

Abstract
Pleomorphic xanthoastrocytoma (PXA) is a rare astrocytoma predominantly affecting children and young adults. We performed comprehensive genomic characterization on a cohort of 67 patients with histologically defined PXA (n = 53, 79%) or anaplastic PXA (A‐PXA, n = 14, 21%), including copy number analysis (ThermoFisher Oncoscan, n = 67), methylation profiling (Illumina EPIC array, n = 43) and targeted next generation sequencing (n = 32). The most frequent alterations were CDKN2A/B deletion (n = 63; 94%) and BRAF p.V600E (n = 51, 76.1%). In 7 BRAF p.V600 wild‐type cases, alternative driver alterations were identified involving BRAF, RAF1 and NF1. Downstream phosphorylation of ERK kinase was uniformly present. Additional pathogenic alterations were rare, with TERT, ATRX and TP53 mutations identified in a small number of tumors, predominantly A‐PXA. Methylation‐based classification of 46 cases utilizing a comprehensive reference tumor allowed assignment to the PXA methylation class in 40 cases. A minority grouped with the methylation classes of ganglioglioma or pilocytic astrocytoma (n = 2), anaplastic pilocytic astrocytoma (n = 2) or control tissues (n = 2). In 9 cases, tissue was available from matched primary and recurrent tumors, including 8 with anaplastic transformation. At recurrence, two tumors acquired TERT promoter mutations and the majority demonstrated additional non‐recurrent copy number alterations. Methylation class was preserved at recurrence. For 62 patients (92.5%), clinical follow‐up data were available (median follow‐up, 5.4 years). Overall survival was significantly different between PXA and A‐PXA (5‐year OS 80.8% vs. 47.6%; P = 0.0009) but not progression‐free survival (5‐year PFS 59.9% vs. 39.8%; P = 0.05). WHO grade remained a strong predictor of overall survival when limited to 38 cases defined as PXA by methylation‐based classification. Our data confirm the importance of WHO grading in histologically and epigenetically defined PXA. Methylation‐based classification may be helpful in cases with ambiguous morphology, but is largely confirmatory in PXA with well‐defined morphology.
Keywords: astrocytoma, glioma, methylation, pleomorphic xanthoastrocytoma
Introduction
Pleomorphic xanthoastrocytoma (PXA) is a rare astrocytic neoplasm predominantly affecting children and young adults. PXA is defined histologically by the presence of large pleomorphic, spindled and lipidized cells, numerous eosinophilic granular bodies and dense pericellular reticulin (13). PXA has a relatively favorable prognosis, but a higher tendency to recur than other pediatric low‐grade gliomas, leading to its designation as a WHO grade II neoplasm. Up to one third of PXA show features of anaplasia, characterized by increased mitotic activity and at times necrosis, which is associated with decreased overall survival (16). As such, anaplastic PXA (A‐PXA) is designated WHO grade III in the 2016 WHO Classification of Central Nervous System Tumors (12).
PXAs are characterized by genetic alterations of the mitogen‐activated protein kinase (MAPK) pathway. The majority harbor BRAF p.V600E mutation, which has been identified in 60‐78% of tumors (13). Other MAPK alterations have been identified in a smaller fraction of PXA, including BRAF insertion/deletion mutations (29), fusions involving BRAF or RAF1 (15, 28) and mutation of NF1 (4). The majority of PXA also harbor homozygous deletion of CKDN2A/B. Initial studies identified this alteration in approximately 60% of cases (39) while more recent reports have found a higher incidence (>85%) (27, 36). PXAs show frequent additional copy number changes, most commonly whole chromosome or whole chromosomal arm gains and losses. Recurrent alterations reported across multiple studies include loss −9 or −9p, and loss −22; recurrent gains include +7, +5, +20 and +21 (27, 36, 39). The genetic alterations underlying recurrence and anaplasia are not well understood. A recent study of 19 PXA patients, including 15 A‐PXA, identified TERT promoter alterations in 47% of A‐PXA. Mutations of ATRX, PTEN, TP53 and BCOR/BCORL1 were also identified in single cases of A‐PXA (27). Additionally, genomic analyses have been performed on a small number of matched primary and recurrent tumors undergoing anaplastic transformation. These studies identified complex changes in chromosomal copy number as well as acquisition of mutations in TERT, ARID1A and NF1 upon recurrence and anaplastic transformation (27, 36).
PXA can present a diagnostic challenge as it shares overlapping genomic features with multiple other tumor types. BRAF p.V600E is a common alteration in many low‐grade glial and glioneuronal tumors (31). Concurrent MAPK alterations and CDKN2A/B deletion is also frequent in epithelioid glioblastoma (1, 19, 20, 24), pediatric secondary high‐grade gliomas (25) and the recently described anaplastic astrocytoma with piloid features (30). Additionally, as genome‐wide methylation‐based classification is increasingly utilized in the diagnosis of brain tumors (6), multiple studies have identified tumors with a “PXA‐like” methylation profile, including epithelioid glioblastoma (15), astroblastoma (23) and a subset of pediatric gliomas (24). The precise relationship of these tumors to PXA and their long‐term prognosis remains uncertain. Methylation‐based classification is not able to differentiate between PXA and A‐PXA (6).
To more fully understand the genomics of PXA, its long‐term prognosis and its relationship to other MAPK‐altered tumors, we performed a comprehensive genomic analysis of a cohort of 67 patients with histologically defined PXA and correlated the findings with long‐term clinical follow‐up. Our results confirm that the majority of PXA are characterized by CDKN2A/B deletion and MAPK activation and that WHO grading remains an important prognostic factor in PXA.
Materials and Methods
Patient cohort
We identified 67 patients who underwent resection of a PXA or A‐PXA with pathology reviewed Mayo Clinic (n = 30), Johns Hopkins Hospital (JHH) (n = 5) or University of Bonn (n = 32).
All studies were conducted in accordance with the ethical standards of the Declaration of Helsinki. Studies were approved by the Institutional Review Boards of Mayo Clinic and JHH. The use of clinical data and biologic material is in conformity with the ethics regulation standards of the University of Bonn Medical Center. Cases reviewed at University of Bonn included patients in German pediatric low‐and high‐grade glioma trials. All tumors underwent histologic review with confirmation of diagnosis by at least two neuropathologists (C.G, T.P, F.R. and R.V.). A‐PXA was defined by the presence of 5 or more mitotic figures per 10 high‐power fields (field size of 0.23 mm2) consistent with 2016 WHO criteria (12). A subset of 29 patients from the Mayo Clinic/JHH cohort was included in previous clinical (16) and genomic studies (36). One patient had an apparent clinical diagnosis and family history of neurofibromatosis (case 63) without other manifestations or known germline testing. One patient (case 22) harbored a germline deletion of CKDN2A/B and presented with multiple, bilateral PXAs.
Immunohistochemistry
Immunohistochemistry was performed on FFPE sections using antibodies directed against p16INK4a (mouse monoclonal anti‐p16, clone E6H4, Roche Diagnostics, Mannheim, Germany) and BRAF V600E (mouse monoclonal anti‐BRAF V600E, clone VE1, Roche Diagnostics, Mannheim, Germany) using a Ventana Benchmark XT Immunostainer (Roche Ventana, Darmstadt, Germany). Immunohistochemistry for phospho‐ERK was studied as described before (32); a detailed method is provided in the supplementary methods.
Genome‐wide chromosomal copy number analysis
DNA was extracted from 5‐micron formalin‐fixed paraffin embedded (FFPE) tissue sections using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) as described (17). Genomic copy number losses and gains were identified with a molecular inversion probe array (MIP) (OncoScan CNV Plus Array, Thermo‐Fisher Scientific, Waltham, MA, USA). The array covers frequent copy number changes across 900 cancer genes. Using at least 80 ng tumor DNA, MIP was performed as previously described (38). Briefly, all probes contain two genomic homology regions each flanking a SNP site. Gaps were filled and ligated after annealing to the DNA, followed by digestion of remaining non‐circularized probes by exonucleases. After cleavage the now inverted probes were amplified by PCR using universal primers, followed by labeling with fluorescent molecules and hybridization to oligonucleotide chip arrays. Raw data were analyzed using the Nexus Copy Number 8.0 Discovery Edition software (BioDiscovery, El Segundo, CA, USA). The manufacturer’s SNP‐FASST2 segmentation algorithm was used to make copy number and loss of heterozygosity estimations. Whole chromosomes were counted as gained or lost when at least 90% of the probe signals from both chromosome arms were above or below the defined threshold as described previously (14). For acrocentric chromosomes (13, 14, 15, 21, 22) only q‐arm changes were counted as whole chromosomal gains or losses. We applied GISTIC (Genomic Identification of Significant Targets in Cancer) to distinguish significant chromosomal aberrations from random background (3).
Targeted Sequencing
Targeted next generation sequencing was performed at Mayo Clinic using the clinically validated Neuro‐Oncology Expanded DNA Panel (https://www.mayocliniclabs.com/test‐catalog/Overview/603047). The DNA subpanel assesses for alterations in 150 genes associated with CNS tumors and utilizes a QIAseq Targeted DNA custom amplicon‐based panel with molecular bar code technology to allow traceability of PCR artifacts/duplicates. Sequencing was performed using an Illumina HiSeq 2500 (Illumina, Inc., San Diego, CA; paired‐end, 2 × 151). The RNA subpanel was performed in a subset of cases lacking BRAF p.V600E or other MAPK alteration. The RNA panel utilizes a QIAseq Targeted RNAscan custom amplicon‐based panel designed to detect 104 known gene‐gene fusions and novel fusion transcripts, including BRAF‐KIAA1549 fusion. Paired‐end 231/71 sequencing was performed on an Illumina MiSeq instrument. A gene list is included in the supplementary methods. DNA and RNA panel data were processed through custom bioinformatics pipelines developed to detect single nucleotide variants and small insertions/deletions (< 50 base pairs) with at least 15% variant allelic frequency and gene fusion events with at least 10 fusion transcripts, respectively. Variants with < 15 supporting molecules were excluded. DNA and RNA sequence alterations were visualized using Alamut Visual (Interactive Biosoftware, Rouen, France) and integrative genome browser (IGV) (34), respectively. DNA sequence alterations classified as benign based on publicly available data from genetic databases and literature were excluded.
TERT promoter mutation analysis
Targeted analysis of the TERT promoter was performed by pyrosequencing or digital droplet PCR (ddPCR). Prosequencing analysis of the TERT promotor hotspots (C228 and C250) was performed as previously described (11). ddPCR was performed using two TaqMan assays for specific for TERT C228T and C250T (Biorad, Hercules CA). PCR products were analyzed on a QX200 droplet reader (Biorad) and analyzed with QuantaSoft software (Biorad). Further details are available in supplementary methods.
Genome‐wide methylation profiling
Genome‐wide methylation profiling was performed in 35 patients from the Mayo Clinic/JHH Hopkins and 23 patients from the German cohort using Illumina Infinium Human MethylationEPIC BeadChip (850K). The methylation data were preprocessed using the minfi package (v.1.28.4) (2) in R (http://www.r‐project.org, version 3.5.3). Quality control was performed by examining the detection P‐value and calculating the median log (base2) intensities for methylated and unmethylated signals for each array. Samples with detection P‐value less than 0.01 and unmethylated and methylated median intensity values above 10 were carried forward for subsequent analysis. Beta density plots for all samples before and after normalization were also examined. After the quality control process, 46 samples were carried forward in the subsequent methylation analysis.
Functional normalization (8) with NOOB background correction and dye‐bias normalization (35) was performed. Probe filtering was performed after normalization. Specifically, probes located on sex chromosomes, containing nucleotide polymorphism (dbSNP132 Common) within five base pairs of and including the targeted CpG‐site or mapping to multiple sites on hg19 (allowing for one mismatch), as well as cross reactive probes were removed from analysis.
Unsupervised analysis of methylation data was performed using t‐distributed stochastic neighbor embedding (t‐SNE) using Rtsne (v.0.15) in comparison to the published Heidelberg reference cohort (6). In brief, principal components were calculated using the 5000 most variably methylated CpG probes, as measured by the standard deviation (SD) of the probe‐level beta values across samples. The top 13 principal components which explained more than 99% of the total variance across all samples were used as input in the t‐SNE analysis.
Mate‐pair sequencing
Mate‐pair sequencing is a whole‐genome protocol that detects structural and copy number variation. DNA extraction and mate pair library preparation were performed as previous described (26). Briefly, DNA was isolated using the Qiagen Puregene extraction kit and processed using the Illumina Nextera Mate Pair library preparation kit and sequenced on the Illumina HiSeq 4000. DNA was fragmented at 2‐5 kb fragments and sequenced from both ends. The sequencing data were mapped to the reference genome (GRCh38) using BIMA and the output was analyzed using SVAtools (7, 18). Junctions and CNAs were graphically illustrated using genome and junction plots as previously described (10).
Statistical analyses
Patient and clinicopathologic characteristics were summarized with frequencies and percentages or medians and ranges, as appropriate. Comparisons between groups (i.e., PXA II vs. A‐PXA) were performed with Fisher’s exact (categorical data) or Wilcoxon rank‐sum tests (ordinal data). Progression was defined as the first recurrence or death caused by disease (whichever occurred earlier). Progression‐free survival (PFS) and overall survival (OS) were summarized using the Kaplan‐Meier method (5‐year survival) and were compared between groups with likelihood ratio tests from Cox proportional hazards regression models. Survival estimates and hazard ratios were reported along with 95% confidence intervals (CI). P‐values less than 0.05 were considered statistically significant. All analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC) or R.
Results
Patient cohort characteristics
Characteristics of the overall cohort of 67 patients diagnosed with PXA or A‐PXA are shown in Figure 1 and detailed in Supporting Table S1. Median age at initial diagnosis was 20.3 years (range: 5.6 to 73.1 years), with 28 tumors (41.8%) occurring in pediatric patients (<18 years of age) and 39 (58.2%) in adults (Figure S1). The vast majority of tumors occurred in a supratentorial location (n = 64), most frequently the temporal lobe (n = 32), with rare (n = 3) posterior fossa tumors. A similar proportion of patients were female (n = 37, 55.2%) and male (n = 30, 44.8%). At primary diagnosis, the majority of patient were diagnosed with PXA grade II (n = 53, 79.1%) vs. A‐PXA (n = 14, 20.9%). There were no significant differences in age or gender between patients diagnosed with PXA vs. A‐PXA (Table S1). Patients from the German cohort were younger relative to the Mayo/JHH cohort (median age: 15.4 years vs. 21.9 years) and more frequently diagnosed as PXA grade II (96.9% vs. 62.9%) (Table S1). Overall, our cohort reflects the typical demographics of PXA patients (13).
Figure 1.

Clinical and molecular characteristics of the patient cohort. BRAF p.V600E mutations were identified by targeted sequencing or (●) immunohistochemistry (IHC) only. TERT promoter mutations were identified by targeted sequencing or ddPCR. CDKN2A/B deletion and whole chromosome gains and losses were derived from MIP data. Genome‐wide methylation data were analyzed against the Heidelberg reference brain tumor cohort (6). Methylation class was defined by unsupervised analysis using t‐SNE dimensionality reduction and utilizing the Heidelberg online classification tool (classifier version v11b4). A calibrated Heidelberg score of >0.9 (●) was considered a definitive match to the reference methylation class. ANA PA, anaplastic pilocytic astrocytoma; LGG‐GG, low‐grade glioma, ganglioglioma; LGG‐PA/GG‐ST, low‐grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma; CONT INFLAM, control tissue, inflammatory tumor microenvironment; CONT REACT, control tissue, reactive tumor microenvironment; GBM RTK II, glioblastoma, IDH wild‐type, subclass RTK II; GBM G34, glioblastoma, IDH wild‐type, H3.3 G34 mutant; PLEX PED B, plexus tumor, subclass pediatric B; GBM MES, glioblastoma, IDH wild‐type, subclass mesenchymal; NA, not available; LTF, lost to follow‐up.
Genomic analysis
In 65 cases, genomic analyses were performed on specimens obtained at initial resection, while 2 cases had tissue available only at first recurrence, including one tumor that had undergone anaplastic transformation. Tumors were assessed for the BRAF p.V600E mutation by targeted sequencing (n = 59) or IHC alone (n = 8) (Figure 1). Overall, 51 tumors (76.1%) were positive for BRAF p.V600E, including PXA (79.2%) and A‐PXA (64.3%) (Table S2). All tumors were analyzed for genome‐wide copy number by molecular inversion probe (MIP) technology (Figure S2). The most frequent alteration was loss of CDKN2A/B, present in the vast majority of cases (n = 63; 94%). In 53 cases, CDKN2A/B showed homozygous deletion. Ten cases showed at least hemizygous loss of CDKN2A/B, but in a subset of six cases, the deletion could not be unequivocally interpreted as hemizygous vs. homozygous loss because of DNA quality, small deletion size or tumor cellularity. To further evaluate this finding, IHC for p16 was performed in 33 cases. In cases with CDKN2A/B homozygous deletion, a complete loss of p16 protein expression was observed in 22 (of 23) cases while 1 case showed protein loss only in a subset of cells. In 6 (of 6) tumors with hemizygous or questionable hemi/homozygous deletion, p16 expression was also lost. Expression was retained in 3 (of 4) tumors lacking CDKN2A/B deletion.
By genome‐wide chromosomal copy number analysis (MIP), most cases also demonstrated additional chromosomal gains/losses, predominantly involving whole chromosomes or chromosomal arms (Figure S2). The most frequent whole chromosome gains were gain +7 (n = 17), +5 (n = 12), +12 (n = 10), +21 (n = 10), +2 (n = 10) and +20 (n = 9). The most frequent losses were loss −9 (n = 24), −22 (n = 9) and −13 (n = 8). Tumors tended to show a predominance of either chromosomal gains or losses, but this pattern was not associated with tumor grade or other molecular features. Loss −10 was less common (n = 6) and no tumors demonstrated concurrent gain +7/loss −10, characteristically found in glioblastoma (5). No focal amplifications (including TERT) were identified. A‐PXA showed a significantly higher number of whole chromosome CNAs than PXA grade II (median 5.5 vs. 1.0; P = 0.004). Gain +7, +20 and +21 were more frequent in A‐PXA (Table S2).
To further characterize the pattern of CNAs, mate‐pair sequencing was performed in 6 cases with available frozen tumor tissue (3 PXA and 3 A‐PXA) (Figure S3). In 5 cases, CDKN2A/B deletion resulted from a small interstitial deletion, while case 51 showed a more complex intrachromosomal rearrangement of 9p. Genome‐wide, chromosomal translocations were not identified in two cases while three cases showed a small number of translocations that did not result in relevant gene fusions. In contrast, case 45 demonstrated a complex pattern consistent with chromothripsis and chromoplexis (Figure S4). This involved multiple chromosomes, with an intrachromosomal event of 3p resulting in ATG7‐RAF1 fusion, an alteration previously reported in an A‐PXA (28).
In 31 cases (21 PXA; 10 A‐PXA), DNA was available in suitable quality to perform targeted next generation sequencing of 150 CNS tumor associated genes. In an additional 32 cases, targeted sequence analysis of the TERT promoter was performed by pyrosequencing or digital droplet PCR (ddPCR). In most PXA grade II cases, BRAF p.V600E was the sole pathogenic alteration identified by NGS (Figure 1). Of the BRAF p.V600 wild‐type PXAs, two cases harbored previously reported BRAF activating insertion/deletion mutations: BRAF p.V504_R506dup and BRAF p.L485_P490delinsF, the latter identified in conjunction with a splice site mutation in NF1 (29). ATG7‐RAF1 fusion was present in two cases, including case 45 characterized by mate‐pair sequencing. Thus, alternative MAPK pathway alterations were identified in all PXA analyzed by targeted NGS. A single PXA harbored a TERT promoter mutation and another single tumor an inactivating mutation in PIK3C2G. Alterations in ATRX, TP53 or PTEN were not identified in PXA grade II.
Of 10 A‐PXA analyzed by targeted NGS testing, 7 harbored a BRAF p.V600E mutation. Additional mutations were identified in 5 (of 7) BRAF‐mutant A‐PXA, involving TERT (n = 1), ATRX (n = 2), TP53 (n = 1) and PIK3C2G (n = 1). The TP53 mutation (c.783‐2A > G) involved the splice acceptor site and has been previously reported. Overall, targeted TERT promoter analysis identified mutations in 2 of 14 (14.3%) A‐PXA. Three A‐PXA harbored an unusual molecular profile, characterized by NF1 mutation in a background of marked chromosomal aneuploidy (Figure S5). Case 62 showed an NF1 p.L844F mutation, a point mutation in PTEN and an apparently triploid genome, with complex superimposed CNAs. Cases 63 and 64 harbored truncating mutations in NF1 and showed an almost identical CNA profile, characterized by a near haploid genome. Review of tumor histology confirmed the diagnosis of A‐PXA in these cases (Figure S5). Case 62 showed predominance of large, pleomorphic, lipidized cells and prominent perivascular inflammation. Cases 63 and 64 showed more classic morphology, with necrosis (both cases) and microvascular proliferation (case 64).
In nine cases, BRAF p.V600 was wild‐type, but tissue could not be further analyzed by NGS. As a surrogate for MAPK pathway activation, phospho‐ERK immune staining was performed in 5 BRAF p.V600 negative cases and in 10 cases with known MAPK alterations. pERK was positive in all tumors examined, supporting MAPK pathway activation as a hallmark of PXA.
Methylation‐based classification
Tumor classification of 46 cases (31 PXA; 15 A‐PXA) was performed based on genome‐wide methylation data obtained from the Illumina MethylationEPIC array. Two tumors analyzed at recurrence/anaplastic progression only are included with A‐PXA. These tumors were classified against a comprehensive reference data set (6) by two independent methods: an unsupervised t‐SNE (t‐distributed stochastic neighbor embedding) analysis and the Heidelberg supervised brain tumor classifier, version v11b4 (6) (https://www.molecularneuropathology.org/mnp). t‐SNE analysis was performed using a subset of 14 reference classes (Figure 2A); inclusion of all reference classes did not substantially alter classification (not shown). Among 46 cases, both methods consistently classified 38 cases with the PXA (n = 36), hemispheric pilocytic astrocytoma and ganglioglioma (n = 1) and ganglioglioma (n = 1) reference methylation classes.
Figure 2.
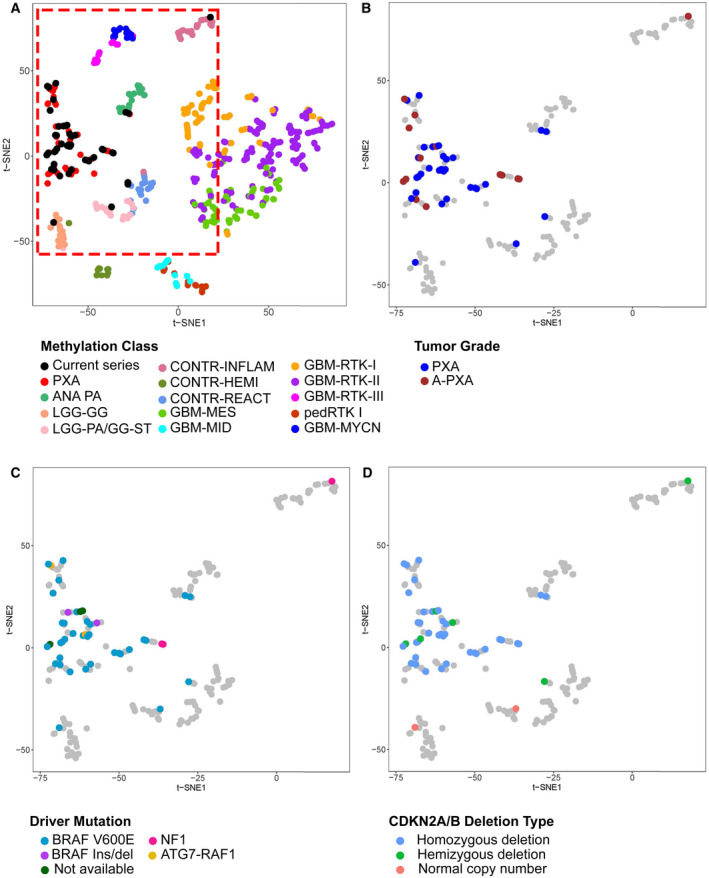
Genome‐wide methylation profiling and methylation‐based classification of PXA. (A) Clustering of PXA relative to published reference methylation classes. Analysis was performed using the top 5000 most differentially methylated probes with t‐SNE visualization. Current series denotes cases from our cohort. (B‐D) Grouping of current cases relative to (B) tumor grade, (C) genetic driver alteration and (D) CDKN2A/B deletion type. Reference cases are shown in grey within the area denoted in red in panel A. Reference methylation classes included: PXA, (anaplastic) pleomorphic xanthoastrocytoma; ANA PA, anaplastic pilocytic astrocytoma; LGG‐GG, low‐grade glioma, ganglioglioma; LGG‐PA/GG‐ST, low‐grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma; CONT‐INFLAM, control tissue, inflammatory tumor microenvironment; CONT‐HEMI, control tissue, hemispheric cortex; CONT‐REACT, control tissue, reactive tumor microenvironment; glioblastoma IDH wild‐type subgroups: GBM‐MES, mesenchymal subclass; GBM‐MID, midline subclass; GBM‐RTK‐I subclass; GBM RTK‐II subclass; GBM‐RTK‐III subclass; pedRTK I subclass; GBM‐MYCN subclass. Two cases analyzed at only recurrence/anaplastic transformation are included with A‐PXA.
By unsupervised t‐SNE, most cases grouped with PXA (n = 40) and the remaining with the classes anaplastic pilocytic astrocytoma (n = 2), hemispheric pilocytic astrocytoma and ganglioglioma (n = 1), ganglioglioma (n = 1) or control tissues (n = 2). The anaplastic pilocytic astrocytoma reference group utilized in this analysis contains 21 tumors (6), which subsequently all clustered with the newly defined entity anaplastic astrocytoma with piloid features (30) (D. Capper, personal communication). Of the tumors grouping with PXA, there was no apparent subgrouping related to WHO grade, genomic driver alterations or CDKN2A/B deletion status (Figure 2B‐D). Analysis using the Heidelberg methylation brain tumor classifier (v11b4) available as an online tool yielded a definitive assignment to a reference class (defined by a calibrated score ≥ 0.9) for 26 tumors (56.5%) (Table S3). The number of definitively matched tumors was higher for PXA grade II (n = 20, 64.5%) than for A‐PXA (n = 6, 40.0%). Overall, for 38 tumors (82.6%), PXA was the highest scoring reference class, including 24 (52.2%) with scores ≥ 0.9. Only two tumors showed a high score for other low‐grade glioma reference classes, consistent with the t‐SNE analysis (Figure 1). Overall, in the majority of cases, methylation profiling was concordant with the histologic diagnosis of PXA.
For cases with discordant classification by methylation profiling, tumor histology was re‐reviewed (Figure S6). All were considered to remain in keeping with PXA. Neither of the two cases matching to other low‐grade glioma methylation classes (case 1 and 2) demonstrated a definitive ganglion cell component and were more cellular and pleomorphic than typical of pilocytic astrocytoma. The two cases grouping with anaplastic pilocytic astrocytoma (case 23 and 41) were both cerebellar PXA grade II, with characteristic morphology. The two cases matching to the “control tissue” methylation classes (cases 4 and 62) both showed solid tumor histologically. Although an inflammatory component was apparent in case 62, the CNA profile supported high tumor cell content (Figure S5B).
Genomic characterization of matched primary and recurrent samples
In 9 cases, genomic analyses were performed on matched primary and recurrent tumors, including 2 cases at both first and second recurrences (Figure 3). Case 38 received radiotherapy prior repeat resection; the remaining cases did not receive radiation or chemotherapy prior to recurrence. Tumors underwent anaplastic transformation on recurrence in 8 cases while one patient presented initially with an A‐PXA. Recurrent tumors were analyzed by MIP genome‐wide copy number analysis (n = 9), TERT promoter analysis (n = 9) and methylation profiling (n = 6). The majority of tumors showed an increase in complexity at recurrence, with acquisition of multiple additional CNAs (Figure 3A). The changes were largely non‐recurrent, although gains of chromosomes 2 (n = 2) and 7 (n = 2) as well as loss of chromosome 10 (n = 2) were identified in multiple tumors at recurrence. In two cases, TERT promoter mutations were acquired at recurrence (case 22 and 38). For the 6 matched samples analyzed by methylation profiling, the methylation class was stable for tumors grouping with the PXA (n = 5) and anaplastic pilocytic astrocytoma methylation class (n = 1) (Figure 3C).
Figure 3.
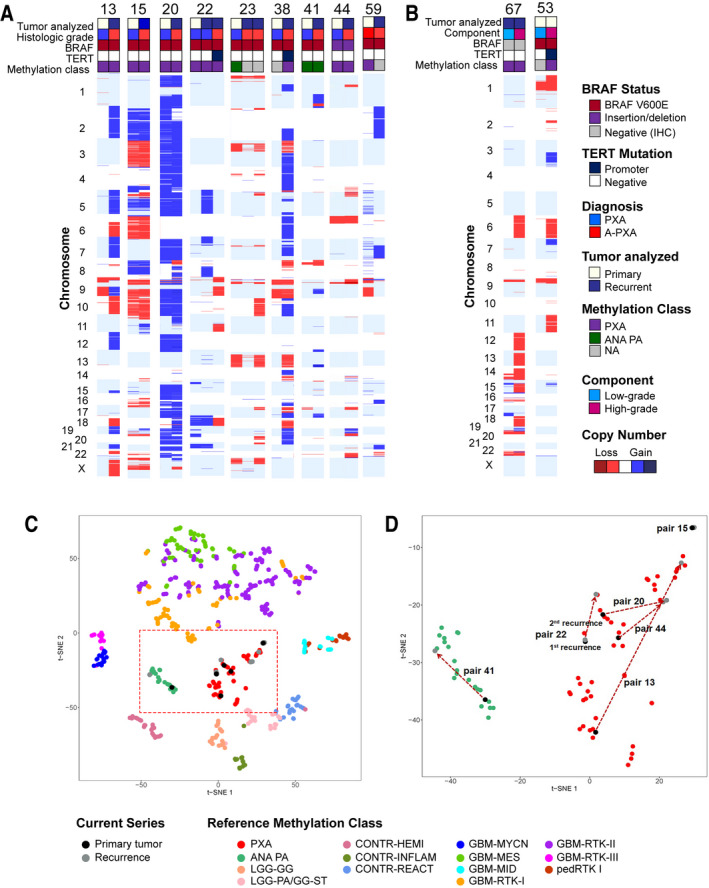
Genomic analysis of matched primary and recurrent tumors. (A) Genomic characterization of 9 matched primary and recurrent tumors. In 8 cases, tumors underwent anaplastic transformation upon recurrence. (B) Analysis of distinct areas of low‐grade and high‐grade morphology in A‐PXA. DNA was extracted and analyzed independently from histologically low‐grade and high‐grade areas within the same tumor. Tumors were analyzed by chromosomal microarray and targeted sequencing of BRAF and TERT. (C and D) Genome‐wide methylation profiling of matched primary and recurrent tumors. Matched tumors were analyzed in comparison to the 14 reference methylation classes included in Figure 2. (D) Details of tumor pairs within the PXA and anaplastic pilocytic astrocytoma (ANA PA) methylation class. NA, not available.
In two cases, tissue was available from matched low‐ and high‐grade components within the same tumor. Additional CNAs were present in the high‐grade component of both tumors, including loss of chromosome 6 (Figure 3B). Additionally, TERT mutation was identified only in the high‐grade component of case 53. Methylation class was preserved between the low‐ and high grade components of case 67 (Figure S7). Overall, our results suggest that on recurrence or anaplastic transformation, the methylation class is preserved. The majority of tumors acquire additional, largely non‐recurrent CNAs and a subset acquires TERT promoter mutations.
Association of histology and genomic features with patient outcome
Follow‐up was available for 62 patients, with a median follow‐up period of 5.4 years (Table S4). Overall, histologic WHO grade was a strong predictor of patient overall survival (P < 0.001). A trend toward decreased progression‐free survival was also present, but this did not reach statistical significance (P = 0.05) (Figure 4A,B). For PXA, five‐year progression‐free survival was 59.9% (95% CI: 45.0%, 74.8%) and overall survival was 80.8% (95% CI: 68.8%, 92.9%). For A‐PXA, five‐year progression‐free survival was 39.3% (95% CI: 12.7%, 65.9%) and overall survival 47.6% (95% CI: 20.1%, 75.1%). Limiting survival analysis to the 38 cases that were confidentially assigned to the PXA methylation class by methylation‐based classification showed consistent results (Figure 4C,D); the presence of anaplastic features remained strongly associated with overall survival (P = 0.002) with a non‐significant trend toward decreased progression‐free survival (P = 0.09) in those PXA defined by methylation‐based classification.
Figure 4.
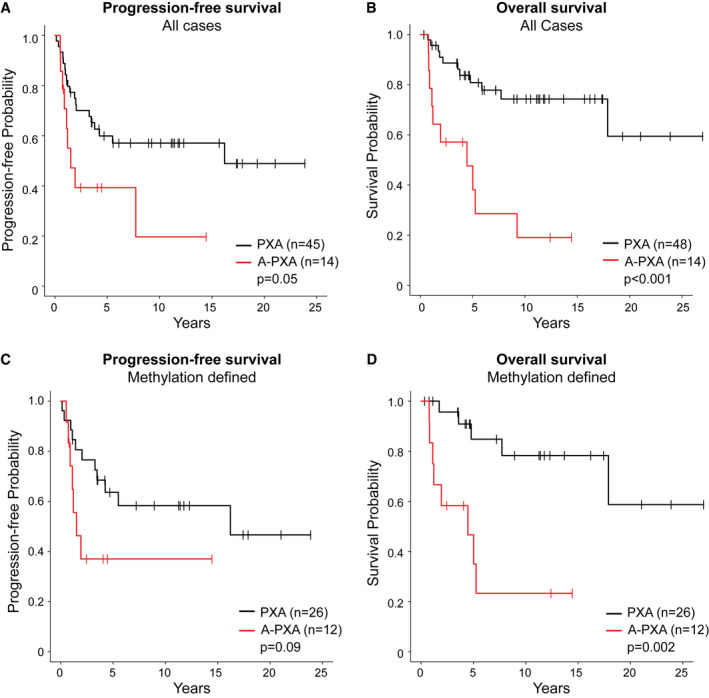
Patient survival relative to histologic grade. Kaplan‐Meir curves showing (A) progression‐free survival and (B) and overall survival across all cases with available follow‐up. (C) Progression‐free survival and (D) overall survival limited to 38 tumors grouping with PXA by methylation profiling.
The presence of necrosis was also strongly associated shorter overall survival (P = 0.0003, Table S4). Five‐year overall survival was 86.3% for tumors with no necrosis (95% CI: 74.9, 97.9) vs. 43.8% for tumors with necrosis (95% CI: 20.5, 67.0). However, given a strong association of necrosis with elevated mitotic activity and relatively small number of cases, we were unable to reliably evaluate necrosis and mitotic index as independent predictors of survival. Associations between patient survival and molecular genetic features were less robust. Neither the total number of whole chromosome CNAs nor any individual whole chromosome gain or loss showed a significant association with overall survival (Table S4). CDKN2A/B deletion was not associated with overall survival (P = 0.18), although the power of this analysis was very limited as only three cases in our series lacked CDKN2A/B deletion and had clinical follow‐up available. TERT promoter mutations were rare (n = 3), but showed strong association with shorter overall survival (P = 0.002), with no patients alive at five years. An insufficient number of cases harbored other molecular alterations, including ATRX, TP53 and NF1 mutations, to correlate with patient survival.
Discussion
This cohort of PXA patients is unique for the number of cases and the detailed correlation of clinical, histologic and genomic features with long‐term clinical follow‐up. The study confirms that the vast majority of PXA are characterized genetically by the presence of CDKN2A/B deletion in combination with MAPK pathway alterations (Figure 1). While BRAF p.V600E was the most frequent driver alteration (n = 51, 76.1%), we also identified previously reported alterations of BRAF (29) and RAF1 (28). Three A‐PXA showed an unusual genomic profile, characterized by NF1 mutation and marked aneuploidy (Figure S5), including two confirmed by methylation‐based classification. NF1 has been described as a somatic mutation in PXA (4) and PXA rarely occur in the setting of neurofibromatosis type 1, the majority low‐grade (37). However, to our knowledge, this molecular profile has not been previously reported in A‐PXA.
Multiple studies have identified CDKN2A/B deletion to be a frequent genomic alteration in PXA (20, 27, 36, 39). However, the frequency at which it has been observed has varied across studies, from approximately 60% (20, 39) up to 100% (27). While this may in part reflect variability across patient cohorts, it is also likely to result from technical differences in the methods used to detect CDKN2A/B deletion. In our series, CDKN2A/B loss was present in 63 cases (94%), although in 10 cases the loss was hemizygous or could not be unequivocally interpreted as hemi‐ vs. homozygous deletion. This variability in detection sensitivity has important implications as CDKN2A/B becomes increasingly utilized as a diagnostic and prognostic marker. The MIP technology used in this study represents a high‐resolution, quantitative method for the assessment of allele‐specific copy numbers in FFPE archival tissue. As CDKN2A/B deletion was present in the vast majority of our cases, we are unable to assess its prognostic significance in PXA. Only three tumors in our series lacked CDKN2A/B deletion and had clinical follow‐up available, although none of these tumors recurred. In other tumor types, including pediatric gliomas (21) and IDH‐mutant astrocytomas (33) CDKN2A/B deletion is an adverse prognostic factor. However, in PXA, it appears to be a defining molecular alteration likely representing an early pathogenic event, present in both low‐grade and anaplastic PXA.
In comparison to PXA grade II, A‐PXA showed a higher number of whole chromosome CNAs (Table S2) and a subset harbored additional pathogenic alterations, including mutations of TERT, ATRX and TP53 (Figure 1). These findings are comparable to a recently published genetic characterization of 15 A‐PXA from UCSF (27). However, this series reported a high frequency of TERT alterations, identified in 47% (7 of 15) of A‐PXA (27). Two of these tumors harbored TERT amplification rather than promoter mutation, an alteration we did not observe. In our cohort, TERT promoter mutations were uncommon, identified in 2.0% of PXA grade II and 14.3% of A‐PXA. A difference between the UCSF series and our series is that the majority of the A‐PXA in the UCSF series (9 of 15, 60%) were analyzed at recurrence (27) and TERT alterations were more frequent in recurrent tumors (5 of 9; 56%). No details were provided regarding adjuvant treatments patients received prior to recurrence. Additionally, 2 (of 9) tumors in our series (Figure 3) and 1 (of 4) tumors in the UCSF series acquired TERT mutation at recurrence, suggesting that TERT promoter mutations may be a late event associated with recurrence and anaplastic transformation. A high frequency of TERT alterations was also identified in a group of PXA utilized as a reference in comparison to epithelioid glioblastoma (20). In this cohort, TERT promoter mutations were observed in 19% of PXA grade II and 36% of A‐PXA. The clinicopathologic features of this cohort have not been independently reported. Overall, our findings suggest that additional CNAs and pathogenic mutations are present in A‐PXA, including TERT promoter mutations. However, no single recurrent alteration seems to underlie anaplastic transformation.
For the majority of cases in our series, methylation‐based classification was concordant with the histologic diagnosis of PXA (Figure 2, Table S3). In only two cases both unsupervised t‐SNE clustering and supervised analysis using the Heidelberg classifier unequivocally reassigned tumors to other entities. Both were PXA grade II by histology, which matched to other low‐grade glioma methylation groups (pilocytic astrocytoma or ganglioglioma). Intriguingly, these were the only two cases submitted for methylation profiling that lacked CDKN2A/B deletion. By unsupervised clustering, an additional two PXA cases grouped with the methylation class of anaplastic pilocytic astrocytoma (Figure 2). The cases included in this reference group cluster with the subsequently described anaplastic astrocytoma with piloid features (30). Notably, these were the only two cerebellar tumors analyzed, suggesting a possible influence of tumor site on the methylation profile. While methylation profiling was largely concordant with histology, the Heidelberg classifier did not uniformly provide a definitive diagnosis, with low‐confidence calls (calibrated score < 0.9) in 34.4% of cases overall and 60.0% of A‐PXA (Table S3). This is in keeping with a recently published series, in which 44% of pediatric low‐grade gliomas similarly showed low‐confidence calls using this tool (9). Across our series, methylation profiling largely confirmed the diagnosis of PXA and did not provide additional information likely to alter clinical management. In fact, its inability to distinguish low‐ and high‐grade PXA represents a significant limitation of methylation‐based classification in this setting.
Within this cohort, WHO grade was the most significant prognostic factor. The presence of anaplastic features (defined by > 5 mitoses per 10 high‐power fields (area of 0.23 mm2) was a strong predictor of patient overall survival (P < 0.001) with a trend toward decreased progression‐free survival (P = 0.05) (Figure 4). This was consistent even when limited to those 38 tumors that could be defined as PXA both by histology and methylation‐based classification (Figure 4C,D). These findings parallel those of a recent study showing the importance of histology in determining prognosis of pediatric gliomas with a PXA‐like methylation signature (9). Although identified in a small number of cases, our findings also suggest that TERT mutation may be associated with poor patient outcome. Each of the three patients with TERT‐mutant tumors at initial resection died, including one patient with a PXA grade II (Figure 1). In keeping with this, the UCSF case series, which identified TERT promoter mutations in 47% of A‐PXA, reported an extremely high rate of recurrence (12 of 13 patients) with very short recurrence‐free survival (median 1.3 years) (27). In contrast, in our series, 5‐year recurrence free survival for A‐PXA was 40.9%. Larger series with long‐term follow‐up are needed to clarify the role of TERT promoter mutation in PXA.
Our study was restricted to PXA with well‐defined histologic features. As such, methylation‐based classification was largely confirmatory. A number of recent studies have reported tumors that can be assigned to the PXA methylation class but lack its characteristic morphology, including epithelioid glioblastoma (20), astroblastoma (22) and a subset of pediatric gliomas (23). While the relationship of these tumors to PXA remains incompletely understood, our results emphasize the importance of careful histologic examination and correlation with long‐term follow‐up. While “PXA‐like” tumors may have more favorable outcomes than IDH‐wild‐type glioblastoma, it is premature to assume that all will have outcomes analogous to PXA.
In summary, our results confirm that the vast majority of PXA harbor CDKN2A/B deletion in conjunction with alterations of the MAPK pathway. In both histologically and epigenetically defined PXA, tumor WHO grade remains a strong predictor of patient survival. Methylation profiling may be useful in tumors with ambiguous morphology, but serves largely to confirm the diagnosis in cases with classic histology.
Conflict of Interest
The authors have no conflict of interest to declare.
Supporting information
Figure S1. Age and gender distribution of PXA patients. Age at first diagnosis is shown for 67 patients. M, Male; F, Female.
Figure S2. Copy number alterations in PXA. (A) Genome‐wide copy number alterations. (B) Copy number alterations of 9p, highlighting homozygous deletions of CDKN2A/B. Case order matches that of Figure 1. cnLOH, copy neutral loss of heterozygosity.
Figure S3. Structural rearrangements determined by mate‐pair sequencing. Genome U‐plots are shown with chromosomes plotted in order of size as denoted on the y‐axes. Copy number gains are shown in blue and losses in red. Structural rearrangements are depicted as connecting lines, with unbalanced translocations in magenta, balanced translations in dark green, transposons in light green, and complex or ambiguous rearrangements in orange. For unbalanced translocations, red or blue ends denote reads mapped to reverse or forward strand, respectively.
Figure S4. Structural rearrangements of case 45. (A) Genome plot of case 45. Complex structural rearrangements were identified, including chromoplexis involving chromosomes 6, 13, 16, and 22. (B) Junction plot demonstrating ATG7‐RAF1 fusion. The fusion involved intronic regions resulting in fusion of ATG7 exons 1‐18 with RAF1 exons 11‐17.
Figure S5. Histology and copy number profiles of NF1‐mutant A‐PXA. (A) Morphology of NF1‐mutant A‐PXA. Case 62 showed a predominance of large, pleomorphic and variably lipidized cells, with prominent perivascular inflammation. Cases 63 and 64 showed prominent spindled morphology, admixed with pleomorphic and scattered xanthomatous cells. All cases demonstrated > 5 mitoses per 10 high‐power fields. Case 63 also showed necrosis and case 42 both necrosis and microvascular proliferation. H&E photomicrographs shown at 200x (top panel; scale bar = 100 μM) and 400x (bottom, scale bar = 50 μM) magnification. (B) Genome‐wide copy number profiles of NF1‐mutant A‐PXA. Case 62 showed an apparently triploid genome with many additional complex CNVs. Cases 63 and 64 showed a nearly identical pattern of CNVs, with a near haploid genotype and homozygous deletion of CDKN2A/B.
Figure S6. Histology of PXA clustering with other reference groups. Cases 1 and 2 demonstrated pleomorphic, low‐grade gliomas. No apparent ganglion cell component was identified by immunohistochemistry (not shown). Case 2 showed numerous eosinophilic granular bodies and was more cellular than typical of pilocytic astrocytoma. Cases 4 and 62 showed cellular tumor consistent with PXA. Cases 41 and 23 were cerebellar tumors with spindled and pleomorphic components. Case 40 is shown at initial resection and at recurrence. The recurrent tumor was high‐grade and unrecognizable as a PXA, with small cell morphology, necrosis, and microvascular proliferation. H&E photomicrographs shown at 200x (left panel) and 400x (right panel) for each case.
Figure S7. t‐SNE of distinct low‐ and high‐grade areas of case 67. Histologically low‐grade (grey) and high‐grade (black) components from case 67 were analyzed independently by methylation profiling. Reference methylation classes included: PXA, (anaplastic) pleomorphic xanthoastrocytoma; ANA PA, anaplastic pilocytic astrocytoma; LGG‐GG, low‐grade glioma, ganglioglioma; LGG‐PA/GG‐ST, low‐grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma; CONT‐INFLAM, control tissue, inflammatory tumor microenvironment; CONT‐HEMI, control tissue hemispheric cortex; CONT‐REACT, control tissue, reactive tumor microenvironment; glioblastoma IDH wildtype subgroups: GBM‐MES, mesenchymal subclass; GBM‐MID, midline subclass; GBM‐RTK‐I subclass; GBM RTK‐II subclass; GBM‐RTK‐III subclass; pedRTK I subclass; GBM‐MYCN subclass.
Table S1‐S4
Table S1. Characteristics of patient cohort.
Table S2. Association of genomic alterations with tumor grade.
Table S3. Tumor classification by genome‐wide methylation profiling.
Table S4. Association of clinical, histologic, and genomic characteristics with patient overall survival.
Supplementary Material
Acknowledgments
This work was supported by Deutsche Kinderkrebsstiftung (German Children Cancer Foundation) grants DKS 2006.03, 2009.19, 2011.01 and 2014.17 to TP. Funding was provided by Mayo Clinic by Mayo Department of Laboratory Medicine and Pathology to CG.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Alexandrescu S, Korshunov A, Lai SH, Dabiri S, Patil S, Li R et al (2016) Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas‐same entity or first cousins? Brain Pathol 26:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aryee MJ, Jaffe AE, Corrada‐Bravo H, Ladd‐Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D et al (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A 104:20007–20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ et al (2013) Exomic sequencing of four rare central nervous system tumor types. Oncotarget 4:572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB et al (2018) cIMPACT‐NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH‐wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol 136:805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation‐based classification of central nervous system tumours. Nature 555:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drucker TM, Johnson SH, Murphy SJ, Cradic KW, Therneau TM, Vasmatzis G (2014) BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics 30:1627–1679. [DOI] [PubMed] [Google Scholar]
- 8. Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ et al (2014) Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 15:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fukuoka K, Mamatjan Y, Tatevossian R, Zapotocky M, Ryall S, Stucklin AG et al (2020) Clinical impact of combined epigenetic and molecular analysis of pediatric low grade gliomas. Neuro‐oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gaitatzes A, Johnson SH, Smadbeck JB, Vasmatzis G (2018) Genome U‐Plot: a whole genome visualization. Bioinformatics 34:1629–1634. [DOI] [PubMed] [Google Scholar]
- 11. Gessi M, van de Nes J, Griewank K, Barresi V, Buckland ME, Kirfel J et al (2014) Absence of TERT promoter mutations in primary melanocytic tumours of the central nervous system. Neuropathol Appl Neurobiol. 40:794–797. [DOI] [PubMed] [Google Scholar]
- 12. Giannini C, Paulus W, Louis DN, Liberski PP, Figarella‐Branger D, Capper D (2016) Anaplastic pleomorphic xanthoastrocytoma. In: WHO Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 98–99. International Agency for Research on Cancer: Lyon. [Google Scholar]
- 13. Giannini C, Paulus W, Louis DN, Liberski PP, Figarella‐Branger D, Capper D (2016) Pleomorphic xanthoastrocytoma. In: WHO Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 94–97. International Agency for Research on Cancer: Lyon. [Google Scholar]
- 14. Goschzik T, Schwalbe EC, Hicks D, Smith A, Zur Muehlen A, Figarella‐Branger D et al (2018) Prognostic effect of whole chromosomal aberration signatures in standard‐risk, non‐WNT/non‐SHH medulloblastoma: a retrospective, molecular analysis of the HIT‐SIOP PNET 4 trial. Lancet Oncol 19:1602–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsiao SJ, Karajannis MA, Diolaiti D, Mansukhani MM, Bender JG, Kung AL, Garvin JH Jr (2017) A novel, potentially targetable TMEM106B‐BRAF fusion in pleomorphic xanthoastrocytoma. Cold Spring Harb Mol Case Stud 3:a001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ida CM, Rodriguez FJ, Burger PC, Caron AA, Jenkins SM, Spears GM et al (2015) Pleomorphic xanthoastrocytoma: natural history and long‐term follow‐up. Brain Pathol 25:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ida CM, Vrana JA, Rodriguez FJ, Jentoft ME, Caron AA, Jenkins SM, Giannini C (2013) Immunohistochemistry is highly sensitive and specific for detection of BRAF V600E mutation in pleomorphic xanthoastrocytoma. Acta Neuropathol Commun 1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson SH, Smadbeck JB, Smoley SA, Gaitatzes A, Murphy SJ, Harris FR et al (2018) SVAtools for junction detection of genome‐wide chromosomal rearrangements by mate‐pair sequencing (MPseq). Cancer Genet 221:1–18. [DOI] [PubMed] [Google Scholar]
- 19. Kleinschmidt‐DeMasters BK, Aisner DL, Birks DK, Foreman NK (2013) Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol 37:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korshunov A, Chavez L, Sharma T, Ryzhova M, Schrimpf D, Stichel D et al (2018) Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol 28:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R et al (2017) Therapeutic and prognostic implications of BRAF V600E in pediatric low‐grade gliomas. J Clin Oncol 35:2934–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lehman NL, Usubalieva A, Lin T, Allen SJ, Tran QT, Mobley BC et al (2019) Genomic analysis demonstrates that histologically‐defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mackay A, Burford A, Molinari V, Jones DTW, Izquierdo E, Brouwer‐Visser J et al (2018) Molecular, pathological, radiological, and immune profiling of non‐brainstem pediatric high‐grade glioma from the HERBY Phase II randomized trial. Cancer Cell 33:829–842.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsumura N, Nakajima N, Yamazaki T, Nagano T, Kagoshima K, Nobusawa S et al (2017) Concurrent TERT promoter and BRAF V600E mutation in epithelioid glioblastoma and concomitant low‐grade astrocytoma. Neuropathology 37:58–63. [DOI] [PubMed] [Google Scholar]
- 25. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M et al (2015) BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high‐grade glioma. J Clin Oncol 33:1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murphy SJ, Cheville JC, Zarei S, Johnson SH, Sikkink RA, Kosari F et al (2012) Mate pair sequencing of whole‐genome‐amplified DNA following laser capture microdissection of prostate cancer. DNA Res 19:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Bastian BC et al (2019) The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol 29:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Jin LW et al (2016) Activating NRF1‐BRAF and ATG7‐RAF1 fusions in anaplastic pleomorphic xanthoastrocytoma without BRAF p. V600E mutation. Acta Neuropathol 132:757–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pratt D, Camelo‐Piragua S, McFadden K, Leung D, Mody R, Chinnaiyan A et al (2018) BRAF activating mutations involving the beta3‐alphaC loop in V600E‐negative anaplastic pleomorphic xanthoastrocytoma. Acta Neuropathol Commun 6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DE et al (2018) Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136:273–291. [DOI] [PubMed] [Google Scholar]
- 31. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 32. Schulte SL, Waha A, Steiger B, Denkhaus D, Dorner E, Calaminus G et al (2016) CNS germinomas are characterized by global demethylation, chromosomal instability and mutational activation of the Kit‐, Ras/Raf/Erk‐ and Akt‐pathways. Oncotarget 7:55026–55042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F et al (2018) Novel, improved grading system(s) for IDH‐mutant astrocytic gliomas. Acta Neuropathol 136:153–166. [DOI] [PubMed] [Google Scholar]
- 34. Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Triche TJ Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD (2013) Low‐level processing of illumina infinium DNA methylation beadarrays. Nucleic Acids Res 41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vaubel RA, Caron AA, Yamada S, Decker PA, Eckel Passow JE, Rodriguez FJ et al (2018) Recurrent copy number alterations in low‐grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol 28:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vizcaino MA, Caccamo DV, Fox E, Rodriguez FJ (2014) Pleomorphic xanthoastrocytoma: report of two cases with unconventional clinical presentations. Clin Neuropathol 33:380–387. [DOI] [PubMed] [Google Scholar]
- 38. Wang Y, Cottman M, Schiffman JD (2012) Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet 205:341–355. [DOI] [PubMed] [Google Scholar]
- 39. Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B et al (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088–1097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Age and gender distribution of PXA patients. Age at first diagnosis is shown for 67 patients. M, Male; F, Female.
Figure S2. Copy number alterations in PXA. (A) Genome‐wide copy number alterations. (B) Copy number alterations of 9p, highlighting homozygous deletions of CDKN2A/B. Case order matches that of Figure 1. cnLOH, copy neutral loss of heterozygosity.
Figure S3. Structural rearrangements determined by mate‐pair sequencing. Genome U‐plots are shown with chromosomes plotted in order of size as denoted on the y‐axes. Copy number gains are shown in blue and losses in red. Structural rearrangements are depicted as connecting lines, with unbalanced translocations in magenta, balanced translations in dark green, transposons in light green, and complex or ambiguous rearrangements in orange. For unbalanced translocations, red or blue ends denote reads mapped to reverse or forward strand, respectively.
Figure S4. Structural rearrangements of case 45. (A) Genome plot of case 45. Complex structural rearrangements were identified, including chromoplexis involving chromosomes 6, 13, 16, and 22. (B) Junction plot demonstrating ATG7‐RAF1 fusion. The fusion involved intronic regions resulting in fusion of ATG7 exons 1‐18 with RAF1 exons 11‐17.
Figure S5. Histology and copy number profiles of NF1‐mutant A‐PXA. (A) Morphology of NF1‐mutant A‐PXA. Case 62 showed a predominance of large, pleomorphic and variably lipidized cells, with prominent perivascular inflammation. Cases 63 and 64 showed prominent spindled morphology, admixed with pleomorphic and scattered xanthomatous cells. All cases demonstrated > 5 mitoses per 10 high‐power fields. Case 63 also showed necrosis and case 42 both necrosis and microvascular proliferation. H&E photomicrographs shown at 200x (top panel; scale bar = 100 μM) and 400x (bottom, scale bar = 50 μM) magnification. (B) Genome‐wide copy number profiles of NF1‐mutant A‐PXA. Case 62 showed an apparently triploid genome with many additional complex CNVs. Cases 63 and 64 showed a nearly identical pattern of CNVs, with a near haploid genotype and homozygous deletion of CDKN2A/B.
Figure S6. Histology of PXA clustering with other reference groups. Cases 1 and 2 demonstrated pleomorphic, low‐grade gliomas. No apparent ganglion cell component was identified by immunohistochemistry (not shown). Case 2 showed numerous eosinophilic granular bodies and was more cellular than typical of pilocytic astrocytoma. Cases 4 and 62 showed cellular tumor consistent with PXA. Cases 41 and 23 were cerebellar tumors with spindled and pleomorphic components. Case 40 is shown at initial resection and at recurrence. The recurrent tumor was high‐grade and unrecognizable as a PXA, with small cell morphology, necrosis, and microvascular proliferation. H&E photomicrographs shown at 200x (left panel) and 400x (right panel) for each case.
Figure S7. t‐SNE of distinct low‐ and high‐grade areas of case 67. Histologically low‐grade (grey) and high‐grade (black) components from case 67 were analyzed independently by methylation profiling. Reference methylation classes included: PXA, (anaplastic) pleomorphic xanthoastrocytoma; ANA PA, anaplastic pilocytic astrocytoma; LGG‐GG, low‐grade glioma, ganglioglioma; LGG‐PA/GG‐ST, low‐grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma; CONT‐INFLAM, control tissue, inflammatory tumor microenvironment; CONT‐HEMI, control tissue hemispheric cortex; CONT‐REACT, control tissue, reactive tumor microenvironment; glioblastoma IDH wildtype subgroups: GBM‐MES, mesenchymal subclass; GBM‐MID, midline subclass; GBM‐RTK‐I subclass; GBM RTK‐II subclass; GBM‐RTK‐III subclass; pedRTK I subclass; GBM‐MYCN subclass.
Table S1‐S4
Table S1. Characteristics of patient cohort.
Table S2. Association of genomic alterations with tumor grade.
Table S3. Tumor classification by genome‐wide methylation profiling.
Table S4. Association of clinical, histologic, and genomic characteristics with patient overall survival.
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.