

Abstract
Recent studies have highlighted a potential role of genetic and epigenetic variation in the development of Alzheimer’s disease. Application of the CRISPR‐Cas genome‐editing platform has enabled investigation of the functional impact that Alzheimer’s disease‐associated gene mutations have on gene expression. Moreover, recent advances in the technology have led to the generation of CRISPR‐Cas–based tools that allow for high‐throughput interrogation of different risk variants to elucidate the interplay between genomic regulatory features, epigenetic modifications, and chromatin structure. In this review, we examine the various iterations of the CRISPR‐Cas system and their potential application for exploring the complex interactions and disruptions in gene regulatory circuits that contribute to Alzheimer’s disease.
Keywords: Alzheimer's disease (AD), CRISPR, epigenetics, genetics, genome editing
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that affects approximately 50 million people worldwide and carries a significant public health burden (44). The disorder is characterized by the aggregation of amyloid beta (Aβ) plaques and neurofibrillary tangles of hyperphosphorylated tau protein in the cerebral cortex and limbic brain regions, which in turn cause neuronal cell death and thus, cognitive impairment (12). Although the pathophysiology of AD has been well described, current therapeutic strategies only temporarily alleviate symptoms and, to date, no disease‐modifying treatments that target the underlying causes are available.
Owing to advances in genomic technology, a number of large‐scale genome‐wide association studies (GWAS) and epigenome wide association studies (EWAS) have successfully been undertaken in AD in recent years, which are extensively reviewed elsewhere in this mini‐symposium series (2, 45, 67, 77). Regarding its genetic heritability, the autosomal dominant inheritance of disease‐causing mutations in the amyloid precursor protein (APP), presenilin (PSEN) 1 and 2 genes results in the early onset familial form of AD, which accounts for less than 5% of total AD cases (60). In contrast, the majority of AD cases are sporadic, which is associated with a number of common genetic risk factors that individually only slightly contribute to disease development. However, the accumulation of these common risk variants has a significant impact on disease risk (79). The vast majority of AD GWAS loci reside in non‐coding regions of the genome and combined with the polygenicity of sporadic AD this severely complicates the functional characterization of risk variants. Similarly, as epigenetic variation is tissue and cell‐type specific and as AD EWAS have to be undertaken on post‐mortem brain tissue, it is not possible to determine whether disease‐associated epigenetic changes are causal, or are simply secondary to the disease process. Furthermore, the relationship between epigenetic variation and gene expression is situation‐dependent and largely still needs to be explored in the context of AD. Taken together, further research is now required to elucidate the downstream functional consequence of AD‐associated genetic and epigenetic variation, which could then allow their translation to novel treatments.
The Genome Editing Toolbox
In the past decade, functional genomics has been transformed by the emergence of various different gene editing tools, including zinc‐finger nucleases (ZFNs), transcription activator‐like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeat (CRISPR)‐CRISPR‐associated protein 9 (Cas9) technology. Genome engineering approaches like ZFNs and TALENs use Fokl endonuclease domains fused to transcription factor derived DNA‐binding modules to specifically modify a locus (3, 29). TALEN technology significantly improved upon the feasibility of gene editing experiments pioneered by the ZFNs. However, despite its DNA binding specificity at a single nucleotide level and immense targeting capacity, TALEN guided gene editing can be costly and time intensive (29). In contrast, the CRISPR‐Cas9 system offers a simple, cost‐effective, multifunctional platform that has led to the generation of new model systems in vitro and in vivo in various research areas (23, 83). As a result, this technology has the realistic potential to elucidate the functional consequence of genetic and epigenetic signatures in AD, thereby establishing whether these associations are secondary to pathogenesis or causal in disease development. In this review, we discuss the recent advances in CRISPR technology, its existing application to other neurodegenerative disorders and the limitations of such approaches.
The CRISPR‐Cas system is an adaptive immune mechanism used by prokaryotes to intercept foreign nucleic acids (81). The type II CRISPR‐Cas9 identified in Streptococcus pyogenes is the most commonly used of the bacterial CRISPR systems and effectively achieves DNA cleavage at a genomic locus of interest, which is illustrated in Figure 1A. The ribonucleoprotein Cas9 forms a complex with a synthetic single guide RNA (sgRNA) strand through a scaffold sequence and recognizes its target via a ~20 nucleotide long spacer sequence adjacent to the Protospacer Adjacent Motif (PAM) (14). Upon target binding, the Cas9 protein undergoes a conformational change that places the nuclease domains RuvC and HNH on opposite strands to induce a double‐strand break (DSB) within the target DNA. The cleaved DNA may then be repaired by the error‐prone non‐homologous end joining (NHEJ) pathway, which causes nucleotide insertions or deletions at the DSB site that can knockout a gene through premature stop codon formation. It is possible to introduce specific nucleotide modifications using the homology directed repair (HDR) pathway where a homologous DNA donor template is provided and its sequence is incorporated into the target DNA during repair at a low efficiency (47, 58).
Figure 1.
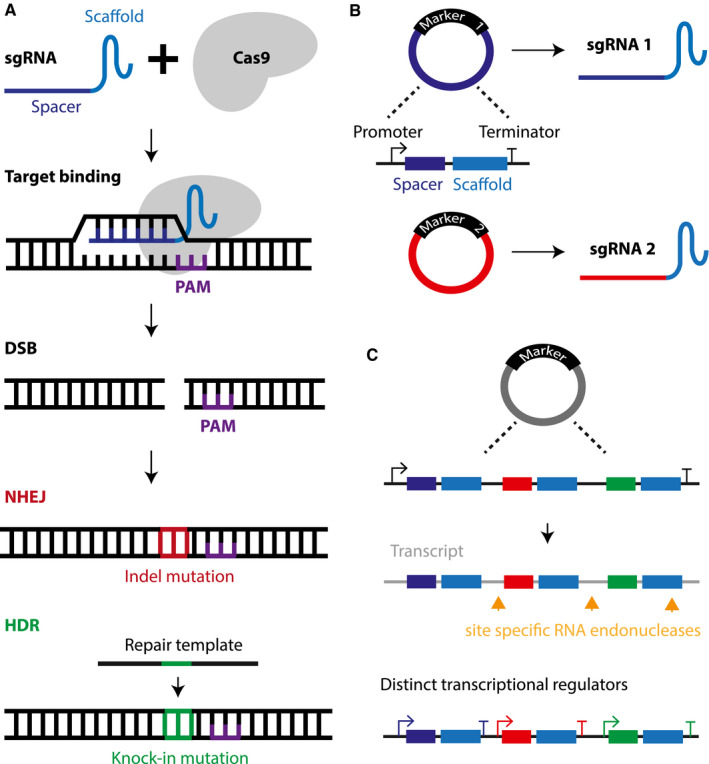
A. The Cas9 nuclease of the clustered regularly interspaced short palindromic repeat (CRISPR) gene editing platform recognizes a DNA sequence of interest via the single guide (sg) RNA that forms a complex with Cas9. The target DNA must contain the protospacer adjacent motif (PAM) sequence for the Cas9‐sgRNA complex to bind and induce a double strand break (DSB). The damaged DNA is then repaired by the non‐homologous end joining (NHEJ) pathway, which is error‐prone and may produce insertion or deletion (Indel) mutations that in turn can cause a gene knockout by the formation of premature stop codons. Alternatively, a precise gene knock‐in can be generated by harnessing the homology directed repair pathway (HDR), which requires a homologous DNA template. B. Multiplexing is facilitated by the short construct size of the plasmid carrying the sgRNA sequence and the various selection markers than can be applied to identify successfully transfected cell populations. C. Multiple sgRNA sequences can be integrated into a single plasmid and the different sgRNA are then delivered via post‐translational processing of the mRNA transcript by site specific RNA endonucleases or distinct promoter sequences for each sgRNA sequence.
A major advantage of CRISPR technology is its inherent ability for multiplex genome engineering. Previous gene editing tools required repeated delivery of large expression constructs coupled with numerous selection markers such as fluorescent reporter or antibiotic resistance genes to generate a population of cells carrying multiple modified loci. The labor intensive nature of these methodologies limited the development of isogenic control cell lines and disease gene knockout models suitable for modeling a polygenic disease. With the introduction of sgRNA, Cas9 complexes can be directed to different loci simultaneously without the need for designing specific DNA targeting domains, which is depicted in Figure 1B (9). Moreover, the experimental drawbacks from repeated construct delivery and selection can be averted with “all‐in‐one” vectors that carry multiple sgRNA expression cassettes on a single plasmid and are visualized in Figure 1C. Transcriptional regulation is then guided by several independent promoters or linked sgRNA sequences under the control of one promoter, where the transcript requires post‐translational processing to deliver the various sgRNA (30, 35, 62). Therefore, the concurrent modification of multiple disease‐associated loci in the context of sporadic AD may elucidate the interplay between numerous GWAS identified genes. The power of this tool culminates in its ability to determine the functional consequences of genetic variation in a high‐throughput fashion. As such, CRISPR‐Cas9 technology holds the key to dissect the etiology of complex diseases such as AD.
Attacking the Disease at its Source: Targeting the Genetic Mutations Causing AD
As discussed elsewhere in this Mini‐Symposium (2), genomics plays a significant role in AD pathogenesis, particularly in familial AD where autosomal dominant mutations in the APP, PSEN1, and PSEN2 genes are known to cause enhanced production of the Aβ1‐42 peptide. Briefly, Aβ production begins with the aberrant cleavage of the transmembrane protein APP by the beta‐secretase BACE1. A subsequent cleavage through the gamma secretase complex, where the protein products of PSEN1 and PSEN2 are located, yields the Aβ peptide with these monomers accumulating into the characteristic amyloid plaques of AD (6, 65). Although only a small fraction of AD cases result from these autosomal dominant mutations, the pathological hallmarks of familial AD and sporadic AD are identical and thus, familial AD is ideally suited for studying the consequence of amyloid pathology using the CRISPR‐Cas9 system.
A number of studies have used the CRISPR‐Cas9 system in familial AD patient induced pluripotent stem cell (iPSC) lines to generate isogenic controls without the causal mutation and this process is depicted in Figure 2. Ortiz‐Virumbrales et al used CRISPR‐Cas9 in iPSC neurons derived from a patient carrying the PSEN2 N141I mutation to correct the autosomal dominant mutation, which led to the normalization of the Aβ42/40 ratio (50). The generation of gene‐corrected isogenic control iPSC lines from patients has also been performed in patient lines bearing the PSEN1 mutations A79V and L150P and constitutes a useful in vitro disease model to investigate both the mechanisms underlying familial AD and the consequence of Aβ pathology (54, 56). In the APP gene, the Swedish mutation is a well characterized, amyloidogenic double mutation that occurs adjacent to the BACE1 cleavage site in APP and causes a two amino acid substitution where lysine and methionine become asparagine and leucine. The resulting increase in secreted Aβ40 and Aβ42 from this mutation leads to severe amyloid pathology (63). Using patient derived fibroblasts carrying the Swedish mutation, Gyorgy et al recently disrupted the cleavage site and observed a 60% decrease in Aβ production (22). Furthermore, the mutation was also targeted in both cultured primary cortical neurons and in vivo using Tg2576 adult mice that carry the APPSW mutation via adeno‐associated virus mediated delivery of the Cas9 and sgRNA constructs into the hippocampus. Although disruption of the Swedish mutation in the form of single base pair insertions was observed, the editing efficiency was significantly limited by the Tg2576 mouse model, which carries around 100 copies of the transgene in each neuron (22). To avoid the restrictions of such models, Park et al instead targeted BACE1, using self‐assembling nanocomplexes of the amphiphilic R7L10 peptide to package the Cas9 and gRNA constructs (51). Injection of the nanocomplexes into the hippocampus of the 5xFAD transgenic AD mouse model showed an approximately 70% decrease in BACE1 expression. This was accompanied by an up to 15% reduction in hippocampal plaque burden and correlated with improvements in behavioral tests (51). Together, these strategies demonstrate the capability of CRISPR‐Cas9 to modulate gene expression in patient derived iPSC lines and reduce pathology in transgenic mouse models of familial AD, by successfully manipulating the amyloidogenic pathway. However, non‐dominantly inherited forms of AD constitute the majority of AD cases and stem from a multiplicity of different risk factors that require alternative methodologies to develop an effective therapeutic option for these cases. A potentially protective mutation was identified in the 3’UTR of the APP gene and induced in APP knock‐in mouse zygotes with CRISPR‐Cas9 mediated deletions. The 3’UTR disruption substantially reduced APP expression at both the transcriptional and translational levels (49). Such a targeted screening strategy of rare variants in human populations can be used to identify protective AD mutations. Indeed, similar novel approaches have been utilized by Sun et al, where the C‐terminus of the APP gene was targeted to prevent endosomal trafficking of the protein, and thus aberrant cleavage by β‐secretase and decreased Aβ secretion was observed in both iPSCs and mouse derived cells (74).
Figure 2.

Interrogation of AD‐associated gene mutations using iPSC technology and CRISPR‐Cas9 gene editing to generate isogenic (ie, same genetic background) control cell lines. iPSC cells derived from a patient affected by familial AD (FAD) carry the pathogenic mutation, which is corrected by CRISPR‐Cas9. The mutation corrected iPSCs are used to model disease development during differentiation into specific cell types of the brain (eg, neurons). Introduction of the pathogenic mutation in iPSC cells derived from a healthy patient by CRISPR‐Cas9 may be applied to demonstrate recapitulation of disease pathogenesis in vitro when compared to the isogenic controls. The CRISPR‐Cas9 platform enables the study of gene interaction networks and environmental conditions in AD.
In terms of genetic risk factors for AD, the most significantly associated gene with sporadic AD is the ε4 isoform of the polymorphic Apolipoprotein E (APOE) gene. The major three alleles differ by single amino acid substitutions of cysteine to arginine that cause conformational changes in the protein and modify its ability to interact with APOE receptors and Aβ. Although our understanding of APOE ε4’s role in AD development is limited, it is known that carriers of the ε4 allele have a significantly increased risk of developing AD in comparison to those with the more common ε3 allele, whereas the rare ε2 variant confers reduced risk (10, 36, 40). To elucidate APOE ε4’s role in sporadic AD, Lin et al generated an isogenic iPSC APOE ε4 line from an APOE ε3 line, which had been modified to harbor homozygous APOE ε4 alleles through a HDR‐mediated CRISPR‐Cas9 edit where the repair template carried the single amino acid substitution (Cys112Arg) to yield APOE ε4 (39). After differentiating the APOE ε3 and APOE ε4 iPSC lines into different brain cell types, they demonstrated impaired Aβ clearance in astrocytes and microglia, and increased Aβ42 secretion in neurons, in the APOE ε4 line compared to its APOE ε3 isogenic counterpart. Furthermore, when they used the same methodology to convert APOE ε4 to APOE ε3 in iPSCs generated from sporadic AD patients, they observed reduced extracellular uptake and accumulation of Aβ (39). Consequently, this study establishes the impact of the APOE ε4 allele on cell‐type specific alterations and highlights the potential applications of CRISPR‐Cas9 technology to elucidate the functional consequence of AD‐associated variants. Although this tool has been rapidly implemented to study the role of specific genes in AD (eg, APOE, APP, PSEN1, and PSEN2), sporadic AD has a considerable polygenic component, with common risk variants often found within non‐coding regions of the genome, which makes it more complicated to understand their functional impact on cellular pathways. Furthermore, the presence of protective alleles such as APOE ε2 and the existence of other (non‐genetic) risk factors further complicates the modeling of disease phenotype. Finally, it is difficult to model treatment development in vivo, as post‐mitotic neurons are unsuitable for CRISPR approaches that use HDR mediated gene modification where cell division must occur. Despite the promising results from modeling the functional consequence of AD‐associated variants in single genes, it still remains to be seen whether CRISPR‐Cas9 is the optimal technology for modeling all the aspects of a complex disease such as sporadic AD. Indeed, emulation of the gradual‐, age‐related processes, and various environmental influences on short living cells in cell culture is challenging. Nonetheless, the platform has experienced incredible development in recent years and disease‐causing genes are not the only targets that can now be investigated with this technology.
New Frontiers for Gene Editing in AD
Engineering of the Cas9 nuclease has transformed the CRISPR system into a DNA recognition complex capable of modifying gene expression independent of its ability to induce DSBs. Inactivation of the RuvC and HNH nuclease domains by point mutations in SpCas9 (D10A and H840A, respectively) result in a catalytically deficient Cas9 nuclease (dCas9) whose only function is to bind a DNA sequence complementary to the sgRNA (57). Additionally, recombinant DNA technology has advanced CRISPR‐Cas9 into a modular platform that enables the fusion of dCas9 with enzymatic effector domains. As a result, a vast dCas9 toolbox with diverse modifiers has been engineered to elicit targeted gene regulation, among many other applications. Although experiments harnessing these tools to explore AD‐associated genetic and epigenetic variation are yet to be published, other neurological conditions have been investigated with CRISPR‐Cas9 in that context, thereby establishing the technology as the ideal platform for future investigations.
In CRISPR interference (CRISPRi), transcription repressor domains such as the Kruppel associated box (KRAB) are tethered to dCas9 and effectively cause gene silencing when targeting promoter regions such as the transcription start site (TSS) by inducing heterochromatin formation (21). In cultured hippocampal neurons, Zheng and colleagues achieved more than 90% knock down efficiency in genes essential to neurotransmission with dCas9‐KRAB and consequently, reduced neurotransmitter release (85). Moreover, by multiplexing their dCas9‐KRAB and sgRNA constructs, inactivation of five genes (Syt1, Vamp2, Snap25, Stx1a, and Stx1b) was achieved in the mouse hippocampus, thereby demonstrating the feasibility to multiplex CRISPRi and modulate the expression of multiple genes, which would lend itself well to polygenic neuronal disorders such as AD (85).
Conversely, CRISPR activation (CRISPRa) uses specific activation systems such as the dCas9‐VPR protein, which consists of the three transcription factors VP64, p65, and Rta joined to dCas9. This tripartite synthetic activator may initiate transcription when guided to the promoter regions of the target gene (5). Similarly, Savell et al recently sought to multiplex gene expression activation of immediate early genes (Fos, Fosb, and Egr1) involved in neuronal function (64). Simultaneous induction of target genes was observed in striatal cultures when pooled lentiviruses were applied. Next, to establish the specificity of CRISPRa, they manipulated specific mRNA transcript levels by targeting the unique upstream promoter regions within the BDNF gene, which has a vital role in diverse neuronal processes. sgRNAs were devised to selectively induce expression of the transcript variants BDNF‐I and BDNF‐IV in hippocampal neurons, and upregulation of both the respective variant mRNA levels and total BDNF mRNA was observed (64). Thus, this approach can be used to directly characterize the functional roles of individual mRNA transcripts, which would be an incredibly powerful tool to investigate transcriptional dysregulation in AD‐associated genes.
Introduction of exogenous transcriptional regulators has proven to be an excellent way to study endogenous gene expression and investigate its dysfunction in disease (25). The field of epigenetics could particularly benefit from the CRISPR platform, since the mechanistic role of aberrant epigenetic modification is poorly understood and the previously available tools lacked feasibility or have global effects on the epigenome (27, 29, 73). With the addition of dCas9, the epigenetic landscape of complex disease can now be interrogated by several different modifiers that are specific to epigenetic marks. Modulating histone acetylation to elicit transcriptional activation can be achieved by tethering dCas9 to the catalytic core domain p300 of histone acetyltransferase, which causes histone acetylation H3Lys27 at endogenous loci and has been shown to transactivate gene expression at promoters and distal enhancers (24). Other epigenetic marks such as DNA methylation can be modified as well via fusion of dCas9 to the catalytic domain of Ten‐eleven translocation methyl cytosine dioxygenase 1 (TET1), which demethylates DNA and may result in targeted activation of gene expression (82). For example, hypermethylation of the CGG expansion mutation in the FMR1 gene leads to transcriptional repression that is responsible for fragile X syndrome. Liu and colleagues were able to reinstate FMR1 expression by demethylating the mutation with dCas9‐TET1 in vitro and in vivo, thereby demonstrating the applicability of such tools to study the causality of disease‐associated DNA methylation events (41). Likewise, dCas9 coupled with the catalytic domain of DNA methyltransferase 3A (DNMT3A) has been used to cause targeted DNA methylation of CpG islands within promoter regions, which resulted in decreased transcript levels and thus, gene silencing (80). To improve upon the methylation editing efficiency, repeating peptide arrays were added to the fusion constructs, which in turn recruit multiple catalytic domains fused to antibodies that elicit a far more significant change in methylation across a region of interest. Pioneered by the SunTag system, derivatives of this technology demonstrate more than 90% demethylation in several tested loci when TET1 was combined with the system and the introduction of DNMT3A can methylate a 4.5kb genomic region to silence HOXA5 gene expression (26, 48). The epigenetic dCas9 toolbox has expanded rapidly and the technologies described herein are illustrated in Figure 3. Although experiments specifically focusing on modeling epigenetic changes in AD models are yet to be published, there is considerable potential for their application in the context of AD, which are discussed in the following section.
Figure 3.
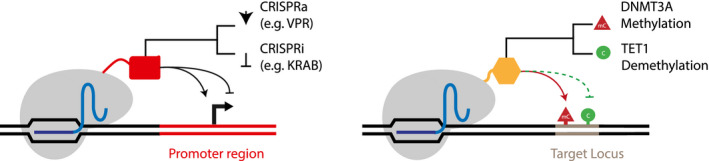
The CRISPR Cas9 platform enables targeted gene regulation via deactivation of the catalytic domain in the Cas9 nuclease (dCas9) and fusion to an enzymatic domain. Transcriptional activation (CRISPRa) may be achieved by a tripartite effector protein termed VPR (VP64, p65, Rta) that is targeting the promoter region of a gene, whereas transcriptional inactivation (CRISPRi) requires transcriptional repressor domains such as the Krüppel‐associated box (KRAB). DNA methylation can be altered by the DNA methyl transferase 3A (DNMT3A) enzyme that adds methyl groups to cytosines or the Ten‐eleven translocation methylcytosine dioxygenase 1 (TET1) enzyme, which removes DNA methylation at the target locus.
Solving the Epigenetic Puzzle of AD: In Search of the Causal Mechanisms
As reviewed elsewhere in this Mini‐Symposium, a growing number of EWAS investigating DNA methylation patterns in AD have been published in recent years (77). Interestingly, many of the differentially methylated loci reported in AD seem to be consistently observed across different cohorts. The first large‐scale EWAS in AD, published in 2014 by Lunnon et al and De Jager et al reported a number of overlapping CpG sites between studies, featuring, for example, genes such as ANK1, CDH23, and RHBDF2, which were significantly associated with differences in mRNA expression (13, 42, 43). These loci and a number of others have since been replicated by many other studies (37, 67, 69, 70). However, one caveat of the EWAS approach is the inability to determine whether the epigenetic variation seen in AD actually leads to disease, or is simply a consequence of the ongoing pathology. With the CRISPR‐Cas9 system, the functional consequence of these associations could now be investigated by inducing AD‐associated methylation levels at specific loci, such as ANK1, both in vitro and in vivo. However, it is worth noting that neuronal processes are driven by complex regulatory networks of gene expression and particularly in neurodegeneration, the dysregulation caused by epigenetic alterations is unlikely to be unidirectional. Given the many differentially methylated loci in AD, the true power of the CRISPR‐Cas9 technology emerges from its ability to be multiplexed.
High‐throughput functional screening of genes and regulatory elements is made possible by extensive sgRNA libraries that can knockout vast amounts of targets in the genome and annotate both the coding and non‐coding regions within it, which is illustrated in Figure 4. Coupling of these genome‐wide pooled CRISPR‐Cas9 screens with single‐cell RNA sequencing enables the interrogation of higher order interactions and is exemplified by Perturb‐Seq (15). In this method, the effects of CRISPR‐based perturbations on gene targets and cell states are analyzed to infer complex phenotypes such as transcription factor regulation by dendritic cells in response to an immunogenic compound like lipopolysaccharide. The identity of the perturbation is encoded in a guide barcode on the sgRNA and single‐cell RNA sequencing tags each cell with a unique molecular identifier, thereby associating the genetic modification with their respective transcriptional profiles. As a result, next generation sequencing of the sgRNA library facilitates the study of individual gene or epistatic effects on cell behavior and this could be a valuable tool for the AD field. The ability to perform CRISPR‐pooled screens with epigenetic modifiers opens up a variety of experimental designs that could be readily applied to AD. The CRISPR‐Cas9‐based epigenomic regulatory element screening (CERES) technique pioneered by Klann and colleagues effectively identifies regulatory element activity in the native chromosomal context and uses CRISPRi and CRISPRa to carry out loss‐of‐function and gain‐of‐function screens (32, 33). Applying such high‐content functional annotation in the context of AD may deliver insight into the regulatory elements that determine cellular phenotype in disease.
Figure 4.

High‐throughput genetic screens are made possible by sgRNA libraries. Pools of oligonucleotides are cloned into plasmids and packaged into a lentiviral library encoding the sgRNAs. Cas9 or dCas9 expressing cells are then transduced with the sgRNA library at a multiplicity of infection that delivers one sgRNA construct per cell. This methodology enables annotation of the coding and non‐coding genome via screening of functional and regulatory elements based on a selected phenotype. Selection is facilitated by markers such as fluorescent stains or reporters that are associated with the phenotype and identify the cell population of interest via fluorescent activated cell sorting (FACS). Alternatively, exposure to a chemical compound during cell culture may select for a cellular trait of interest such as vulnerability or resistance to the compound. Coupling of these processes with next generation sequencing to quantitate sgRNA enrichment across cells may determine the genes controlling the phenotype of interest.
Considering the epigenetic variation seen in AD, a straightforward application of the combinatorial approaches to the CRISPR platform would be the multiplexed emulation of the AD methylomic signature in a non‐AD patient‐derived cell line model, which is outlined in Figure 5. By modifying the methylation status of the numerous differentially methylated positions that have been robustly reported across various EWAS studies in AD and subsequently couple this with deep epigenomic and transcriptomic profiling, one could potentially infer the causality of loci via the acquisition of an AD‐like cellular phenotype. However, it is worth noting that some of these disease‐associated loci are intergenic, occurring outside of coding regions or the TSS. Therefore, although it is possible to induce the combinatorial methylomic signatures in vitro, understanding the regulatory consequence of epigenetic network alterations in the context of genome topology requires further thought. Hence, the development of epigenome editing strategies targeting the regulatory units termed topologically associated domains. Gene‐enhancer interactions are mediated by CTCF insulator proteins that bind specific sites in the DNA to generate chromosomal loop structures and separate distal enhancers from promoters, which prevents the expression of neighboring genes (59). In cancer, insulator interactions are often compromised by DNA methylation of CTCF binding sites, which leads to oncogene activation (18). Epigenome editing via dCas9‐DNMT3A has been shown to disrupt CTCF insulators, thereby simulating disease associated insulator loss events, which informs the mechanism behind epigenetic dysregulation of the chromatin structure (17, 75). Given the complexity behind the various regulatory layers that direct neurodevelopment and function, dissecting the impact of AD‐associatclinical implementationed epigenetic variation ultimately includes interrogation of the genome’s three‐dimensional organization in disease. Recently, the assay for transposase‐accessible chromatin (ATAC) was paired with high‐throughput single‐cell knockout screening to investigate genome‐wide chromatin states. Known as Perturb‐ATAC, application of this method disrupted trans‐regulatory factors such as transcription factors and non‐coding RNAs, which identified roles in nucleosome positioning. Furthermore, epistatic relationships were mapped between transcription factors of regulatory modules uncovered in keratinocyte differentiation, which enables the prediction of genetic interactions through the co‐localization and expression of transcriptions factors (61). Perturb‐ATAC provides an effective tool to elucidate the interplay between the epigenetic factors that modulate the chromatin state, which in turn determine cellular phenotypes.
Figure 5.
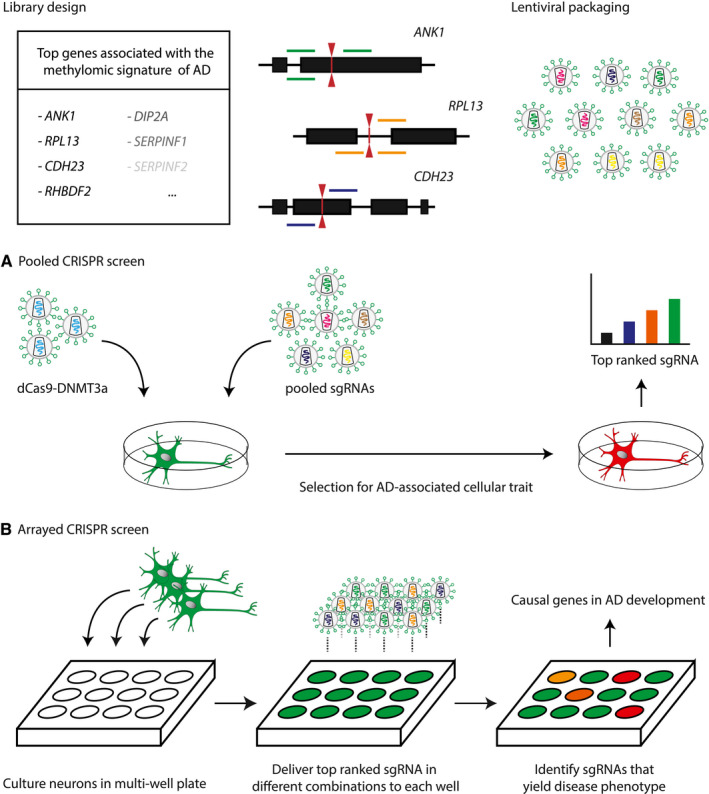
A. Application of a pooled CRISPR screen to determine the phenotypic impact of epigenetic variation in AD. The sgRNA library is designed according to the EWAS identified hits with multiple sgRNA for each locus of interest. The pooled oligonucleotides are packaged into a lentiviral library and transduced into a non‐AD primary cell line of interest (eg, neurons) stably expressing dCas9 coupled to an editor that induces the DNA methylomic signature of AD. The cell population of interest is selected according to an AD‐associated cellular trait such as amyloid plaque formation that can be identified via fluorescent tagging of gene expression. Thereafter, next generation sequencing is used to determine the targeted genes whose expression was modified, which delivers insight into the epigenetic mechanism underlying AD development. B. In an arrayed screen, neurons are placed in individual wells and transduced with distinct sgRNAs. Each well may receive a set of sgRNAs targeting one or multiple genes. Using a combinatorial approach, synergistic relationships between epigenetic loci can be identified. Thus, the genes and epigenetic interactions necessary to develop the AD‐associated phenotype may be determined by the array screen.
Application of such methods would focus the interpretation of EWAS identified genes and separate causal epigenetic alterations from those secondary to disease development. However, not all cellular traits of interest can be evaluated using a pooled screen and the individual contributions of epigenetically modified loci to AD remain unclear. To further query the subset of potential genes contributing to AD at a high throughput level, arrayed CRISPR screens may be applied that take advantage of multi‐well plates and validate the functional role each gene may have in the selected phenotype, which is further illustrated in Figure 5B (71). Although this technology is yet to be applied in AD, a proof‐of‐concept experiment harnessed array screening to enable longitudinal study of CRISPRi effects in iPSC derived neurons, thereby paving the way for future applications in neurodegeneration (76). In conclusion, investigating the causality of epigenetic variation in sporadic AD is a daunting task that is hindered by the complex regulatory interactions that affect gene expression. Nonetheless, the CRISPR‐Cas9 system provides a myriad of different solutions to interrogate disease‐associated epigenetic loci. Its capacity for high content screening of regulatory elements will drive the identification of affected cellular pathways and determine which role DNA methylomic signatures and other epigenetic marks play in the etiology of AD.
Considerations and Challenges When Utilizing CRISPR to Study AD
The CRISPR platform has established itself as the gold‐standard method for gene editing and its capacity for technological advancement seems boundless. Nevertheless, experimental issues remain that prevent the tools from fully realizing their potential. Concerns about the specificity of CRISPR nucleases such as Cas9 permeate the critical reception of many studies (23, 84). Off‐target activity has been evaluated in numerous different CRISPR applications and their findings substantiate the lack of specificity inherent to the system. Upon targeted editing of genes, a considerable amount of unintended mutations and epigenetic modifications may occur both in coding and non‐coding regions of the genome, which leads to genomic instability and hinders clinical implementation (11, 20, 38). However, recent advances in off‐target prediction models such as the GuideScan specificity score have been shown to accurately determine off target activity in pooled CRISPR screens and filter out confounding sgRNA designs to improve upon specificity (52, 78). Moreover, orthologues of the Cas9 nuclease differ in their substrate specificities with variations in target sequence length and PAMs, which provides alternative effector enzymes to not only diminish off‐target activity, but also increase on‐target specificity when the genomic region of interest is challenging (4). In addition to specialized sgRNA designs, numerous studies are dedicated toward the elimination of off‐target mutations, thereby implying a rapid development of novel techniques to combat this issue (34).
A major contributing factor to off‐target effects is the constitutive expression of the Cas9 nuclease and sgRNA even after target gene modification has occurred. Strict spatiotemporal control over expression is not only necessary to avoid unintended modifications, but also plays an important role in complex neurodevelopmental and behavioral experiments where precisely timed induction of gene modification is crucial to the study design. Importantly, in vivo delivery of Cas9 constructs has been shown to produce an immune response in mice, thereby impeding therapeutic development (7, 66). Dynamic control of Cas9 function is now possible via inducible systems that can be triggered by light or small molecules (16, 55). Doxycycline mediated induction of Cas9 transcription is a popular method for expression control that doesn’t sacrifice its editing efficiency unlike other systems. However, it does suffer from significant leakiness, which leads to Cas9 expression in the absence of doxycycline (72). Recently, Chylinski et al made a radical leap in inducible systems in the form of CRISPR‐Switch (SgRNA With Induction/Termination by Cre Homologous recombination) (8). This is a novel method that achieves rapid induction and termination of gene editing through gRNA expression modulation, whilst avoiding potential immunogenicity in vivo and demonstrating no detectable leakiness. Strikingly, the system enables sequential editing of two loci via tightly controlled consecutive expression of different gRNAs and was used to determine the order of mutagenic events necessary for glioma tumorigenesis (8). Consequently, implementation of this technique in combinatorial approaches within the AD field will facilitate the deductive identification of key players in the molecular mechanisms underlying disease etiology.
Despite all the advancements in CRISPR‐Cas9 technology, the field of AD research still lacks proper in vivo and in vitro models of disease that capture both its polygenic nature and other non‐genetic factors. Current transgenic mouse models rely on human sequences bearing autosomal dominant mutations to recapitulate aspects of AD pathology such as amyloid plaques and no murine models carrying the multiple modified loci representative of sporadic AD have yet been developed (19, 31). iPSCs have also greatly contributed toward our understanding of autosomal dominant forms of AD, where assessment of a gene variant’s functional impact is facilitated by modified isogenic control cell lines. Furthermore, iPSC lines can also be derived from sporadic AD patients bearing a high polygenic load for AD. However, there are certain limitations to using iPSCs to model epigenomic changes in the context of age‐related disorders such as AD. For example, the epigenome of patient derived iPSCs is transformed by the process of inducing pluripotency, and recent studies have shown that iPSC‐derived neurons display a fetal epigenetic age even when they resemble mature neurons morphologically (28). As such careful consideration needs to be given to the cell population being used to model DNA methylomic signatures in AD. Finally, as epigenetic patterns are cell type‐specific, multiplex epigenetic editing to model disease should be performed in different brain cell types (eg, microglia, neurons, and astrocytes), with consideration given as to which disease‐associated loci should be altered in each given population.
Future Perspectives
The publishing rate of new CRISPR methodologies is truly astounding and their various capabilities open up a raft of exciting possibilities for investigating AD etiology. The primary focus of this review has been on the potential application of the CRISPR‐Cas9 platform for elucidating the functional consequence of AD‐associated genomic and epigenomic variation. However, given the rapid growth of this area, there are many more existing techniques that may have utility, but were not discussed herein. In fact, these strategies could also have genuine relevance to the comprehension of genetic and epigenetic mechanisms in AD, the range of which extends beyond the scope of this mini‐symposium and a detailed review thereof can be found here (53). Nonetheless, recent research highlights are worth mentioning that encapsulate the impact this platform will have on the future of research in the field of neurodegeneration. For instance, a small molecule inhibitor of Cas9 was identified by a high‐throughput CRISPR‐Cas9 screen that enables dose and temporal control of Cas9 activity (46). Furthermore, “prime editing” is a revolutionary new CRISPR‐Cas9 method that enables precision genome editing without the need of creating a DSB at the target locus, thereby avoiding the pitfalls of HDR whilst demonstrating a higher specificity through lower off‐target effects (1). Such methods promise an exciting time for functional genetics and the prospects of uncovering the spatiotemporal pattern of molecular mechanisms driving AD pathology will continue to grow rapidly. Ultimately, as both genetic and epigenetic factors contribute to the development of AD, combinatorial approaches of CRISPR‐Cas9 will help to elucidate the role of genetic and epigenetic risk variants in disease.
References
- 1. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM et al (2019) Search‐and‐replace genome editing without double‐strand breaks or donor DNA. Nature 576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bertram L, Tanzi RE (2020) Genomic mechanisms in Alzheimer’s disease. Brain Pathol 30:966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carroll D (2011) Genome engineering with zinc‐finger nucleases. Genetics 188:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cebrian‐Serrano A, Davies B (2017) CRISPR‐Cas orthologues and variants: optimizing the repertoire, specificity and delivery of genome engineering tools. Mamm Genome 28:247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer EPR et al (2015) Highly efficient Cas9‐mediated transcriptional programming. Nat Methods 12:326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, Xu HE (2017) Amyloid beta: Structure, biology and structure‐based therapeutic development. Acta Pharmacol Sin 38:1205–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chew WL, Tabebordbar M, Cheng JKW, Mali P, Wu EY, Ng AHM et al (2016) A multifunctional AAV‐CRISPR‐Cas9 and its host response. Nat Methods 13:868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chylinski K, Hubmann M, Hanna RE, Yanchus C, Michlits G, Uijttewaal ECH et al (2019) CRISPR‐Switch regulates sgRNA activity by Cre recombination for sequential editing of two loci. Nat Commun 10, 5454. 10.1038/s41467-019-13403-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923. [DOI] [PubMed] [Google Scholar]
- 11. Corrigan‐Curay J, O’Reilly M, Kohn DB, Cannon PM, Bao G, Bushman FD et al (2015) Genome editing technologies: defining a path to clinic. Mol Ther 23: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 19:R12–R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L et al (2014) Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 17:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA et al (2011) CRISPR RNA maturation by trans‐encoded small RNA and host factor RNase III. Nature 471:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby‐Arnon L et al (2016) Perturb‐Seq: dissecting molecular circuits with scalable single‐cell RNA profiling of pooled genetic screens. Cell 167:1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G et al (2015) Inducible in vivo genome editing with CRISPR‐Cas9. Nat Biotechnol 33:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flavahan WA, Drier Y, Johnstone SE, Hemming ML, Tarjan DR, Hegazi E et al (2019) Altered chromosomal topology drives oncogenic programs in SDH‐deficient GISTs. Nature 575:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer‐Rachamimov AO et al (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foidl B, Humpel C (2020) Can mouse models mimic sporadic Alzheimer’s disease? Neural Regen Res 15:401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nat Biotechnol 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE et al (2013) CRISPR‐mediated modular RNA‐guided regulation of transcription in eukaryotes. Cell 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. György B, Lööv C, Zaborowski MP, Takeda S, Kleinstiver BP, Commins C et al (2018) CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early‐onset Alzheimer’s disease. Mol Ther Nucleic Acids 11:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heidenreich M, Zhang F (2016) Applications of CRISPR‐Cas systems in neuroscience. Nat Rev Neurosci 17:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA (2015) Epigenome editing by a CRISPR‐Cas9‐based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho SM, Hartley BJ, Flaherty E, Rajarajan P, Abdelaal R, Obiorah I et al (2017) Evaluating synthetic activation and repression of neuropsychiatric‐related genes in hiPSC‐derived NPCs, neurons, and astrocytes. Stem Cell Reports 9:615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang Y‐H, Su J, Lei Y, Brunetti L, Gundry MC, Zhang X et al (2017) DNA epigenome editing using CRISPR‐Cas SunTag‐directed DNMT3A. Genome Biol 18. 10.1186/s13059-017-1306-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hull EE, Montgomery MR, Leyva KJ (2016) HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and inflammatory diseases. Biomed Res Int 2016:1–15. 10.1155/2016/8797206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo C, Lancaster MA, Castanon R, Nery JR, Knoblich JA, Ecker JR. (2016) Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain. Cell Reports 17: 3369–3384. 10.1016/j.celrep.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA (2014) Multiplex CRISPR/Cas9‐based genome engineering from a single lentiviral vector. Nucleic Acids Res 42:e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kitazawa M, Medeiros R, LaFerla FM (2012) Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Des 18:1131–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB et al (2017) CRISPR‐Cas9 epigenome editing enables high‐throughput screening for functional regulatory elements in the human genome. Nat Biotechnol 35:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klann TS, Crawford GE, Reddy TE, Gersbach CA (2018) Screening regulatory element function with CRISPR/Cas9‐based epigenome editing. Methods Mol Biol 1767: 447–480. [DOI] [PubMed] [Google Scholar]
- 34. Kocak DD, Josephs EA, Bhandarkar V, Adkar SS, Kwon JB, Gersbach CA (2019) Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat Biotechnol 37:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kurata M, Wolf NK, Lahr WS, Weg MT, Kluesner MG, Lee S et al (2018) Highly multiplexed genome engineering using CRISPR/Cas9 gRNA arrays. PLoS One 13:e0198714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert JC, Ibrahim‐Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al (2013) Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lardenoije R, Roubroeks JAY, Pishva E, Leber M, Wagner H, Iatrou A et al (2019) Alzheimer’s disease‐associated (hydroxy)methylomic changes in the brain and blood. Clin Epigenetics 11:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin L, Liu Y, Xu F, Huang J, Daugaard TF, Petersen TS et al (2018) Genome‐wide determination of on‐target and off‐target characteristics for RNA‐guided DNA methylation by dCas9 methyltransferases. Gigascience 7. 10.1093/gigascience/giy011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin Y‐T, Seo J, Gao F, Feldman HM, Wen H‐L, Penney J et al (2018) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC‐derived brain cell types. Neuron 98:1141–1154.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein e and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J et al (2018) Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172:979–992.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lord J, Cruchaga C (2014) The epigenetic landscape of Alzheimer’s disease. Nat Neurosci 17:1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M et al (2014) Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci 17:1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prince M, Wimo A, Guerchet M, Ali G‐C, Wu Y‐T, Prina M, World Alzheimer Report (2015) The global impact of dementia: an analysis of prevalence, incidence, cost and trends. Alzheimers’s Dis Int 2015:1–87. [Google Scholar]
- 45. Ma Y, Klein H, De Jager PL (2020) Considerations for integrative multi‐omic approaches to explore Alzheimer’s disease mechanisms. Brain Pathol 30:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maji B, Gangopadhyay SA, Lee M, Shi M, Wu P, Heler R et al (2019) A high‐throughput platform to identify small‐molecule inhibitors of CRISPR‐Cas9. Cell 177:1067–1079.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al (2013) RNA‐guided human genome engineering via Cas9. Science 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K et al (2016) Targeted DNA demethylation in vivo using dCas9‐peptide repeat and scFv‐TET1 catalytic domain fusions. Nat Biotechnol 34:1060–1065. [DOI] [PubMed] [Google Scholar]
- 49. Nagata K, Takahashi M, Matsuba Y, Okuyama‐Uchimura F, Sato K, Hashimoto S et al (2018) Generation of App knock‐in mice reveals deletion mutations protective against Alzheimer’s disease‐like pathology. Nat Commun 9. 10.1038/s41467-018-04238-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ortiz‐Virumbrales M, Moreno CL, Kruglikov I, Marazuela P, Sproul A, Jacob S et al (2017) CRISPR/Cas9‐correctable mutation‐related molecular and physiological phenotypes in iPSC‐derived Alzheimer’s PSEN2N141I neurons. Acta Neuropathol Commun 5. 10.1186/s40478-017-0475-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park H, Oh J, Shim G, Cho B, Chang Y, Kim S et al (2019) In vivo neuronal gene editing via CRISPR–Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat Neurosci 22:524–528. [DOI] [PubMed] [Google Scholar]
- 52. Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, Ventura A (2017) GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol 35:347–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pickar‐Oliver A, Gersbach CA (2019) The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol 20:490–507. 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pires C, Schmid B, Petræus C, Poon A, Nimsanor N, Nielsen TT et al (2016) Generation of a gene‐corrected isogenic control cell line from an Alzheimer’s disease patient iPSC line carrying a A79V mutation in PSEN1. Stem Cell Res 17:285–288. [DOI] [PubMed] [Google Scholar]
- 55. Polstein LR, Gersbach CA (2015) A light‐inducible CRISPR‐Cas9 system for control of endogenous gene activation. Nat Chem Biol 11:198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Poon A, Schmid B, Pires C, Nielsen TT, Hjermind LE, Nielsen JE et al (2016) Generation of a gene‐corrected isogenic control hiPSC line derived from a familial Alzheimer’s disease patient carrying a L150P mutation in presenilin 1. Stem Cell Res 17:466–469. [DOI] [PubMed] [Google Scholar]
- 57. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013) Repurposing CRISPR as an RNA‐γuided platform for sequence‐specific control of gene expression. Cell 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Reitz C, Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 88:640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rubin AJ, Parker KR, Satpathy AT, Qi Y, Wu B, Ong AJ et al (2019) Coupled single‐cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176:361–376.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T (2014) Multiplex genome engineering in human cells using all‐in‐one CRISPR/Cas9 vector system. Sci Rep 4. 10.1038/srep05400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B et al (2017) APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36:2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Savell KE, Bach SV, Zipperly ME, Revanna JS, Goska NA, Tuscher JJ et al (2019) A neuron‐optimized CRISPR/dCas9 activation system for robust and specific gene regulation. eNeuro 6. 10.1523/ENEURO.0495-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Simhadri VL, McGill J, McMahon S, Wang J, Jiang H, Sauna ZE (2018) Prevalence of pre‐existing antibodies to CRISPR‐associated nuclease Cas9 in the USA population. Mol Ther Methods Clin Dev 10:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smith AR, Mill J, Lunnon K (2020) The molecular etiology of Alzheimer’s disease. Brain Pathol 30:964–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smith RG, Hannon E, De Jager PL, Chibnik L, Lott SJ, Condliffe D et al (2018) Elevated DNA methylation across a 48‐kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimer’s Dement 14:1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith RG, Pishva E, Shireby G, Smith AR, Roubroeks JAY, Hannon E et al (2020) Meta‐analysis of epigenome‐wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. BioxRiv. 10.1101/2020.02.28.957894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith AR, Smith RG, Pishva E, Hannon E, Roubroeks JAY, Burrage J et al (2019) Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology‐associated epigenetic variation in Alzheimer’s disease. Clin Epigenetics 11:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. So RWL, Chung SW, Lau HHC, Watts JJ, Gaudette E, Al‐Azzawi ZAM et al (2019) Application of CRISPR genetic screens to investigate neurological diseases. Mol Neurodegener 14. 10.1186/s13024-019-0343-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. de Solis CA, Ho A, Holehonnur R, Ploski JE (2016) The Development of a viral mediated CRISPR/Cas9 system with doxycycline dependent gRNA expression for inducible in vitro and in vivo genome editing. Front Mol Neurosci 9. 10.3389/fnmol.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stresemann C, Lyko F (2008) Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer 123:8–13. [DOI] [PubMed] [Google Scholar]
- 74. Sun J, Carlson‐Stevermer J, Das U, Shen M, Delenclos M, Snead AM et al (2019) CRISPR/Cas9 editing of APP C‐terminus attenuates β‐cleavage and promotes α‐cleavage. Nat Commun 10. 10.1038/s41467-018-07971-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tarjan DR, Flavahan WA, Bernstein BE (2019) Epigenome editing strategies for the functional annotation of CTCF insulators. Nat Commun 10. 10.1038/s41467-019-12166-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D et al (2019) CRISPR interference‐based platform for multimodal genetic screens in human iPSC‐derived neurons. Neuron 104:239–255.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Van den Hove DLA, Riemens RJM, Koulousakis P, Pishva E (2020) Epigenome‐wide association studies in Alzheimer’s disease; achievements and challenges. Brain Pathol 30:978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tycko J, Wainberg M, Marinov GK, Ursu O, Hess GT, Ego BK et al (2019) Mitigation of off‐target toxicity in CRISPR‐Cas9 screens for essential non‐coding elements. Nat Commun 10. 10.1038/s41467-019-11955-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, Julg B et al (2016) Repurposing the CRISPR‐Cas9 system for targeted DNA methylation. Nucleic Acids Res 44:5615–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA‐guided genetic silencing systems in bacteria and archaea. Nature 482:331–338. [DOI] [PubMed] [Google Scholar]
- 82. Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W et al (2016) A CRISPR‐based approach for targeted DNA demethylation. Cell Discov 2. 10.1038/celldisc.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zarei A, Razban V, Hosseini SE, Tabei SMB (2019) Creating cell and animal models of human disease by genome editing using CRISPR/Cas9. J Gene Med 21:e3082. [DOI] [PubMed] [Google Scholar]
- 84. Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH (2015) Off‐target effects in CRISPR/Cas9‐mediated genome engineering. Mol Ther Nucleic Acids 4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zheng Y, Shen W, Zhang J, Yang B, Liu YN, Qi H et al (2018) CRISPR interference‐based specific and efficient gene inactivation in the brain. Nat Neurosci 21:447–454. [DOI] [PubMed] [Google Scholar]