

Abstract
Background
The Plasmodium sexual gametocyte stages are the only transmissible form of the malaria parasite and are thus responsible for the continued transmission of the disease. Gametocytes undergo extensive functional and morphological changes from commitment to maturity, directed by an equally extensive control program. However, the processes that drive the differentiation and development of the gametocyte post-commitment, remain largely unexplored. A previous study reported enrichment of H3K36 di- and tri-methylated (H3K36me2&3) histones in early-stage gametocytes. Using chromatin immunoprecipitation followed by high-throughput sequencing, we identify a stage-specific association between these repressive histone modifications and transcriptional reprogramming that define a stage II gametocyte transition point.
Results
Here, we show that H3K36me2 and H3K36me3 from stage II gametocytes are associated with repression of genes involved in asexual proliferation and sexual commitment, indicating that H3K36me2&3-mediated repression of such genes is essential to the transition from early gametocyte differentiation to intermediate development. Importantly, we show that the gene encoding the transcription factor AP2-G as commitment master regulator is enriched with H3K36me2&3 and actively repressed in stage II gametocytes, providing the first evidence of ap2-g gene repression in post-commitment gametocytes. Lastly, we associate the enhanced potency of the pan-selective Jumonji inhibitor JIB-04 in gametocytes with the inhibition of histone demethylation including H3K36me2&3 and a disruption of normal transcriptional programs.
Conclusions
Taken together, our results provide the first description of an association between global gene expression reprogramming and histone post-translational modifications during P. falciparum early sexual development. The stage II gametocyte-specific abundance of H3K36me2&3 manifests predominantly as an independent regulatory mechanism targeted towards genes that are repressed post-commitment. H3K36me2&3-associated repression of genes is therefore involved in key transcriptional shifts that accompany the transition from early gametocyte differentiation to intermediate development.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13072-021-00393-9.
Keywords: H3K36me2, H3K36me3, Histone, Malaria, Plasmodium, Gametocyte, Epigenetics, Histone post-translational modifications, Histone demethylation
Background
Malaria remains a serious threat to public health in most of the developing world and is responsible for millions of deaths annually [1]. Nevertheless, progress is being made toward the global eradication of the disease. Malaria eradication relies on preventing the transmission of Plasmodium parasites between human hosts, facilitated by the mosquito vector. In the human host, malaria parasites exist either as the asexual proliferative stages, responsible for the symptoms of malaria, or as sexually differentiated, transmissible gametocytes [2, 3]. The extended 10- to 12-day process of gametocyte development is characterised by morphologically distinct stages (I–V) and is unique to the human malaria parasite, Plasmodium falciparum. Mature gametocytes are the only stage that can be transmitted by the mosquito vector and as such, the process of gametocytogenesis is an attractive target for the development of transmission-blocking strategies [4–6].
The stage transitions within the P. falciparum parasite life cycle are driven by global transcriptomic reprogramming that is tightly controlled by complex transcriptional and post-transcriptional regulation [7–9]. Additionally, epigenetic mechanisms are known to establish and maintain transcriptional programs that support asexual parasite proliferation and have been suggested to do the same during gametocytogenesis [10]. The histone post-translational modification (hPTM) landscape in P. falciparum parasites is dynamic with each life cycle stage characterised by a unique pattern of hPTMs, suggested to be foundational in establishing the specialised transcriptional program for the stage [11]. Accordingly, several hPTMs are confirmed to be functionally relevant for asexual parasite proliferation [12]. For example, H3K4me3, H3K8ac and H3K9ac are involved in the activation of stage-specific gene sets that are linked to proliferation and the maintenance of the euchromatic genome that is characteristic of asexual parasites [13–17]. By contrast, H3K36me2 has been proposed to be globally repressive in asexual parasites [18] and while more frequently associated with broad maintenance functions such as the conservation of genomic integrity, H3K36me2 has also been shown to direct transcriptional repression in other eukaryotes [19–21]. H3K36me3 is also associated with gene silencing and controls the expression of multi-gene families that encode invasion and exported proteins, thereby contributing to the parasite’s extensive capacity for phenotypic plasticity [22–24]. H3K36me2&3 are both well-documented regulators of eukaryotic cellular differentiation and development [19, 25, 26].
During proliferation, certain gametocyte-specific genes are silenced by their association with heterochromatin protein 1 (HP1) that is recruited by enrichment of H3K9me3 at these loci [27, 28]. Sexual differentiation requires the release of ap2-g (a member of the ApiAP2 transcription factor family, that is the master switch for gametocyte commitment [29]) from H3K9me3/HP1-mediated heterochromatin, resulting in a transcriptional environment that drives commitment to gametocytogenesis [29–32]. However, only a handful of studies have examined the functional relevance of epigenetic mechanisms in post-commitment gametocyte development [28, 30, 33]. Aside from the demonstrated role of H3K9me3/HP1 in heterochromatin rearrangement for stage-specific gene sets during gametocytogenesis [5], the function of hPTMs in generating and maintaining gametocyte-relevant transcriptional programs during sexual development have not been studied.
The dynamic nature of the hPTM landscape during gametocyte development and departure from the patterns observed during asexual proliferation suggest that hPTMs may be involved in stage-specific gene expression in gametocytes as has been demonstrated for asexual parasites [10, 12, 16, 28, 30, 34, 35]. Indeed, the concurrent shifts in the hPTM and transcriptomic landscapes associated with morphological transitions in gametocytes support this idea [11, 36]. Of particular interest to us was the striking peak abundance of H3K36me2&3 unique to stage II gametocytes that corresponds to the transcriptomic and morphological changes associated with the transition from early differentiation (stage I) to intermediate (stage II/III) development [11, 36]. We sought to interrogate the functional relevance of these hPTMs in gametocytogenesis [11]. To do so, we performed chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) on three distinct gametocyte populations and integrated the results with data from other ChIP-seq and gene expression profiling studies. We provide comprehensive genome-wide maps of the H3K36me2&3 occupancy during gametocytogenesis and show H3K36me2&3 enrichment in stage II gametocytes is involved in the transcriptional reprogramming underlying the transition from early gametocyte differentiation to intermediate development [36]. Additionally, we assessed the effects of chemically inhibiting histone demethylases (HDMs) on H3K36me2&3 levels and gene expression during early gametocyte development. Using histone methylation profiling and whole transcriptome analysis, we demonstrate the link between histone demethylation and the enhanced potency of the pan-selective Jumonji inhibitor, JIB-04 in gametocytes [37, 38]. This paper provides the first association between H3K36me2&3 and gene regulation in gametocytes and indicates that H3K36me2&3-associated gene repression is involved in key transcriptional shifts that accompany the transition from early gametocyte differentiation to intermediate development.
Results
H3K36me2&3 occupancy is dynamic in P. falciparum gametocytes
To investigate the functional role of H3K36me2&3 in P. falciparum stage II gametocytes [11], we performed ChIP-seq on three distinct gametocyte populations spanning early gametocyte development and classified these as “pre-stage II”, “stage II” and “post-stage II” gametocytes. Gametocyte populations were morphologically binned into specific stages (Additional file 1: Figure S1A) resulting in a pre-stage II population (60 ± 4% asexual parasites; 40 ± 4% and 0.3 ± 0.4% stage I and II gametocytes, respectively) (Fig. 1a). The stage II population was enriched for stage II gametocytes (74 ± 3.2%, 9 ± 6% stage I and 17 ± 2.5% stage III gametocytes) without any asexual parasites present. Post-stage II samples consisted mostly of stage III/IV gametocytes (90 ± 6%; 10 ± 6% stage II gametocytes).
Fig. 1.

H3K36me2&3 have dynamic patterns of occupancy during P. falciparum early and intermediate gametocyte development. a The stage compositions (%) of gametocyte populations used for the ChIP-seq study, classified as pre-stage II (pre-II), stage II (stg II) and post-stage II (post-II) as determined microscopically. Data are from two independent biological repeats, ± S.E. b Plot of Pearson correlation coefficients for the genome-wide occupancy of H3K36me2&3 (me2 and me3) obtained for each gametocyte population with data representing two independent biological repeats. The black box highlights the increased positive correlation between H3K36me2&3 in stage II gametocytes. c H3K36me2&3 occupancy (average log2-transformed ChIP/input) over chromosome 12 for each gametocyte population. Data are representative of two independent biological replicates except for the pre-II gametocytes with a single replicate. Turquoise and purple represent regions where the hPTMs are present (log2ChIP/input ratios > 0) and dark grey represents regions of depletion (log2ChIP/input ratios ≤ 0). The light grey shadowed areas highlight stage II gametocyte-specific sites of H3K36me2&3 enrichment exemplified in a central region of the chromosome 12 (1250–1290 kb) with the positions of genes indicated below the occupancy tracks
We performed ChIP-seq on these populations with ChIP-grade α-H3K36me2&3 antibodies with demonstrated specificity towards these histone marks from various organisms [39–43] as well as against P. falciparum [22, 44]. Nevertheless, we performed an independent validation of the selected antibodies with the results demonstrating specific detection of P. falciparum histone H3 with either H3K36me2 or H3K36me3 (Additional file 1: Figure S1B). ChIP-seq was performed on two independent biological replicates for each sample except for the pre-stage II gametocytes that only had one, with the resultant data for replicate samples well correlated (e.g. Pearson correlation, r2 = 0.93 and r2 = 0.94 between H3K36me2 biological replicates in stage II and post-stage II samples, respectively, Additional file 2: Table S1). We detected H3K36me2 and H3K36me3 individually in each of the sampled gametocyte populations and observed a strong positive correlation between the two hPTMs only in the stage II gametocytes (Pearson correlation, r2 = 0.9, Fig. 1b), and not the pre- or post-stage II gametocytes (Fig. 1b, Additional file 1: Figure S1C). Given that a single nucleosome would not contain di- and tri-methylated H3K36 concurrently, this likely reflects that one of these hPTMs is an intermediate of the other with our samples consisting of a combined pool of nucleosomes with either methylation state.
Our ChIP-seq results show that dynamic patterns of H3K36me2&3 is evident with “low–high–low” abundance profiles associated with pre-stage II, stage II and post-stage II gametocyte populations, respectively (Fig. 1c), as confirmed independently by ChIP-qPCR (Additional file 1: Figure S2). H3K36me2&3 is mostly absent in pre-stage II gametocytes and occupancy in stage II gametocytes is found preferentially in intergenic regions and upstream of gene coding sequences, and the levels of the hPTMs dissipate in post-stage II gametocytes (Fig. 1c). Given the unique abundance of H3K36me2&3 in the stage II gametocytes and the congruency of this with our previous proteomics data [11], our downstream analyses focussed on the preferential association of these hPTMs with stage II gametocytes.
Stage II gametocytes have a unique pattern of H3K36me2&3 enrichment
To determine the genome-wide positioning of H3K36me2&3, we interrogated the hPTM occupancy spanning each of P. falciparum genes for which sequencing data were obtained (Additional file 2: Table S2, Fig. 2a). H3K36me2&3 occupancy profiles of are distinctly uniform across the intergenic non-coding regions in stage II gametocytes, contrasting with more variable patterns in the pre- and post-stage II gametocytes where these marks are mostly depleted (Fig. 2a). In stage II gametocytes, H3K36me2&3 are concentrated 1.5 kb upstream of transcription start sites (TSSs, as defined previously [45]), with occupancy values (log2ChIP/input) peaking at > 0.2 and > 0.17, respectively, contrasting with a depletion of these hPTMs in coding regions (CDRs) (occupancy values of < 0.1 Fig. 2a).
Fig. 2.

Stage II gametocytes have unique patterns of H3K36me2&3 occupancy and enrichment associated with genes. a H3K36me2&3 occupancy (average log2 ChIP/input binned into 50 bp regions) spanning 1.7 kb upstream and 2.5 kb downstream of transcriptional start sites (TSS) of 5206 P. falciparum genes (PlasmoDB genome annotation, v39) in pre-stage II (pre-II), stage II (stg II) and post-stage II (post-II) gametocyte populations. Data are representative of the average occupancy obtained for two independent biological repeats, except for pre-stage II gametocytes with only one. Genes were rank ordered individually in each heatmap. Summary plots of the H3K36me2&3 (turquoise and purple, respectively) occupancy in each of the gametocyte stages with ribbons representing ± SE. The light grey shadowed region indicates regions of increased occupancy in the stage II gametocytes. b Clustered column plots specifying the number of H3K36me2&3-enriched genes (i.e. log2 ChIP/input ≥ 0.2 and ≥ 0.15, respectively) with genes robustly enriched (i.e. log2 ChIP/input ≥ 0.5) represented by patterned bars. C Numbers of genes with H3K36me2&3 enrichment (MACS2 peaks, q-value < 0.05; present in both replicates) in stage II and post-stage II gametocytes. No peaks were detected for pre-stage II gametocytes. The proportion of genes with enrichment 1.5 kb upstream of TSSs is represented by the darker shade compared to the proportion of genes with enrichment in the coding regions (CDRs). d Log2-transformed ChIP/input ratio tracks for H3K36me2&3 in stage II and post-stage II gametocytes for a region on chromosome 8 to exemplify significant enrichment (MACS2 peaks present in both biological replicates, q-value < 0.05) in stage II gametocytes. Called peaks are indicated by the horizontal black bars
The prominent abundance of H3K36me2&3 in stage II gametocytes translates to 2480 and 2691 genes occupied by H3K36me2 and -me3, respectively, with > 95% overlap present between these genes (i.e. above average log2ChIP/input value in stage II gametocytes, Fig. 2b). Within these genes, ~ 17% showed significant enrichment for H3K36me2&3 (significant MACS2 peaks, q-value < 0.05, present in both biological replicates) (Fig. 2c, Additional file 2: Table S3). For H3K36me2, 734 peaks were identified, corresponding to 327 genes with enrichment 1.5 kb upstream of TSSs and 233 genes with this hPTM enriched in CDRs (Fig. 2c). Similarly, H3K36me3 was identified at 569 sites across the genome in stage II gametocytes, equating to the enrichment upstream and in the CDRs of 252 and 154 genes, respectively (Fig. 2c). This enrichment is evident in stage II gametocytes only, with < 100 genes similarly enriched at each of these sites in post-stage II gametocytes (Fig. 2c) and none in pre-stage II gametocytes. The enrichment of H3K36me2&3 upstream of TSSs displays localised overlap in the presence of these marks (Fig. 2d). Since the presence of H3K36me2 as a transient intermediate in the formation of H3K36me3 has been reported elsewhere [22, 46–48] and cannot be excluded here, downstream analyses were performed on the combined set of enriched genes without distinguishing between the di- and tri-methylated state.
H3K36me2&3 are associated with post-commitment transcriptional regulation in stage II gametocytes
We categorised H3K36me2&3-enriched genes based on the location of the hPTMs (i.e. exclusively upstream of TSSs, exclusively in CDR or both) and compared these gene sets to their transcription profiles during gametocytogenesis, using a gene expression time-course dataset previously generated in our lab on the same clonal strain of P. falciparum NF54 parasites [34]. Corresponding expression profiles were obtained for days 2–6 of development (majority stage II gametocytes) for 92% of the H3K36me2&3-enriched genes (Additional file 2: Table S4). A clear variation in the expression profiles of the genes was present and associated with different sites of H3K36me2&3 enrichment. Only genes with H3K36me2&3 exclusively enriched in CDRs (37%) showed a slight increase in transcript abundance in stage II gametocytes (Fig. 3a), but is unclear if this increased abundance is relevant to gametocyte differentiation, as the level is not as prominent as in asexual parasites [22, 36].
Fig. 3.
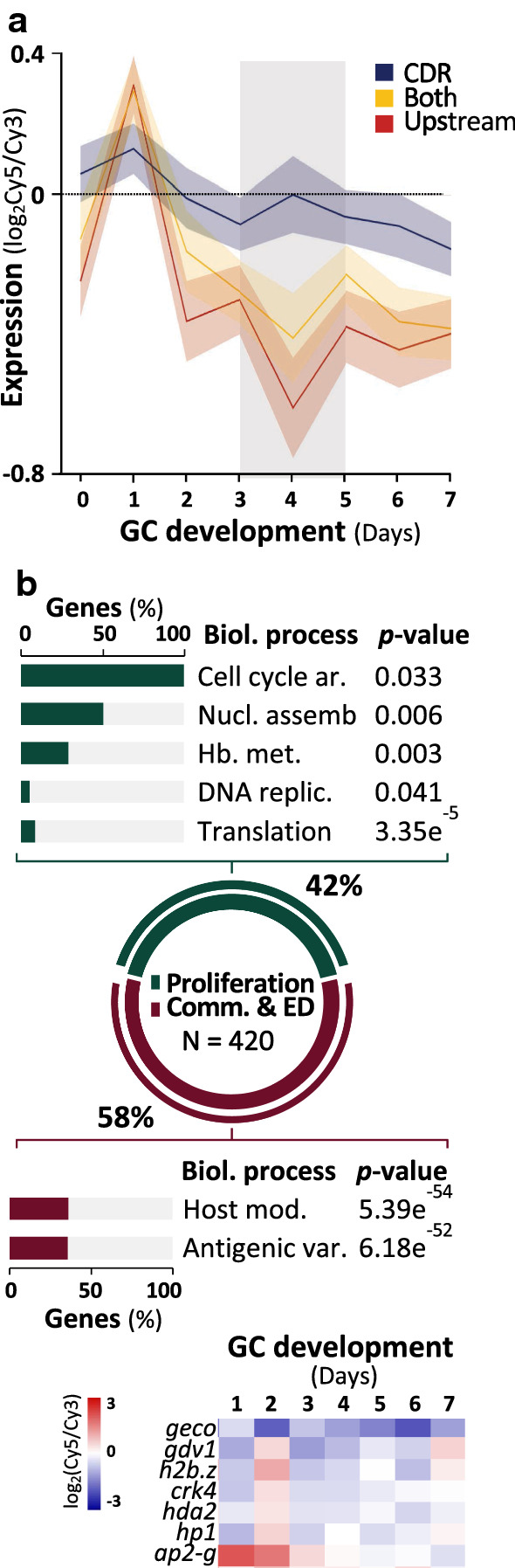
H3K36me2&3 are associated with transcriptional regulation in stage II gametocytes. a Average transcript abundance (log2Cy5/Cy3) on days 0–7 of gametocyte (GC) development [36] for genes with H3K36me2&3 enrichment in coding regions (CDR, blue), 1.5 kb upstream of TSSs (TSS, red) or both (gold) with ribbons representing ± S.D. They grey shadowed region highlights days on which H3K36me2&3-associated transcriptional changes are most prominent. b The 420 H3K36me2&3-enriched and repressed genes were grouped into those associated with proliferation (178 genes, green) that are significantly associated (P < 0 .05) with the biological processes indicated. The number of enriched genes is represented as a % of all background genes associated with the biological processes. Legend: ar. = arrest; nucl. = nucleosome; assemb. = assembly; replic. = replication; mod. = modification; var. = variation. Full descriptions of gene names and their PlasmoDB IDs are available in Additional file 2: Table S4
The majority of H3K36me2&3-enriched genes (63%) are strongly repressed during stage II gametocytes, with the hPTMs present exclusively upstream of the TSSs in 342 of the 417 repressed genes (Fig. 3a). We find that 42% of these repressed genes are typically involved in processes associated with asexual proliferation, including those significantly associated with translation (P = 3.35e−5), haemoglobin metabolism (P = 0.003), cell cycle arrest (P = 0.033) and DNA replication (P = 0.041) (Fig. 3b, Additional file 2: Table S5). Furthermore, more than half of these are essential for asexual parasite development [49]. However, only 6% of these genes that are associated with H3K36me2&3 enrichment in asexual parasites [22], indicating that H3K36me2&3 enrichment in stage II gametocytes is targeted towards a distinct subset of asexual parasite-related genes that are specifically repressed in early gametocytogenesis.
We investigated the overlap of the H3K36me2&3-enriched genes in stage II gametocytes with genes previously described to be related to the processes of sexual commitment and early gametocyte differentiation [27, 30, 31, 36, 50–54]. A large proportion (58%, 242 genes) of the H3K36me2&3-enriched and repressed genes are linked to these processes (Fig. 3b, Additional file 2: Table S6). These include genes significantly associated with antigenic variation (P = 6.18e−52) and host cell modification processes (P = 5.39e−54, Fig. 3b, Additional file 2: Table S5), with a particularly strong repression seen for the stevor multi-gene family. H3K36me2&3-associated repression is present for early gametocyte markers (e.g. geco, PF3D7_1253000; pfg14-744, PF3D7_1477300 and pfg14-748, PF3D7_1477700) [51, 52, 55, 56] and key regulators of commitment and gametocytogenesis: hp1 (PF3D7_1220900), histone deacetylase 2 (hda2, PF3D7_1008000), gametocyte development protein 1 (gdv1, PF3D7_0935400) and the ApiAP2 transcription factor, ap2-g (PF3D7_1222600, Fig. 3b) [28, 29, 53, 54, 57]. Sexual commitment is also associated with displacement of HP1 from 15 heterochromatic loci [30], and results in the expression of ap2-g, leading to the transcriptional cascade that drives sexual commitment [28, 29, 32, 57]. We therefore looked at the proportion of the H3K36me2&3-enriched and repressed genes in our dataset (242 genes involved in commitment) that are targets of AP2-G (31) or are transcribed following the displacement of HP1 [30]. We find that only 18% of the H3K36me2&3-enriched genes are activated by AP2-G or HP1 depletion during commitment, implying the H3K36me2&3-associated transcriptional repression of commitment-related genes in stage II gametocytes are irrespective of mechanisms by which they were activated in the preceding commitment steps. Importantly, 53% of the HP1-depleted genes that are expressed and drive commitment are enriched with these H3K36me2&3 in stage II gametocytes. As all of these genes remain depleted of HP1 [30], yet are associated with a sharp reduction in transcript abundance in the stage II gametocytes [36], their repression can be ascribed to their enrichment with H3K36me2&3. In the stage II gametocytes, H3K36me2&3 are therefore involved in the transcriptional repression of genes that drive commitment and early differentiation once their products become obsolete in post-commitment, developing gametocytes.
H3K36me2&3-associated transcriptional repression is largely independent of other regulatory mechanisms
Previous studies described other gene regulatory mechanisms that function during gametocytogenesis [30, 58], including HP1-mediated heterochromatin expansion throughout gametocyte development, resulting in the silencing of stage-specific gene sets [30]. Crucially, within our stage II gametocyte data, we identify that only 9% of the H3K36me2&3-enriched and repressed genes are also occupied by HP1 in stage II/III gametocytes (Fig. 4a), including those associated with knob-formation (e.g. kahsp40, PF3D7_0201800 and emp3, PF3D7_0201900) and protein export (e.g. hyp). These may therefore be regulated cooperatively by the two mechanisms. A second AP2 transcription factor, AP2-G2 is essential to gametocyte development beyond stage III and in asexual parasites, AP2-G2 represses a subset of genes through an association with H3K36me3 [58]. Only 33% of the H3K36me2&3-enriched genes in our data are also bound by AP2-G2 during intermediate gametocyte development (Fig. 4a), indicating that the transcriptional repression associated with the H3K36me3-AP2-G2 interaction also occurs for these genes in the early gametocytes. These include the genes encoding GDV1 that evicts HP1 from heterochromatic loci prior to commitment [53] and CCT (CTP:phosphocholine cytidylyltransferase, PF3D7_1316600), involved in the synthesis of PC from an alternative substrate under the lysoPC-depleted conditions that cue commitment [50, 59]. This indicates that the interaction between H3K36me2&3 and AP2-G2 may repress these important commitment-related genes once gametocytogenesis has been established.
Fig. 4.

H3K36me2&3-associated transcriptional repression is largely independent of other regulatory mechanisms. a The H3K36me2&3-enriched and repressed gene set identified here for stage II gametocytes was compared to those previously determined to be repressed by HP1-mediated heterochromatin formation (+ HP1, gold) or by AP2-G2 (blue) in the stage II/III gametocytes. Certain genes from each of these categories are exemplified in the gene expression heatmaps [36]. b Log2-transformed ChIP/input ratio tracks of H3K36me2 (turquoise) and H3K36me3 (purple) occupancy for jmjc1, hp1 and ap2-g in pre-stage II, stage II and post-stage II gametocytes, with the coding sequences of each represented by the green, maroon and orange blocks, respectively. Other genes are indicated by grey blocks. Dashed grey boxes represent regions of differential occupancy between the three stages and horizontal black bars indicate called peaks representing regions associated with H3K36me2&3 enrichment in stage II gametocytes. Full descriptions of gene names and their PlasmoDB IDs are available in Additional file 2: Table S4
However, the majority of the H3K36me2&3-enriched and repressed genes in stage II gametocytes are not associated with any other regulatory mechanisms and is likely regulated by a novel repressive mechanism involving the stage II gametocyte-specific enrichment of H3K36me2&3. This includes lysoPC synthesis-associated genes (e.g. acs2, PF3D7_0301000: acs4, PF3D7_1372400; acs7 PF3D7_1200700 and cept, PF3D7_0628300) and several epigenetic and transcriptional regulators (e.g. hda2; set1, PF3D7_0629700 and alba4, PF3D7_1347500; Fig. 4a). Importantly, ap2-g, hp1 and a Jumonji lysine demethylase (KDM) jmjc1 (PF3D7_0809900, Fig. 4a), are enriched for H3K36me2&3 upstream of TSSs in stage II gametocytes, but not in pre- and post-stage II gametocytes (Fig. 4b), with only ap2-g additionally having enrichment within the CDR. This enrichment of these regulatory proteins with H3K36me2&3 provides the first insights into the mechanism by which these genes are “switched off” after commitment to allow gametocyte development to proceed. Furthermore, the H3K36me2&3 enrichment and repression of jmjc1 is of interest given its homology with the H3K36-specific, lysine demethylase 2 (kdm2) family [44]. As such, the repression of jmjc1 may be present as means of safeguarding the integrity of H3K36me2&3 enrichment and the associated transcriptional repression in the stage II gametocytes. Taken together, these results implicate H3K36me2&3 as a novel mechanism of transcriptional repression in stage II gametocytes that is largely independent of other mechanisms described to date.
Inhibition of H3K36 demethylation by JIB-04 is associated with altered patterns of transcription in P. falciparum gametocytes
To investigate the functional relevance of enzymes involved in deposition (histone methyltransferases, HMT) and removal (KDMs) of H3K36me2&3, we interrogated inhibition of selected enzymes on H3K36 methylation levels on days associated with stage II gametocyte development. In asexual parasites, the HMT SET2 (PF3D7_1322100) methylates H3K36 [44, 60], but is comparatively poorly expressed during gametocytogenesis (Fig. 5a). Although knockout lines of set2 are available [22, 60], these do not produce gametocytes and therefore could not be used to interrogate the involvement of SET2 in H3K36 methylation stage II gametocytes. Three Jumonji KDMs of P. falciparum (jmjc1 (PF3D7_0809900), jmjc2 (PF3D7_0602800) and jmj3 (PF3D7_1122200)) are transcribed in stage II gametocyte development, strongly suggesting that at least one of these enzymes demethylates H3K36 (Fig. 5a). We used inhibitors of the KDMs and could show that JIB-04 (pan-selective) and ML324 (targeting KDM4, PfJMJ3), both with activity against gametocytes [38, 61, 62], caused hypermethylation of H3K36 (~ 13-fold, Fig. 5b, Figure S3A). This was not seen for GSK-J4 (inhibiting KDM6, H3K27me3 selective) or PCPA-2 (targeting LSD1) (Fig. 5b) [63, 64]. This indicates that one of the P. falciparum Jumonji KDMs demethylate H3K36me2&3 after they peak in abundance in stage II gametocytes.
Fig. 5.

Chemical inhibition of histone demethylase activity leads to the disruption of normal gene expression in gametocytes a Expression profiles (log2Cy5/Cy3) of genes encoding the P. falciparum Jumonji-C domain-containing demethylases (jmjc1, jmjc2 and jmj3) and the H3K36-specific methyltransferase, set2 on days 0 to 7 of gametocyte (GC) development [36], with the grey shadowed region representing days of increased transcript abundance of the demethylases that are associated with stage II gametocytes. b H3K36me2&3 abundance on day 4 of gametocyte development following treatment (24 h, 5 µM) with the Jumonji demethylase inhibitors JIB-04, ML324 or GSK-J4 or the lysine specific demethylase 1 (LSD1) inhibitor 2-PCPA expressed as fold changes (treated/parallel untreated control gametocytes) of relative density units (RDU). c Functional classification of differentially expressed genes (log2 fold change ≥ 0.5 in either direction) with decreased (blue) or increased (red) transcript abundance in response to the inhibition of histone demethylase activity by JIB-04. The % of differentially expressed genes assigned to each biological category is shown. The proportion (%) of differentially expressed genes s that had H3K36me2/3 occupancy (light grey) or enrichment (dark grey) in the stage II gametocytes (data representative of two independent biological repeats) are shown on the two inner most segments. Transcript levels in the untreated and JIB-04 treated gametocytes are exemplified for certain differentially expressed genes. d Comparison of differentially expressed genes (blue and pink) in gametocytes (GC) treated with JIB-04 in this study with differentially expressed genes in JIB-04 treated asexual parasites (shown in grey, [38]), immature gametocytes (imGC) treated with the G9a-specific inhibitor BIX-0129 [69] and imGC treated with the HDAC inhibitor Trichostatin A [70]. Full descriptions of gene names and their PlasmoDB IDs are available in Additional file 2: Table S4
We were particularly interested in the transcriptional effects of JIB-04 in gametocytes. As such, we performed genome-wide transcriptional profiling of stage II gametocytes treated with JIB-04 for 24 h (Additional file 3: Table S7). JIB-04-treated gametocytes displayed differential expression of ~ 13% of the genome (711 and 696 decreased and increased abundance, respectively, Fig. 5c, Additional file 1: Figure S3B), similar to the restricted effect induced by this inhibitor in asexual parasites [38]. Differentially expressed genes with decreased transcript abundance are associated with various processes including chromatin organisation (3%), gene expression (12%) and transport (6%), each of which have confirmed roles in gametocyte differentiation and development [30, 36, 51, 53–55, 65–67] (Fig. 5c), but are also seen in asexual parasites [38] (Fig. 5d). Interestingly, we also find an increase in the transcript levels of jmjc2 and the histone acetyltransferase-encoding genes, gcn5 (PF3D7_0823300) and myst (PF3D7_1118600) (Fig. 5c) [14, 68], which may reflect an attempt to counteract the abnormal histone methylation patterns induced by JIB-04. While JIB-04-treated gametocytes do not share common differentially expressed genes with the G9a-specific HMT inhibitor, BIX-01294 [69], some overlap is present with HDAC inhibition by trichostatin A [70]. This is in line with other studies which showed similarities in the gene expression signatures of JIB-04 and TSA in cancer cell lines [71].
Of the genes differentially expressed due to JMJ inhibition, 25% are also enriched for H3K36me2&3 and whilst this confirms the involvement in JMJ activity and removal of H3K36 methylation marks, we cannot exclude that other marks, e.g. H3K9me is not also involved. Indeed, the use of this general Jumonji HDM inhibitor highlights the importance of normal histone methylation patterns for transcriptional reprogramming during gametocyte differentiation and development that involves H3K36 methylation.
Discussion
Understanding the gene regulatory mechanisms that drive differentiation and development in P. falciparum gametocytes is essential for the discovery and advancement of novel malaria transmission-blocking strategies [72]. Here, we indicate a dynamic yet stage-specific nature of H3K36me2&3 enrichment in stage II gametocytes, associated transcriptional reprogramming occurring post-commitment that is required to drive gametocyte development in P. falciparum.
Our approach allowed for the delineation of the genomic regions dynamically occupied by H3K36me2&3 during gametocytogenesis. We demonstrate that the location-dependent outcome of H3K36me2&3 deposition to either intergenic regions or CDRs in regulating gene expression is retained in P. falciparum gametocytes. The stage II gametocyte-specific abundance of H3K36me2&3 manifests as a wide-spread, yet largely intergenic enrichment that results chiefly in transcriptional repression, congruent with the characteristic patterns of these hPTMs when functioning repressively in other organisms, particularly during cellular differentiation [73–81]. CDR enrichment is associated with transcriptionally permissive genes, similar to other eukaryotes [22, 82, 83]. The distribution of H3K36me2&3 enrichment and the associated transcriptional effects in stage II gametocytes aligns with the SET2-mediated deposition of H3K36me2&3 in the CDRs of active var genes in asexual parasites, while enrichment of this hPTM upstream of the TSSs silences these genes [22, 60]. These similar patterns of H3K36me2&3-associated transcriptional repression in stage II gametocytes and presence of transcript levels for set2 during early development [36], suggests this HMT is the most likely candidate enzyme of H3K36 methylation in gametocytes. The proposed shared regulatory mechanisms of H3K36 methylation between asexual parasites and gametocytes are supported by similar hypermethylation of this PTM in both asexual parasites [38] and here in gametocytes, by either pan-reactive JMJ inhibitors (like JIB-04) or even with the JMJC2-specific inhibitor, ML324.
Accumulating evidence demonstrates that epigenetic regulators are essential drivers of the transcriptional reprogramming necessary for cellular differentiation [73, 84–86]. Accordingly, for P. falciparum gametocyte differentiation, we indicate a role of H3K36me2&3 in governing transcriptional shifts that coincide with the transition from early differentiation to intermediate gametocyte development. The H3K36me2&3-associated repression of genes involved in asexual proliferation-specific processes (e.g. DNA replication), does indeed attest to the influential role of H3K36me2&3 at this transition point. Additionally, the enrichment of genes that are upregulated under conditions of lysoPC depletion links H3K36me2&3 with the parasite’s earliest responses to the environmental cue for sexual commitment [50]. Specifically, we distinguish an exclusive association between H3K36me2&3 and the post-commitment repression identified previously for hp1 and ap2-g [36, 87] and as such, provide the first insights into the mechanisms that govern the regulation of these genes in differentiated gametocytes [36, 87]. While we find that H3K36me2&3 function largely independently of other regulatory mechanisms, we do find an association between these hPTMs and AP2-G2 regulation in gametocytes as reported previously for asexual parasites [58] as well as a link between H3K36me2&3 and the formation of heterochromatin in early gametocytes in accordance with similar observations in other organisms [75, 88, 89].
Conclusions
This work highlights the crucial nature of the epigenetic mechanisms underlying the vast transcriptional reprogramming associated with P. falciparum gametocyte differentiation [11, 30, 35, 36]. Here, we demonstrated an association between H3K36me2&3 and the repression of proliferation- and commitment-specific transcripts once they become obsolete in differentiated gametocytes. This marks H3K36me2&3 as important hallmarks of a transition point in gametocyte development from early differentiation to intermediate development. We conclude that the transcriptional repression associated with H3K36me2&3 enrichment occurs largely independent of other mechanisms described to date for P. falciparum parasites. Importantly, we identify an exclusive association between H3K36me2&3 enrichment and hp1 and ap2-g (25, 31), thereby providing the first insights into the mechanisms governing the regulation of these genes in the post-commitment, terminally differentiated gametocytes. The findings of this study make substantial contributions to the understanding of epigenetics in P. falciparum gametocytes which until recently had remained largely unexplored.
Methods
Parasite culturing
P. falciparum NF54 asexual parasite cultures were maintained in vitro at 37 °C in human erythrocytes at a 5% haematocrit (ethics approval obtained from the University of Pretoria Research Ethics Committee, Health Sciences Faculty 506/2018) and synchronised using 5% D-sorbitol as previously described [90, 91]. P. falciparum gametocytes were induced as previously described [92–94] through a combination of nutrient starvation and a drop in haematocrit from synchronous (≥ 95% ring stages) asexual parasite cultures (0.5% parasitaemia, 6% haematocrit). Media (RPMI-1640, 23.8 mM Na2CO3, 0.024 mg/ml gentamycin, 25 mM HEPES, 0.2 mM hypoxanthine and 0.5% (w/v) Albumax II) was replaced 48 h after initiation. The haematocrit was reduced to 4% 72 h after initiation (hereafter referred to as day 0) and maintained as such throughout gametocyte development with daily replacement of glucose-enriched (20 mM) media. Residual asexual parasites were removed by supplementing glucose-enriched media with 50 mM N-acetylglucosamine (Sigma-Aldrich) from day 1 of development. Gametocytes were sampled on days 2, 4 and 7 post-induction as the pre-stage II, stage II and post-stage II samples, respectively. Two independent cultures were used as biological replicates for each ChIP-seq and ChIP-qPCR experiment. Parasite proliferation and gametocyte differentiation were monitored daily by microscopic evaluation of Giemsa-stained smears.
Antibody validation
The specificity of commercially obtained ChIP-grade rabbit anti-H3K36me2 (Abcam, ab9049) and anti-H3K36me3 (Abcam, ab9050) antibodies (same batch numbers as previously validated for specificity against H3K36me2&3, [22, 34, 39–41, 43]) was tested using modified and unmodified synthetic peptides (Genscript) of the P. falciparum 3D7 histone H3 sequence as per ENCODE standards (www.encodeproject.org; Additional file 4: Table S8). Modified peptides were di- or tri-methylated on K9 or K36 each with a corresponding unmodified K9 (ARTKQTARKSTAGKAPRKQ) and K36 (ARKSAPISAGIKKPHRYRPG) peptide. Nitrocellulose membranes spotted with 25 ng of each peptide were blocked for 30 min in blocking buffer (5% milk powder in TBS-t (50 mM Tris (pH 7.5), 150 mM NaCl and 0.1% (v/v) tween-20) and incubated with primary antibodies (1:5000) against H3K36me2 or H3K36me3 overnight followed 1 h incubation with secondary antibody (1:10,000 goat anti-rabbit IgG conjugated to HRP). Chemiluminescent signal (Pierce Super Signal West Pico PLUS Chemiluminescent Substrate) was quantified by densitometry using ImageJ analysis software [95].
Chromatin immunoprecipitation
ChIP was performed as previously described [31] with modifications. Gametocytes (1–3% gametocytaemia, fixed in 1% formaldehyde for 10 min at 37 °C followed by 0.125 mM glycine quenching) were released from erythrocytes using 0.1% (w/v) saponin. Thereafter, the gametocytes were resuspended in lysis buffer (10 mM Hepes (pH 7.9), 10 mM KCl, 0.1 mM EDTA (pH 8.0), 0.1 mM EGTA (pH 8.0)) and lysed with 0.25% Nonidet P-40 and douncing. Nuclei were then resuspended in Covaris shearing buffer (0.1% sodium dodecyl sulfate (SDS), 10 mM Tris (pH 8.1), 1 mM EDTA) and sonicated with the conditions: 5% duty cycle, 75 W peak incident power, 200 cycles per burst for a total treatment time of 300 s using a M220 ultrasonicator (Covaris). The sonicated chromatin was then pre-cleared using Protein A/G magnetic beads (Millipore 16–663) and aside from a small quantity kept separately as input material, chromatin was incubated overnight with 3 µg of anti-H3K36me2 or anti-H3K36me3. Reversal of cross-linking was achieved by adding 0.2 M NaCl followed by RNAseA and ProteinaseK treatment. DNA was purified using the Qiagen MinElute kit and used to prepare DNA libraries for sequencing and qPCR validation.
Library preparation and sequencing
DNA libraries were prepared for sequencing as previously described [31]. End-repair and A-tailing of DNA fragments were achieved using NEBNext End Repair Enzyme Mix (#E6051A) and 3′-5′ exo Klenow fragment (#E6054A). Indexed adaptors (NEXTflex DNA-Seq barcoded adaptors) were ligated (NEB Quick Ligase, #M2200L) to fragments that were then size selected (250 bp) using 0.7 × AMPure XP beads (Beckman Coulter). The selected fragments were amplified using KAPA HiFi with dNTPs, and NEXTflex primer mix (#NOVA-514107–96). The PCR products were purified (0.9 × AMPure XP beads) and quantified using the Qubit fluorometer HS DNA kit. Sequencing was carried out on an Illumina HiSeq 2500.
ChIP-qPCR
ChIP-qPCR was performed on the same material used for ChIP-seq with primers listed in Additional file 4: Table S9. Using serial dilutions of 3D7 genomic DNA all primers were determined to be > 90% efficient and specific, evidenced by single peaks on the melting curves. ChIP-qPCR data were obtained using the Applied Biosystems 7500 Real-Time PCR machine and SDS v1.4 and analysed using the ΔΔCt method. Values are expressed as fold enrichment of immunoprecipitated to input DNA, averaged for two biological replicates for H3K36me2&3 in stage II and post-stage II gametocytes. Pre-stage II gametocyte data represents only 1 replicate as insufficient DNA was obtained for the second replicate samples.
Detection of changes in histone methylation
Gametocytes were treated with JIB-04 (5 µM) on days 2, 3 and 4 of development and sampled 24 h later. Histones were extracted as described before [17] with minor modifications. Nuclei were extracted by Dounce homogenisation in hypotonic lysis buffer (10 mM Tris–HCl (pH 8), 0.25 M sucrose, 3 mM MgCl2, 0.2% (v/v) Nonidet P-40) and protease inhibitors (Roche)). Histones were isolated from nuclei resuspended in Tris buffer (10 mM Tris (pH 8.0), 0.8 M NaCl, 1 mM EDTA and protease inhibitors) by overnight acid-extraction (0.25 M HCl, with rotation at 4 °C) and subsequent precipitated with 20% TCA. Histone pellets were washed with acetone, reconstituted in dddH2O and spotted quantitatively (100 ng per sample) on nitrocellulose membranes. Membranes were submerged in blocking buffer for 30 min followed by 1 h incubation with α-H3K36me2 (Abcam, ab9049) or α-H3K36me3 (Abcam, ab9050) primary antibody dilutions (1:5000 in TBS-t). Membranes were washed three times in TBS-t and then incubated with goat α-rabbit IgG antibody conjugated to HRP (1:10000) for 1 h. Chemiluminescent signal (Pierce SuperSignal West Pico PLUS Chemiluminescent Substrate) was quantified with ImageJ analysis software [95].
DNA microarrays
DNA microarrays (60-mer, Agilent Technologies, USA) based on the full P. falciparum genome as previously described [96] were used to assess global transcriptomic changes in gametocytes treated with JIB-04. Day 2 and day 3 gametocyte cultures (1–3% gametocytaemia, 4% haematocrit) were treated with 5 µM JIB-04 (Cayman Chemicals) for 24 h followed by isolation of gametocytes using 0.01% (w/v) saponin. Total RNA was isolated with a combination of TRIzol (Sigma-Aldrich, USA) and phenol–chloroform extraction and subsequently used to synthesise cDNA as previously described [96] for the untreated and JIB-04 treated day 2 and day 3 gametocyte samples. Sample cDNA was labelled with Cy5 dye (GE Healthcare, USA) prior to hybridisation to arrays with an equal amount (350–500 ng) of Cy3-labelled (GE Healthcare, USA) reference pool containing equal amounts of cDNA from each gametocyte sample and mixed stage 3D7 asexual parasites. After hybridisation, the slides were scanned on a G2600D (Agilent Technologies, USA) scanner and normalised signal intensities for each oligo were extracted using the GE2_1100_Jul11_no_spikein protocol and Agilent Feature Extractor Software (v 11.5.1.1) as described before [96].
Data analysis: ChIP-seq
ChIP-seq data analysis was performed according to ENCODE guidelines [97] and as described previously [31, 58]. Sequence read quality was determined using FastQC [98] prior to analysis and adapter sequences were removed using Trimmomatic (v0.32.3) [99]. Reads were mapped to the P. falciparum 3D7 genome (v39 obtained from PlasmoDB) and duplicate and low-quality reads filtered using BWA-MEM (v0.4.1) [100] and SAMtools (v1.1.2) [101], respectively. Correlations between corresponding biological replicates were determined prior to subsequent analysis. The deepTools suite (v3.1.2.0.0) [102] was used to plot the average occupancy of hPTMs (plotProfile and plotHeatmap tools) and to generate bigwig files that were viewed in IGV [103]. Genome-wide occupancy was calculated and is reported as log2-transformed ChIP/input ratios averaged for 1 kb bins. Occupancy 1.5 kb upstream of gene TSSs (TSSs as defined by Adjalley et al., 2016 [45]; PlasmoDB, genome release v39) and within the CDR was calculated from log2ChIP/input ratios over 50 bp bins averaged across these regions. MACS2 [104] was used with the broad peak option (q-value < 0.05; ‘-broad—cutoff 0.1′) to call peaks as described by others [31, 58, 105]. As two biological replicates were used for each experiment, only regions enriched in both biological replicates were used for all subsequent analyses. These were identified and annotated using the Multiple Intersect function and ClosestBED, respectively, from the BEDtools suite [106]. All results are representative of data averaged for two biological replicates unless otherwise stated. GO enrichment analyses were performed with PlasmoDB using a P-value cut-off ≤ 0.05.
Data analysis: DNA microarrays
Microarray signal intensities that passed Genepix standard background filters (P < 0.01), were normalised with Robust-spline for within-array and G-quantile for between slide normalisation in R (v3.2.3, www.r-project.org) using the limma and marray packages. The fit of a linear model was used to obtain log2-transformed expression values (log2Cy5/Cy3). Genes with log2-transformed FC ≥ 0.5 in either direction were defined as differentially expressed in the JIB-04-treated gametocytes. Visualisation of differentially expressed genes was performed using TIGR MeV with functional classification of genes to significantly associated biological processes (P-values ≤ 0.05) performed using PlasmoDB (v39, www.plasmodb.org).
Supplementary Information
Additional file 1. Figures and corresponding legends for Figures S1–S3 as a supplement to main text and figure.
Additional file 2: Table S1. Pearson correlation of genome-wide read counts binned into 1 kb for corresponding biological replicates used for ChIP-seq. Table S2. Log2-transformed ChIP/input values for H3K36me2&3 occupancy upstream of 5602 P. falciparum genes in pre-stage II, stage II and post-stage II gametocytes. Table S3. Sites of H3K36me2&3 enrichment that were present in both biological replicates as determined by peak calling for stage II gametocytes. Table S4. Expression values on days 0 to 10 of gametocyte development for the gene nearest to each significant peak. Table S5. Gene ontology analysis results for biological processes significantly associated with H3K36me2&3-enriched gene sets in stage II gametocytes. Table S6. H3K36me2&3-enriched genes in stage II gametocytes previously identified as commitment- or early differentiation-related or associated with HP1, AP2-G2 or AP2-G2 in other studies [27, 30, 31, 36, 50–54, 58].
Additional file 3: Table S7. Gene expression values and log2fold changes from microarray experiments for the untreated and JIB-04 treated gametocytes on day 2 and 3 with associated functional categories for each gene.
Additional file 4: Table S8. Sequence information of peptides used for antibody validation. Table S9. Primer sequences used in ChIP-qPCR validation.
Abbreviations
- CDR
Coding region
- ChIP-seq
Chromatin immunoprecipitation followed by sequencing
- FC
Fold change
- GO
Gene ontology
- H3K36me2&3
Histone 3 lysine 36 di- and tri-methylation
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylase
- HMT
Histone methyltransferase
- HP1
Heterochromatin protein 1
- hPTM
Histone post-translational modification
- JMJC
Jumonji-C
- KDM
Lysine demethylase
- LSD1
Lysine-specific demethylase 1
- lysoPC
Lysophosphatidylcholine
- PC
Phosphatidylcholine
- TSS
Transcription start site
Authors' contributions
JC conducted the experimental work and data analysis and JR provided technical assistance. GJ and LO provided training and assistance with laboratory experiments. Experiments were conducted in the laboratories of ML and LMB. LMB conceptualised the work and supervised the project. JC and LMB wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by the South African Medical Department of Science and Innovation and National Research Foundation South African Research Chairs Initiative Grant (LMB UID: 84627).
Availability of data and materials
The ChIP-seq datasets generated and supporting the conclusions of this article are deposited in the NCBI Sequence Read Archive, accession number GSE163432 available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163432. DNA microarray data are available from the Gene Expression Omnibus repository, accession number GSE163189, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163189. Microarray time-course data [36] were obtained from the Gene Expression Omnibus (GEO) under the accession number GSE104889 and presented as expression values (log2Cy5/Cy3) in heatmaps generated using TIGR MeV. Additional data sets pertaining to H3K9me3/HP1 in early gametocytes (GSE102695) [30], AP2-G binding sites (GSE120448 and SRP091939) [31] and AP2-G2 binding sites [58] were obtained from GEO and SRA [30, 31, 35]. The panel of commitment- and development-specific genes was compiled from supplementary data files associated with previous publications [27, 30, 31, 36, 50–54]. Data pertaining to the transcriptome analysis of JIB-04 in asexual parasites, BIX-01294 and TSA were obtained from the supplementary files associated with [38, 69, 70].
Declarations
Ethics approval and consent to participate
The human erythrocytes used in this study were collected from healthy volunteers with due approval from the University of Pretoria Health Sciences Ethics Committee (506/2018).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World malaria report . 20 years of global progress and challenges. Geneva: World Health Organization; 2020. p. 2020. [Google Scholar]
- 2.Bruce MC, Alano P, Duthie S, Carter R. Commitment of the malaria parasite Plasmodium falciparum to sexual and asexual development. Parasitology. 1990;100(Pt 2):191–200. doi: 10.1017/S0031182000061199. [DOI] [PubMed] [Google Scholar]
- 3.Josling GA, Llinas M. Sexual development in Plasmodium parasites: knowing when it's time to commit. Nat Rev Microbiol. 2015;13(9):573–587. doi: 10.1038/nrmicro3519. [DOI] [PubMed] [Google Scholar]
- 4.Bancells C, Llora-Batlle O, Poran A, Notzel C, Rovira-Graells N, Elemento O, et al. Revisiting the initial steps of sexual development in the malaria parasite Plasmodium falciparum. Nat Microbiol. 2019;4(1):144–154. doi: 10.1038/s41564-018-0291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josling GA, Williamson KC, Llinas M. Regulation of Sexual Commitment and Gametocytogenesis in Malaria Parasites. Annu Rev Microbiol. 2018;72:501–519. doi: 10.1146/annurev-micro-090817-062712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrows JN, Duparc S, Gutteridge WE, van Huijsduijnen R, Kaszubska W, Macintyre F, et al. New developments in anti-malarial target candidate and product profiles. Malar J. 2017;16(1):26. doi: 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui L, Lindner S, Miao J. Translational regulation during stage transitions in malaria parasites. Ann N Y Acad Sci. 2015;1342:1–9. doi: 10.1111/nyas.12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horrocks P, Wong E, Russell K, Emes RD. Control of gene expression in Plasmodium falciparum - ten years on. Mol Biochem Parasitol. 2009;164(1):9–25. doi: 10.1016/j.molbiopara.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Llinas M, Deitsch KW, Voss TS. Plasmodium gene regulation: far more to factor in. Trends Parasitol. 2008;24(12):551–556. doi: 10.1016/j.pt.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Cortes A, Deitsch KW. Malaria Epigenetics. Cold Spring Harb Perspect Med. 2017;7:7. doi: 10.1101/cshperspect.a025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coetzee N, Sidoli S, van Biljon R, Painter H, Llinas M, Garcia BA, et al. Quantitative chromatin proteomics reveals a dynamic histone post-translational modification landscape that defines asexual and sexual Plasmodium falciparum parasites. Sci Rep. 2017;7(1):607. doi: 10.1038/s41598-017-00687-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duffy MF, Selvarajah SA, Josling GA, Petter M. Epigenetic regulation of the Plasmodium falciparum genome. Brief Funct Genomics. 2014;13(3):203–216. doi: 10.1093/bfgp/elt047. [DOI] [PubMed] [Google Scholar]
- 13.Bartfai R, Hoeijmakers WA, Salcedo-Amaya AM, Smits AH, Janssen-Megens E, Kaan A, et al. H2A.Z demarcates intergenic regions of the Plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog. 2010;6(12):e1001223. doi: 10.1371/journal.ppat.1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui L, Miao J, Furuya T, Li X, Su XZ, Cui L. PfGCN5-mediated histone H3 acetylation plays a key role in gene expression in Plasmodium falciparum. Eukaryot Cell. 2007;6(7):1219–1227. doi: 10.1128/EC.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Rubio JJ, Gontijo AM, Nunes MC, Issar N, Hernandez Rivas R, Scherf A. 5' flanking region of var genes nucleate histone modification patterns linked to phenotypic inheritance of virulence traits in malaria parasites. Mol Microbiol. 2007;66(6):1296–1305. doi: 10.1111/j.1365-2958.2007.06009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salcedo-Amaya AM, van Driel MA, Alako BT, Trelle MB, van den Elzen AM, Cohen AM, et al. Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. Proc Natl Acad Sci USA. 2009;106(24):9655–9660. doi: 10.1073/pnas.0902515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trelle MB, Salcedo-Amaya AM, Cohen AM, Stunnenberg HG, Jensen ON. Global histone analysis by mass spectrometry reveals a high content of acetylated lysine residues in the malaria parasite Plasmodium falciparum. J Proteome Res. 2009;8(7):3439–3450. doi: 10.1021/pr9000898. [DOI] [PubMed] [Google Scholar]
- 18.Karmodiya K, Pradhan SJ, Joshi B, Jangid R, Reddy PC, Galande S. A comprehensive epigenome map of Plasmodium falciparum reveals unique mechanisms of transcriptional regulation and identifies H3K36me2 as a global mark of gene suppression. Epigenetics Chromatin. 2015;8:32. doi: 10.1186/s13072-015-0029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venkatesh S, Workman JL. Set2 mediated H3 lysine 36 methylation: regulation of transcription elongation and implications in organismal development. Wiley Interdiscip Rev Dev Biol. 2013;2(5):685–700. doi: 10.1002/wdev.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, et al. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008;6(11):e277. doi: 10.1371/journal.pbio.0060277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22(5):1298–1306. doi: 10.1128/MCB.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang L, Mu J, Zhang Q, Ni T, Srinivasan P, Rayavara K, et al. PfSETvs methylation of histone H3K36 represses virulence genes in Plasmodium falciparum. Nature. 2013;499(7457):223–227. doi: 10.1038/nature12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Rubio JJ, Mancio-Silva L, Scherf A. Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host Microbe. 2009;5(2):179–190. doi: 10.1016/j.chom.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol. 2008;62:445–470. doi: 10.1146/annurev.micro.61.080706.093134. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152(3):642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong MPM, Ng, R. K. Resetting Cell Fate by Epigenetic Reprogramming. In: Logie CaK, T. A., editor. Chromatin and Epigenetics: IntechOpen; 2020. p. 165–206.
- 27.Flueck C, Bartfai R, Volz J, Niederwieser I, Salcedo-Amaya AM, Alako BT, et al. Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLoS Pathog. 2009;5(9):e1000569. doi: 10.1371/journal.ppat.1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brancucci NMB, Bertschi NL, Zhu L, Niederwieser I, Chin WH, Wampfler R, et al. Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host Microbe. 2014;16(2):165–176. doi: 10.1016/j.chom.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Kafsack BF, Rovira-Graells N, Clark TG, Bancells C, Crowley VM, Campino SG, et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature. 2014;507(7491):248–252. doi: 10.1038/nature12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraschka SA, Filarsky M, Hoo R, Niederwieser I, Yam XY, Brancucci NMB, et al. Comparative heterochromatin profiling reveals conserved and unique epigenome signatures linked to adaptation and development of malaria parasites. Cell Host Microbe. 2018;23(3):407–420. doi: 10.1016/j.chom.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Josling GA, Russell TJ, Venezia J, Orchard L, van Biljon R, Painter HJ, et al. Dissecting the role of PfAP2-G in malaria gametocytogenesis. Nat Commun. 2020;11(1):1503. doi: 10.1038/s41467-020-15026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha A, Hughes KR, Modrzynska KK, Otto TD, Pfander C, Dickens NJ, et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 2014;507(7491):253–257. doi: 10.1038/nature12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bunnik EM, Polishko A, Prudhomme J, Ponts N, Gill SS, Lonardi S, et al. DNA-encoded nucleosome occupancy is associated with transcription levels in the human malaria parasite Plasmodium falciparum. BMC Genomics. 2014;15:347. doi: 10.1186/1471-2164-15-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cui L, Miao J. Chromatin-mediated epigenetic regulation in the malaria parasite Plasmodium falciparum. Eukaryot Cell. 2010;9(8):1138–1149. doi: 10.1128/EC.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bunnik EM, Cook KB, Varoquaux N, Batugedara G, Prudhomme J, Cort A, et al. Changes in genome organization of parasite-specific gene families during the Plasmodium transmission stages. Nat Commun. 2018;9(1):1910. doi: 10.1038/s41467-018-04295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Biljon R, van Wyk R, Painter HJ, Orchard L, Reader J, Niemand J, et al. Hierarchical transcriptional control regulates Plasmodium falciparum sexual differentiation. BMC Genomics. 2019;20(1):920. doi: 10.1186/s12864-019-6322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanheer LN, Zhang H, Lin G, Kafsack BFC. Activity of Epigenetic Inhibitors against Plasmodium falciparum Asexual and Sexual Blood Stages. Antimicrob Agents Chemother. 2020;64:7. doi: 10.1128/AAC.02523-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthews KA, Senagbe KM, Notzel C, Gonzales CA, Tong X, Rijo-Ferreira F, et al. Disruption of the Plasmodium falciparum life cycle through transcriptional reprogramming by inhibitors of Jumonji Demethylases. ACS Infect Dis. 2020;6(5):1058–1075. doi: 10.1021/acsinfecdis.9b00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, et al. The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol Cell. 2018;70(2):371–379. doi: 10.1016/j.molcel.2018.02.027. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Wang Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J Biol Chem. 2017;292(28):11951–11959. doi: 10.1074/jbc.M117.788224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DiFiore JV, Ptacek TS, Wang Y, Li B, Simon JM, Strahl BD. Unique and shared roles for histone H3K36 methylation states in transcription regulation functions. Cell Rep. 2020;31(10):107751. doi: 10.1016/j.celrep.2020.107751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu B, Liu Y, Wang B, Luo Q, Shi J, Gan J, et al. The transcription factor OsSUF4 interacts with SDG725 in promoting H3K36me3 establishment. Nat Commun. 2019;10(1):2999. doi: 10.1038/s41467-019-10850-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y, Gu L, Li GM. H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J Biol Chem. 2018;293(20):7811–7823. doi: 10.1074/jbc.RA118.002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui L, Fan Q, Cui L, Miao J. Histone lysine methyltransferases and demethylases in Plasmodium falciparum. Int J Parasitol. 2008;38(10):1083–1097. doi: 10.1016/j.ijpara.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adjalley SH, Chabbert CD, Klaus B, Pelechano V, Steinmetz LM. Landscape and Dynamics of Transcription Initiation in the Malaria Parasite Plasmodium falciparum. Cell Rep. 2016;14(10):2463–2475. doi: 10.1016/j.celrep.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, et al. H3K36me2/3 binding and DNA binding of the DNA Methyltransferase DNMT3A PWWP domain both contribute to its chromatin interaction. J Mol Biol. 2019;431(24):5063–5074. doi: 10.1016/j.jmb.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 47.Mauser R, Kungulovski G, Keup C, Reinhardt R, Jeltsch A. Application of dual reading domains as novel reagents in chromatin biology reveals a new H3K9me3 and H3K36me2/3 bivalent chromatin state. Epigenetics Chromatin. 2017;10(1):45. doi: 10.1186/s13072-017-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sui P, Shi J, Gao X, Shen WH, Dong A. H3K36 methylation is involved in promoting rice flowering. Mol Plant. 2013;6(3):975–977. doi: 10.1093/mp/sss152. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360:6388. doi: 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brancucci NMB, Gerdt JP, Wang C, De Niz M, Philip N, Adapa SR, et al. Lysophosphatidylcholine regulates sexual stage differentiation in the human malaria parasite Plasmodium falciparum. Cell. 2017;171(7):1532–1544. doi: 10.1016/j.cell.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eksi S, Haile Y, Furuya T, Ma L, Su X, Williamson KC. Identification of a subtelomeric gene family expressed during the asexual-sexual stage transition in Plasmodium falciparum. Mol Biochem Parasitol. 2005;143(1):90–99. doi: 10.1016/j.molbiopara.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 52.Eksi S, Morahan BJ, Haile Y, Furuya T, Jiang H, Ali O, et al. Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLoS Pathog. 2012;8(10):e1002964. doi: 10.1371/journal.ppat.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filarsky M, Fraschka SA, Niederwieser I, Brancucci NMB, Carrington E, Carrio E, et al. GDV1 induces sexual commitment of malaria parasites by antagonizing HP1-dependent gene silencing. Science. 2018;359(6381):1259–1263. doi: 10.1126/science.aan6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikadai H, Shaw Saliba K, Kanzok SM, McLean KJ, Tanaka TQ, Cao J, et al. Transposon mutagenesis identifies genes essential for Plasmodium falciparum gametocytogenesis. Proc Natl Acad Sci USA. 2013;110(18):E1676–E1684. doi: 10.1073/pnas.1217712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silvestrini F, Lasonder E, Olivieri A, Camarda G, van Schaijk B, Sanchez M, et al. Protein export marks the early phase of gametocytogenesis of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2010;9(7):1437–1448. doi: 10.1074/mcp.M900479-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morahan BJ, Strobel C, Hasan U, Czesny B, Mantel PY, Marti M, et al. Functional analysis of the exported type IV HSP40 protein PfGECO in Plasmodium falciparum gametocytes. Eukaryot Cell. 2011;10(11):1492–1503. doi: 10.1128/EC.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coleman BI, Skillman KM, Jiang RHY, Childs LM, Altenhofen LM, Ganter M, et al. A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe. 2014;16(2):177–186. doi: 10.1016/j.chom.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh S, Santos JM, Orchard LM, Yamada N, van Biljon R, Painter HJ, et al. The PfAP2-G2 transcription factor is a critical regulator of gametocyte maturation. Mol Microbiol. 2020;00:1–20. doi: 10.1111/mmi.14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Contet A, Pihan E, Lavigne M, Wengelnik K, Maheshwari S, Vial H, et al. Plasmodium falciparum CTP:phosphocholine cytidylyltransferase possesses two functional catalytic domains and is inhibited by a CDP-choline analog selected from a virtual screening. FEBS Lett. 2015;589(9):992–1000. doi: 10.1016/j.febslet.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 60.Ukaegbu UE, Kishore SP, Kwiatkowski DL, Pandarinath C, Dahan-Pasternak N, Dzikowski R, et al. Recruitment of PfSET2 by RNA polymerase II to variant antigen encoding loci contributes to antigenic variation in P. falciparum. PLoS Pathog. 2014;10(1):e1003854. doi: 10.1371/journal.ppat.1003854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coetzee N, von Gruning H, Opperman D, van der Watt M, Reader J, Birkholtz LM. Epigenetic inhibitors target multiple stages of Plasmodium falciparum parasites. Sci Rep. 2020;10(1):2355. doi: 10.1038/s41598-020-59298-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reader J, Van der Watt ME, Taylor D, Le Manach C, Mittal N, Ottilie S, et al. Multistage and transmission-blocking targeted antimalarials discovered from the open-source MMV Pandemic Response Box. BioRxiv. 2020. [DOI] [PMC free article] [PubMed]
- 63.Sengoku T, Yokoyama S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011;25(21):2266–2277. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46(14):4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 65.Tiburcio M, Silvestrini F, Bertuccini L, Sander AF, Turner L, Lavstsen T, et al. Early gametocytes of the malaria parasite Plasmodium falciparum specifically remodel the adhesive properties of infected erythrocyte surface. Cell Microbiol. 2013;15(4):647–659. doi: 10.1111/cmi.12062. [DOI] [PubMed] [Google Scholar]
- 66.Volz JC, Bartfai R, Petter M, Langer C, Josling GA, Tsuboi T, et al. PfSET10, a Plasmodium falciparum methyltransferase, maintains the active var gene in a poised state during parasite division. Cell Host Microbe. 2012;11(1):7–18. doi: 10.1016/j.chom.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 67.Santos JM, Josling G, Ross P, Joshi P, Orchard L, Campbell T, et al. Red blood cell invasion by the malaria parasite is coordinated by the PfAP2-I transcription factor. Cell Host Microbe. 2017;21(6):731–741. doi: 10.1016/j.chom.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miao J, Fan Q, Cui L, Li J, Li J, Cui L. The malaria parasite Plasmodium falciparum histones: organization, expression, and acetylation. Gene. 2006;369:53–65. doi: 10.1016/j.gene.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 69.Ngwa CJ, Kiesow MJ, Orchard LM, Farrukh A, Llinas M, Pradel G. The G9a histone methyltransferase inhibitor BIX-01294 modulates gene expression during Plasmodium falciparum gametocyte development and transmission. Int J Mol Sci. 2019;20:20. doi: 10.3390/ijms20205087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ngwa CJ, Kiesow MJ, Papst O, Orchard LM, Filarsky M, Rosinski AN, et al. Transcriptional Profiling Defines Histone Acetylation as a Regulator of Gene Expression during Human-to-Mosquito Transmission of the Malaria Parasite Plasmodium falciparum. Front Cell Infect Microbiol. 2017;7:320. doi: 10.3389/fcimb.2017.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. doi: 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Delves MJ. Plasmodium cell biology should inform strategies used in the development of antimalarial transmission-blocking drugs. Future Med Chem. 2012;4(18):2251–2263. doi: 10.4155/fmc.12.182. [DOI] [PubMed] [Google Scholar]
- 73.Yuan S, Natesan R, Sanchez-Rivera FJ, Li J, Bhanu NV, Yamazoe T, et al. Global regulation of the histone mark H3K36me2 underlies epithelial plasticity and metastatic progression. Cancer Discov. 2020;10(6):854–871. doi: 10.1158/2159-8290.CD-19-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suzuki S, Kato H, Suzuki Y, Chikashige Y, Hiraoka Y, Kimura H, et al. Histone H3K36 trimethylation is essential for multiple silencing mechanisms in fission yeast. Nucleic Acids Res. 2016;44(9):4147–4162. doi: 10.1093/nar/gkw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chantalat S, Depaux A, Hery P, Barral S, Thuret JY, Dimitrov S, et al. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res. 2011;21(9):1426–1437. doi: 10.1101/gr.118091.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44(4):609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122(4):517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 78.Rao B, Shibata Y, Strahl BD, Lieb JD. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol. 2005;25(21):9447–9459. doi: 10.1128/MCB.25.21.9447-9459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sinha I, Buchanan L, Ronnerblad M, Bonilla C, Durand-Dubief M, Shevchenko A, et al. Genome-wide mapping of histone modifications and mass spectrometry reveal H4 acetylation bias and H3K36 methylation at gene promoters in fission yeast. Epigenomics. 2010;2(3):377–393. doi: 10.2217/epi.10.18. [DOI] [PubMed] [Google Scholar]
- 80.Woo H, Dam Ha S, Lee SB, Buratowski S, Kim T. Modulation of gene expression dynamics by co-transcriptional histone methylations. Exp Mol Med. 2017;49(4):e326. doi: 10.1038/emm.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu SF, Zhang H, Cairns BR. Genes for embryo development are packaged in blocks of multivalent chromatin in zebrafish sperm. Genome Res. 2011;21(4):578–589. doi: 10.1101/gr.113167.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang C, Zhu B. Roles of H3K36-specific histone methyltransferases in transcription: antagonizing silencing and safeguarding transcription fidelity. Biophys Rep. 2018;4(4):170–177. doi: 10.1007/s41048-018-0063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13(2):115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berr A, McCallum EJ, Menard R, Meyer D, Fuchs J, Dong A, et al. Arabidopsis SET DOMAIN GROUP2 is required for H3K4 trimethylation and is crucial for both sporophyte and gametophyte development. Plant Cell. 2010;22(10):3232–3248. doi: 10.1105/tpc.110.079962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee K, Park OS, Jung SJ, Seo PJ. Histone deacetylation-mediated cellular dedifferentiation in Arabidopsis. J Plant Physiol. 2016;191:95–100. doi: 10.1016/j.jplph.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 86.He C, Chen X, Huang H, Xu L. Reprogramming of H3K27me3 is critical for acquisition of pluripotency from cultured Arabidopsis tissues. PLoS Genet. 2012;8(8):e1002911. doi: 10.1371/journal.pgen.1002911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Llora-Batlle O, Michel-Todo L, Witmer K, Toda H, Fernandez-Becerra C, Baum J, et al. Conditional expression of PfAP2-G for controlled massive sexual conversion in Plasmodium falciparum. Sci Adv. 2020;6(24):eaaz5057. doi: 10.1126/sciadv.aaz5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285(34):26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451(7179):734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 90.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 91.Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65(3):418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 92.Carter R, Ranford-Cartwright L, Alano P. The culture and preparation of gametocytes of Plasmodium falciparum for immunochemical, molecular, and mosquito infectivity studies. Methods Mol Biol. 1993;21:67–88. doi: 10.1385/0-89603-239-6:67. [DOI] [PubMed] [Google Scholar]
- 93.Reader J, Botha M, Theron A, Lauterbach SB, Rossouw C, Engelbrecht D, et al. Nowhere to hide: interrogating different metabolic parameters of Plasmodium falciparum gametocytes in a transmission blocking drug discovery pipeline towards malaria elimination. Malar J. 2015;14:213. doi: 10.1186/s12936-015-0718-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Biljon R, Niemand J, van Wyk R, Clark K, Verlinden B, Abrie C, et al. Inducing controlled cell cycle arrest and re-entry during asexual proliferation of Plasmodium falciparum malaria parasites. Sci Rep. 2018;8(1):16581. doi: 10.1038/s41598-018-34964-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Painter HJ, Altenhofen LM, Kafsack BF, Llinas M. Whole-genome analysis of Plasmodium spp. Utilizing a new agilent technologies DNA microarray platform. Methods Mol Biol. 2013;923:213–219. doi: 10.1007/978-1-62703-026-7_14. [DOI] [PubMed] [Google Scholar]
- 97.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Andrews S. FastQC: a quality control tool for high throughput sequence data 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 99.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42(Web Server issue):W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zaidan NZ, Sridharan R. HP1gamma regulates H3K36 methylation and pluripotency in embryonic stem cells. Nucleic Acids Res. 2020;48(22):12660–12674. doi: 10.1093/nar/gkaa1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Figures and corresponding legends for Figures S1–S3 as a supplement to main text and figure.
Additional file 2: Table S1. Pearson correlation of genome-wide read counts binned into 1 kb for corresponding biological replicates used for ChIP-seq. Table S2. Log2-transformed ChIP/input values for H3K36me2&3 occupancy upstream of 5602 P. falciparum genes in pre-stage II, stage II and post-stage II gametocytes. Table S3. Sites of H3K36me2&3 enrichment that were present in both biological replicates as determined by peak calling for stage II gametocytes. Table S4. Expression values on days 0 to 10 of gametocyte development for the gene nearest to each significant peak. Table S5. Gene ontology analysis results for biological processes significantly associated with H3K36me2&3-enriched gene sets in stage II gametocytes. Table S6. H3K36me2&3-enriched genes in stage II gametocytes previously identified as commitment- or early differentiation-related or associated with HP1, AP2-G2 or AP2-G2 in other studies [27, 30, 31, 36, 50–54, 58].
Additional file 3: Table S7. Gene expression values and log2fold changes from microarray experiments for the untreated and JIB-04 treated gametocytes on day 2 and 3 with associated functional categories for each gene.
Additional file 4: Table S8. Sequence information of peptides used for antibody validation. Table S9. Primer sequences used in ChIP-qPCR validation.
Data Availability Statement
The ChIP-seq datasets generated and supporting the conclusions of this article are deposited in the NCBI Sequence Read Archive, accession number GSE163432 available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163432. DNA microarray data are available from the Gene Expression Omnibus repository, accession number GSE163189, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163189. Microarray time-course data [36] were obtained from the Gene Expression Omnibus (GEO) under the accession number GSE104889 and presented as expression values (log2Cy5/Cy3) in heatmaps generated using TIGR MeV. Additional data sets pertaining to H3K9me3/HP1 in early gametocytes (GSE102695) [30], AP2-G binding sites (GSE120448 and SRP091939) [31] and AP2-G2 binding sites [58] were obtained from GEO and SRA [30, 31, 35]. The panel of commitment- and development-specific genes was compiled from supplementary data files associated with previous publications [27, 30, 31, 36, 50–54]. Data pertaining to the transcriptome analysis of JIB-04 in asexual parasites, BIX-01294 and TSA were obtained from the supplementary files associated with [38, 69, 70].