

Abstract
Altered autophagy accompanied by abnormal autophagic (rimmed) vacuoles detectable by light and electron microscopy is a common denominator of many familial and sporadic non‐inflammatory muscle diseases. Even in the era of next generation sequencing (NGS), late‐onset vacuolar myopathies remain a diagnostic challenge. We identified 32 adult vacuolar myopathy patients from 30 unrelated families, studied their clinical, histopathological and ultrastructural characteristics and performed genetic testing in index patients and relatives using Sanger sequencing and NGS including whole exome sequencing (WES). We established a molecular genetic diagnosis in 17 patients. Pathogenic mutations were found in genes typically linked to vacuolar myopathy (GNE, LDB3/ZASP, MYOT, DES and GAA), but also in genes not regularly associated with severely altered autophagy (FKRP, DYSF, CAV3, COL6A2, GYG1 and TRIM32) and in the digenic facioscapulohumeral muscular dystrophy 2. Characteristic histopathological features including distinct patterns of myofibrillar disarray and evidence of exocytosis proved to be helpful to distinguish causes of vacuolar myopathies. Biopsy validated the pathogenicity of the novel mutations p.(Phe55*) and p.(Arg216*) in GYG1 and of the p.(Leu156Pro) TRIM32 mutation combined with compound heterozygous deletion of exon 2 of TRIM32 and expanded the phenotype of Ala93Thr‐caveolinopathy and of limb‐girdle muscular dystrophy 2i caused by FKRP mutation. In 15 patients no causal variants were detected by Sanger sequencing and NGS panel analysis. In 12 of these cases, WES was performed, but did not yield any definite mutation or likely candidate gene. In one of these patients with a family history of muscle weakness, the vacuolar myopathy was eventually linked to chloroquine therapy. Our study illustrates the wide phenotypic and genotypic heterogeneity of vacuolar myopathies and validates the role of histopathology in assessing the pathogenicity of novel mutations detected by NGS. In a sizable portion of vacuolar myopathy cases, it remains to be shown whether the cause is hereditary or degenerative.
Keywords: autophagy, FSHD, glycogenin 1, muscular dystrophy, myofibrillar myopathy, next generation sequencing (NGS), Pompe disease, sarcotubular myopathy, TRIM32, vacuolar myopathy
Introduction
Vacuolar myopathies are characterized by autophagic vacuoles containing myeloid structures, degrading organelles and other cytoplasmic contents that are detectable by muscle biopsy on the light microscopical (rimmed vacuoles) and/or ultrastructural level. Typical genetic causes include deficiencies in lysosomal enzymes. These defects disturb the lysosomal pathway which is a key element of the autophagy machinery (70), exemplified by glycogen storage disease II (GSD II; Pompe disease) caused by lysosomal alpha glucosidase deficiency, Danon disease caused by mutation of the lysosomal membrane receptor LAMP2 and X‐linked myopathy with excessive autophagy (XMEA) caused by mutation of the a V‐ATPase lysosomal proton channel (9, 38).
Alternatively, defects in the ubiquitin‐proteasome system (UPS) that normally degrades non‐functioning, misfolded ubiquitylated proteins may result in the activation of autophagy and subsequent protein degradation in lysosomes, leading to vacuolar myopathy. The degradative capacity of the UPS may also be overloaded by the accumulation of defective myofibrillar proteins. Such distinct protein aggregates combined with rimmed vacuoles are a characteristic feature of myofibrillar myopathies (85). A vacuolar component may also be present in myopathies that are not caused by mutations in these typical pathways. For example, we previously described prominent vacuolar myopathy in five patients with an unusual facioscapulohumeral muscular dystrophy (FSHD) phenotype (74). Furthermore, drugs which lead to alterations of the autophagic pathway such as chloroquine may cause vacuolar myopathy (64).
Thus, altered autophagy accompanied by abnormal autophagic vacuoles and protein aggregates is a common denominator of many familial and sporadic non‐inflammatory muscle diseases. Hence, even in the era of next generation sequencing (NGS), vacuolar myopathies remain diagnostically challenging especially in late‐onset cases without clear family history. Here, we present results of a comprehensive clinical, neuropathological and genetic workup of a cohort of adult vacuolar myopathy patients. We define morphological features useful to differentiate between different causes and discuss the value of NGS in finding a diagnosis.
Methods
Patients
In this study, we included 32 patients from 30 unrelated families with suspected or genetically confirmed myopathies who had presented at the Dept. of Neurology, Klinikum Kassel (Kassel, Germany) from 2001 to 2017 and had exhibited vacuolar muscle biopsy pathology. Patients with a diagnosis of inclusion body myositis were excluded. Genetic and clinical findings without reference to our biopsy results have already been published for P5.1 (98), P6 (49), P7 (85), P9 (57) and P10 (85).
Clinical examination
In the 32 index patients as well as in 36 additional family members a detailed neurological examination was carried out on several occasions by a neurologist with neuromuscular specialization (AF). In all 68 individuals (patients and family members) serum creatine kinase (CK) levels were measured repeatedly. Needle electromyography (EMG) investigation was carried out in all but two index patients and in four of the family members. In selected cases we performed nerve conduction studies. In 27 of the 32 index patients and in three of the family members muscle MRI of the lower extremities and of the pelvic girdle was performed using axial and coronal T1, T2 and STIR sequences.
Muscle biopsy
Open muscle biopsies were obtained for diagnostic purposes from mildly to moderately affected muscles based on the clinical features and MRI findings. In four cases, additional sural nerve biopsy was performed. Unfixed (cryostat), formaldehyde‐fixed (paraffin) and glutaraldehyde‐fixed (semi‐ and ultrathin) sections were stained using standard histological, enzyme histochemical and immunohistochemical methods (65). Electron microscopy (EM) was performed in 28 out of 32 cases using a Philips CM10 transmission electron microscope; in four patients glutaraldehyde‐fixed muscle tissue for EM was not available. To validate the pathogenicity of detected genetic variants light and electron micrographs were revisited and in selected cases additional targeted immunohistochemistry and further EM was performed. A more detailed description of our neuropathological workflow is provided in Suppl. Methods.
Genetic testing
Written informed consent for genetic testing was obtained from all patients and family members involved in this study. Targeted gene analysis via Sanger sequencing was performed mainly at the Institute of Human Genetics, Würzburg. NGS‐based neuromuscular panel diagnostics was carried out mainly at the Centre for Genomics and Transcriptomics CeGaT (Tübingen, Germany). Custom‐design in‐solution hybridization (Agilent SureSelect, Agilent) was used to select panel genes. Over the course of this study, the panel was expanded several times ranging from 98 genes (version 2a) to 317 genes (version 4). Sequencing was performed on SOLiD s5500xl systems (Life Technologies) or Illumina HiSeq2500 and HiSeq4000 systems to an average read depth between 750× and 1400× per sample. SOLiD color‐space reads were aligned and variants called using lifescope (Life Technologies). HiSeq reads were trimmed (cutadapt 1.4.1 or Skewer 0.1.116) and aligned using bwa mem (0.7.2) against the hg19 reference genome. PCR duplicates were removed (samtools 0.1.18), and only uniquely aligned reads kept for variant calling (CeGaT proprietary software). Variants were called using samtools and VarScan (2.3.5 or 2.4.2). One case was diagnosed via NGS‐neuromuscular panel diagnostics at the Medical Genetics Centre, MGZ (Munich, Germany) that used Agilent SureSelectXT, IlluminaR sequencing technology. Patients and their relatives with non‐conclusive genetic workup were followed‐up for further examination and whole exome sequencing (WES). WES was performed at the Institute of Human Genetics, RWTH Aachen University Hospital. For whole WES enrichment, the Nextera Rapid Capture Exome (v1.2, Illumina, SanDiego, CA, USA) was applied. The exome library was sequenced on a NextSeq500Sequencer with 2x75 cycles on a high‐output flow cell. De‐multiplexing and fastq file generation was done using bcl2fastq2 (v.2.2, Ilumina, San Diego, CA, USA). The generation of the analyzed "bam,” “FastQ” and “vcf” files was carried out with an in‐house pipeline based on SeqMule (v.1.2.6.) (reference genome: hg19). The raw data were analyzed using the variant annotation software kggseq (v1, http://grass.cgs.hku.hk/limx/kggseq/). Variants with a minor allele frequency (MAF) of >0.75% in relevant databases (ie, EXAC, dbSNP, 1000G), synonymous variants and intronic variants that do not affect the canonical splice‐sites were not considered. Bioinformatics pathogenicity assessment (PolyPhen‐2, MutationTaster, SIFT and CADD) was performed, considering that this method only indicates a possible pathogenicity. Mean coverage depth was app. 75X. Segregation analysis was carried out in 29 of the family members.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article and its additional files.
Results
General clinical and genetic findings
For an overview of the clinical features of the patients including CK levels, see Tables 1 and 2. EMG revealed myopathic patterns in 17 index patients, pathogenic, but not clearly distinguishable patterns in 11 and neurogenic and normal patterns in two patients. MRI imaging of the legs showed focal fatty infiltration in 21 patients, diffuse changes in four and widespread fatty degeneration of all leg muscles in two patients. Of the 36 family members examined, 13 were clearly affected and 19 unaffected. Four family members had isolated hyperCKemia and the clinical classification was difficult.
Table 1.
Clinical and genetic characteristics of 17 patients with identified genetic cause for vacuolar myopathy.
| Patient | M/F | Gene | Mutation | Status | Segr. | AAO (y) | AAF/AAL (Y) | Muscle weakness at first/last examination | CK, U/L | Additional clinical features | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Face | Neck | Trunk | UL prox. | UL dist. | LL prox. | LL dist. | ||||||||||
| 1.1 | M | TRIM 32 | NM_012210.3: c.467T>C, p.(Leu156Pro), rs145907585; deletion exon 2 | c.het. | + | 27 | 31 | (+) | − | + | − | − | + | + | 926 | Unathletic since childhood, hypertrophy of quadriceps |
| 43 | (+) | − | + | + | − | +++ | + | |||||||||
| 1.2 | M | TRIM 32 | see Patient 1.1 | c.het | + | 36 | 48 | − | − | − | (+) | − | + | + | 1500 | Atrophy of hamstrings/LL, w/c at 53y |
| 54 | − | − | − | (+) | − | +++ | + | |||||||||
| 2 | M | FKRP | NM_024301.4: c.826C>A, p.(Leu276Ile) | hom. | na | 44 | 53 | − | − | − | ++ | − | ++ | − | 747 | Respiratory muscle weakness, w/c at 58y |
| 68 | − | e++ | − | ++ | − | +++ | + | |||||||||
| 3 | M | FKRP | NM_024301.4: c.C826C>A, p.(Leu276lle); c.1486T>A, p.(*496Argext*21) | c.het. | + | 31 | 32 | − | − | − | − | − | ++ | + | 1775 | Cardiomyopathy, cardioverter defibrillator implant at 37y |
| 41 | − | − | − | (+) | − | ++ | + | |||||||||
| 4 | F | DYSF | NM_003494.3: c.265C>T, p.(Arg89*) | hom. | na | 30 | 34 | − | − | − | − | + | ++ | ++ | 10 036 | Parents are cousins, Pakistani descent |
| 37 | − | − | − | − | + | ++ | ++ | |||||||||
| 5.1 | F | DYSF | NM_003494.3: c.864_865insA, p.(Ser289Ilefs*39); c.3196_3197ins6bp, p.(Glu1065_Gly1066ins2a.a.) | c.het | + | 19 | 20 | − | − | − | ++ | − | ++ | − | 7770 | Trendelenburg's sign bilateral |
| 5.2 | F | DYSF | see Patient 5.1 | c.het | + | 22 | 22 | − | − | − | − | − | + | − | 7370 | − |
| 6 | M | SMCHD1 | NM_015295.2: c.1433G>A, p.(Gly478Glu); 11% methylation of D4Z4 | het. | + | 43 | 44 | + | − | + | + | − | − | − | 458 | Scapular winging R>L, atrophy of pectoralis/shoulder girdle |
| D4Z4 | ||||||||||||||||
| 48 | + | − | + | ++ | − | ++ | − | |||||||||
| 7 | M | DES | NM_001927.3: c.1049G>C, p.(Arg350Pro) | het. | na | 39 | 40 | − | − | − | − | − | ++ | − | 1031 | Pes cavus, quadriceps spared |
| 48 | − | ++ | + | + | − | ++ | ++ | |||||||||
| 8 | M | DES | NM_001927.3: c.1049G>C, p.(Arg350Pro) | het. | na | 41 | 46 | − | − | + | − | − | +++ | + | 2695 | Left leg atrophy, pes cavus, quadriceps spared |
| 52 | − | − | + | (+R) | + | +++ | ++ | |||||||||
| 9 | F | MYOT | NM_006790.2: c.179C>T, p.(Ser60Phe) | het. | na | 60 | 63 | − | − | − | − | − | − | + | 262 | Hypesthesia feet, respiratory weakness, w/c at 72y |
| 75 | − | − | − | − | − | − | ++ | |||||||||
| 10 | F | LDB3 (ZASP) | NM_001080116.1: c.494C>T, p.(Ala165Val) | het. | + | 46 | 49 | − | f+ | − | − | + L | +R | +R | 172 | Myalgia, dysphagia |
| 62 | − | f+ | − | − | + | + | ++ | |||||||||
| 11 | F | COL6A2 | NM_001849.3: c.1861G>A, p.(Asp621Asn) | het. | + | 8 | 23 | + | f+ | − | + | + | + | + | 482 | Contractures of arm flexors |
| 12 | M | GAA | NM_000152.4: c.‐32‐13T>G intronic; c.1796C>A, p.(Ser599Tyr) | c.het | na | 40 | 52 | − | − | + | + | − | + | − | 919 | Scapular winging on R, nocturnal BiPAP therapy at 52y |
| 13 | M | GYG1 | NM_004130.3: c.164_165delTT, p.(Phe55*); c.646C>T, p.(Arg216*) | c.het. | + | 49 | 54 | − | − | − | − | − | (+) | ++ | 306 | Severe foot drop gait |
| 58 | − | − | − | − | − | + | ++ | |||||||||
| 14 | F | GNE | NM_005476.5: c.1760T>C, p.(Ile587Thr) | hom. | na | 23 | 27 | − | − | − | − | − | ++ | + | 276 | Turkish descent, w/c at 36y, sparing of quadriceps |
| 44 | − | − | − | + | + | +++ | +++ | |||||||||
| 15 | M | CAV3 | NM_033337.2: c.277G>A, p.(Ala93Thr) | het. | + | 54 | 55 | − | − | − | − | − | − | − | 960 | Hypesthesia/reduced vibration sense R foot, gout |
| 56 | − | − | − | − | − | − | − | |||||||||
Table 1: For novel mutations the reference SNP ID number is given if available. Table 2: Protein sequences of variants of unclear significance are given. Further details are provided in Table S1.
Abbreviations: 1.1/1.2 = brothers; 5.1/5.2 = sisters; AAF = age at first examination (years); AAL = age at last examination if available (years); AAO = age at onset (years); BiPAP = Biphasic Positive Airway Pressure; c.het = compound heterozygous; CK = creatine kinase (maximum level of repeated measures given); CK normal range (males <170 U/L; females <145 U/L); het. = Heterozygous; hom. = homozygous; e = extension; F = female; f = flexion; L = left; LL dist. = lower limb distal muscles; LL prox. = lower limb proximal muscles; M = male; na = not available; R = right; Segr. = Segregation, + variant segregates within family; UL dist. = upper limb distal muscles; UL prox. = upper limb proximal muscles; w/c = wheelchair bound; y = (years); Muscle weakness was rated according to severity: − no weakness; (+) = mild weakness; + = mild to moderate weakness; ++ = moderate to severe weakness; +++ = plegia; + = variant segregates within family.
Table 2.
Clinical and genetic characteristics of 15 patients without definite identified genetic cause for vacuolar myopathy.
| Patient | M/F | Gene | Variants of unclear significance see Table S1 | Status | AAO (y) | AAF/AAL (Y) | Muscle weakness at first/last examination | CK, U/L | Additional clinical features | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Face | Neck | Trunk | UL prox. | UL dist. | LL prox. | LL dist. | |||||||||
| 16 | M | DYSF | c.2105C>T, p.(Thr702Met) | het. | 7 | 56 | − | − | − | − | +L | − | ++ | 67 | Marked pes cavus, chloroquine therapy for systemic lupus erythematosus |
| DYSF | c.3115C>T, p.(Arg1039Trp) | het. | |||||||||||||
| KIF1B | c.5038C>T, p.(Arg1680Cys) | ||||||||||||||
| 17 | M | TTN | c.13984A>G, p.(Ile4662Val) | het. | 51 | 52 | − | − | − | − | − | − | e+ | 700 | Reduced vibration sense on ankles, hypesthesia R big toe, repetitive discharges in EMG |
| 59 | − | − | − | − | − | − | ++ | ||||||||
| 18 | M | − | − | − | 36 | 56 | − | − | − | (+) | e++ | + | − | 1803 | Finger extension paresis first symptom, six family members affected with camptocormia |
| 61 | − | − | − | (+) | e++ | ++ | e+ | ||||||||
| 19 | M | SYNE2 | c.9854A>T, p.(Asp3285Val) | het. | 55 | 59 | − | − | − | − | − | − | ++ | 767 | Hypertrophy of calves, dysesthesia on feet |
| 72 | − | − | − | − | − | − | ++ | ||||||||
| 20 | M | RYR | c.6617C>T, p.(Thr2206Met) | het. | 30 | 30 | − | − | − | − | − | − | − | 2971 | Presented with rhabdomyolysis, CK levels chronically elevated |
| CAPN3 | c.549delA, p.(Thr184Argfs*36) | het. | 36 | − | − | − | − | − | − | − | |||||
| 21 | M | RYR1 | c.10511C>T, p.(Ser3504Phe) | het. | 47 | 52 | − | − | + | + | − | ++ | − | 8006 | Follicular non‐Hodgkin lymphoma, chemotherapy, Ro52 and SRP autoantibody positive, familial hyperCKemia |
| SYNE2 | c.6194T>C, p.(Met2065Thr) | het. | 55 | − | − | + | + | − | ++ | − | |||||
| 22 | F | DMD | c.4233+2C>T | het. | 59 | 63 | − | − | + | − | − | + | − | 78 | Camptocormia on walking, Trendelenburg's sign on L |
| TTN | see Table S1 | 70 | − | − | ++ | − | − | + | − | ||||||
| 23 | M | SYNE1 | c.1402C>T, p.(Arg468Trp) | het. | 51 | 53 | − | − | − | +L | − | + | + | 373 | Myalgia, raised lactate levels, reduced vibration sense on ankles, waddling gait |
| DNM2 | c.2026C>T, p.(Pro676Ser) | het. | 64 | − | − | − | +L | − | + | + | |||||
| PDYN | c.276C>G, p.(Tyr92*) | het. | |||||||||||||
| 24 | M | − | − | 50 | 63 | − | − | − | (+) | − | − | +L | 397 | Swedish descent, reduced vibration sense on ankles, lumbar spinal canal stenosis | |
| 72 | − | − | − | (+) | − | − | ++ | ||||||||
| 25 | M | DYSF | c.3175C>T, p.(Arg1059Cys) | het. | 20 | 21 | − | − | − | − | e++ | − | − | 1131 | Severe muscle cramps, first symptom moderate paresis of hand/finger extension |
| TTN | c.4396T>C, p.(Phe1466Leu) | het. | 59 | − | e ++ | − | − | e++ | − | e++ | |||||
| 26 | M | SYNE2 | c.13156C>G, p.(Gln4386Glu) | het. | 33 | 54 | − | − | − | − | − | − | − | 396 | Muscle cramps in calves, myalgia |
| ITGA7 | c.1693G>A, p.(Val565Met) | het. | 58 | − | − | − | − | − | − | − | |||||
| 27 | F | − | − | − | 24 | 37 | + | f++ | + | + | + | + | ++ | 1362 | Dysarthria after bronchoscopy, w/c at 42y, myalgia |
| 45 | + | f++ | + | + | + | ++ | +++ | ||||||||
| 28 | M | − | − | − | 44 | 54 | − | − | − | +R | − | ++ | + | 778 | Waddling gait, back pain, pes cavus, claw toes, bilateral Trendelenburg's sign |
| 55 | − | − | − | + | − | ++ | + | ||||||||
| 29 | M | ANO5 | c.155A>G, p.(Asn52Ser) | het. | 56 | 57 | − | − | − | − | − | +L | − | 1255 | Atrophy of left thigh, pes cavus bilateral |
| 59 | − | − | − | − | − | +L | + L | ||||||||
| 30 | M | − | − | − | 52 | 54 | − | − | + | ++ | − | + | e++ | 725 | Scapular winging, atrophy of upper arms, foot drop gait, abdominal muscle paresis, hypesthesia on fingers/toes |
| 55 | − | e+ | ++ | ++ | + | + | +++ | ||||||||
Table 1: For novel mutations the reference SNP ID number is given if available. Table 2: Protein sequences of variants of unclear significance are given. Further details are provided in Table S1.
Abbreviations: 1.1/1.2 = brothers; 5.1/5.2 = sisters; AAF = age at first examination (years); AAL = age at last examination if available (years); AAO = age at onset (years); BiPAP = Biphasic Positive Airway Pressure; c.het = compound heterozygous; CK = creatine kinase (maximum level of repeated measures given); CK normal range (males <170 U/L; females <145 U/L); het. = Heterozygous; hom. = homozygous; e = extension; F = female; f = flexion; L = left; LL dist. = lower limb distal muscles; LL prox. = lower limb proximal muscles; M = male; na = not available; R = right; Segr. = Segregation, + variant segregates within family; UL dist. = upper limb distal muscles; UL prox. = upper limb proximal muscles; w/c = wheelchair bound; y = (years); Muscle weakness was rated according to severity: − no weakness; (+) = mild weakness; + = mild to moderate weakness; ++ = moderate to severe weakness; +++ = plegia; + = variant segregates within family.
In 11 families Sanger sequencing established a genetic diagnosis: Five families were identified with the autosomal recessive (AR) disorders LGMD2B, LGMD2i (LGMDR9, new nomenclature following OMIM) and GSDII, harboring mutations in DYSF, FKRP and GAA, respectively. Four families were diagnosed with autosomal dominant (AD) myofibrillar myopathy caused by mutations in MYOT, LDB3/ZASP and DES. One index patient each showed heterozygous mutations in GNE (hereditary inclusion body myopathy) and SMCHD1, the latter combined with a FSHD permissive haplotype in a digenic manner (FSHD2). Panel NGS identified heterozygous mutations in two patients in COL6A2 (Bethlem myopathy) and CAV3, respectively. Novel compound heterozygous mutations were found in TRIM32 (tripartite motif‐containing protein 32) and GYG1 by NGS in two separate families and were confirmed by segregation analysis, leading to the diagnoses of LGMD2H (LGMDR8, sarcotubular myopathy) and GSD XV, respectively. In 15 patients no definite pathogenic mutation causing vacuolar myopathy was found including one patient who was eventually diagnosed with chloroquine myopathy. WES performed in 12 of these cases yielded variants of unknown significance (Tables 2 and S1), but did not clarify the diagnosis.
General histopathological findings
Sixteen patients showed moderate (affecting <10%), 10 marked (10%–50%) and six severe (>50% of the muscle fibers in the sections examined) myopathic changes including myofibrillar disintegration, non‐subsarcolemmal nuclei, atrophy, basophilia and necrosis. By light microscopy, 16 biopsies exhibited numerous (>10) (rimmed) vacuoles, 10 biopsies had light (1, 2, 3) to moderate (3, 4, 5, 6, 7, 8, 9, 10) vacuolar defects in the sections examined; in six patients a considerable vacuolar myopathy characterized by large abnormal autophagic vacuoles was evident on the ultrastructural level only (Table 3). In 18 cases, there was additional neurogenic muscle fiber atrophy. Sural nerve biopsy revealed a predominantly axonal neuropathy with minor demyelinating component in three patients and a predominantly demyelinating neuropathy in one patient.
Table 3.
Histopathological and ultrastructural findings in 32 patients with vacuolar myopathy.
| Patient | Gene/Cause | Biopsied muscle | Age at biopsy (Y) | LM (rimmed) vacuoles | EM autophagic vacuoles | Exocytosis | Sarcopl. tubulofilaments | Nuclear tubulofilaments | Z‐band streaming | Sarcopl. granulofilaments | Myofibril. disintegration | Nemaline rods | Glycogen deposits | Lipofuscin deposits | Sarcolem. invaginations | Sarcolemmal defects | Myonuclear lobulations | Abnormal chromatin | Mitochondrial inclusions | Mitochondrial defects | SR/T‐tub. proliferation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1.1 | TRIM32 | pLL | 31 | +++ | +++ | − | − | − | − | − | +++ | − | − | ++ | +++ | − | +++ | +++ | − | + | ++ |
| 1.2 | TRIM32 | dLL | 36 | +++ | |||||||||||||||||
| 2 | FKRP | pLL | 53 | ++ | +++ | − | − | − | + | − | +++ | − | + | − | − | − | − | − | ++ | ++ | ++ |
| 3 | FKRP | nk | 40 | +++ | +++ | − | +++ | +++ | ++ | − | ++ | ++ | − | − | − | − | ++ | ++ | − | ++ | + |
| 4 | DYSF | dLL | 34 | + | + | − | − | − | +++ | − | +++ | − | − | − | + | ++ | − | − | − | − | ++ |
| 5.1 | DYSF | pLL | 20 | ++ | +++ | +++ | − | − | − | − | ++ | − | + | − | + | +++ | − | − | − | + | ++ |
| 5.2 | DYSF | pLL | 23 | ++ | +++ | ++ | − | − | + | − | ++ | − | + | + | − | +++ | + | − | − | − | + |
| 6 | SMCHD1 | nk | 44 | +++ | ++ | − | − | − | + | − | − | − | + | − | − | − | ++ | − | − | ++ | ++ |
| 7 | DES | pLL | 40 | +++ | +++ | − | +++ | − | ++ | +++ | +++ | ++ | + | − | − | − | ++ | ++ | − | ++ | − |
| 8 | DES | dLL | 46 | +++ | |||||||||||||||||
| 9 | MYOT | dLL | 63 | ++ | +++ | ||||||||||||||||
| 10 | LDB3/ZASP | dLL | 49 | − | +++ | − | + | − | +++ | ++ | +++ | − | + | − | + | + | ++ | + | + | + | + |
| 11 | COL6A2 | nk | 23 | − | ++ | − | − | − | + | − | − | − | − | − | − | − | − | − | ++ | + | − |
| 12 | GAA | pLL | 52 | +++ | +++ | − | − | − | − | − | − | − | +++ | ++ | − | − | ++ | − | − | ++ | − |
| 13 | GYG1 | nk | 54 | + | +++ | − | ++ | ++ | ++ | − | +++ | − | ++ | − | − | − | ++ | − | − | + | ++ |
| 14 | GNE | nk | 29 | +++ | |||||||||||||||||
| 15 | CAV3 | dLL | 56 | − | +++ | − | − | − | − | − | + | − | − | − | − | − | − | − | − | − | ++ |
| 16 | nk/CHQ | dLL | 57 | +++ | +++ | − | − | − | − | − | ++ | − | − | +++ | − | − | − | − | − | ++ | ++ |
| 17 | nk | pLL | 52 | +++ | +++ | − | +++ | +++ | − | − | ++ | − | − | − | − | − | − | ++ | − | ++ | − |
| 18 | nk | nk | 57 | +++ | +++ | − | − | − | + | − | − | − | − | ++ | − | − | − | − | +++ | ++ | − |
| 19 | nk | dLL | 63 | + | +++ | − | ++ | − | ++ | +++ | ++ | − | ++ | + | − | − | − | − | − | ++ | ++ |
| 20 | nk | nk | 30 | +++ | +++ | − | ++ | − | − | − | +++ | − | + | − | − | − | − | − | − | ++ | − |
| 21 | nk | pLL | 52 | − | +++ | +++ | − | − | + | − | + | − | − | − | − | − | − | − | − | + | − |
| 22 | nk | pLL | 65 | ++ | +++ | − | − | − | − | − | + | − | + | +++ | − | − | − | − | − | ++ | − |
| 23 | nk | pLL | 58 | +++ | +++ | − | − | − | − | − | ++ | ++ | ++ | − | − | − | − | − | +++ | ++ | +++ |
| 24 | nk | pLL | 68 | + | ++ | − | − | − | + | − | + | − | − | − | − | − | − | + | − | ++ | ++ |
| 25 | nk | nk | 58 | + | + | − | − | − | ++ | ++ | +++ | − | + | ++ | ++ | − | ++ | + | ++ | ++ | ++ |
| 26 | nk | pLL | 55 | − | + | − | − | − | + | − | + | − | − | − | − | − | − | − | − | + | − |
| 27 | nk | pUL | 37 | +++ | +++ | ++ | ++ | +++ | − | − | ++ | − | + | − | − | − | − | − | − | ++ | − |
| 28 | nk | dLL | 53 | − | ++ | − | + | − | − | − | + | − | + | − | − | − | − | + | +++ | +++ | − |
| 29 | nk | nk | 57 | +++ | +++ | − | +++ | +++ | − | − | +++ | − | + | − | − | − | ++ | ++ | − | ++ | − |
| 30 | nk | pUL | 55 | +++ | +++ | − | +++ | +++ | ++ | − | +++ | − | − | − | − | − | − | ++ | − | ++ | ++ |
Histopathological features are graded according to frequency: − none, + rare, ++ often, +++ frequent. In patients 1.2, 8, 9 and 14 electron microscopical data were not available.
Abbreviations: CHQ = chloroquine intake; dLL = distal lower limb; dUL = distal upper limb; EM = electron microscopical; LM = light microscopical; myofibril. = myofibrillar; nk = not known; pLL = proximal lower limb; pUL = proximal upper limb; sarcopl. = sarcoplasmatic; sarcolem. = sarcolemmal; SR/T‐tub. = sarcoplasmic reticulum/T‐tubule system; (y) = years.
Vacuolar myopathies with identified pathogenic mutation
GNE myopathy (Hereditary inclusion body myopathy)
P15 who was of Turkish descent had difficulty in climbing stairs at age 23 and was wheelchair bound 13 years later. She developed high‐grade paraparesis and plegia of hip flexion and of foot dorsi‐ and plantarflexion, while quadriceps function was largely spared. Muscle biopsy showed severe vacuolar myopathy with rimmed vacuoles often extending along several sarcomeres (Figure 1A). Sanger sequencing revealed the homozygous GNE c.1760T>C, p.(Ile587Thr) mutation of known pathogenicity (20).
Figure 1.
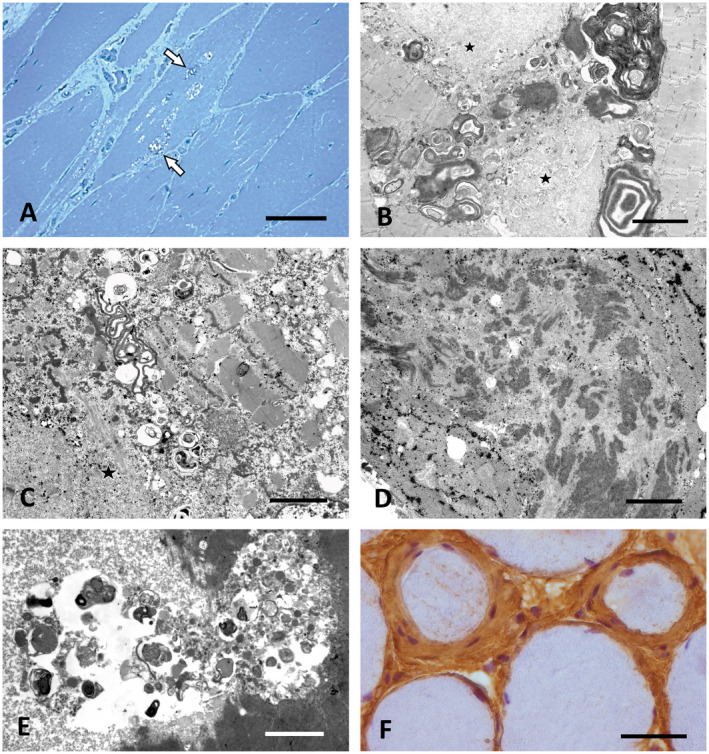
Characteristic morphological findings in vacuolar myopathies. A. Prominent vacuoles containing granular osmiophilic material (arrows) in GNE myopathy (P14). Semithin section, toluidine blue. Scale bar = 40 µm. B,C. Large osmiophilic membranous and granular inclusions indicative of altered autophagy combined with granulofilamentous material and other myofibrillar breakdown products (asterisks) in desminopathy (P7). EM. Scale bar in B = 3 µm, in C = 1.5 µm. D. Filamentous bundles characteristic for ZASPopathy in P10. EM. Scale bar = 3 µm. E. Large autophagic vacuole containing pleomorphic granular and membranous material in a case of Bethlem myopathy (P11) caused by COL6A2 mutation. EM. Scale bar = 2 µm. F. Prominent endomysial fibrosis with concentric accumulation of collagen and of fibroblasts around individual muscle fibers in P11. Paraffin section, Coll VI immunohistochemistry (brown). Scale bar = 30 µm.
Myofibrillar myopathy
In four cases, the diagnosis was myofibrillar myopathy of which P9 and P10 presented with weakness of the distal lower legs, whereas P7 and P8 showed predominant weakness of the iliopsoas, sparing of the quadriceps, and initially preserved lower leg function. In each of the muscle biopsies, rimmed vacuoles were found along with alterations of Z‐band architecture and accumulations of granulofilamentous material (Figure 1B,C). In P10 groups of filamentous bundles previously found to be characteristic for ZASPopathy (12) were frequently encountered (Figure 1D). Confirming this association, Sanger sequencing identified the pathogenic heterozygous LDB3/ZASP mutation c.494C>T, p.(Ala165Val) (83) in this case. The same method revealed heterozygous mutations of known pathogenicity in MYOT (P9: c.179C>T, p.(Ser60Phe)) (82) and in DES (c.1049G>C, p.(Arg350Pro)), the latter leading to a limb‐girdle syndrome in P7 and P8, which is a known phenotype of Arg350Pro‐desminopathy (99).
COL6A2 (Bethlem myopathy)
P11 reported generalized muscle weakness since childhood and showed contractures of the finger and elbow flexors. MRI of the legs revealed an outside rim of fatty infiltration of the vastus lateralis muscle contrasted by relative sparing of the central part. The biopsy showed large autophagic vacuoles filled with membranous and granular osmiophilic material (Figure 1E). There was focally accentuated endomysial fibrosis. Collagen VI immunohistochemistry revealed concentric accumulations of collagen and of fibroblasts around individual muscle fibers (Figure 1F). The prominent flexor contractions combined with the morphological alterations and the typical MRI features (58) raised the suspicion of Bethlem myopathy. NGS panel testing for COL6A1, COL6A2 and COL6A3 revealed the heterozygous c.1861G>A, p.(Asp621Asn) COL6A2 gene mutation of known pathogenicity (by alternative nomenclature p.(Asp620Asn), GenBank accession No.: M34571) (76). Moderate mitochondrial changes, such as globoid inclusions, were present (not shown), confirming previous observations in Bethlem myopathy (103). Segregation analysis revealed the same COL6A2 mutation in the mother, who did not complain of muscle weakness, but showed mild muscular atrophy on MRI and chronic hyperCKaemia (up to 1100 U/L).
TRIM32 (Sarcotubular myopathy)
P1.1 suffered from weakness of the leg and back muscles starting at age 27 and developed plegia of hip flexion and knee extension over 16 years. His older brother P1.2 who showed similar symptoms at a later age was wheelchair bound at the age of 53. Initial biopsy analysis yielded marked dystrophic features. Linkage analysis of the family including the healthy parents and three additional healthy brothers suggested TRIM32 as a candidate gene. Panel NGS of DNA of P1.1 revealed the heterozygous c.467T>C, p.(Leu156Pro) TRIM32 variant (rs145907585) of unknown significance. This is a rare variant with a minor allele frequency <0.01% that is rated as pathogenic by Mutation Taster and Polyphen 2 and is located within a highly conserved region (GERP 5.36). By quantitative RTqPCR, an additional heterozygous deletion of exon 2, the only coding exon of TRIM32, was detected. Subsequent analysis of new semithin sections of the biopsies of P1.1 and P1.2 revealed rimmed vacuoles and many vacuoles located in rows in markedly affected muscle fibers (Figure 2A,B). In electron micrographs vacuoles were associated with autophagic membranous and tubular structures and focal myofibrillar disintegration (Figure 2C,D), a pattern typical for sarcotubular myopathy (46, 62). Gene panel NGS also yielded the heterozygous MYH7 variant c.4258C>T, p(Arg1420Trp) which has been described previously in patients with a cardiomyopathy (97). However, the limb‐girdle phenotype, the AR mode of inheritance in this family and the lack of morphological alterations typical for MYH7 mutations such as myosin storage/hyaline bodies and cores/minicores or fiber type disproportion (25, 90) argued against the diagnosis of MYH7‐related myopathy. Moreover, subsequent analysis of DNA of the affected brother P1.2 showed the same TRIM32 genotype as found in P1.1. The five unaffected family members mentioned above underwent segregation analysis: The father carried the heterozygous p.(Leu156Pro) TRIM32 variant, while the mother and another brother harbored the TRIM32 deletion, confirming that one normal copy of TRIM32 is sufficient for proper gene function. Two other brothers harboured neither the exon 2 deletion nor the p.(Leu156Pro) mutation. We conclude that the combination of the p.(Leu156Pro) TRIM32 variant and the deletion of exon 2 cause LGMD2H (LGMDR8).
Figure 2.
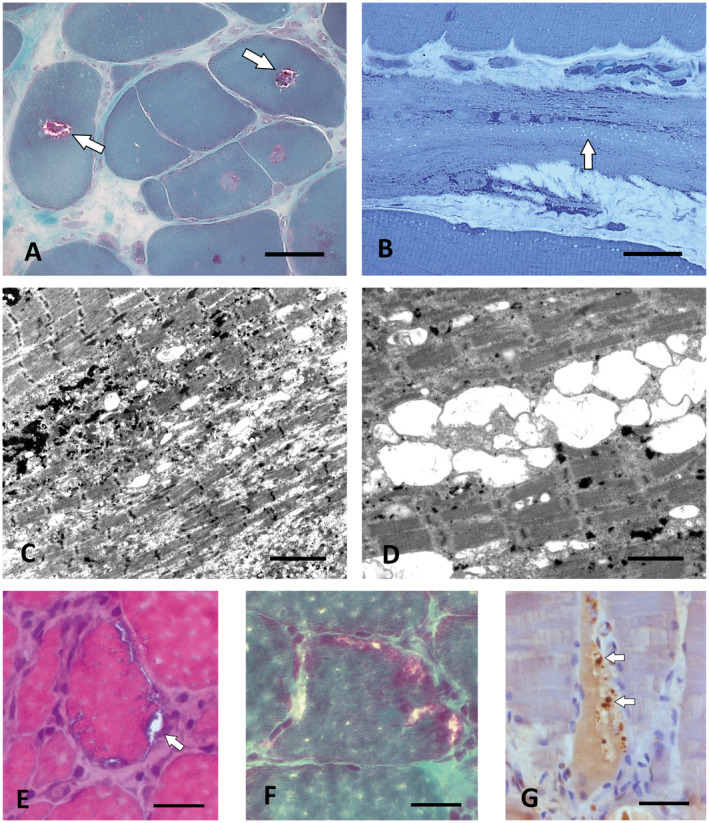
Rimmed vacuoles in sarcotubular myopathy and FSHD2. A. Central rimmed vacuoles (white arrows) in P1.2 with compound heterozygous TRIM32 mutations. Cryostat section, Gömöri trichrome. Scale bar = 30 µm. B. Numerous smaller empty vacuoles arranged in rows typical for sarcotubular myopathy in P1.1, the brother of P1.2 who carried the same compound heterozygous TRIM32 mutations as P1.2. Semithin section, toluidine blue; scale bar = 30 µm. C,D. By EM, the small vacuoles in the biopsy of P1.1. At least partially correspond to dilations of the sarcoplasmic reticulum. Scale bar in C = 4 µm, in D = 2 µm. E. Muscle biopsy of the FSHD2 patient P6 showing subsarcolemmal rimmed vacuoles (arrow). Cryostat section, H&E. Scale bar = 20 µm. F. Subsarcolemmal rimmed vacuoles in the same biopsy as in (E). Cryostat section, Gömöri trichrome. Scale bar = 20 µm. G. The autophagic vacuoles in P6 contain pTDP‐43‐immunoreactive granular material (arrows). Paraffin section. Scale bar = 30 µm.
FSHD2
P6 presented with facial weakness, atrophy of the shoulder girdle muscles and scapular winging. Light microscopy showed several muscle fibers with rimmed vacuoles (Figure 2E,F), some of which contained pTDP‐43‐immunoreactive aggregates (Figure 2G), and moderate focal endomysial accumulations of LCA immunoreactive cells including a small number of CD8 positive T‐cells. By EM, frequent strongly lobulated or fragmented myonuclei and focal proliferation of the sarcoplasmic reticulum and T‐system were found (not shown). After genetic exclusion of FSHD1 (contraction of the D4Z4 repeat unit on chromosome 4q) Sanger sequencing revealed the heterozygous c.1433G>A, p.(Gly478Glu) SMCHD1 mutation associated with a 11% hypomethylation pattern of the D4Z4 region; this digenic inheritance is typical for FSHD2 (49).
FKRP
P2 and P3 initially presented with severe proximal paresis of the legs. Proximal arm weakness was severe in P2 while P3 had relatively well‐preserved arm function. The sister of P2 had died of respiratory muscle failure at the age of 39 caused by an undiagnosed muscle disease. P2 developed wheelchair dependency and respiratory muscle weakness 14 years after disease onset. P3 received an implantable cardioverter‐defibrillator at age 37 caused by an episode of ventricular tachycardia and suspected cardiomyopathy. In the muscle biopsy of P2, subsarcolemmal proliferations of enlarged mitochondria projected deeply into many muscle fibers and were associated with marked proliferations of the T‐system and focal myofibrillar degradation (not shown). Autophagic vacuoles containing membranous, myelin‐like material were frequently seen immediately below the sarcolemma. Similar predominantly subsarcolemmal autophagic vacuoles were also found in the biopsy of P3 and were associated with cytoplasmic and nuclear accumulations of 12–18 nm tubulofilaments (Figure 3A,B). Because of the limb‐girdle distribution of muscle weakness and the AR inheritance pattern in both families, targeted Sanger sequencing of FKRP was performed, revealing the most frequent FKRP mutation c.826C>A, p.(Leu276Ile) in P2 in a homozygous state (7). P3 was compound heterozygous for this mutation combined with the c.1486T>A, p.(*496Argext*21) mutation that abolishes a stop codon and leads to an extension of the protein by 21 amino acids (19). In line with the genetic findings, alpha‐dystroglycan immunohistochemistry yielded absent reactivity; the immunoblot showed a markedly reduced intensity of the alpha‐dystroglycan band and an additional band of reduced molecular weight (not shown).
Figure 3.
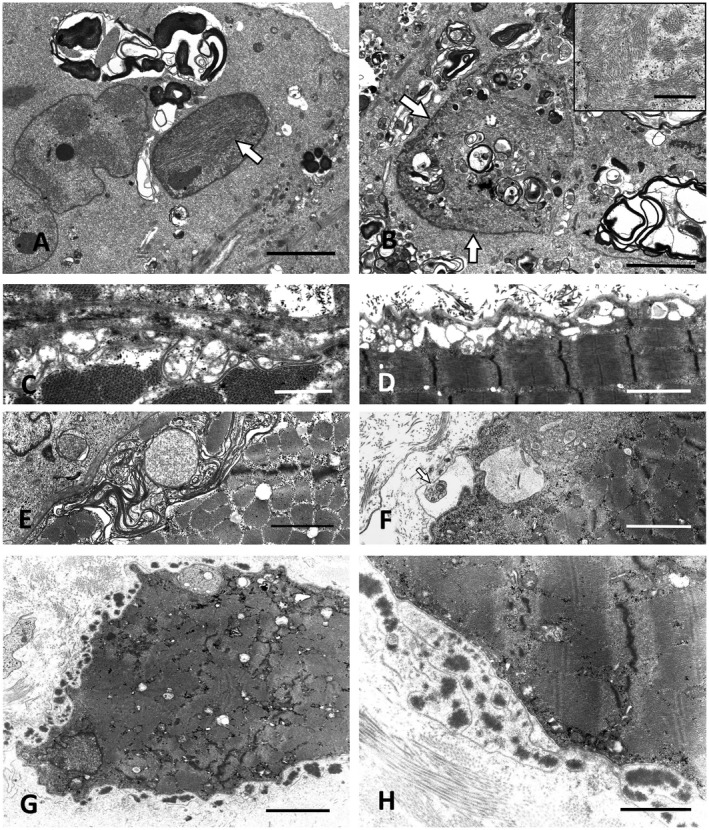
Ultrastructural pathology of FKRP, CAV3 and DYSF mutation cases. A. Prominent autophagic vacuoles combined with intranuclear tubulofilamentous inclusions (white arrow) in LGMD2i (LGMDR9) caused by compound heterozygous FKRP mutations (P3). Scale bar = 3 µm. B. P3: Membranous autophagic material accumulating within a myonucleus (arrows) and in the perinuclear sarcoplasm. Scale bar = 3 µm. Inset: High power view of sarcoplasmic tubulofilaments. Scale bar = 0.5 µm. C,D. Subsarcolemmal vacuoles and proliferations of membranes in a case with CAV 3 mutation (P15). Scale bar in C = 1 µm and D = 2 µm. E. Similar subsarcolemmal membranous proliferations in LGMD2B caused by compound heterozygous DYSF mutations (P5.2). Scale bar = 2 µm. F. Exocytosis of membranous autophagic material in the dysferlinopathy patient P5.1, the sister of P5.2. Scale bar = 2 µm. G,H. Large extracellular electron‐dense globules with abundant amorphous extracellular material surrounded by surplus basal lamina in the biopsy of P5.1. Scale bar in G = 10 µm, in H = 1.5 µm.
Caveolin‐3
P15 presented with hypesthesia of the feet, reduced vibration sense of the ankles and claw toes. No paresis was detected. Needle EMG of the right tibialis anterior muscle was normal while nerve conduction velocity and muscle action potentials were moderately reduced. CK was elevated to 960 U/L. Nerve biopsy showed a chronic, slowly progressive, predominantly axonal neuropathy with mild demyelination (not shown). Muscle biopsy revealed neurogenic muscular atrophy combined with myopathic changes. Gene Panel NGS detected the heterozygous c.277G>A, p.(Ala93Thr) (alternatively p.(Ala92Thr), GenBank accession No.: AF043101.1) CAV3 mutation which has previously been linked to rippling muscle disease (45). The p.(Ala93Thr) variant segregated with the polyneuropathy phenotype and mildly elevated CK levels (up to 270 U/L) in the patient’s mother. However, neither the index patient nor his mother showed muscle rippling. To further examine the pathogenicity of this CAV3 mutation, we performed immunoblotting and EM analyses. The Caveolin 3 band was absent in the immunoblot. By EM, caveolae at the plasma membrane were found to be largely absent, whereas irregular foci of filamentous material attached to the cytoplasmic face of the sarcolemma were frequently seen, resembling previous ultrastructural observations in Ala93Thr‐caveolinopathy (45). In addition, subsarcolemmal proliferations of membranes, often forming loops and vacuolar structures, were prominent in several muscle fibers; they probably correspond to abnormal extensions of the sarcoplasmic reticulum (Figure 3C,D).
Dysferlin
Three young female patients, one of consanguineous Pakistani descent (P4) and two sisters (P5.1 and P5.2), presented with limb‐girdle phenotype and severely elevated CK levels (up to 10 000 U/L). Muscle biopsies showed focal vacuolar alterations in all patients. Dysferlin immunoreactivity was absent in cryostat sections, in line with the absence of dysferlin protein bands in immunoblots of each of the three biopsies. In the biopsies of P5.1 and P5.2, caveolin‐3 and β‐dystroglycan staining was reduced. Ultrastructurally, many small subsarcolemmal vacuoles associated with defects of the plasma membrane and membranous proliferations were found (Figure 3E). Frequently, degenerated globular dense material within redundant loops of basal lamina typical for dysferlinopathy (84) was seen. These features were combined with signs of abnormal autophagy and exocytosis described already by Selcen et al (84) such as subsarcolemmal vacuoles with pleomorphic material in contact with the extracellular space and with abundant amorphous osmiophilic extracellular deposits surrounded by free basal lamina (Figure 3F–H). The diagnosis of dysferlinopathy was confirmed by Sanger sequencing: P5.1 and P5.2 harbored the compound heterozygous mutations c.864_865insA, p.(Ser289Ilefs*39) and c.3196_3197ins6bp, p.(Glu1065_Gly1066ins2a.a.) in DYSF (NM_003494.3) as reported previously (98). The former causes a frameshift and leads to premature termination of the protein; the latter leads to insertion of two amino acids without frameshift. P4 carried the homozygous mutation c.265C>T, p.(Arg89*) that leads to a premature stop codon and has been reported previously to cause dysferlinopathy in a compound heterozygous state (44, 98).
Glycogen storage diseases
P12 presented with mild proximal arm and leg paresis and respiratory muscle weakness requiring nocturnal BiPAP therapy at the age of 52. Biopsy revealed many PAS‐positive vacuoles in numerous muscle fibers suggestive of GSD. By EM (Figure 4A), the extensive glycogen deposits were associated with autophagic vacuoles and abnormal lipofuscin as well as considerable mitochondrial pathology, consistent with GSD II (24, 51). Alpha‐1,4‐glucosidase activity was reduced to 0.9 µmol/l/h (normal >3.3 µmol/l/h). Sanger sequencing detected the intronic mutation c.‐32‐13T>G, which is present in most late‐onset GSD II cases and gives rise to alternatively spliced transcripts with deletion of the first coding exon (34), combined with the c.1796C>A, p.(Ser599Tyr) missense mutation of known pathogenicity (69).
Figure 4 1.
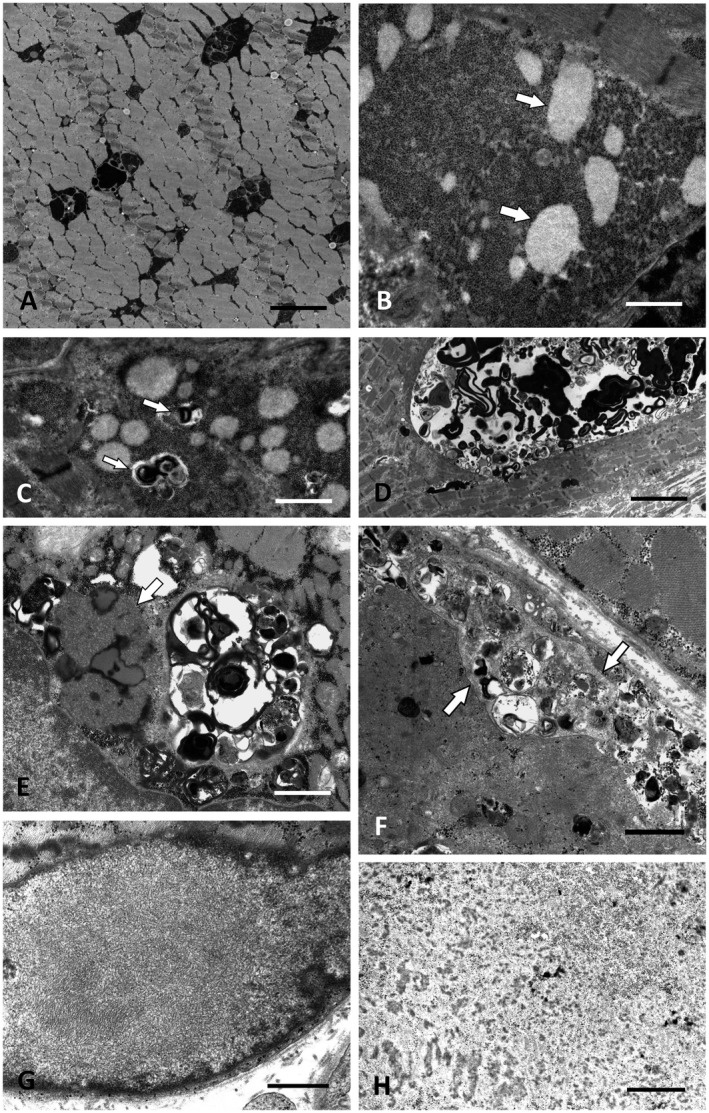
Ultrastructure of GSD II, GSD XV, chloroquine myopathy and cases without identifiable gene defect. A. Intermyofibrillar deposits of excess glycogen in GSD II (Pompe disease; P12). Scale bar = 3 µm. B. Muscle biopsy of P13 with novel compound heterozygous GYG1 (GSD XV) mutations showing excess glycogen with distinct round clusters of less osmiophilic fine granules, possibly representing partially degraded glycogen (arrows). Scale bar = 2 µm. C. P13: Autophagic vacuoles (arrows) associated with the glycogen deposits. Scale bar = 2.5 µm. D. P13: Large intermyofibrillar autophagic vacuole. Scale bar = 8 µm. E. Autophagic vacuoles combined with extensive accumulations of lipofuscin and curvilinear material (arrow) typical for chloroquine myopathy (P16). Scale bar = 2 µm. F. Arrows marking the membranous borders of exocytosed material between the plasma membrane and the basal lamina in P21. However, no mutation in LAMP‐2 or any other myopathy gene was found in this patient by WES. Scale bar = 2 µm. G. Distinct OPMD‐like tubulofilaments within a myonucleus in P27, who did not harbor a detectable PABPN1 repeat expansion or any other mutation in a myopathy gene. Scale bar = 0.5 µm. H. Deposition of granulofilamentous material in P19 suggested the diagnosis of a desminopathy/myofibrillar myopathy, but no defect in any of the relevant known genes was detected by WES. Scale bar = 0.5 µm.
P13 presented with severe foot drop and mild hip flexion paresis starting at age 50. Muscle biopsy revealed numerous large autophagic vacuoles, cytoplasmic and intranuclear accumulation of tubulofilamentous material, myonuclear degeneration, mitochondrial alterations and focal myofibrillar breakdown. In several muscle fibers, large abnormal, partially membrane‐bound accumulations of glycogen granules were associated with the autophagic vacuoles. Within the glycogen accumulations, round clusters of finely granular material which was less osmiophilic than normal glycogen were frequent. Some of these clusters were fully or partially engulfed by a membrane (Figure 4B–D). NGS panel detected the previously undescribed compound heterozygous mutations c.164_165delTT, p.(Phe55*) in exon 3 and c.646C>T, p.(Arg216*) in exon 6 of GYG1. The clinically unaffected adult son and the healthy maternal aunt of the patient were heterozygous for p.(Arg216*), in line with a compound heterozygous disease process in the index patient.
Vacuolar myopathy associated with chloroquine therapy
P16 presented with left hand weakness, severe foot drop, marked bilateral pes cavus and claw toes. He reported problems with his feet since childhood; there had been similar complaints of ambulation by his father, sister and nephew. CK levels were normal. Panel NGS for CMT and myopathy genes yielded variants of unknown significance in KIF1B and DYSF (Table S1). Muscle biopsy revealed severe vacuolar myopathy with numerous rimmed vacuoles combined with extensive accumulations of lipofuscin and of abundant curvilinear material (Figure 4E). Prominent build‐up of lipofuscin and of curvilinear material are typical for chloroquine myopathy (64). When asked explicitly, the patient then reported that he had taken chloroquine for several years to treat systemic lupus erythematosus.
Unclassified cases and variants of unknown significance
P21 exhibited proximal leg weakness and had a history of follicular non‐Hodgkin lymphoma and chemotherapy. Anti‐Ro52 and anti‐SRP antibodies were found in the blood, but muscle biopsy did not reveal any signs of myositis. His son and his brother had moderately elevated CK‐levels. Granular and diffuse intermyofibrillar LAMP2 and dysferlin immunoreactivity was enhanced mainly in damaged fibers (not shown). There was focal degradation of myofibrils with large quantities of partially membrane‐bound autophagic material in a predominantly subsarcolemmal position. Often such material was also found in the extracellular space between plasma membrane and basal lamina (Figure 4F). These findings of marked autophagy and exocytosis resembled Danon disease caused by LAMP2 mutation, which was, however, genetically excluded. WES did not yield any relevant alterations except for the c.10511C>T, p.(Ser3504Phe) RYR1 variant of unknown significance which segregated with the elevated CK levels in the family.
Biopsies of P17, P27, P29 and P30 showed marked inclusion body myopathy combined with prominent sarcoplasmic as well as myonuclear 12–18 nm tubulofilamentous inclusions. In P17, myopathic changes were accompanied by severe neurogenic atrophy. P27 was particular in that EM revealed many subsarcolemmal autophagic vacuoles that were continuous with the extracellular space indicating ongoing exocytosis. In addition, accumulations of sarcoplasmic 12–18 nm tubulofilaments were combined with intranuclear tubulofilaments of smaller diameter reminiscent of oculopharyngeal muscular dystrophy (OPMD) (92) (Figure 4G). However, no trinucleotide expansion was found in PABPN1, and WES did not detect any relevant mutation neither in this patient (P27) nor in P17. In addition, clinical features in P27 only partly matched those found in OPMD: facial symptoms were mild, ptosis was lacking and limb weakness was pronounced rendering the patient wheelchair bound at the age of 42 years. In P29 and P30 WES could not be performed; extensive Sanger sequencing of several candidate genes only revealed the heterozygous variant c.155A>G, p.(Asn52Ser) in ANO5 in P29, but no second mutation including ANO5 exon deletions or duplications (by MLPA) could be detected.
Three patients (P18, P23 and P28) showed focal myofibrillar breakdown combined with autophagic vacuoles and prominent accumulations of abnormal mitochondria often containing multiple paracrystalline inclusions. No pathogenic variants were found using WES in any of these cases. In P18, this was surprising because he is member of a large family with an autosomal dominant trait including six additional affected family members in three generations. The index patient had distinct high‐grade paresis of finger extension, whereas leg weakness developed at a much later stage. One of his brothers had been wheelchair bound at the age of 40 and had died at 58 years of an undiagnosed muscle disease. Other affected family members had difficulty in maintaining an erect posture, which correlated with fatty degeneration of spinal muscles on radiological imaging. In the biopsy of P23, nemaline bodies were found repeatedly, albeit not as frequent as usually seen in sporadic late‐onset nemaline myopathy (SLONM) (78). In addition, muscle weakness was only slowly progressive in this case; there was no rapid progression leading to respiratory weakness within 1–5 years after onset or axial weakness or dysphagia which is often encountered in SLONM cases (78).
P20 presented with rhabdomyolysis and showed extensive myofibrillar degradation associated with large autophagic vacuoles and sarcoplasmic accumulations of 12–18 nm tubulofilaments. CK levels remained severely elevated 6 years after first presentation. Genetic analysis including WES revealed the heterozygous RYR1 sequence variant c.6617C>T, p.(Thr2206Met) which has been linked to AD malignant hyperthermia (55) and AR muscular dystrophy (2), but was found in the unaffected mother of P20 as well. In addition, the recurrent c.549delA, p.(Thr184Argfs*36) CAPN3 frameshift mutation was found (rs80338800), but only in a heterozygous state. Copy number variations of CAPN3 exons were ruled out by MLPA analysis.
Distinct weakness of finger and hand extensors was the initial symptom in P25 and consecutive weakness of toe, foot and neck extensors evolved. Muscle biopsy showed predominantly subsarcolemmal myofibrillar alterations often reminiscent of cores as well as granulofilamentous inclusions, sarcolemmal reduplication and large tubular aggregates. P19 presented with distal leg paresis and showed myofibrillar alterations and granulofilamentous inclusions similar to P25 (Figure 4H). P22 showed weakness of the paraspinal and gluteal muscles. In her biopsy, we found numerous autophagic vacuoles along with markedly increased lipofuscin. P24 and P26 exhibited considerable vacuolar myopathy without any other specific histopathologic features. No pathogenic variant was found in any of these patients by WES. In P24 who was of Swedish descent and in P19 sural nerve biopsy showed an additional neuropathy.
In all cases with significantly elevated CK that could not be assigned to a genetically defined entity (n = 6), dystrophinopathy was ruled out caused by the absence of an X‐chromosomal trait, predominantly distal muscle involvement, normal dystrophin (Dys1‐3) in immunohistochemistry and immunoblot and a prominent vacuolar component.
Discussion
In healthy cells potentially harmful cellular contents such as protein aggregates or damaged organelles are eliminated by selective autophagy. Defective autophagic processes result in poor protein quality control, limited availability of free amino acids, disrupted metabolic homeostasis and inefficient stress response triggering compensatory mechanisms that lead to excess autophagy (38). Altered, upregulated autophagy involves a decline in the selectiveness of this process that now takes place in an indiscriminate manner (16), in line with the phenotype of vacuolar myopathies. In these disorders, normal, tightly regulated autophagy is disturbed, leading to the accumulation of autophagic substrate in large, membrane‐bound vacuoles. Muscle fibers are particularly susceptible to alterations of autophagy, because they constantly accumulate autophagic substrate caused by high protein turnover and exposure to mechanical stress (38). In addition, dysfunctional mitochondria that undergo mitophagy accumulate at high numbers in muscle fibers during aging (18). The study of muscle biopsies, thus, offers a unique opportunity to examine tissue patterns of altered autophagy in human patients.
The patients of our cohort fall into four major groups: In the first group characteristic clinical and biopsy features guided targeted sequencing, and thus led to a swift definite diagnosis in the GNE mutation, GSDII and myofibrillar myopathy cases. In two patients with myofibrillar myopathy, extensive granulofilamentous inclusions and prominent filamentous bundles detected by EM specifically suggested the diagnoses of desminopathy and ZASPopathy, respectively. These diagnoses were subsequently confirmed by targeted sequencing.
Biopsy findings expand the phenotype of known mutations
In the second group, markedly disturbed autophagy was linked to known pathogenic mutations in genes not regularly associated with autophagy alterations. Such changes were prominent in P2 and P3 with LGMD2i (LGMDR9) caused by the p.(*496Argext*21)/p.(Leu276Ile) and p.(Leu276Ile)/ p.(Leu276Ile) FKRP mutations (Figure 3A,B). They are consistent with the known function of FKRP in the glycosylation of sarcolemmal proteins and correlate with previous findings by others in an FKRP mouse model (11) and in two FKRP patients homozygous for p.(Leu276Ile) (73). The severe mitochondrial changes in P3 have not been described before in LGMD2i (LGMDR9).
Dysferlin is involved in membrane repair and induces sarcolemma resealing following damage (10). Dysferlin mutations lead to marked sarcolemmal defects combined with characteristic extracellular electron‐dense globules detectable by EM (84), as confirmed in P5.1 and 5.2. Associated with the membrane defects, we also confirmed subsarcolemmal autophagic vacuoles and extracellular deposits of membranous cell debris, consistent with the hypothesis that dysferlin mutations interfere with normal autophagy (22, 84).
Caveolin‐3 is a major component of caveolae (59). P15 and his affected mother carried the heterozygous p.(Ala93Thr) CAV3 mutation previously associated with AD rippling muscle disease, RMD (45), but did not show muscle rippling. EM analysis revealed a marked reduction of caveolae, large subsarcolemmal vacuoles and focal proliferations of the sarcoplasmic reticulum, findings typical for caveolinopathy (5, 6, 59). Polyneuropathy has been described by us before in RMD caused by the c.80G>A, p.(Arg27Gln) CAV3 mutation; in functional assays this mutation diminished signaling of the nerve growth factor receptor TrkA, potentially accounting for the manifestation of neuropathy (5). Hence, the present cases expand the spectrum of CAV3 mutations leading to neuromyopathy (45, 102).
Collagen VI contributes to the anchoring of the muscle fiber basal lamina in the extracellular matrix (47). Defective autophagy has been observed in a COL6A2 myopathy mouse model, along with decreased levels of beclin‐1 and Bnip3 in human Bethlem myopathy patients (30). The marked vacuolar changes detectable in P11 harboring the known pathogenic AD COL6A2 mutation p.(Asp621Asn) (76) support the notion that altered autophagy is a relevant pathomechanism of collagen VI‐related myopathy. Similar to the prominent core/rod pathology suggestive of altered proteostasis reported by us in a patient with COL6A3 mutation (13), the exact sequence of events leading to these changes remains unclear.
Previously, we have described five unrelated patients with genetically confirmed FSHD1 whose biopsies showed vacuolar myopathy with rimmed vacuoles (74). To our knowledge, we here report the first case of genetically confirmed FSHD2 associated with markedly vacuolar myopathy. Again, the pathomechanism leading to the vacuolar features is unclear.
Biopsy confirms the pathogenicity of novel mutations detected by NGS
The third group included cases with novel mutations that were classified as pathogenic based on the clinical findings including segregation analysis and on the biopsy results. GYG1 mutations cause AR GSD XV (33, 54, 61). The novel p.(Phe55*) and p.(Arg216*) GYG1 mutations in P13 each lead to premature termination of the coding sequence. The disease phenotype was only expressed by the index patient, but not by the heterozygous family members, thus confirming that one functional copy of the GYG1 gene is sufficient. Glycogenin‐1 is thought to be a primer of glycogen synthesis. As reported already in GSD XV cases and in glycogenin‐1 KO mice (91), glycogen accumulated in the muscle fibers of P13. Of note, the signs of altered autophagy were particularly prominent (Figure 4D). Accumulation of finely granular material, probably of smaller glycogen granules, was a further distinguishing feature of our case (Figure 4B,C). Interestingly, smaller than usual (partially degraded?) glycogen granules have already been shown to occur in GSD II muscle fibers (21).
TRIM32 is an E3 ubiquitin ligase located in the Z‐band (53). All LGMD2H (LGMDR8) mutations reported so far are located within the NHL domain of TRIM32 which is critical for the recognition of protein targets for ubiquitination (27, 53). Two brothers with clinical LGMD phenotype (P1.1. and P1.2) showed a deletion of exon 2, the only coding exon of TRIM32, and the sequence variant p.(Leu156Pro), resulting in a change of leucine to the structurally unrelated proline in a highly conserved region, but not within the NHL domain. These results indicate that mutation in TRIM32 outside of the NHL domain can cause sarcotubular myopathy/LGMD2H/LGMDR8. Notably, our initial histopathology of the muscle biopsy of P1.1 had not shown the typical vacuolar, sarcotubular expansions of the sarcoplasmic reticulum combined with myofibrillar disintegration (46, 79). However, subsequent semithin sections and EM examination of archival material established the diagnosis of sarcotubular myopathy, and thus confirmed the functional relevance of the p.(Leu156Pro) mutation. This focal occurrence of the sarcotubular alterations confirms previous observations by others (62). Our morphological workup did not yield any evidence for pathogenicity of the detected MYH7 sequence variant, in line with the limb‐girdle phenotype of the index patient. Thus, highlighted by this case, repeated and extended morphological examination may eventually settle an uncertain genetic diagnosis.
Non‐inflammatory vacuolar myopathies with negative WES
In the fourth group consisting of 15 patients including the chloroquine myopathy patient, no pathogenic gene defects were detected by broad genetic screening and NGS panel analysis. In 12 of these cases, additional WES was inconclusive. Trio WES analysis would be a more powerful and promising approach to identify causal mutations, however, especially in patients with late‐onset disease, parental samples are often not available. Several of these patients showed characteristic pathology such as OPMD‐like intranuclear filamentous inclusions, prominent exocytosis or granulofilamentous deposits. It remains unclear whether such cases can at some point be assigned to mutations that are not within the sequenced coding regions or to copy number variations or other structural alterations of the genome. Alternatively, at least some of them might represent sporadic degenerative myopathies of non‐genetic origin which share characteristic features of autophagy impairment. This is reminiscent of neurodegenerative disorders such as Alzheimer´s disease, which are also age‐related and characterized by abnormal autophagy, comprising early‐onset, often genetic and late‐onset, often non‐genetic forms.
Classification of cases according to frequency and extent of the vacuolar changes
Table 4 provides an alternative classification of non‐inflammatory vacuolar myopathies, distinguishing myopathies with a consistent vacuolar phenotype from those with only occasional vacuolar changes. In the latter, abnormal autophagic vacuoles are not found regularly at high numbers and may in some cases only be detected by electron microscopy. Still, we believe that it is important to keep in mind when reading biopsies that signs of considerable autophagy impairment may also occur in the second group. Moreover, these morphological findings indicate that altered autophagy contributes to the pathophysiological chain of events in these disorders.
Table 4.
Differential diagnosis of non‐inflammatory vacuolar myopathies.
| Disease entities | Gene defects/Causes | Cases of this study, References | Additional prominent or special features |
|---|---|---|---|
| Myopathies with a consistent vacuolar phenotype | |||
| Inherited causes | |||
| Myofibrillar myopathy | DES | P7, P8, Weihl 2015 (100), Claeys 2008 (12) | Granulofilamentous aggregates |
| CRYAB | Selcen 2003 (81), (12) | Granulofilaments, sandwich formations | |
| MYOT | P9, Selcen 2004 (82), (12) | Tubulofilaments, Filamentous bundles | |
| LDB3/ZASP | P10, Selcen 2005 (83), (12) | Filamentous bundles | |
| FLNC ‐ rod domain | Kley 2012 (42), Fürst 2013 (28) | ||
| FHL1 | Schessl 2009 (77), Selcen 2011 (80) | Reducing bodies | |
| BAG3 | Semmler 2014 (85) | ||
| TTN ‐ exon 344 (HMERF) | Pfeffer 2014 (68), Palmio 2019 (67) | ||
| Glycogen storage disease | GAA (GSD II, Pompe) | P12, Lewandowska 2008 (51) | Excess lipofuscin |
| AGL (GSD III) | DiMauro 1979 (17), Laforêt 2019 (48) | ||
| Lysosomal myopathy | LAMP2 (Danon) | Danon 1981 (15), Murakami 1995 (63) | Exocytosis, sarcolemmal folding |
| VMA21 (XMEA) | Ramachandran 2013 (72) | ||
| Neuronal ceroid lipofuscinosis | CLN3 | Cortese 2014 (14), Radke 2018 (71) | |
| Limb‐girdle muscular dystrophy | DNAJB6 (LGMDD1) | Sarparanta 2012 (75), Harms 2012 (32) | |
| hnRNPDL (LGMDD3) | Berardo 2019 (4) | ||
| Distal myopathy with rimmed vacuoles | GNE (HIBM) | Nonaka 1981 (66), Kayashima 2002 (39) | |
| TIA1 (Welander) | Hackman 2013 (31) | ||
| TTN ‐ exon 363 (Udd) | Udd 1993 (93) | ||
| OPDM, gene not known | Uyama 1998 (95) | ||
| Neuromyopathy/vacuolar myopathy and motor neurone disease | HSPB8 (dHMN2, CMT2L) | Ghaoui 2016 (29), Al‐Tahan 2019 (1) | |
| VCP (IBMPFD, ALS) | Weihl 2009 (101), Johnson 2010 (36) | Inflammatory infiltration | |
| SQSTM1/p62 | Bucelli 2015 (8), Fecto 2011 (23) | ||
| hnRNPA2B1, hnRNPA1 | Kim 2013 (41), Izumi 2015 (35) | ||
| MATR3 (VCPDM, ALS) | Senderek 2009 (86), Johnson 2014 (37) | ||
| Hypokalaemic periodic paralysis | CACNA1S, SCN4A | Lehmann‐Horn 2004 (50) | Sarcoplasmic reticulum vacuoles |
| Acquired causes | |||
| Amphiphilic cationic drugs | Chloroquine etc. | Sieb 2004 (87) | Excess lipofuscin |
| Chronic hypokalaemia | Alcohol abuse, diuretics | Khurana 1977 (40) | |
| Myopathies with occasional vacuolar changes | |||
| Inherited causes | |||
| Muscular dystrophy | DYSF (LGMD2B) | P4, P5.1, P5.2, Selcen 2001 (84) | Sarcolemmal/basal lamina defects, exocytosis |
| FKRP (LGMD 2i/R9) | P2, P3, Reilich 2006 (73) | ||
| TRIM32 (LGMD 2H/R8) | P1.1, P1.2, Schoser 2005 (79) | Sarcotubular alterations | |
| TCAP (LGMD2G) | Moreira 1997 (60), Vainzof 2002 (96) | ||
| PABPN1 (OPMD) | Tomé 1980 (92), Uyama 1996 (94) | Intranuclear thin tubulofilaments | |
| D4Z4 (FSHD1) | Reilich 2010 (74) | ||
| SMCHD1, D4Z4 (FSHD2) | P6 | Inflammatory infiltration | |
| Bethlem myopathy | COL6A2 | P11 | Mitochondrial alterations |
| Glycogen storage disease | GBE1 (GSD IV) | Nolte 2008 (65) | Polyglucosan bodies |
| GYG1 (GSD XV) | P13, Moslemi 2010 (61) | Clusters of pale glycogen granules | |
| Myosin‐related myopathy | MYH2 | Tajsharghi 2002 (89) | |
| MYH7 (Laing) | Fiorillo 2016 (26) | Hyaline bodies, cores/minicores | |
| Nemaline‐body myopathy | ACTA1 | Liewluck 2019 (52) | |
| CAV‐3‐related myopathy | CAV3 | P15, Brauers 2017 (6) | Subsarcolemmal vacuoles/membrane loops, neuromyopathy |
| Acquired causes | |||
| SLONM | MGUS, HIV | Sieb 1997 (88), Schnitzler 2017 (78) | |
| Endocrine disorders | Thyrotoxicosis | Korényi‐Both 1981 (43) | |
| Acromegaly | Mastaglia 1973 (56) | ||
| Hyperaldosteronism | Atsumi 1979 (3) | ||
Abbreviations: ALS = amyotrophic lateral sclerosis; CMT2L = Charcot‐Marie‐Tooth type 2L; dHMN2 = distal hereditary motor neuronopathy type IIa; HIBM = hereditary inclusion body myopathy; HIV = human immunodeficiency virus; HMERF = hereditary myopathy with early respiratory failure; IBMPFD = inclusion body myopathy associated with Paget's disease of bone and frontotemporal dementia; MGUS = monoclonal gammopathy of undetermined significance; OPDM = oculopharyngodistal myopathy; SLONM = sporadic late‐onset nemaline myopathy; VCPDM = vocal cord and pharyngeal weakness with distal myopathy; XMEA = X‐linked myopathy with excessive autophagy.
Finally, Table 4 includes a group of interesting monogenetic disorders that has emerged in recent years. In these diseases, mutations in genes involved in proteostasis and/or RNA processing such as HSPB8, VCP, SQSTM1/p62, hnRNPA2B1, hnRNPA1 and MATR3 may lead to either vacuolar myopathy or motor neurone disease, or combinations thereof (“neuromyopathy”). Hence the study of myopathic skeletal muscle should provide valuable insight into pathophysiological mechanisms that are also relevant for common neurodegenerative diseases.
Declarations
Ethics approval and consent to participate
Informed written consent was available from all patients for open biopsy and diagnostic genetic testing. The analysis of the diagnostic material and data for the purpose of this study including publication has been approved by the Ethical Committee of RWTH Aachen University Hospital (Ruling No. EK 127/18).
Competing interests
The authors declare that they have no competing interests.
Author contributions
A. Ferbert and J. Weis collected the majority of cases described in this study. K. Knop and W. Brück provided further cases. Neurological workup was performed by A. Ferbert, D. Mair and K. Knop. J. Weis, S. Nikolin, D. Mair, F.B. Koenig, S. Zechel and W. Brück performed the light microscopical neuropathological studies. Electron microscopy was performed by J. Weis. Genetic studies were performed by S. Biskup, W. Kress, A. Abicht, K. Eggermann and I. Kurth. S. Tey contributed to the preparation of the NGS and to the analysis of WES data. D. Mair compiled all data and wrote the manuscript. J. Weis and A. Ferbert revised the manuscript. All authors discussed results and commented on the manuscript.
Supporting information
Table S1. Variants of unknown significance in neuromuscular genes in 15 patients with no identified genetic cause of vacuolar myopathy.
Suppl. Methods. Workflow of neuropathological and diagnostic workup.
Acknowledgments
We thank Mrs. A. Knischewski, Mrs. C. Krude, Mrs. H. Mader and Dr. I. Katona for technical assistance and support. Dr. I. S. Zechel is a member of the European Reference Network for Rare Neuromuscular Diseases (ERN‐Euro NMD). J. Weis is a member of the German Muscular Dystrophy, Motor Neuron Disease and Charcot‐Marie‐Tooth Disease Networks (MD‐Net, NMD‐Net and CMT‐Net, respectively). This study was funded by the German Ministry of Science and Education (BMBF‐MND‐Net; Funds 360644), the German Research Foundation (INST 948/41‐1 FUGG, 2018‐2019), the German Myopathy Society (DGM) and the Interdisciplinary Centre for Clinical Research, RWTH Aachen University (IZKF Aachen, N7‐4) to J. Weis.
Contributor Information
Dorothea Mair, Email: dam1g13@soton.ac.uk, Email: jweis@ukaachen.de.
Joachim Weis, Email: dam1g13@soton.ac.uk, Email: jweis@ukaachen.de.
References
- 1. Al‐Tahan S, Weiss L, Yu H, Tang S, Saporta M, Vihola A et al (2019) New family with HSPB8‐associated autosomal dominant rimmed vacuolar myopathy. Neurol Genet 5:e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amburgey K, Bailey A, Hwang JH, Tarnopolsky MA, Bonnemann CG, Medne L et al (2013) Genotype‐phenotype correlations in recessive RYR1‐related myopathies. Orphanet J Rare Dis 8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atsumi T, Ishikawa S, Miyatake T, Yoshida M (1979) Myopathy and primary aldosteronism: electronmicroscopic study. Neurology 29:1348–1353. [DOI] [PubMed] [Google Scholar]
- 4. Berardo A, Lornage X, Johari M, Evangelista T, Cejas C, Barroso F et al (2019) HNRNPDL‐related muscular dystrophy: expanding the clinical, morphological and MRI phenotypes. J Neurol 266:2524–2534. [DOI] [PubMed] [Google Scholar]
- 5. Brauers E, Dreier A, Roos A, Wormland B, Weis J, Krüttgen A (2010) Differential effects of myopathy‐associated caveolin‐3 mutants on growth factor signaling. Am J Pathol 177:261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brauers E, Roos A, Kollipara L, Zahedi RP, Beckmann A, Mohanadas N et al (2017) The Caveolin‐3 G56S sequence variant of unknown significance: muscle biopsy findings and functional cell biological analysis. Proteomics Clin Appl 11:1600007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA et al (2001) Mutations in the fukutin‐related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 10:2851–2859. [DOI] [PubMed] [Google Scholar]
- 8. Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM et al (2015) SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 85:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castets P, Frank S, Sinnreich M, Rüegg MA (2016) “Get the balance right”: pathological significance of autophagy perturbation in neuromuscular disorders. J Neuromuscul Dis 3:127–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cenacchi G, Fanin M, de Giorgi LB, Angelini C (2005) Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J Clin Pathol 58:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan YM, Keramaris‐Vrantsis E, Lidov HG, Norton JH, Zinchenko N, Gruber HE et al (2010) Fukutin‐related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet 19:3995–4006. [DOI] [PubMed] [Google Scholar]
- 12. Claeys KG, Fardeau M, Schröder R, Suominen T, Tolksdorf K, Behin A et al (2008) Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul Disord 18:656–666. [DOI] [PubMed] [Google Scholar]
- 13. Claeys KG, Schrading S, Bozkurt A, Friedrich‐Freksa A, Pallua N, Kuhl C et al (2012) Myopathy with lobulated fibers, cores, and rods caused by a mutation in collagen VI. Neurology 79:2288–2290. [DOI] [PubMed] [Google Scholar]
- 14. Cortese A, Tucci A, Piccolo G, Galimberti CA, Fratta P, Marchioni E et al (2014) Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology 82:2072–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH (1981) Lysosomal glycogen storage disease with normal acid maltase. Neurology 31:51–57. [DOI] [PubMed] [Google Scholar]
- 16. Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19:349–364. [DOI] [PubMed] [Google Scholar]
- 17. DiMauro S, Hartwig GB, Hays A, Eastwood AB, Franco R, Olarte M et al (1979) Debrancher deficiency: neuromuscular disorder in 5 adults. Ann Neurol 5:422–436. [DOI] [PubMed] [Google Scholar]
- 18. Drake JC, Yan Z (2017) Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J Physiol 595:6391–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Driss A, Noguchi S, Amouri R, Kefi M, Sasaki T, Sugie K et al (2003) Fukutin‐related protein gene mutated in the original kindred limb‐girdle MD 2I. Neurology 60:1341–1344. [DOI] [PubMed] [Google Scholar]
- 20. Eisenberg I, Grabov‐Nardini G, Hochner H, Korner M, Sadeh M, Bertorini T et al (2003) Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat 21:99. [DOI] [PubMed] [Google Scholar]
- 21. Engel AG, Hirschhorn R, Huie ML (2004) Acid maltase deficiency. In: Myology: Basic and Clinical, 3rd edn. Engel AG, Franzini‐Armstrong C (eds), pp. 1559–1586. McGraw‐Hill: New York. [Google Scholar]
- 22. Fanin M, Nascimbeni AC, Angelini C (2014) Muscle atrophy, ubiquitin‐proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve 50:340–347. [DOI] [PubMed] [Google Scholar]
- 23. Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W et al (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68:1440–1446. [DOI] [PubMed] [Google Scholar]
- 24. Feeney EJ, Austin S, Chien YH, Mandel H, Schoser B, Prater S et al (2014) The value of muscle biopsies in Pompe disease: identifying lipofuscin inclusions in juvenile‐ and adult‐onset patients. Acta Neuropathol Commun 2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferbert A, Zibat A, Rautenstrauß B, Kress W, Hügens‐Penzel M, Weis J et al (2016) Laing distal myopathy with a novel mutation in exon 34 of the MYH7 gene. Neuromuscul Disord 26:598–603. [DOI] [PubMed] [Google Scholar]
- 26. Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F et al (2016) MYH7‐related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis 11:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frosk P, Weiler T, Nylen E, Sudha T, Greenberg CR, Morgan K et al (2002) Limb‐girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3‐ubiquitin‐ligase gene. Am J Hum Genet 70:663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fürst DO, Goldfarb LG, Kley RA, Vorgerd M, Olivé M, van der Ven PFM (2013) Filamin C‐related myopathies: pathology and mechanisms. Acta Neuropathol 125:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghaoui R, Palmio J, Brewer J, Lek M, Needham M, Evilä A et al (2016) Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 86:391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E et al (2010) Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16:1313–1320. [DOI] [PubMed] [Google Scholar]
- 31. Hackman P, Sarparanta J, Lehtinen S, Vihola A, Evilä A, Jonson PH et al (2013) Welander distal myopathy is caused by a mutation in the RNA‐binding protein TIA1. Ann Neurol 73:500–509. [DOI] [PubMed] [Google Scholar]
- 32. Harms MB, Sommerville RB, Allred P, Bell S, Ma D, Cooper P et al (2012) Exome sequencing reveals DNAJB6 mutations in dominantly‐inherited myopathy. Ann Neurol 71:407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hedberg‐Oldfors C, Mensch A, Visuttijai K, Stoltenburg G, Stoevesandt D, Kraya T et al (2018) Polyglucosan myopathy and functional characterization of a novel GYG1 mutation. Acta Neurol Scand 137:308–315. [DOI] [PubMed] [Google Scholar]
- 34. Huie ML, Chen AS, Tsujino S, Shanske S, DiMauro S, Engel AG, Hirschhorn R (1994) Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (‐13T–>G) mutation in a majority of patients and a novel IVS10 (+1GT–>CT) mutation. Hum Mol Genet 3:2231–2236. [DOI] [PubMed] [Google Scholar]
- 35. Izumi R, Warita H, Niihori T, Takahashi T, Tateyama M, Suzuki N et al (2015) Isolated inclusion body myopathy caused by a multisystem proteinopathy—linked hnRNPA1 mutation. Neurol Genet 1:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, van Deerlin VM, Trojanowski JQ et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jokl EJ, Blanco G (2016) Disrupted autophagy undermines skeletal muscle adaptation and integrity. Mamm Genome 27:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kayashima T, Matsuo H, Satoh A, Ohta T, Yoshiura K‐i, Matsumoto N et al (2002) Nonaka myopathy is caused by mutations in the UDP‐N‐acetylglucosamine‐2‐epimerase/N‐acetylmannosamine kinase gene (GNE). J Hum Genet 47:77–79. [DOI] [PubMed] [Google Scholar]
- 40. Khurana R, Kalyanaraman K (1977) Hypokalemic vacuolar myopathy of chronic alcoholism. A histological and histochemical study. Dis Nerv Syst 38:287–289. [PubMed] [Google Scholar]
- 41. Kim HJ, Kim NC, Wang Y‐D, Scarborough EA, Moore J, Diaz Z et al (2013) Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kley RA, Serdaroglu‐Oflazer P, Leber Y, Odgerel Z, van der Ven PFM, Olivé M et al (2012) Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain 135:2642–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korényi‐Both A, Korényi‐Both I, Kayes BC (1981) Thyrotoxic myopathy. Pathomorphological observations of human material and experimentally induced thyrotoxicosis in rats. Acta Neuropathol 53:237–248. [DOI] [PubMed] [Google Scholar]
- 44. Krahn M, Béroud C, Labelle V, Nguyen K, Bernard R, Bassez G et al (2009) Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum Mutat 30:345–75. [DOI] [PubMed] [Google Scholar]
- 45. Kubisch C, Schoser BG, von Düring M, Betz RC, Goebel H, Zahn S et al (2003) Homozygous mutations in caveolin‐3 cause a severe form of rippling muscle disease. Ann Neurol 53:512–520. [DOI] [PubMed] [Google Scholar]
- 46. Kudryashova E, Wu J, Havton LA, Spencer MJ (2009) Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet 18:1353–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuo H‐J, Maslen CL, Keene DR, Glanville RW (1997) Type VI Collagen anchors endothelial basement membranes by interacting with type IV Collagen. J Biol Chem 272:26522–26529. [DOI] [PubMed] [Google Scholar]
- 48. Laforêt P, Inoue M, Goillot E, Lefeuvre C, Cagin U, Streichenberger N et al (2019) Deep morphological analysis of muscle biopsies from type III glycogenesis (GSDIII), debranching enzyme deficiency, revealed stereotyped vacuolar myopathy and autophagy impairment. Acta Neuropathol Commun 7:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Larsen M, Rost S, El Hajj N, Ferbert A, Deschauer M, Walter MC et al (2015) Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet 23:808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lehmann‐Horn F, Rüdel R, Jurkat‐Rott K (2004) Nondystrophic myotonias and periodic paralyses. In: Myology: Basic and Clinical, 3rd edn. Engel AG, Franzini‐Armstrong C (eds), pp. 1257–1300. McGraw‐Hill: New York. [Google Scholar]
- 51. Lewandowska E, Wierzba‐Bobrowicz T, Rola R, Modzelewska J, Stepień T, Lugowska A et al (2008) Pathology of skeletal muscle cells in adult‐onset glycogenosis type II (Pompe disease): ultrastructural study. Folia Neuropathol 46:123–133. [PubMed] [Google Scholar]
- 52. Liewluck T, Niu Z, Moore SA, Alsharabati M, Milone M (2019) ACTA1‐myopathy with prominent finger flexor weakness and rimmed vacuoles. Neuromuscul Disord 29:388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Locke M, Tinsley CL, Benson MA, Blake DJ (2009) TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet 18:2344–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Malfatti E, Nilsson J, Hedberg‐Oldfors C, Hernandez‐Lain A, Michel F, Dominguez‐Gonzalez C et al (2014) A new muscle glycogen storage disease associated with glycogenin‐1 deficiency. Ann Neurol 76:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Manning BM, Quane KA, Ording H, Urwyler A, Tegazzin V, Lehane M et al (1998) Identification of novel mutations in the ryanodine‐receptor gene (RYR1) in malignant hyperthermia: genotype‐phenotype correlation. Am J Hum Genet 62:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mastaglia FL (1973) Pathological changes in skeletal muscle in acromegaly. Acta Neuropathol (Berl) 24:273–286. [DOI] [PubMed] [Google Scholar]
- 57. McNeill A, Birchall D, Straub V, Goldfarb L, Reilich P, Walter MC et al (2009) Lower limb radiology of distal myopathy due to the S60F myotilin mutation. Eur Neurol 62:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mercuri E, Lampe A, Allsop J, Knight R, Pane M, Kinali M et al (2005) Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord 15:303–310. [DOI] [PubMed] [Google Scholar]
- 59. Minetti C, Bado M, Broda P, Sotgia F, Bruno C, Galbiati F et al (2002) Impairment of caveolae formation and T‐System disorganization in human muscular dystrophy with caveolin‐3 deficiency. Am J Pathol 160:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moreira ES, Vainzof M, Marie SK, Sertié AL, Zatz M, Passos‐Bueno MR (1997) The seventh form of autosomal recessive limb‐girdle muscular dystrophy is mapped to 17q11‐12. Am J Hum Genet 61:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moslemi A‐R, Lindberg C, Nilsson J, Tajsharghi H, Andersson B, Oldfors A (2010) Glycogenin 1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med 362:1203–1210. [DOI] [PubMed] [Google Scholar]
- 62. Müller‐Felber W, Schlotter B, Töpfer M, Ketelsen U‐P, Müller‐Höcker J, Pongratz D (1999) Phenotypic variability in two brothers with sarcotubular myopathy. J Neurol 246:408–411. [DOI] [PubMed] [Google Scholar]
- 63. Murakami N, Goto Y‐I, Itoh M, Katsumi Y, Wada T, Ozawa E, Nonaka I (1995) Sarcolemmal indentation in cardiomyopathy with mental retardation and vacuolar myopathy. Neuromuscul Disord 5:149–155. [DOI] [PubMed] [Google Scholar]
- 64. Neville HE, Maunder‐Sewry CA, McDougall J, Sewell JR (1979) Chloroquine‐induced cytosomes with curvilinear profiles in muscle. Muscle Nerve 2:376–381. [DOI] [PubMed] [Google Scholar]
- 65. Nolte KW, Janecke AR, Vorgerd M, Weis J, Schröder JM (2008) Congenital type IV glycogenosis: the spectrum of pleomorphic polyglucosan bodies in muscle, nerve, and spinal cord with two novel mutations in the GBE1 gene. Acta Neuropathol 116:491–506. [DOI] [PubMed] [Google Scholar]
- 66. Nonaka I, Sunohara N, Ishiura S, Satoyoshi E (1981) Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci 51:141–155. [DOI] [PubMed] [Google Scholar]
- 67. Palmio J, Leonard‐Louis S, Sacconi S, Savarese M, Penttilä S, Semmler A‐L et al (2019) Expanding the importance of HMERF titinopathy: new mutations and clinical aspects. J Neurol 266:680–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pfeffer G, Barresi R, Wilson IJ, Hardy SA, Griffin H, Hudson J et al (2014) Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J Neurol Neurosurg Psychiatry 85:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pittis MG, Donnarumma M, Montalvo ALE, Dominissini S, Kroos M, Rosano C et al (2008) Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum Mutat 29:27–36. [DOI] [PubMed] [Google Scholar]
- 70. Platt FM, Boland B, van der Spoel AC (2012) The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 199:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Radke J, Koll R, Gill E, Wiese L, Schulz A, Kohlschütter A et al (2018) Autophagic vacuolar myopathy is a common feature of CLN3 disease. Ann Clin Transl Neurol 5:1385–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N et al (2013) VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol 125:439–457. [DOI] [PubMed] [Google Scholar]
- 73. Reilich P, Petersen JA, Vielhaber S, Mawrin C, Schneider‐Gold C, Sommer C et al (2006) LGMD 2I due to the common mutation 826C>A in the FKRP gene presenting as myopathy with vacuoles and paired‐helical filaments. Acta Myol 25:73–76. [PubMed] [Google Scholar]
- 74. Reilich P, Schramm N, Schoser B, Schneiderat P, Strigl‐Pill N, Müller‐Höcker J et al (2010) Facioscapulohumeral muscular dystrophy presenting with unusual phenotypes and atypical morphological features of vacuolar myopathy. J Neurol 257:1108–1118. [DOI] [PubMed] [Google Scholar]
- 75. Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M et al (2012) Mutations affecting the cytoplasmic functions of the co‐chaperone DNAJB6 cause limb‐girdle muscular dystrophy. Nat Genet 44:450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Scacheri PC, Gillanders EM, Subramony SH, Vedanarayanan V, Crowe CA, Thakore N et al (2002) Novel mutations in collagen VI genes: expansion of the Bethlem myopathy phenotype. Neurology 58:593–602. [DOI] [PubMed] [Google Scholar]
- 77. Schessl J, Taratuto AL, Sewry C, Battini R, Chin SS, Maiti B et al (2009) Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain 132:452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schnitzler LJ, Schreckenbach T, Nadaj‐Pakleza A, Stenzel W, Rushing EJ, van Damme P et al (2017) Sporadic late‐onset nemaline myopathy: clinico‐pathological characteristics and review of 76 cases. Orphanet J Rare Dis 12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schoser BGH, Frosk P, Engel AG, Klutzny U, Lochmüller H, Wrogemann K (2005) Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann Neurol 57:591–595. [DOI] [PubMed] [Google Scholar]
- 80. Selcen D, Bromberg MB, Chin SS, Engel AG (2011) Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology 77:1951–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Selcen D, Engel AG (2003) Myofibrillar myopathy caused by novel dominant negative alpha B‐crystallin mutations. Ann Neurol 54:804–810. [DOI] [PubMed] [Google Scholar]
- 82. Selcen D, Engel AG (2004) Mutations in myotilin cause myofibrillar myopathy. Neurology 62:1363–1371. [DOI] [PubMed] [Google Scholar]
- 83. Selcen D, Engel AG (2005) Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol 57:269–276. [DOI] [PubMed] [Google Scholar]
- 84. Selcen D, Stilling G, Engel AG (2001) The earliest pathologic alterations in dysferlinopathy. Neurology 56:1472–1481. [DOI] [PubMed] [Google Scholar]
- 85. Semmler A‐L, Sacconi S, Bach JE, Liebe C, Bürmann J, Kley RA et al (2014) Unusual multisystemic involvement and a novel BAG3 mutation revealed by NGS screening in a large cohort of myofibrillar myopathies. Orphanet J Rare Dis 9:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Senderek J, Garvey SM, Krieger M, Guergueltcheva V, Urtizberea A, Roos A et al (2009) Autosomal‐dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 84:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sieb JP (2004) Myopathies due to drugs, toxins and nutritional deficiency. In: Myology: Basic and Clinical, 3rd edn. Engel AG, Franzini‐Armstrong C (eds), pp. 1693–1712. McGraw‐Hill: New York. [Google Scholar]
- 88. Sieb JP, VonOertzen J, Tolksdorf K, Dörfler P, Kappes‐Horn K, Jerusalem F (1997) Sporadic adult‐onset distal myopathy with rimmed vacuoles, 15–18 nm tubulofilaments and extensive rod formation. J Neurol Sci 146:81–84. [DOI] [PubMed] [Google Scholar]
- 89. Tajsharghi H, Thornell L‐E, Darin N, Martinsson T, Kyllerman M, Wahlström J, Oldfors A (2002) Myosin heavy chain IIa gene mutation E706K is pathogenic and its expression increases with age. Neurology 58:780–786. [DOI] [PubMed] [Google Scholar]
- 90. Tajsharghi H, Thornell L‐E, Lindberg C, Lindvall B, Henriksson K‐G, Oldfors A (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 54:494–500. [DOI] [PubMed] [Google Scholar]
- 91. Testoni G, Duran J, García‐Rocha M, Vilaplana F, Serrano AL, Sebastián D et al (2017) Lack of glycogenin causes glycogen accumulation and muscle function impairment. Cell Metab 26:256–266. [DOI] [PubMed] [Google Scholar]
- 92. Tomé FM, Fardeau M (1980) Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol (Berl) 49:85–87. [DOI] [PubMed] [Google Scholar]
- 93. Udd B, Partanen J, Halonen P, Falck B, Hakamies L, Heikkilä H et al (1993) Tibial muscular dystrophy. Late adult‐onset distal myopathy in 66 Finnish patients. Arch Neurol 50:604–608. [DOI] [PubMed] [Google Scholar]
- 94. Uyama E, Nohira O, Chateau D, Tokunaga M, Uchino M, Okabe T et al (1996) Oculopharyngeal muscular dystrophy in two unrelated Japanese families. Neurology 46:773–778. [DOI] [PubMed] [Google Scholar]
- 95. Uyama E, Uchino M, Chateau D, Tomé FMS (1998) Autosomal recessive oculopharyngodistal myopathy in light of distal myopathy with rimmed vacuoles and oculopharyngeal muscular dystrophy. Neuromuscul Disord 8:119–125. [DOI] [PubMed] [Google Scholar]
- 96. Vainzof M, Moreira ES, Suzuki OT, Faulkner G, Valle G, Beggs AH et al (2002) Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta 1588:33–40. [DOI] [PubMed] [Google Scholar]
- 97. Vdriest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ (2004) Comprehensive analysis of the beta‐myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:602–610. [DOI] [PubMed] [Google Scholar]
- 98. Walter MC, Braun C, Vorgerd M, Poppe M, Thirion C, Schmidt C et al (2003) Variable reduction of caveolin‐3 in patients with LGMD2B/MM. J Neurol 250:1431–1438. [DOI] [PubMed] [Google Scholar]
- 99. Walter MC, Reilich P, Huebner A, Fischer D, Schröder R, Vorgerd M et al (2007) Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult‐onset, dominant myopathies are associated with the desmin mutation R350P. Brain 130:1485–1496. [DOI] [PubMed] [Google Scholar]
- 100. Weihl CC, Iyadurai S, Baloh RH, Pittman SK, Schmidt RE, Lopate G et al (2015) Autophagic vacuolar pathology in desminopathies. Neuromuscul Disord 25:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Weihl CC, Pestronk A, Kimonis VE (2009) Valosin‐containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto‐temporal dementia. Neuromuscul Disord 19:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP (2004) Caveolinopathies: mutations in caveolin‐3 cause four distinct autosomal dominant muscle diseases. Neurology 62:538–543. [DOI] [PubMed] [Google Scholar]
- 103. Zamurs LK, Idoate MA, Hanssen E, Gomez‐Ibañez A, Pastor P, Lamandé SR (2015) Aberrant mitochondria in a Bethlem myopathy patient with a homozygous amino acid substitution that destabilizes the collagen VI α2(VI) chain. J Biol Chem 290:4272–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Variants of unknown significance in neuromuscular genes in 15 patients with no identified genetic cause of vacuolar myopathy.
Suppl. Methods. Workflow of neuropathological and diagnostic workup.
Data Availability Statement
The dataset supporting the conclusions of this article is included within the article and its additional files.