unfolded proteins, and physical expansion of the ER (26). Failure of these cellular programs to restore ER homeostasis results in the induction of apoptosis through transcription factors such as CHOP. As a component of the adaptive phase of the UPR, molecular chaperones such as FKBP13 are important for promoting cell survival through the de novo folding and assembly of proteins, refolding of misfolded proteins, and facilitation of proteolytic degradation (16).
Each cellular subpopulation of the lung has a potential role in either promoting or protecting against the fibrotic remodeling process, and many of these roles remain poorly characterized. Because chaperones such as FKBP13 are often ubiquitously expressed, the phenotype resulting from its global deficiency depends on the net effect on all cell types. Loss of chaperones in matrix- and profibrotic cytokine–producing cell types such as fibroblasts and M2 macrophages may help to impair the function of these pathological cells. Conversely, loss of the same chaperones in epithelial cells can increase their susceptibility to damage from external insults. For example, loss of calreticulin, FKBP65, and HSP47 impairs myofibroblast function because of their role in type 1 collagen biosynthesis (5, 27, 28), whereas GRP78 deficiency has been shown to promote M2-macrophage apoptosis (17). HSP70 has been shown to prevent epithelial cells from undergoing TGF-β1–induced epithelial–mesenchymal transition, and its overexpression in mice protects against bleomycin-induced pulmonary fibrosis (29). Here, we report evidence that the complete loss of FKBP13 has a net fibrotic effect on the development of pulmonary fibrosis. We further explored the impact of this loss on epithelial-cell injury and cellular infiltrates into the lung tissue, both of which have been shown to correlate with the extent and severity of fibrosis in the bleomycin model (30, 31).
Disturbance of the bronchial and alveolar epithelium through apoptosis has been observed in human IPF lung tissue and is considered to be an initial event in acute lung injury that leads to the subsequent fibrotic response (21, 22, 32). There is now increasing evidence that prolonged ER stress can induce these apoptotic processes in alveolar epithelial cells (33–35). In human IPF lung tissues, the amount of FKBP13 in fibrotic regions correlated with various markers of ER stress and apoptosis, indicating a potential involvement of this chaperone in these pathways. We observed here that a deficiency in FKBP13 increased the sensitivity of mice to low doses of bleomycin induced pulmonary fibrosis, suggesting that the epithelium is more susceptible to injury. Consistent with this, we observed an increase in apoptotic epithelial cells in the FKBP13−/− lung tissue at both Day 7 and Day 21 after bleomycin administration. Given its role as a molecular chaperone, we postulate that the loss of FKBP13 in our model results in a loss of the protein-folding reserve that normally protects epithelial cells from bleomycin-induced ER stress. The upregulation of FKBP13 in both human and mouse fibrotic lung tissue is likely a physiological response mediated by the adaptive UPR to augment this protein-folding reserve. This is consistent with the findings of Jeong and colleagues (2017) (16), who found that knockdown of FKBP13 in plasma cells resulted in cell death through an apoptotic ER stress response mediated by CHOP. At higher concentration of bleomycin, the buffer provided by this extra protein-folding reserve may become exhausted, resulting in epithelial injury.
The immune cells that infiltrate the lung tissue during bleomycin injury lead to the generation of proinflammatory cytokines, which is followed by a switch at around Day 9 to profibrotic signaling. To investigate whether the exacerbated fibrotic response was in part due to an increased cellular infiltration of FKBP13−/− lungs, we examined the inflammatory profile of the BALF. In response to the low dose of bleomycin, both strains displayed cellular infiltration and significantly elevated IL-6 at Day 7 over their respective control animals, although the concentration in the FKBP13−/− mice were twofold greater than those of the WT mice. We have previously shown that IL-6 potentiates the profibrotic phenotype of M2 macrophages, driving the fibrosis resulting from bleomycin injury (36). TGF-β1 concentration were also elevated in the knockout mice at Day 7, and TGF-β1 is known to stimulate myofibroblast proliferation and activation, as indicated by the increased α-SMA staining. In addition to the more robust cellular infiltration seen in the knockout mice, the loss of FKBP13 may alter the intrinsic functioning of the responding immune cells. Our single-cell RNA-sequencing analysis found elevated FKBP13 expression in plasma cells, whereas histopathological analysis revealed positive staining of the protein in alveolar macrophages. The elevated cytokine concentrations in FKBP13−/− mice may be the result of unopposed cytokine production by these cells. It has been shown in plasma cells that FKBP13 acts to restrict IgA production to maintain ER homeostasis and promote cell survival (16), and a similar mechanism may therefore be involved in regulating the output of cytokines. Furthermore, the milder but significant acute inflammation seen in the WT mice suggests that these animals still experienced some degree of lung injury in response to bleomycin but were able to resolve the injury without progressing to the fibrotic stage. The increased resistance of epithelial cells to apoptosis and the subdued immune response may both contribute to the protective effect of FKBP13 in WT mice.
The bleomycin model is characterized by an ability of the afflicted mice to recover and undergo resolution of fibrosis. Several mechanisms of pulmonary fibrosis resolution have been proposed in the literature, including the degradation of the fibrotic extracellular matrix, removal of myofibroblasts, reepithelialization, and removal of inflammatory cells such as profibrotic macrophages (23). At the higher bleomycin dose of 0.06 U, both WT and FKBP13−/− mice displayed a similar increase in lung-elastance measurements at Day 21, implying that although the WT mice are more resilient to the lower dose of bleomycin, this buffer is exhausted at the higher dose, resulting in the same extent of fibrosis and stiffening. At Day 50, lung elastance returned to baseline in the WT mice but remained elevated in the FKBP13−/− mice, suggesting that the loss of FKBP13 impairs the resolution of fibrosis in the bleomycin model.
A limitation of our study is the lack of prognostic data on the patients with IPF who were enrolled. Pulmonary function tests were conducted within a median of 48 days from the date of biopsy. Additional data on patient survival and serial pulmonary function testing results may aid in further study of the prognostic relevance of FKBP13. To further investigate the pathophysiological role of FKBP13 in pulmonary fibrosis, next steps would involve in vitro experiments to delineate the cell-specific effects of FKBP13 knockdown as well as the generation of tissue-specific knockouts in the epithelial or myeloid lineage to stratify the effects on epithelial injury and inflammation. Finally, validation of the correlation between FKBP13 and FVC in peripheral blood could increase the clinical applicability of this potential biomarker.
In conclusion, our data suggest that FKBP13 is upregulated and plays a protective role in the pathogenesis of pulmonary fibrosis. Mice lacking this chaperone are more susceptible to injury from lower doses of bleomycin. Because FKBP13 is predominantly expressed in lung epithelial cells, we postulate that the loss of FKBP13 reduces the ability these cells to respond to ER stress, priming them for more injury and apoptosis in response to pulmonary insults. In human IPF lung tissues, the amount of FKBP13 is associated with clinical parameters of disease severity. Although unlikely to be a pharmacological target because of its protective role, next steps should involve validation of FKBP13 as a potential biomarker for the progression of fibrotic interstitial lung diseases.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank the Central Animal Facility and McMaster Immunology Research Centre’s Core Histology Facility (Mary Jo Smith) for their exceptional technical training and service; Jane Ann Smith (McMaster University, Hamilton, ON, Canada) for her exceptional technical assistance with the laboratory animal work; Jennifer Wattie (St. Joseph’s Healthcare, Hamilton, ON, Canada) for technical help in performing flexiVent lung-function assessments; Christine Mader (Farncombe Metagenomics Facility, McMaster University, Hamilton, ON, Canada) for help with NanoString experiments; and Pavithra Parthasarathy, James Murphy, Dr. Carl Richards, Dr. Martin Stämpfli, Dr. Jeffrey Dickhout, and Dr. Jack Gauldie for their support and constructive discussions.
Footnotes
Supported by the Ontario Graduate Scholarship and the Canadian Institute of Health Research (CIHR) Doctoral Award (E.A.A.).
Author Contributions: Conception and design: V.T., E.A.A., A.N., J.-C.C., N.H., J.K., M. Vaughan, J.M., M.R.J.K., and K.A. Data acquisition and interpretation: V.T., E.A.A., A.A., M. Vierhout, S.N., M.K.P., S.A., O.M., K.T., S.D.R., T.Y., P.-S.B., P.S.K., A.D.-G., and A.N. Drafting and revision of manuscript: V.T., E.A.A., A.A., M. Vierhout, S.N., M.K.P., S.A., O.M., A.D.-G., J.K., J.M., M.R.J.K., and K.A.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0121OC on November 30, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 2.Wei J, Rahman S, Ayaub EA, Dickhout JG, Ask K. Protein misfolding and endoplasmic reticulum stress in chronic lung disease. Chest. 2013;143:1098–1105. doi: 10.1378/chest.12-2133. [DOI] [PubMed] [Google Scholar]
- 3.Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest. 2018;128:64–73. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 5.Staab-Weijnitz CA, Fernandez IE, Knüppel L, Maul J, Heinzelmann K, Juan-Guardela BM, et al. FK506-binding protein 10, a potential novel drug target for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2015;192:455–467. doi: 10.1164/rccm.201412-2233OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Z, Cai H, Zhu H, Toque H, Zhao N, Qiu C, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK)/calcineurin signaling is a novel pathway regulating intracellular calcium accumulation which might be involved in ventricular arrhythmias in diabetic cardiomyopathy. Cell Signal. 2014;26:2591–2600. doi: 10.1016/j.cellsig.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Duran I, Martin JH, Weis MA, Krejci P, Konik P, Li B, et al. A chaperone complex formed by HSP47, FKBP65, and BiP modulates telopeptide lysyl hydroxylation of type I procollagen. J Bone Miner Res. 2017;32:1309–1319. doi: 10.1002/jbmr.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishikawa Y, Holden P, Bächinger HP. Heat shock protein 47 and 65-kDa FK506-binding protein weakly but synergistically interact during collagen folding in the endoplasmic reticulum. J Biol Chem. 2017;292:17216–17224. doi: 10.1074/jbc.M117.802298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lietman CD, Rajagopal A, Homan EP, Munivez E, Jiang M-M, Bertin TK, et al. Connective tissue alterations in Fkbp10−/− mice. Hum Mol Genet. 2014;23:4822–4831. doi: 10.1093/hmg/ddu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nigam SK, Jin YJ, Jin MJ, Bush KT, Bierer BE, Burakoff SJ. Localization of the FK506-binding protein, FKBP 13, to the lumen of the endoplasmic reticulum. Biochem J. 1993;294:511–515. doi: 10.1042/bj2940511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bush KT, Hendrickson BA, Nigam SK. Induction of the FK506-binding protein, FKBP13, under conditions which misfold proteins in the endoplasmic reticulum. Biochem J. 1994;303:705–708. doi: 10.1042/bj3030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Partaledis JA, Berlin V. The FKB2 gene of Saccharomyces cerevisiae, encoding the immunosuppressant-binding protein FKBP-13, is regulated in response to accumulation of unfolded proteins in the endoplasmic reticulum. Proc Natl Acad Sci USA. 1993;90:5450–5454. doi: 10.1073/pnas.90.12.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, et al. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One. 2009;4:e5134. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horimasu Y, Ishikawa N, Iwamoto H, Ohshimo S, Hamada H, Hattori N, et al. Clinical and molecular features of rapidly progressive chronic hypersensitivity pneumonitis. Sarcoidosis Vasc Diffuse Lung Dis. 2017;34:48–57. doi: 10.36141/svdld.v34i1.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa Y, Mizuno K, Bächinger HP. Ziploc-ing the structure 2.0: endoplasmic reticulum-resident peptidyl prolyl isomerases show different activities toward hydroxyproline. J Biol Chem. 2017;292:9273–9282. doi: 10.1074/jbc.M116.772657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong M, Jang E, Choi SS, Ji C, Lee K, Youn J. The function of FK506-binding protein 13 in protein quality control protects plasma cells from endoplasmic reticulum stress-associated apoptosis. Front Immunol. 2017;8:222. doi: 10.3389/fimmu.2017.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayaub EA, Kolb PS, Mohammed-Ali Z, Tat V, Murphy J, Bellaye P-S, et al. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J Pathol. 2016;239:411–425. doi: 10.1002/path.4738. [DOI] [PubMed] [Google Scholar]
- 18.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 23.Cabrera S, Selman M, Lonzano-Bolaños A, Konishi K, Richards TJ, Kaminski N, et al. Gene expression profiles reveal molecular mechanisms involved in the progression and resolution of bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L593–L601. doi: 10.1152/ajplung.00320.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thamsen M, Ghosh R, Auyeung VC, Brumwell A, Chapman HA, Backes BJ, et al. Small molecule inhibition of IRE1α kinase/RNase has anti-fibrotic effects in the lung. PLoS One. 2019;14:e0209824. doi: 10.1371/journal.pone.0209824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu H-S, Liu C-C, Lin J-H, Hsu T-W, Hsu J-W, Su K, et al. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. 2017;7:14272. doi: 10.1038/s41598-017-14612-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, et al. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem. 2007;282:7024–7034. doi: 10.1074/jbc.M609490200. [DOI] [PubMed] [Google Scholar]
- 27.Zimmerman KA, Graham LV, Pallero MA, Murphy-Ullrich JE. Calreticulin regulates transforming growth factor-β-stimulated extracellular matrix production. J Biol Chem. 2013;288:14584–14598. doi: 10.1074/jbc.M112.447243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otsuka M, Shiratori M, Chiba H, Kuronuma K, Sato Y, Niitsu Y, et al. Treatment of pulmonary fibrosis with siRNA against a collagen-specific chaperone HSP47 in vitamin A–coupled liposomes. Exp Lung Res. 2017;43:271–282. doi: 10.1080/01902148.2017.1354946. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka K, Tanaka Y, Namba T, Azuma A, Mizushima T. Heat shock protein 70 protects against bleomycin-induced pulmonary fibrosis in mice. Biochem Pharmacol. 2010;80:920–931. doi: 10.1016/j.bcp.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 30.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polosukhin VV, Degryse AL, Newcomb DC, Jones BR, Ware LB, Lee JW, et al. Intratracheal bleomycin causes airway remodeling and airflow obstruction in mice. Exp Lung Res. 2012;38:135–146. doi: 10.3109/01902148.2012.658595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol. 1998;275:L1192–L1199. doi: 10.1152/ajplung.1998.275.6.L1192. [DOI] [PubMed] [Google Scholar]
- 33.Kamp DW, Liu G, Cheresh P, Kim S-J, Mueller A, Lam AP, et al. Asbestos-induced alveolar epithelial cell apoptosis: the role of endoplasmic reticulum stress response. Am J Respir Cell Mol Biol. 2013;49:892–901. doi: 10.1165/rcmb.2013-0053OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen H, Uhal BD. The unfolded protein response controls ER stress-induced apoptosis of lung epithelial cells through angiotensin generation. Am J Physiol Lung Cell Mol Physiol. 2016;311:L846–L854. doi: 10.1152/ajplung.00449.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nita I, Hostettler K, Tamo L, Medová M, Bombaci G, Zhong J, et al. Hepatocyte growth factor secreted by bone marrow stem cell reduce ER stress and improves repair in alveolar epithelial II cells. Sci Rep. 2017;7:41901. doi: 10.1038/srep41901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ayaub EA, Dubey A, Imani J, Botelho F, Kolb MRJ, Richards CD, et al. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Sci Rep. 2017;7:13281. doi: 10.1038/s41598-017-13511-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.