

Abstract
Zika virus (ZIKV) is a flavivirus that can cause neuropathogenesis in adults and fetal neurologic malformation following the infection of pregnant women. We used a nonhuman primate model, the Indian‐origin Rhesus macaque (IRM), to gain insight into virus‐associated hallmarks of ZIKV‐induced adult neuropathology. We find that the virus causes prevalent acute and chronic neuroinflammation and chronic disruption of the blood‐brain barrier (BBB) in adult animals. ZIKV infection resulted in specific short‐ and long‐term augmented expression of the chemokine CXCL12 in the central nervous system (CNS)of adult IRMs. Moreover, CXCL12 expression persists long after the initial viral infection is apparently cleared. CXCL12 plays a key role both in regulating lymphocyte trafficking through the BBB to the CNS and in mediating repair of damaged neural tissue including remyelination. Understanding how CXCL12 expression is controlled will likely be of central importance in the definition of ZIKV‐associated neuropathology in adults.
Keywords: blood‐brain barrier, CXCL12, flavivirus, nonhuman primate, Zika virus
Introduction
Zika virus (ZIKV), is a neurotropic flavivirus associated with Guillain‐Barre’ syndrome (GBS) in adults and is also well‐known for causing fetal neurologic malformation following infection of pregnant women (5, 51). In addition to causing GBS, which features damage to the protective myelin sheath surrounding axons, ZIKV can cause neuropathology in adults in the form of meningoencephalitis and myelitis (9, 32). The pathogenesis of ZIKV and the host‐pathogen interactions important for the development of these lesions still need to be elucidated.
The blood‐brain barrier (BBB), the boundary between circulatory and CNS tissues, is composed of brain microvascular endothelial cells (BMECs) and supporting associated pericytes and astrocytes. Intercellular tight junction (TJ) and adherens junction (AJ) integrity are important for the maintenance of the intracellular network of MECs that comprises the vascular endothelium. Disruption of the BBB occurs during the pathogenesis of a wide range of infectious, autoimmune and neurodegenerative diseases. Neurotropic flaviviruses, including Japanese Encephalitis virus (JEV) and West Nile virus (WNV), disturb the BBB in adults through the disruption of the BMEC network (28, 43).
The chemokine CXCL12 is a key regulator of both myelin formation during embryogenesis and remyelination following neural damage in the CNS and peripheral nervous system (PNS) in adults (14, 41). CXCL12 facilitates the migration and maturation of oligodendrocyte precursor cells (OPCs) during CNS remyelination (14, 41). In the PNS, CXCL12 may similarly function in Schwann cell migration (16), as CXCL12 is expressed by perisynaptic Schwann cells during the recovery of neuromuscular junctions following damage (36). In addition to serving as a key regulator of neural repair, CXCL12 plays an important function in regulating lymphocyte migration through the BBB. During homeostasis CXCL12 is expressed by BMECs resulting in the spatial restriction of lymphocytes to the microvascular perivascular space and changes in chemokine expression and distribution can lead to neuroinflammation(29, 30). The role of CXCL12 in neural repair, lymphocyte migration into the CNS parenchyma and in multifarious functions during development and immunity, are mediated through interaction of this chemokine with its primary receptor, CXCR4 (55). Stimulation of CXCR4 by CXCL12 results in the activation of an interwoven set of downstream effector pathways (12, 54).
Nonhuman primates (NHPs) are good animal models for recapitulating human neurological disease since they are genetically and physiologically similar to humans and exhibit CNS and PNS elaboration and brain morphology closely resembling that of humans. We have delineated the effect of ZIKV on the neural tissue of 18 adult Indian Rhesus macaques (IRMs). Our data indicate that the virus causes acute and chronic inflammation of neural tissue in adult animals with accompanying damage to the BBB. Moreover, we find that the expression of CXCL12 in the CNS is upregulated both during acute ZIKV infection and, surprisingly, for an extended time after infection. We propose that this chemokine is likely to be important in both long‐term neuropathology and neural repair following ZIKV‐induced damage of the PNS and CNS. Thus, this NHP model is valuable for experimentally deciphering important hallmarks of novel ZIKV‐induced neuropathology in adults and in elucidating molecular mechanisms underlying virus‐induced GBS spectrum disorders in humans.
Materials and Methods
Ethics statement
The Indian origin rhesus macaques (IRMs)(Macaca mulatta) used in this study were housed at the TNPRC. The TNPRC is fully accredited by AAALAC International (Association for the Assessment and Accreditation of Laboratory Animal Care), Animal Welfare Assurance No. A3180‐01. Animals were cared for in accordance with the NRC Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Tulane University (protocols P0336 and P0367). Social housing and interactive enrichment were used for all NHPs used in this study. Animal care staff conduct routine husbandry procedures (e.g., cleaning, feeding and watering) and animal care staff and veterinarians observed animals several times daily for signs of disease, pain and distress and this information was reported to the attending veterinarian through both verbal and written communication in the animal’s health record. The Tulane University IACUC and the Division of Veterinary Medicine have established procedures to minimize pain and distress through several means. The use of preemptive and post‐procedural analgesia is required for procedures that would likely cause more than momentary pain or distress in humans undergoing the same procedure. For minor procedures such as blood collection animals are anesthetized with ketamine hydrochloride (10 mg/kg IM).
Animals, viruses and challenge
ZIKV strain Rio U‐1/2016 (7) was isolated in Rio de Janeiro, Brazil in 2016 (KU926309). Viral challenge stocks were prepared by propagating the virus in Vero cells for two passages post virus isolation(7). The stocks were quantitated by viral plaque assay. The viral stocks were diluted in Leibovitz's L‐15 and SPG media as described previously (13). All animals were challenged via the subcutaneous route, with 104 PFU.
As described in detail in the Results section, 15 of the IRMs used in this study were females infected during pregnancy (26) and three were males. Figure 1 displays the overall timing of infection relative to pregnancy, sample collection and necropsy. It is worth noting that some of the females were euthanized and necropsied near the end of pregnancy (e.g. KE38 and JR20), while others were evaluated for various extended times following parturition (e.g. EI99 and HE27).
Figure 1.

NHP study design. Fifteen female IRMs (26) and three male IRMs were infected with 104 PFU of ZIKV strain Rio‐1. Pregnancy in the Rhesus macaque is divided into three 55‐day trimesters, which are equivalent in developmental landmarks to the trimesters in human pregnancy (6). These trimesters are depicted in the timeline at the top, as are relative times of infection, sample collection and necropsy for the dams. Acute viremia was detected in all animals (Figure S1).
Luminex analysis of CSF cytokines
Prior to assay, thawed serum samples were mixed well and then clarified by adding 120 µL of each to Ultrafree Centrifugal Filters, pore size 0.65 µm (Millipore #UFC30DV00) and centrifuged at 12 000 × g for 4 minutes. Concentrations of cytokines and chemokines present in the serum were quantified using the Life Technologies Cytokine Monkey Magnetic 37‐Plex Panel for Luminex™ Platform (#EPX370‐40045‐901, Thermo Fisher Scientific, Waltham, MA), according to the manufacturer’s instructions. Assay Diluent from the kit was used to reconstitute standards and to prepare standard serial dilutions. All standards, blanks and samples were assayed in duplicate wells. The analytes detected by this panel are indicated in Table S1. Final reactions in the microtiter plates were read on a Bio‐Plex® 200 System (Bio‐Rad Laboratories, Hercules, CA). Results were calculated using the Bio‐Plex Manager™ Software v6.1 (Bio‐Rad).
Tissue sampling and fixation
Regular peripheral blood draws and CSF collections were performed during the course of the study. At the end of the study, all animals underwent a complete necropsy and tissue samples were collected in either Zinc‐buffered formalin, RNA Later, RPMI media, or fresh frozen. Fixed samples were trimmed, processed and embedded in paraffin 2 days after necropsy. Paraffin‐embedded tissues were cut in 5 um sections, adhered to charged glass slides and stained routinely with hematoxylin and eosin or left unstained for immunohistochemical and immunofluorescent staining.
Fibrinogen extravasation
The percent of vessels demonstrating fibrinogen extravasation was determined using immunofluorescence analysis with Alexa Fluor 594‐labeled anti‐fibrinogen and Alexa Fluor 488‐labeled anti‐GLUT‐1 Ab and by running linear plot profiles on the green and red channels of individual vessels and graphing the resulting numerical data in GraphPad as dual overlay histograms. The histograms were then analyzed to determine whether the fibrinogen was above background levels outside of the two primary GLUT‐1 peaks; vessels that displayed this phenotype were considered to be extravasated. A total of 25 vessels were examined from each animal via random imaging, but were required to meet the following criteria; vessels must be less than 10 µm in luminal diameter and no single radius can be more than twice the length of the smallest luminal radius to ensure nearly horizontal cross‐sections. A Zeiss Axio Observer.Z1 fluorescence microscope was used to analyze the fluorescent‐labeled sections. Zeiss AxioVision Release 4.8.2 was used to capture and merge fluorescence images. Adobe Photoshop CS12.1 was also used to merge layers into a single image.
Immunohistochemistry
Immunohistochemistry was performed using anti‐CXCL12. Sections were deparaffinized by incubating them for 1 h at 58‐60°C. After the sections were deparaffinized, they were rehydrated and pretreated for antigen retrieval by microwaving in a citrate‐based Antigen Unmasking Solution. Sections were washed with Tris‐based saline (TBS) contain 0.05% Tween‐20 for 10 minutes, Following, sections were treated with peroxidase blocking solution for 10 minutes. After washing again, sections were incubated with 5% normal goat serum in TBS for 30 minutes. Immediately following the goat serum, sections were incubated with CXCL12 antibody for 1 h at room temperature. After another wash in TBS, sections were incubated with biotinylated secondary antibody for 30 minutes. After washing the sections in TBS, sections were then incubated with avidin‐biotin‐peroxidase Complex for 30 minutes. Following another wash, sections were developed for 10 minutes diaminobenzidine with Mayer’s Hematoxylin used as a nuclear counterstain. Sections were dehydrated and mounted using VectaMount. Using a Nikon Coolscope digital microscope, sections were visualized and photos were captured. ImageJ was used to set a threshold level of staining and the area above this threshold was counted as positive staining with 20 random, 200x, images analyzed per animal.
Immunofluorescence microscopy
Triple‐label immunofluorescence was performed with anti‐CXCL12, GLUT‐1 and CD206. As described above, sections were de‐paraffinized and rehydrated, followed by antigen retrieval. After washing with phosphate‐buffered saline (PBS) containing 0.2% fish skin gelatin(FSG), sections were permeabilized with PBS containing 0.2% FSG and 0.1% Triton X‐100 for 1 h. Following another wash, sections were incubated with 5% normal goat serum in PBS for 30 minutes at room temperature before incubation for 1 h at room temperature with primary antibodies diluted in PBS/FSG. After primary antibody incubation, the sections were washed in PBS/FSG and incubated with an Alexa Fluor 350‐, 488‐, or 594‐ conjugated secondary antibody in PBS/FSG for 1 h at room temperature. The sections were washed with PBS/FSG before the addition of the next primary antibody. After immunofluorescence staining, the sections were treated with 10 mM CuSO4 in 50 mM ammonium acetate buffer for 45 minutes to quench autofluorescence. The sections were rinsed in distilled water and coverslipped with Aqua‐Mount aqueous mounting medium. Confocal images were taken with a Zeiss 880 Laser scanning confocal microscope with a 100x emersion oil objective. ZenBlack and ZenBlue programs were used to capture and merge images.
Measurement of viral RNA load (qRT‐PCR)
Quantitative realtime PCR (qRT‐PCR) was used for the measurement of viral loads, based on a previously validated assay (21, 44). In brief, RNA was extracted from 140 to 1000 µL of frozen fluids, depending on availability, using the QIAamp Viral RNA Mini Kit or QIAamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany). Total nucleic acid was eluted in two centrifugation steps with 40 µL of Buffer AVE each. A qRT‐PCR reaction was then carried out with 20 µL of samples and 10 µL of primer, probes and TaqMan Fast Virus 1‐Step Master Mix (Applied Biosystems, Foster City, CA). We used pre‐combined probe and primers (500 nM primers and 250 nM probe; IDT Technologies, Coralville, IA). The primer and probe sequences were designed to match the sequences of the Brazilian ZIKV isolate KU321639 and were as follows: Primer 1 5′TTGAAGAGGCTGCCAGC3′; Primer 2 5′CCCACTGAACCCCATCTATTG3′; Probe 5′TGAGACCCAGTGATGGCTTGATTGC3′. The probe was double‐quenched (ZEN/Iowa Black FQ) and labeled with the FAM dye (IDT Technologies, Coralville, IA). Ten‐fold serial dilutions of a 401 bp in vitro RNA transcript encoding the ZIKV capsid gene (KU321639) starting at approximately 5 × 105 RNA copies µL−1 were used as standards. Results were reported as the median equivalent viral RNA genomes per ml. The limit of detection was between 12 and 90 viral RNA copies ml−1, depending on the extracted volumes. ZIKV‐positive and ‐negative samples were included in every run.
Results
ZIKV infection causes acute and chronic perivascular neuroinflammation
We inoculated 18 adult IRMs subcutaneously with 104 plaque‐forming units (pfu)/animal of a minimally passaged Brazilian ZIKV isolate (Rio‐U1)(7). An outline of the experimental design is shown in Figure 1. Fifteen of the animals used in the study were females infected during pregnancy and three were adult males. The females were from a previous project focusing on the effect of ZIKV on the fetus (26). Following infection at different times during gestation, these animals generally displayed extended acute viremia compared with males as reported previously (Figure S1), with some animals exhibiting the transmission of virus to amniotic fluid. Infants were delivered by C‐section, or pregnancy was experimentally terminated, or pregnancy ended through ZIKV‐mediated demise. As outlined in Figure 1, necropsy and collection of neural tissue were obtained from the dams at various times before or after parturition or fetal termination. Four of the dams (EL21, ID92, JR20 and JI20) were infected and necropsied 16 or 17 days after infection (acute) and the remainder was maintained for significantly longer periods of time after infection (3.5 to 10 months). The three adult males (HP17, HP87 and JP58) were infected and necropsied after 30 days.
Interestingly, H & E staining of CNS and PNS samples collected at necropsy revealed consistent scant to moderate perivascular inflammation in the meninges along with sporadic additional pathological features in the CNS parenchyma including glial nodules and lymphocytic infiltration (Figure 2). Inflammation was observed in neural tissue both acutely and chronically after infection. Moreover, inflammation was observed regardless of whether the analysis was carried out on neural tissue from pregnant animals or from animals many months after parturition. Table 1 provides a summary of observations from neural tissue in individual animals.
Figure 2.
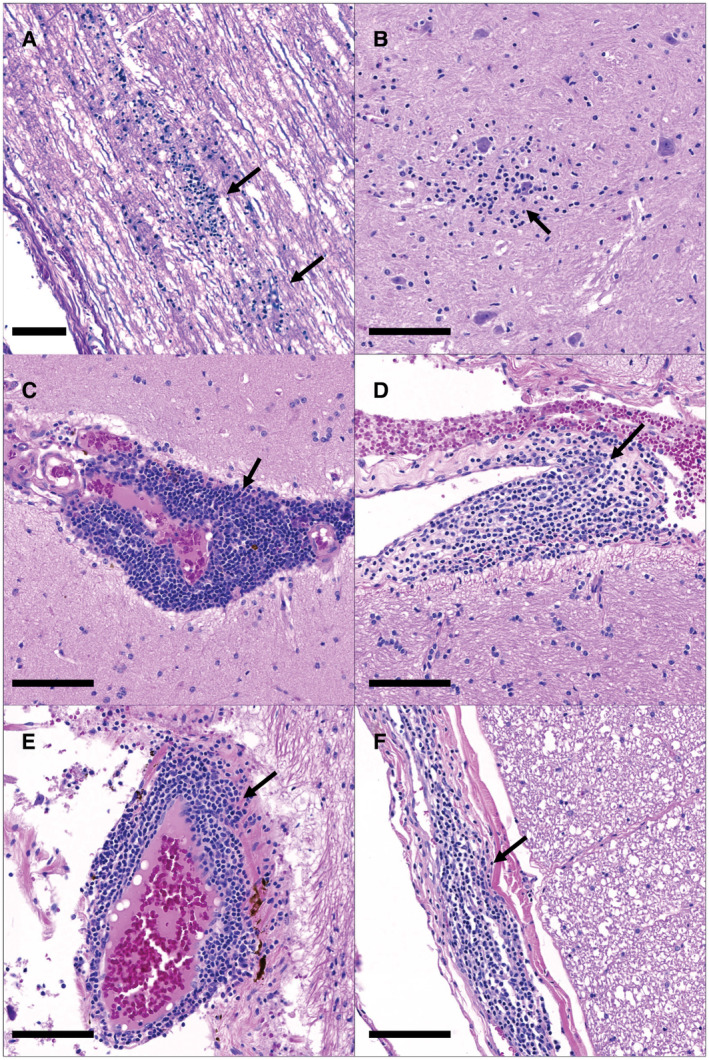
Neuroinflammation in the spinal cord of ID92 (A and B) and the spinal cord and brain of HE27 (C‐F). ID92 had multifocal glial nodules (arrows) within both the white (A) and grey (B) matter of the spinal cord. HE27 had widespread perivascular inflammation (arrows) at multiple levels of the cerebrum (C and D), cerebellum (E) and spinal cord (F). H&E, Bar = 100um.
Table 1.
Summary of observed perivascular inflammation in the CNS and PNS of adult animals infected with ZIKV.
| ID | Days post‐infection | Sciatic nerve | Lumbar cord | Thoracic cord | Cervical cord | Choroid plexus | Brainstem | Cerebellum | Occipital lobe | Temporal lobe | Frontal lobe | Basal Ganglia |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID92 | 16 | + | + | + | ||||||||
| JI20 | 16 | + | ||||||||||
| JR20 | 17 | + | + | + | + | + | + | |||||
| EL21 | 17 | + | + | + | + | |||||||
| JP58 | 30 | + | ||||||||||
| KE38 | 108 | + | ||||||||||
| EC36 | 116 | + | ||||||||||
| KT23 | 139 | + | + | |||||||||
| HE27 | 298 | + | ++ | ++ | + | ++ | ||||||
| JA38 | 306 | + | + | + | ||||||||
| HA73 | 306 | + |
H and E staining of the CNS and PNS was carried out to evaluate perivascular inflammation as in Figure 2. + indicates detectible mild inflammation. ++ indicates moderate inflammation.
ZIKV infection disrupts the adult blood‐brain barrier
Since ZIKV caused perivascular inflammation with accompanying lymphocytic infiltration, this suggested that inflammation is likely to arise through BBB dysregulation. Fibrinogen is a protein normally restricted to serum. Thus, extravasation of fibrinogen into the perivascular space of CNS vessels is indicative of the disruption of the BBB. To detect and quantify ZIKV‐associated extravasated fibrinogen, we performed multi‐label immunofluorescence staining using anti‐fibrinogen Ab. To visualize microvascular endothelial cells, we used anti‐GLUT‐1 Ab. GLUT‐1 is a plasma membrane protein found in abundance on vessel endothelial cells at the BBB (27) (Figure 3A). Using dual overlay histograms of 25 vessels per animal, we quantified the percent of vessels with fibrinogen extravasation in ZIKV‐infected and control groups in both cortical brain tissue and spinal cord (Figure 3B). There was a significant increase in extravasated fibrinogen with infection in both brain and spinal cord (Figure 3C,D). This indicates that the integrity of the microvascular endothelium and the BBB was compromised following ZIKV infection.
Figure 3.
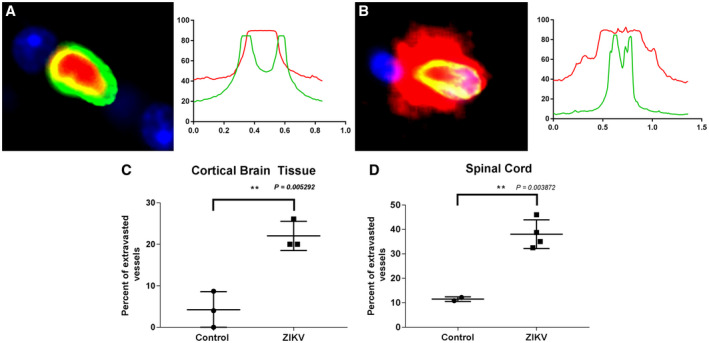
ZIKV disrupts the BBB in adult IRMs as evidenced by an increase in extravasated vessel fibrinogen during infection. Vessel endothelial cells were detected using Alexa Fluor 488‐labeled anti‐GLUT‐1 Ab (green) and fibrinogen detected using Alexa Fluor 594‐labeled anti‐fibrinogen Ab (red). Nuclei were detected with DAPI. A. Cross‐section and immunofluorescence analysis of a stained vessel without extravasation and associated dual overlay histogram. B. Immunofluorescence stained vessel with extravasation and associated dual overlay histogram. Percent of extravasated vessels in cortical brain tissue (C) and spinal cord (D).
ZIKV infection acutely and chronically upregulates CXCL12 in the CNS
We then carried out a series of experiments focusing on the components of the cerebral spinal fluid (CSF) from the infected animals to gain insight into the cause of ZIKV‐induced neural inflammation in adult animals. In our prior study (26), efforts to detect ZIKV RNA in the CSF of infected IRMs were unsuccessful. However, data from other studies indicate that the infection of IRMs can result in the acute transmission of virus to the CNS in many animals in parallel with acute viremia (1, 10, 20). We used RT‐PCR to attempt the detection of virus in the CSF of the males used in the study (HP17, HP87 and JP58). In addition, some CSF samples were available from unrelated studies with ZIKV‐infected male and female IRMs, and we also attempted to detect CSF‐associated ZIKV in these samples. This analysis indicated that virus was present in the CNS/PNS in a majority of the animals during acute infection and was generally, but not always cleared, within 2 weeks (Figure S2).
Neurological disease, including infectious neuropathology and autoimmune polyneuropathy, can result in transient or sustained elevated total protein concentration in the CSF (17). For four animals evaluated during acute infection (HP87, EL21, JI20 and JR20) with available pre‐infection controls there was a significant increase in CSF protein following acute ZIKV infection as evidenced by the comparison of matched preinfection controls with samples collected 2 weeks after infection (Figure 4A). Pre‐infection control samples were not available for the CSF samples taken from animals evaluated for an extended period after infection. Two of the animals (HJ72 and HE27) exhibited that CSF protein levels markedly higher than the samples from uninfected animals consistent with the possibility that infection can result in a longer‐term increase in CSF protein in individual IRMs. However, the CSF samples from seven animals obtained from long times after infection had protein concentrations that did not differ significantly from the preinfection controls.
Figure 4.
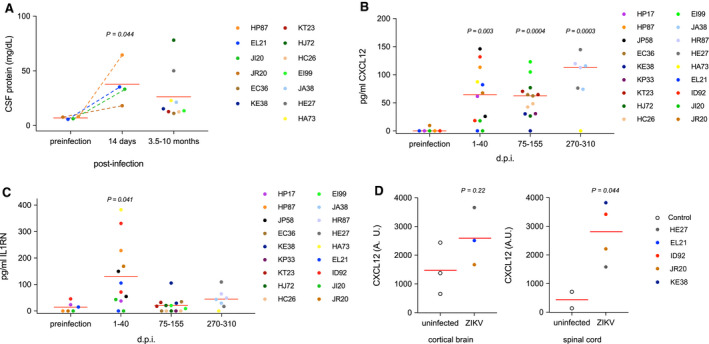
Upregulation of CXCL12 in the CSF of adult IRMs. A. Total protein concentrations in the CSF of acutely and chronically infected IRMs. For acutely infected animals (HP87, EL21, JI20 and JR20) a two‐tailed paired T‐test was used to compare protein concentrations with match pre‐infection samples. For samples derived from later times following infection, comparison with the pre‐infection samples using a two‐tailed unpaired T‐test indicated that the means of the two samples were below the threshold of significance (P = 0.12). B and C. Concentrations of CXCL12 and IL1RN in CSF samples following ZIKV infection, respectively. For purposes of statistical analysis samples were placed in four groups based on time of collection relative to virus infection. P values were determined by comparison to preinfection controls using two‐tailed unpaired T‐tests. Exact times of CSF sample collection are displayed in Figure S4. D. Immunohistochemistry using anti‐CXCL12. CXCL12 was visualized in cortical brain or spinal cord tissue for several uninfected controls and animals acutely or chronically infected with ZIKV. ImageJ was used to quantify the area of CXCL12‐positive staining in 20 random images per animal. A.U. – arbitrary units.
To investigate the mechanism of ZIKV‐induced neural damage and repair, we carried out the Luminex‐based quantification of cytokine levels in the CSF in four female IRMs (EL21, JI20, JR20 and ID92) and three male IRMs (HP17, HP87 and JP58) using a macaque‐specific panel designed to detect cytokines found during infection and inflammation (a list of the cytokine panel is provided in Table S1). Surprisingly, of the 37 cytokines screened and quantified in this initial analysis only two, CXCL12 and IL1RN, were significantly affected by virus infection (Figure S3). CXCL12 is a chemokine important in multiple processes including neural repair and maintenance of BBB integrity. The IL1RN gene product is an indirect negative regulator of CXCL12; binding of virus‐induced type I interferon (IFN) to its receptor, IL1R, triggers CXCL12 expression and the IL1RN gene product binds the IL1R inhibiting IFN binding thereby blocking IFN signal transduction. We extended the analysis to all animals using CSF samples from both short and long time intervals following ZIKV infection by a custom panel capable of quantifying CXCL12 and IL1RN. In accordance with our initial experiment, CXCL12 and IL1RN were both significantly upregulated in the CSF during acute infection (Figure 4B,C). Moreover, CXCL12 concentrations remained strikingly high in the CSF long after infection. In contrast, IL1RN markedly waned following acute infection and returned to levels similar to those observed prior to ZIKV infection.
To determine the location of CXCL12 expression in neural tissue following ZIKV infection, we carried out immunohistochemistry using an anti‐CXCL12 antibody. As expected, the majority of CXCL12 was detected in association with microvascular endothelia, stained with GLUT‐1. Expression by vascular endothelial cells is consistent with the role of CXCL12 in maintaining BBB integrity and restriction of lymphocytes into the CNS parenchyma during homeostasis. Parallel evalutation of uninfected control animals and several of the acutely or chronically ZIKV‐infected animals by semi‐quantitative immunohistochemistry using an anti‐CXCL12 antibody was undertaken to investigate whether the increase in CXCL12 seen in the CSF was mirrored in neural tissue. Surprisingly, we did not see a significant difference in cortical tissue between control and ZIKV‐infected animals. However, expanding the analysis to spinal cord, there was a significant increase in CXCL12 staining in ZIKV infected tissue consistent with the increase in CXCL12 seen in the CSF (Figure 4D). These results show that CXCL12 is dysregulated during Zika virus infection and that the spinal cord is a potentially important site of action of viral infection in adults.
Positive regulators of the expression of CXCL12, as well as a spectrum of additional antimicrobial and proinflammatory cytokines, include tumor necrosis factor (TNF)(19), the proinflammatory cytokine, IL1B (42) and a soluble form of the peptide, CD40LG (22). Thus, TNF, IL1B and CD40LG were included as targets in the custom Luminex panel we used to quantify CXCL12 and IL1RN in Figure 4. Quantification of these potential regulators of CXCL4 indicated that all remained below the limit of detection in the CSF prior to and after ZIKV infection (Figure S5).
Discussion
We have shown that ZIKV causes acute and chronic neural perivascular inflammation and that ZIKV compromises BBB integrity in adult IRMs. Consequently, this is an important NHP model with high potential for elucidating the facets of ZIKV‐induced neuropathology in adult humans.
A striking observation is that ZIKV infection resulted in specific short‐ and long‐term augmented expression of the chemokine CXCL12 in the CNS of adult IRMs, while other cytokines often triggered by viral infections appeared not to be expressed. Consequently, the way in which ZIKV infection induces CXCL12 is likely central to understanding ZIKV‐induced neural damage and repair in adults. CXCL12 plays ubiquitous and diverse tasks in development, immunity and repair including in the CNS. CXCL12 expression results in the recruitment or retention of CXCR4‐effector cells to appropriate sites for function or homeostasis. In this regard, CXCL12 plays two roles pertinent to understanding ZIKV‐associated neuropathogenesis. First, this chemokine has the capacity to regulate the migration of lymphocytes through the BBB and into the CNS parenchyma (29, 30). Entry of lymphocytes across the BBB can be essential for combating viral infection in the CNS but the process can also lead to incidental neural damage through accompanying inflammation. ZIKV infection also results in augmented CXCL12 expression in monocytes(33). Trafficking of CXCL12‐expressing monocytes across the BBB could further contribute to spatial skewing of neural CXCL12 gradient. Second, although we do not yet know the underlying mechanism of protracted CXCL12 expression after apparent viral clearance, we suggest that long‐term CXCL12 expression is needed for the remyelination of virus‐induced damage to the myelin sheath. CXCL12 is important in neural repair, specifically mediating myelin restoration in the adult neural tissue through recruitment and differentiation of oligodendrocyte progenitor cells (OPCs) to effect remyelination in the CNS (14, 41) and in Schwann cell migration during repair of the PNS (16, 36). We do not yet know whether one or both of these CXCL12‐dependent processes are of primary importance for understanding adult ZIKV neuropathology in the IRM.
The molecular mechanism responsible for the induction of CXCL12 expression in the CNS is also unclear as we could not find overt evidence for the upregulation of typical virus‐induced triggers of broad cytokine induction and inflammation including INFβ, TNF and CD40LG. The observation that CXCL12 remains upregulated in the CNS at times long after the initial infection is likely significant for understanding the effect of ZIKV on the adult CNS. Sustained CXCL12 expression may be mediated by a mechanism that narrowly protracts targeted expression without the concomitant expression of other cytokines. Specific post‐transcription expression of CXCL12 is negatively controlled by microRNA‐23a(4). It is possible that ZIKV infection results in reduced expression of this micro RNA in neural tissue. Regardless of the expression mechanism, long‐term maintenance of high CXCL12 levels is likely to reflect a heretofore unrecognized chronic response of the host to ZIKV infection and may indicate the potential for virus‐induced sequelae arising long after infection.
Most of the animals used in our evaluation of ZIKV on the adult CNS were females that had been infected during pregnancy. There does not appear to be an obvious effect of pregnancy on virus‐induced CXCL12 expression in the CNS or on neuroinflammation. CSF samples taken following parturition, or from males, were not significantly different in CXCL12 levels than those obtained during pregnancy (Figure S6). Similarly, there was no obvious correlation between the time of necropsy/histological evaluation revealing inflammation and pregnancy. Nonetheless, it is widely appreciated that pregnancy results in changes in the immune system that promote tolerance for the fetus. Such changes may alter other immune responses to the virus that were not monitored in our study.
We found that ZIKV infection of adult IRMs resulted in neuroinflammation with a primary outcome of high incidence meningitis. As reported here and elsewhere (1, 20), CSF‐associated ZIKV appears to be cleared during acute infection. Consequently, we were surprised that neuroinflammation persisted long after infection and at times when virus is generally considered to be cleared from the CNS and other tissues. Chronic neuroinflammation could indicate that ZIKV replicates and remains in neural tissue at levels below the limit of detection. Alternatively, the virus may trigger a host response marked by persistent neuroinflammation and CXCL12 expression in the absence of virus. In either case, protracted ZIKV‐associated neuropathology has potentially significant clinical ramifications.
African and Asian ZIKV strains are associated with phenotypic differences in both in vitro replication and in vivo pathogenesis (31, 45, 46, 53). The Rio‐U1 ZIKV stock used in this study was a primary stock isolated from a Brazilian patient and is not adapted to cell culture. The virus forms smaller plaques on Vero cell monolayers than other strains. In addition, Rio‐U1 is more pathogenic than other commonly used American strains in AG129 mice (25). It will be interesting to see whether other ZIKV strains elicit identical neuropathology to that described in this study.
While the detailed mechanism of ZIKV‐induced neuropathology in the IRM model remains to be elucidated, the neural damage and repair we observe is likely to overlap with pre‐clinical or clinical ZIKV‐induced GBS spectrum disease in humans. GBS and related disorders that affect the peripheral and central nervous system comprise a continuum of pathologies marked by inflammation and damage to the myelin sheath of neurons. Demyelination can arise from a constellation of genetic and environmental causes (15, 18, 24, 49, 50, 52), including infection by ZIKV (35, 37, 40) and its relative Dengue virus (8, 48).
Diverse neurotropic viruses from multiple virus families gain access to the CNS parenchyma causing disruption of the BBB. Japanese Encephalitis and West Nile viruses are neurotropic viruses of the flavivirus genus, closely related to ZIKV. These viruses can replicate in the CNS, cause encephalitis and measurably affect BBB integrity (3, 11, 23, 34, 39, 47). In contrast, results from an in vitro BBB reconstitution model using a primary virus isolate from Thailand, and an interferon receptor‐deficient murine model using Ugandan and Brazilian isolates suggests that a primary isolate of ZIKV doesn't significantly disrupt the BBB to gain access to the CNS and that long‐term damage to the BBB is minimal (2, 38). Similarly, productive ZIKV infection of human MECs in vitro does not result in cytopathic effects (34). In contrast, our study with the NHP model indicates that ZIKV induces significant disruption of the meningeal BBB both acutely and long after infection. We think that the disruption of the BBB in the IRM model of ZIKV infection is highly likely to faithfully recapitulate pathology and disease mechanisms of human infection and that ZIKV‐induced compromise of the BBB in neurotropic human infections is likely. The lack of BBB disruption in the murine and in vitro reconstitution systems may arise from fundamental differences between these valid and useful but very diverse experimental systems relative to the IRM model. We do not yet know whether BBB disruption is required for CNS access in the IRM or whether BBB damage occurs following viral CNS access and neural ZIKV replication.
Conflict of Interest
The authors have no financial or non‐financial competing interests.
Supporting information
Figure S1. Acute viremia following ZIKV infection. (A) Viremia in eleven of the pregnant females used in the current study. (B) Viremia in the three males used in the study. As reported previously (26), infection of nonpregnant adults results in rapid robust replication of virus as evidenced by quantification of viral RNA in serum with viral clearance from blood after about 7 to 10 days whereas viremia in pregnant females typically persists for longer times.
Figure S2. CSF samples collected at various times from 11 ZIKV‐infected animals was analyzed using ZIKV‐specific RT‐PCR. Virus was detectible in the CSF in seven of the animals. Females are denoted using (x) and males by circles. None of the females used in the experiment were pregnant.
Figure S3. Acute infection of IRMs results in upregulation of CXCL12 (A) and IL1RN (B) in the CSF. At top, samples obtained from 14‐30 days after infection are grouped to facilitate comparison with preinfection controls. P values were determined using paired two‐tailed T‐tests to compare CXCL12 or URN concentrations following infection with that of preinfection controls. At bottom, kinetics of cytokine expression are provided with available CSF samples.
Figure S4. CXCL12 and URN concentrations in the CSF of ZIKV‐infected animals. Samples correspond to those in Figure 4.
Figure S5. Quantification of TNF, IL1B, and CD4OLG in the CSF of ZIKV infected IRMs. See text for description. Samples were divided into three temporal groups to facilitate comparison with Figure 4B,C.
Figure S6. This figure is reconfigured version of Figure 4B. Points denoted with an “x” indicate that the sample was taken from a pregnant animal while points represented by a circle indicate that the sample was taken from a nonpregnant animal.
Table S1. Initial analysis of several animals was carried out using an NHP‐specific panel designed to detect key cytokines (Cytokine/Chemokine/Growth Factor 37 Plex NHP ProcartaPlex Panel). The specific cytokines detected in the panel are listed above.
Acknowledgments
This work was supported by the Bill and Melinda Gates Foundation (OPP1152818) and by support from the National Center for Research Resources and the Office of Research Infrastructure Programs (ORIP) of the NIH through grant P51 OD011104 to the Tulane National Primate Research Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Aid M, Abbink P, Larocca RA, Boyd M, Nityanandam R, Nanayakkara O et al (2017) Zika virus persistence in the central nervous system and lymph nodes of rhesus monkeys. Cell 169:610–620.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alimonti JB, Ribecco‐Lutkiewicz M, Sodja C, Jezierski A, Stanimirovic DB, Liu Q et al (2018) Zika virus crosses an in vitro human blood brain barrier model. Fluids Barriers CNS 15:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al‐Obaidi MMJ, Bahadoran A, Har LS, Mui WS, Rajarajeswaran J, Zandi K et al (2017) Japanese encephalitis virus disrupts blood‐brain barrier and modulates apoptosis proteins in THBMEC cells. Virus Res 233:17–28. [DOI] [PubMed] [Google Scholar]
- 4. Arabanian LS, Fierro FA, Stolzel F, Heder C, Poitz DM, Strasser RH et al (2014) MicroRNA‐23a mediates post‐transcriptional regulation of CXCL12 in bone marrow stromal cells. Haematologica 99:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Araujo AQ, Silva MT, Araujo AP (2016) Zika virus‐associated neurological disorders: a review. Brain 139(Pt 8):2122–2130. [DOI] [PubMed] [Google Scholar]
- 6. Barry PA, Lockridge KM, Salamat S, Tinling SP, Yue Y, Zhou SS et al (2006) Nonhuman primate models of intrauterine cytomegalovirus infection. ILAR J 47:49–64. [DOI] [PubMed] [Google Scholar]
- 7. Bonaldo MC, Ribeiro IP, Lima NS, Dos Santos AA, Menezes LS, da Cruz SO et al (2016) Isolation of Infective Zika Virus from Urine and Saliva of Patients in Brazil. PLoS Negl Trop Dis 10:e0004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carod‐Artal FJ, Wichmann O, Farrar J, Gascon J (2013) Neurological complications of dengue virus infection. Lancet Neurol 12:906–919. [DOI] [PubMed] [Google Scholar]
- 9. Carteaux G, Maquart M, Bedet A, Contou D, Brugieres P, Fourati S et al (2016) Zika virus associated with meningoencephalitis. N Engl J Med 374:1595–1596. [DOI] [PubMed] [Google Scholar]
- 10. Coffey LL, Pesavento PA, Keesler RI, Singapuri A, Watanabe J, Watanabe R et al (2017) Zika virus tissue and blood compartmentalization in acute infection of rhesus macaques. PLoS One 12:e0171148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daniels BP, Holman DW, Cruz‐Orengo L, Jujjavarapu H, Durrant DM, Klein RS (2014) Viral pathogen‐associated molecular patterns regulate blood‐brain barrier integrity via competing innate cytokine signals. MBio 5:e01476‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeVries ME, Kelvin AA, Xu L, Ran L, Robinson J, Kelvin DJ (2006) Defining the origins and evolution of the chemokine/chemokine receptor system. J Immunol 176:401–415. [DOI] [PubMed] [Google Scholar]
- 13. Durbin AP, Karron RA, Sun W, Vaughn DW, Reynolds MJ, Perreault JR et al (2001) Attenuation and immunogenicity in humans of a live dengue virus type‐4 vaccine candidate with a 30 nucleotide deletion in its 3'‐untranslated region. Am J Trop Med Hyg 65:405–413. [DOI] [PubMed] [Google Scholar]
- 14. Durrant DM, Williams JL, Daniels BP, Klein RS (2014) Chemokines referee inflammation within the central nervous system during infection and disease. Adv Med 2014:806741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eldar AH, Chapman J (2014) Guillain Barre syndrome and other immune mediated neuropathies: diagnosis and classification. Autoimmun Rev 13:525–530. [DOI] [PubMed] [Google Scholar]
- 16. Gao D, Sun H, Zhu J, Tang Y, Li S (2018) CXCL12 induces migration of Schwann cells via p38 MAPK and autocrine of CXCL12 by the CXCR4 receptor. Int J Clin Exp Pathol 11:3119–3125. [PMC free article] [PubMed] [Google Scholar]
- 17. Gardner WJ, Spitler DK, Whitten C (1954) Increased intracranial pressure caused by increased protein content in the cerebrospinal fluid; an explanation of papilledema in certain cases of small intracranial and intraspinal tumors, and in the Guillain‐Barre syndrome. N Engl J Med 250:932–936. [DOI] [PubMed] [Google Scholar]
- 18. Guillain G, Barre JA, Strohl A (1999) Radiculoneuritis syndrome with hyperalbuminosis of cerebrospinal fluid without cellular reaction. Notes on clinical features and graphs of tendon reflexes. 1916. Ann Med Interne (Paris) 150:24–32. [PubMed] [Google Scholar]
- 19. Han Y, He T, Huang DR, Pardo CA, Ransohoff RM (2001) TNF‐alpha mediates SDF‐1 alpha‐induced NF‐kappa B activation and cytotoxic effects in primary astrocytes. J Clin Invest 108:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hirsch AJ, Smith JL, Haese NN, Broeckel RM, Parkins CJ, Kreklywich C et al (2017) Zika virus infection of rhesus macaques leads to viral persistence in multiple tissues. PLoS Pathog. 13:e1006219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson BW, Russell BJ, Lanciotti RS (2005) Serotype‐specific detection of dengue viruses in a fourplex real‐time reverse transcriptase PCR assay. J Clin Microbiol 43:4977–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim KW, Cho ML, Kim HR, Ju JH, Park MK, Oh HJ et al (2007) Up‐regulation of stromal cell‐derived factor 1 (CXCL12) production in rheumatoid synovial fibroblasts through interactions with T lymphocytes: role of interleukin‐17 and CD40L‐CD40 interaction. Arthritis Rheum 56:1076–1086. [DOI] [PubMed] [Google Scholar]
- 23. Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J et al (2015) Viral infection of the central nervous system and neuroinflammation precede blood‐brain barrier disruption during japanese encephalitis virus infection. J Virol 89:5602–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lubetzki C, Stankoff B (2014) Demyelination in multiple sclerosis. Handb Clin Neurol 122:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Magnani DM, Rogers TF, Beutler N, Ricciardi MJ, Bailey VK, Gonzalez‐Nieto L et al (2017) Neutralizing human monoclonal antibodies prevent Zika virus infection in macaques. Sci Transl Med 9:eaan8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magnani DM, Rogers TF, Maness NJ, Grubaugh ND, Beutler N, Bailey VK et al (2018) Fetal demise and failed antibody therapy during Zika virus infection of pregnant macaques. Nat Commun 9:1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maher F, Vannucci SJ, Simpson IA (1994) Glucose transporter proteins in brain. FASEB J 8:1003–1011. [DOI] [PubMed] [Google Scholar]
- 28. Mathur A, Khanna N, Chaturvedi UC (1992) Breakdown of blood‐brain barrier by virus‐induced cytokine during Japanese encephalitis virus infection. Int J Exp Pathol 73:603–611. [PMC free article] [PubMed] [Google Scholar]
- 29. McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, Klein RS (2008) Pathological expression of CXCL12 at the blood‐brain barrier correlates with severity of multiple sclerosis. Am J Pathol 172:799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS (2006) CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol 177:8053–8064. [DOI] [PubMed] [Google Scholar]
- 31. McDonald EM, Duggal NK, Brault AC (2017) Pathogenesis and sexual transmission of Spondweni and Zika viruses. PLoS Negl Trop Dis 11:e0005990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mecharles S, Herrmann C, Poullain P, Tran TH, Deschamps N, Mathon G et al (2016) Acute myelitis due to Zika virus infection. Lancet 387:1481. [DOI] [PubMed] [Google Scholar]
- 33. Michlmayr D, Andrade P, Gonzalez K, Balmaseda A, Harris E (2017) CD14(+)CD16(+) monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat Microbiol 2:1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mladinich MC, Schwedes J, Mackow ER (2017) Zika virus persistently infects and is basolaterally released from primary human brain microvascular endothelial cells. MBio 8:00952‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munoz LS, Barreras P, Pardo CA (2016) Zika virus‐associated neurological disease in the adult: guillain‐barre syndrome, encephalitis, and myelitis. Semin Reprod Med 34:273–279. [DOI] [PubMed] [Google Scholar]
- 36. Negro S, Lessi F, Duregotti E, Aretini P, La Ferla M, Franceschi S et al (2017) CXCL12alpha/SDF‐1 from perisynaptic Schwann cells promotes regeneration of injured motor axon terminals. EMBO Mol Med 9:1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oehler E, Watrin L, Larre P, Leparc‐Goffart I, Lastere S, Valour F et al (2014) Zika virus infection complicated by Guillain‐Barre syndrome–case report, French Polynesia, December 2013. Euro Surveill 19:20720. [DOI] [PubMed] [Google Scholar]
- 38. Papa MP, Meuren LM, Coelho SVA, Lucas CGO, Mustafa YM, Lemos Matassoli F et al (2017) Zika virus infects, activates, and crosses brain microvascular endothelial cells, without barrier disruption. Front Microbiol 8:2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pardigon N (2017) Pathophysiological mechanisms of Flavivirus infection of the central nervous system. Transfus Clin Biol 24:96–100. [DOI] [PubMed] [Google Scholar]
- 40. Parra B, Lizarazo J, Jimenez‐Arango JA, Zea‐Vera AF, Gonzalez‐Manrique G, Vargas J et al (2016) Guillain‐barre syndrome associated with Zika virus infection in colombia. N Engl J Med 375:1513–1523. [DOI] [PubMed] [Google Scholar]
- 41. Patel JR, Williams JL, Muccigrosso MM, Liu L, Sun T, Rubin JB, Klein RS (2012) Astrocyte TNFR2 is required for CXCL12‐mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol 124:847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng H, Erdmann N, Whitney N, Dou H, Gorantla S, Gendelman HE et al (2006) HIV‐1‐infected and/or immune activated macrophages regulate astrocyte SDF‐1 production through IL‐1beta. Glia 54:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S (2012) West Nile virus‐induced disruption of the blood‐brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J Gen Virol 93(Pt 6):1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santiago GA, Vergne E, Quiles Y, Cosme J, Vazquez J, Medina JF et al (2013) Analytical and clinical performance of the CDC real time RT‐PCR assay for detection and typing of dengue virus. PLoS Negl Trop Dis 7:e2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simonin Y, Loustalot F, Desmetz C, Foulongne V, Constant O, Fournier‐Wirth C et al (2016) Zika virus strains potentially display different infectious profiles in human neural cells. EBioMedicine 12:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith DR, Sprague TR, Hollidge BS, Valdez SM, Padilla SL, Bellanca SA et al (2018) African and asian zika virus isolates display phenotypic differences both in vitro and in vivo. Am J Trop Med Hyg 98:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suen WW, Prow NA, Hall RA, Bielefeldt‐Ohmann H (2014) Mechanism of West Nile virus neuroinvasion: a critical appraisal. Viruses 6:2796–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Verma R, Sahu R, Holla V (2014) Neurological manifestations of dengue infection: a review. J Neurol Sci 346:26–34. [DOI] [PubMed] [Google Scholar]
- 49. Wakerley BR, Uncini A, Yuki N, GBS Classification Group (2014) Guillain‐Barre and Miller Fisher syndromes–new diagnostic classification. Nat Rev Neurol 10:537–544. [DOI] [PubMed] [Google Scholar]
- 50. Wakerley BR, Yuki N (2015) Mimics and chameleons in Guillain‐Barre and Miller Fisher syndromes. Pract Neurol 15:90–99. [DOI] [PubMed] [Google Scholar]
- 51. White MK, Wollebo HS, David Beckham J, Tyler KL, Khalili K (2016) Zika virus: an emergent neuropathological agent. Ann Neurol 80:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wijdicks EF, Klein CJ (2017) Guillain‐barre syndrome. Mayo Clin Proc 92:467–479. [DOI] [PubMed] [Google Scholar]
- 53. Xia H, Luo H, Shan C, Muruato AE, Nunes BTD, Medeiros DBA et al (2018) An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat Commun 9:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zlotnik A (2006) Involvement of chemokine receptors in organ‐specific metastasis. Contrib Microbiol 13:191–199. [DOI] [PubMed] [Google Scholar]
- 55. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR (1998) Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393:595–599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Acute viremia following ZIKV infection. (A) Viremia in eleven of the pregnant females used in the current study. (B) Viremia in the three males used in the study. As reported previously (26), infection of nonpregnant adults results in rapid robust replication of virus as evidenced by quantification of viral RNA in serum with viral clearance from blood after about 7 to 10 days whereas viremia in pregnant females typically persists for longer times.
Figure S2. CSF samples collected at various times from 11 ZIKV‐infected animals was analyzed using ZIKV‐specific RT‐PCR. Virus was detectible in the CSF in seven of the animals. Females are denoted using (x) and males by circles. None of the females used in the experiment were pregnant.
Figure S3. Acute infection of IRMs results in upregulation of CXCL12 (A) and IL1RN (B) in the CSF. At top, samples obtained from 14‐30 days after infection are grouped to facilitate comparison with preinfection controls. P values were determined using paired two‐tailed T‐tests to compare CXCL12 or URN concentrations following infection with that of preinfection controls. At bottom, kinetics of cytokine expression are provided with available CSF samples.
Figure S4. CXCL12 and URN concentrations in the CSF of ZIKV‐infected animals. Samples correspond to those in Figure 4.
Figure S5. Quantification of TNF, IL1B, and CD4OLG in the CSF of ZIKV infected IRMs. See text for description. Samples were divided into three temporal groups to facilitate comparison with Figure 4B,C.
Figure S6. This figure is reconfigured version of Figure 4B. Points denoted with an “x” indicate that the sample was taken from a pregnant animal while points represented by a circle indicate that the sample was taken from a nonpregnant animal.
Table S1. Initial analysis of several animals was carried out using an NHP‐specific panel designed to detect key cytokines (Cytokine/Chemokine/Growth Factor 37 Plex NHP ProcartaPlex Panel). The specific cytokines detected in the panel are listed above.
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.