

Abstract
Pediatric diffuse midline gliomas are devastating diseases. Among them, diffuse midline gliomas H3K27M‐mutant are associated with worse prognosis. However, recent studies have highlighted significant differences in clinical behavior and biological alterations within this specific subgroup. In this context, simple markers are needed to refine the prognosis of diffuse midline gliomas H3K27M‐mutant and guide the clinical management of patients. The aims of this study were (i) to describe the molecular, immunohistochemical and, especially, chromosomal features of a cohort of diffuse midline gliomas and (ii) to focus on H3K27M‐mutant tumors to identify new prognostic markers. Patients were retrospectively selected from 2001 to 2017. Tumor samples were analyzed by immunohistochemistry (including H3K27me3, EGFR, c‐MET and p53), next‐generation sequencing and comparative genomic hybridization array. Forty‐nine patients were included in the study. The median age at diagnosis was 9 years, and the median overall survival (OS) was 9.4 months. H3F3A or HIST1H3B mutations were identified in 80% of the samples. Within the H3K27M‐mutant tumors, PDGFRA amplification, loss of 17p and a complex chromosomal profile were significantly associated with worse survival. Three prognostic markers were identified in diffuse midline gliomas H3K27M‐mutant: PDGFRA amplification, loss of 17p and a complex chromosomal profile. These markers are easy to detect in daily practice and should be considered to refine the prognosis of this entity.
Keywords: chomosomal profile, diffuse midline gliomas, H3K27M‐mutant, molecular alterations, prognostic markers, translational study
Abbreviations
- aCGH
comparative genomic hybridization array
- AT/RT
atypical teratoid/rhabdoid tumor
- CNS
central nervous system
- DIPG
diffuse intrinsic pontine glioma
- FFPE
formalin‐fixed paraffin‐embedded
- H&E
Hematoxylin and Eosin staining
- IHC
immunohistochemistry
- IQR
interquartile range
- MRI
magnetic resonance imaging
- NGS
next‐generation sequencing
- RTKs
receptor tyrosine kinases
- WHO
World Health Organization
Introduction
Brain tumors are the second most frequent malignancies in children and adolescents 18, 36. Within this heterogeneous group of tumors, diffuse midline gliomas—including diffuse intrinsic pontine gliomas (DIPG)—are highly aggressive, with a median survival ranging from 9 to 12 months 25. Due to their location, complete surgical removal is impossible. Treatment is essentially based on radiation therapy given that successive clinical trials have not identified a benefit from adjuvant or neoadjuvant chemotherapy 1, 14, 20. Unfortunately, radiation therapy remains palliative and only increases survival by 3–4 months 20.
Diagnosis is strongly suspected upon rapidly increasing neurological symptoms associated with an infiltrative midline brain tumor on MRI 14. However, because of the inherent risks of biopsy and the invariably fatal outcome of the disease, the diagnosis has remained only clinical and radiological for years. Thanks to technical improvement and experience of neurosurgeons, stereotactic biopsies of these lesions are now considered as a safe procedure and, in most cases, provide suitable material for histological and molecular studies 14, 33.
In 2012, the identification of histone mutations (K27M in H3F3A, HIST1H3B/C and HIST2H3A/C genes) was a major breakthrough in the knowledge of this disease 10, 23, 31, 42. It led to the recognition of a new entity called “diffuse midline glioma, H3K27M‐mutant” in the latest WHO Classification of Central Nervous System Tumors 41. These mutations are found in diffuse midline gliomas, account for up to 84% of DIPG cases and are associated with worse outcomes 7. Other molecular alterations were next described, affecting genes such as ACVR1, TP53, PDGFRA, PIK3CA and MYC 25.
The mutational landscape of diffuse midline gliomas H3K27M‐mutant has been largely described in the past few years. On the contrary, chromosomal alterations have been less characterized in this specific subgroup. Before the identification of histone mutations in 2012, several studies identified recurrent PDGFRA and EGFR amplifications in pediatric high‐grade gliomas, including DIPG. Segmental aneuploidy was also described, such as +1q, −10q, −13q or −17p 3, 30, 40, 44. However, these studies were mostly based on heterogeneous cohorts that grouped DIPG and pediatric hemispheric high‐grade gliomas together 3, 30. Others were limited to DIPG; however, they did not distinguish H3K27M‐mutant and histone wild‐type gliomas 40, 44. Currently, there are no accurate data about chromosomal alterations in the specific subgroup of diffuse midline gliomas H3K27M‐mutant, despite its worse prognosis among diffuse midline gliomas.
Interestingly, in 2015, Castel et al. first described heterogeneous clinical behavior within diffuse midline gliomas, H3K27M‐mutant 10. HIST1H3B mutant gliomas displayed better prognosis and better response to treatment than H3F3A‐mutant gliomas. Regarding molecular alterations, Castel et al. also described significant differences between these two subgroups based on gene expression and DNA methylation profiling 11. These findings suggest that diffuse midline gliomas H3K27M‐mutant are not as homogeneous as first assumed. In this context, it seems particularly interesting to identify new prognostic markers within this specific subgroup, to more precisely refine their clinical behavior. The aims of this study were (i) to describe the molecular, immunohistochemical and, especially, the chromosomal features of pre‐therapeutic diffuse midline gliomas in an unpublished cohort of children and young adults and (ii) to focus on H3K27M‐mutant tumors to identify new prognostic markers within this subgroup.
Materials and Methods
Study population, samples and clinical data
Patients were retrospectively selected from the records of the Lille University Hospital, France, from 1 January 2001 to 31 March 2017. Included patients were children and young adults who had neurological symptoms and radiological findings (by MRI or CT scan) of an infiltrative tumor of the brainstem (DIPG) or of another midline location (thalamus, third ventricle, cerebellum, spinal cord and pineal gland). All patients underwent pre‐therapeutic stereotactic/surgical biopsy or resection of their tumor. Technical routine procedures were standardized, and tissue samples were snap‐frozen and/or formalin‐fixed and paraffin‐embedded (FFPE). Diagnosis of diffuse glioma (grades II–IV) was histologically confirmed in the Department of Pathology, Lille University Hospital, France, according to the WHO classification validated at the time of diagnosis. Patients were secondarily excluded if the tumor material was insufficient for both molecular and immunohistochemical analyses. The percentage of tumor cells was estimated by a neuropathologist for each sample (FFPE and frozen samples) with H&E staining. A minimal amount of 50% of tumor cells was required to perform molecular analyses. The clinical data collected were sex, date of diagnosis (considered as the date of biopsy/surgery), age at diagnosis, tumor location and date of death/date of last contact. This study was approved by the institutional review board. Informed consent for translational research was obtained for each patient.
DNA and RNA extraction
DNA was extracted from FFPE tumor tissues with the QIAamp DNA FFPE Tissue Kit (Qiagen, Courtaboeuf, France) after removing paraffin with the Deparaffinization Solution (Qiagen). DNA was extracted from frozen tumor tissues with the Prepito DNA Cytopure Kit (PerkinElmer, Waltham, MA, USA). The quantity of extracted DNA was measured using the Quant‐it PicoGreen dsDNA assay Kit (ThermoFisher Scientific, Waltham, MA, USA) on a Xenius XC spectrofluorometer (Safas, Monaco). Total RNA was extracted from FFPE tumor tissue using the RNeasy FFPE Kit (Qiagen) and from frozen tumor tissue with the Nucleospin RNA II Kit (Macherey‐Nagel, Germany). The quantity of extracted RNA was measured with a NanoDrop spectrophotometer (ThermoFisher Scientific).
Next‐generation sequencing (NGS)
Custom‐made panels including targets of interest described in pediatric gliomas were designed using the Ion AmpliSeq Designer Software (ThermoFisher Scientific) to identify somatic mutations (Supplemental Table S1).
Approximately 10 ng of each DNA sample was used as a template to prepare the library according to the manufacturer's instructions. Ampliseq libraries were prepared using the Ion Ampliseq Library Kit 2.0 (ThermoFisher Scientific) and barcoded using the Ion Xpress Barcode Adapters Kit (ThermoFisher Scientific). Quality control and quantification of amplified libraries were performed on the 2200 TapeStation (Agilent Technologies, Santa Clara, CA, USA) using the High Sensitivity D1000 ScreenTape assay. Amplified libraries were normalized and pooled. Pooled libraries were clonally amplified on ion sphere particles (ISPs) by emulsion PCR. Amplification, ISPs enrichment and chip loading were performed on the Ion Chef Instrument with the Ion 540 Kit‐Chef or the Ion PI Hi‐Q Chef Kit (ThermoFisher Scientific). Template ISPs were sequenced on the Ion S5 XL Sequencer or the Ion Proton Sequencer (ThermoFisher Scientific). Each run included a positive control, a negative control and a non‐template control to validate the quality of the assay. Data were analyzed using the Torrent Suite Software v.5.2.2 (ThermoFisher ScientificVariant calling was performed with optimized settings (available on demand)). The variants obtained were annotated with the Variant Effect Predictor and integrated in a homemade database called DVD (Bioinformatics, CHU‐Lille). The lower limit of detection for our NGS assay was 5% mutant allele frequency. A minimum sequencing depth of 300x was required to reliably label the gene status as wild type by our assay. Control sample analysis and quality metrics were checked for each case to avoid false results. All identified variants were checked for correct nomenclature using Alamut Visual v.2.11 (Interactive Biosoftware) and/or Integrative Genomics Viewer. Tumor‐specific variants were defined as those producing a coding change in biologically relevant genes which is not in population databases to exclude polymorphisms (1000 Genomes (http://www.1000genome.org), Exac Browser (http://exac.broadinstitute.org) and dbSNP (Build 137, NCBI)) and which had an observed variant allele frequency consistent with the estimated tumor fraction. Pathogenicity predictions were made using cancer‐specific databases (cbioportal (http://www.cbioportal.org), the Catalogue of Somatic Mutations in Cancer (http://cancer.sanger.ac.uk/cosmic) and prediction programs, such as Polymorphism Phenotyping v2 (Polyphen‐2) and Sorting Tolerant from Intolerant (SIFT), through Alamut Visual v.2.11.0 (Interactive Biosoftware, Rouen, France).
Comparative genomic hybridization array (aCGH)
DNA extracted from frozen tissue and reference DNA (Promega) were digested by restriction enzymes (Rsa1 and Alu1) using the SureTag DNA Labeling Kit (Agilent Technologies). DNA extracted from FFPE samples was already fragmented. Tumor DNA and reference DNA were labeled with cyanine 5‐dUTP and cyanine 3‐dUTP, respectively. Labeled DNA was next purified to eliminate unincorporated nucleotides, and the quality of labeling was measured with a NanoDrop spectrophotometer. Tumor DNA and reference DNA were co‐hybridized onto SurePrint G3 Human CGH Microarrays 8 × 60 k (Agilent Technologies) according to the manufacturer's instructions. After 40 h, microarray slides were washed and scanned on the Agilent SureScan Microarray scanner. Scanned images were then analyzed using CytoGenomics Software v.3.0.2.11 (Agilent Technologies) and the ADM‐2 (aberration detection method‐1) algorithm with a log 2 ratio filter of 0.25 and a threshold of 6.0.
KIAA1549‐BRAF fusion
Cases of midline gliomas, initially diagnosed at the time of biopsy or surgical resection as diffuse glioma grade II, were tested for the presence of the KIAA1549‐BRAF fusion after second histological review. cDNA was synthetized using the SuperScript VILO cDNA Synthesis Kit (ThermoFisher Scientific) according to the manufacturer's instructions. KIAA1549–BRAF fusion variants were amplified by PCR (Polymerase Chain Reaction) with specific pairs of primers flanking fusion points between KIAA1549 and BRAF (primers available on request). PCR products were analyzed with 2% agarose gel electrophoresis.
Tissue microarrays (TMA) and immunohistochemistry (IHC)
Tissue microarrays were constructed using an automated tissue arrayer (MiniCore®, Excilone, France) punching cores of 1 mm from FFPE blocks. TMA blocks contained one to six cores of the most representative tumor areas of 29 cases of diffuse midline glioma. TMA blocks also contain samples employed as external controls: two normal cerebral tissues, two non‐midline diffuse gliomas, eight pilocytic astrocytomas or gangliogliomas and one adult high‐grade glioma. An H&E staining slide of each TMA block was generated to assess their quality. Cases without sufficient material after molecular analyses were not included in TMA, but immunohistochemical study was performed on whole slides. For each immunohistochemical reaction, an external positive control was added on the slide. Two pathologists (CD, FR) independently evaluated the stained slides. A consensus was reached by new examination with a multiple‐head microscope to resolve discrepancies and discussed with a third pathologist when necessary (CAM). The expression of the following markers was analyzed by immunohistochemistry on TMA slides: histone H3 lysine 27 trimethylation (H3K27me3) (1:750, rabbit polyclonal antibody Diagenode), IDH1R132H (1:40, mouse monoclonal antibody, clone H09, Dianova), p53 (1:100, mouse monoclonal antibody, clone DO‐7, Dako), EGFR (1:20, mouse monoclonal antibody, clone 31G7, Invitrogen), c‐MET (prediluted, rabbit monoclonal antibody, clone SP44, Ventana Roche), OLIG2 (1:1000, rabbit monoclonal antibody, clone EP112, Epitomics), INI1 (1:50, mouse monoclonal antibody, clone 25/BAF47, Biosciences) and BRG1 (1:100, rabbit monoclonal antibody, clone EPNCIR111A, Abcam). Stainings were performed using a Ventana Benchmark autostainer (Ventana Medical Systems, Tucson, AZ, USA). Nuclear staining was interpreted as positive for OLIG2 and cytoplasmic staining as positive for IDH1R132H. Weak or moderate nuclear staining was considered as normal expression for p53, whereas total absence of staining or strong and diffuse nuclear staining were considered aberrant. EGFR and c‐MET expression was interpreted as negative or positive using the following score and a threshold of positivity of 150:1 × (% cells 1+) + 2 × (% cells 2+) + 3 × (% cells 3+) with 1, 2 and 3 + corresponding to membrane staining intensity 9. H3K27me3, INI1 and BRG1 nuclear expression was evaluated as lost or maintained.
Histological diagnosis
Initial diagnoses—which were established between 2001 and 2017 at the time of biopsy or surgical resection—were secondarily reviewed according to the 2016 WHO classification 41 using sequencing data (histone or IDH mutation), the presence of the KIAA1549–BRAF fusion transcript and histological characteristics. Therefore, patients were secondarily excluded if their tumor was reclassified as pilocytic astrocytoma or diffuse glioma IDH‐mutant.
Statistical analyses
Categorical variables were expressed as frequency (percentage) and continuous variables as median (range). The overall survival was estimated using the Kaplan–Meier method. In the H3K27M‐mutant subgroup, we compared the distribution of molecular and chromosomal alterations between H3F3A mutant patients and HIST1H3B mutant patients by using Fisher's exact test. We also used Fisher's exact test to compare molecular and chromosomal alterations between H3K27M‐mutant and histone wild‐type subgroups. In the H3K27M‐mutant subgroup, we assessed the association of molecular and chromosomal alterations with overall survival by using the log‐rank test. No statistical comparisons were made for molecular and chromosomal alterations with a frequency lower than five events. Regarding the study sample size, we did not attempt to perform multivariate analyses. Statistical testing was done at the two‐tailed α level of 0.05. Data were analyzed using the SAS software package, release 9.4 (SAS Institute, Cary, NC, USA).
Results
Clinical description of brainstem and midline glioma patients
Of the 65 patients assessed for eligibility, a total of 49 patients were ultimately included in our study (Figure 1). Three tumor samples were identified with IDH1 R132H or IDH2 R172S mutation and were reclassified as diffuse glioma IDH‐mutant. A fusion transcript KIAA1549–BRAF was detected in two other tumors, which were consequently reclassified as pilocytic astrocytomas.
Figure 1.
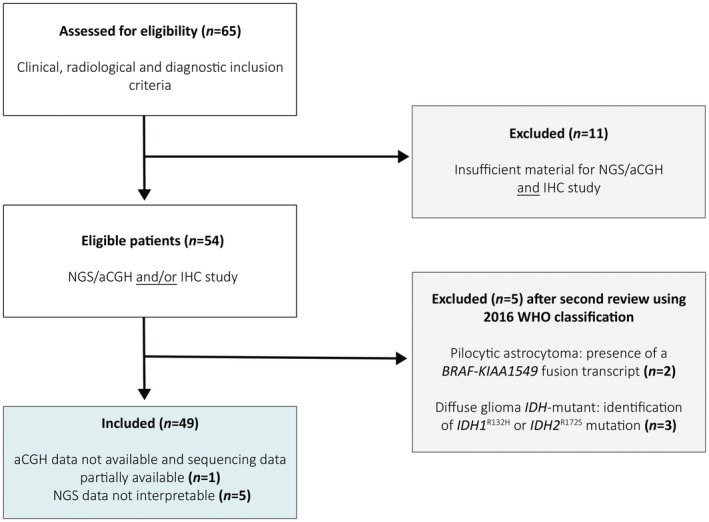
Flow chart of the study. Sixty‐five patients fulfilling the clinical, radiological and diagnostic inclusion criteria were assessed for eligibility. Eleven patients were excluded due to insufficient material for immunohistochemical, molecular and chromosomal studies. After second review, two patients were excluded since their tumors were reclassified as pilocytic astrocytoma after the detection of the fusion transcript KIAA1549‐BRAF. Three patients were excluded because of the identification of an IDH R132H or IDH2 R172S mutation, reclassifying their tumor as a diffuse glioma IDH‐mutant according to the 2016 WHO classification of CNS tumors. A total of forty‐nine patients were included with sufficient material for immunohistochemical analysis. Among these, NGS data were uninterpretable for five patients. For one other patient, sequencing data were only partially available, and aCGH could not be performed due to insufficient tumor material. NGS: next‐generation sequencing; aCGH: comparative genomic hybridization array.
The gender ratio (M/F) was 1.3. The median age at diagnosis was 9 years (1 to 22). At the end of data collection, 89.6% of the patients were deceased (n = 43). The median overall survival (OS) was 9.4 months (IQR 4.6; 17). The overall survival for each patient is detailed in Supplemental Table S2. Thirty‐three tumors (67.3%) were exclusively located in the brainstem, whereas 11 tumors (22.5%) involved another midline location (thalamus, third ventricle, cerebellum, spinal cord and pineal gland). Five other tumors (10.2%) developed both in the brainstem and the thalamus. At initial review—performed between 2001 and 2017 at the time of biopsy or surgical resection—15 tumors (30.6%) were diagnosed as diffuse glioma grade IV, 24 (49.0%) as grade III and ten (20.4%) as grade II. A summary of these data is shown in Table 1.
Table 1.
Clinicopathological characteristics of the 49 patients included in the study. Values are expressed as frequencies (percentage) or median (range).
| Clinical characteristics (n = 49) | |
|---|---|
| Gender | |
| Male | 28 (57.1%) |
| Female | 21 (42.9%) |
| Location | |
| Brainstem exclusively | 33 (67.3%) |
| Other midline location | 11 (22.5%) |
| Brainstem and thalamus | 5 (10.2%) |
| Histological grade at initial review† | |
| IV | 15 (30.6%) |
| III | 24 (49.0%) |
| II | 10 (20.4%) |
| Age at diagnosis (years) | 9 (1 to 22) |
Performed between 2001 and 2017 at the time of biopsy/surgical resection.
Molecular and chromosomal characterization
Full molecular, chromosomal and immunohistochemical results are summarized in Figure 2. NGS data were not interpretable for five patients. For one other patient, sequencing data were only partially available and aCGH could not be performed due to insufficient tumor material.
Figure 2.
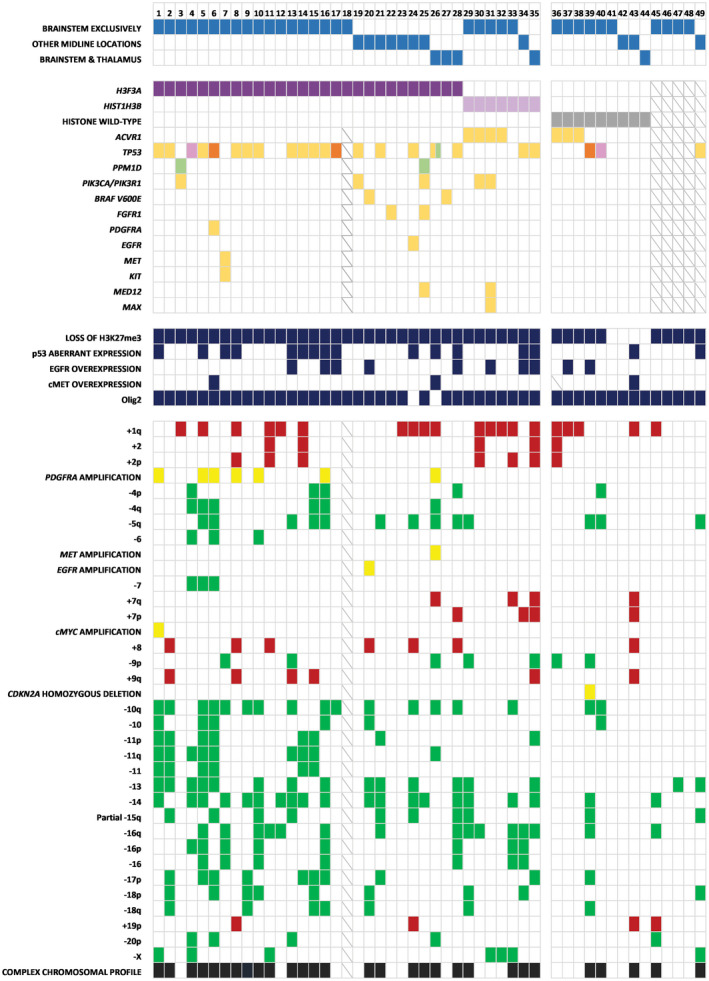
Molecular, chromosomal and immunohistochemical alterations. Each column represents a patient.  H3F3A K27M mutation,
H3F3A K27M mutation,  HIST1H3B K27M mutation,
HIST1H3B K27M mutation,  Histone Wild‐Type,
Histone Wild‐Type,  Unavailable data,
Unavailable data,  Missense variant,
Missense variant,  Frameshift variant,
Frameshift variant,  Splice acceptor variant,
Splice acceptor variant,  Stop gained,
Stop gained,  Segmental chromosomal loss,
Segmental chromosomal loss,  Segmental chromosomal gain, and
Segmental chromosomal gain, and  Focal amplification/deletion.
Focal amplification/deletion.
H3F3A mutation (K27M) was identified in 28 tumors (63.6%), and HIST1H3B mutation (K27M) in seven tumors (15.9%). We did not find any mutations in H3F3B and HIST1H3C. Altogether, only nine tumors (20.5%) had no histone mutation and were consequently defined as histone wild‐type tumors, although not all histone genes were targeted in our study. Loss of H3K27me3 without identification of histone mutation was not considered sufficient for the diagnosis of diffuse midline glioma H3K27M‐mutant, as this entity is strictly defined by the presence of histone mutation, according to the WHO classification of CNS tumors 2016.
Diffuse midline glioma H3K27M‐mutant subgroup
Main molecular and chromosomal alterations
A total of 35 patients (79.5%) were identified with H3F3A or HIST1H3B mutation (Figure 2). In this subgroup, the median age at diagnosis was 9 years (3–22), and the median overall survival was 7.9 months (IQR 3,8; 13,9). Twenty‐three patients (65.7%) had a tumor exclusively located in the brainstem, whereas 8 patients (22.9%) had a tumor in another midline location, as described previously. Four patients had a tumor in both the brainstem and the thalamus (11.4%). Twelve histone‐mutant tumors were initially diagnosed as diffuse glioma grade IV (34.3%), 18 as grade III (51.4%) and 5 as grade II (14.3%). Representative morphologies of these tumors are shown in Supplemental Figure S1.
Twenty‐three patients (65.7%) had a p53 pathway alteration defined as TP53 or PPM1D mutation and/or aberrant p53 expression in IHC. Indeed, PMM1D regulates the p53 cell cycle checkpoint, and mutations in both genes have been found to be mutually exclusive in diffuse midline gliomas 43, 45. As detailed in Figure 2, TP53 mutation was identified in 20 patients (57.1%), and PPM1D mutation was identified in two other patients (5.7%). Only one patient showed p53 aberrant expression in IHC (2.9%), without evidence of TP53 or PPM1D mutation. PIK3CA or PIK3R1 mutations were found in five patients (14.3%), and ACVR1 mutations in four patients (11.8%). BRAF (V600E) mutation co‐occurred with H3F3A K27M mutation in two patients (5.7%). FGFR1 mutations (N546K; K656E) were detected in two patients (5.7%), PDGFRA mutation (C235F) in one patient (2.9%), and EGFR mutation (G598V) in one patient (2.9%) with no EGFR overexpression in IHC. One patient had both KIT (L576P) and MET (C1228Y) mutations. MED12 mutations (L36R; V1428M) were identified in two patients (5.7%) and co‐occurred with MAX (R60Q) mutation in one case (2.9%). The details of these mutations are available in Supplemental Table S3.
INI1 and BRG1 expression was preserved in all tumors, excluding a potential diagnosis of atypical teratoid–rhabdoid tumor (AT/RT). OLIG2 was diffusely positive in all samples except for two tumors. All histone mutant tumors showed a loss of H3K27me3 in immunohistochemistry. EGFR overexpression was found in eight patients (22.9%) with EGFR amplification (2.9%) in one of them. Two patients (5.7%) had a c‐MET overexpression, which was linked to MET amplification in one case (Figure 2).
The most frequent chromosomal alterations were +1q (44.1%), −5q (29.4%), −10q (44.1%), −11q (26.5%), −13 (41.2%), −14 (52.9%), −16q (35.3%) and −17p (26.5%). Interestingly, a complex chromosomal profile (≥5 chromosomes with copy number alterations) was found in 25 patients (70.6%). Moreover, all tumors with loss of 17p (including TP53 locus) were also TP53 mutant. PDGFRA amplification was detected in seven patients (20.6%).
Prognostic markers
In H3K27M‐mutant patients, PDGFRA amplification (P = 0.010), loss of 17p (P = 0.008) and a complex chromosomal profile (P = 0.044) were significantly associated with a shorter overall survival (Figure 3).
Figure 3.
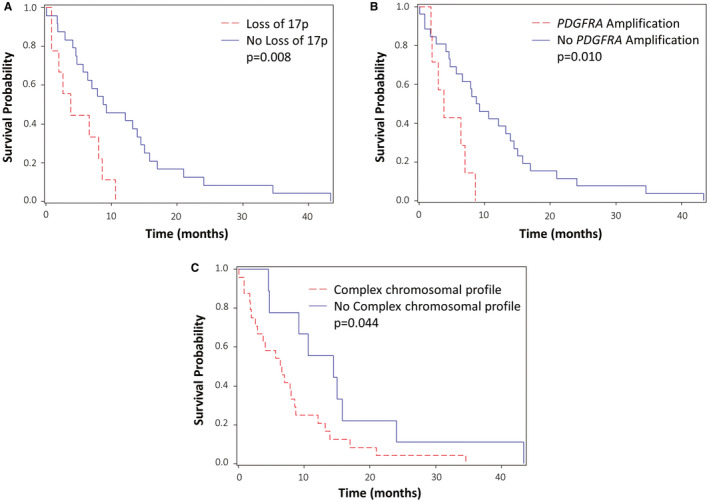
Main prognostic markers for overall survival in H3K27M‐mutant subgroup. A. H3K27M‐mutants with and without loss of 17p. B. H3K27M‐mutants with and without PDGFRA amplification. C. H3K27M‐mutants with and without complex chromosomal profile.
Conversely, neither brainstem locations nor other midline locations were significantly associated with worse overall survival (P = 0.879). EGFR overexpression (P = 0.782), ACVR1 mutation (P = 0.689), TP53 mutation (P = 0.098), +1q (P = 0.215), +2 (P = 0.296), −5q (P = 0.675), −10q (P = 0.695), −13 (P = 0.559), −14 (P = 0.468) and −15q (P = 0.458) were also not significantly associated with shorter survival.
H3F3A vs. HIST1H3B mutant patterns
The median age at diagnosis was 5 years (3–13) for patients with HIST1H3B mutation vs. 11 years (4–22) for patients with H3F3A mutation. The overall survival was lower for H3F3A mutant patients with a median survival of 7.9 months (IQR 2.6; 13.9) vs. 12.1 months (IQR 4.6; 14.5) for HIST1H3B mutant patients. This finding was not statistically significant (P = 0.99). The presence of a tumor in the brainstem was not associated with either H3F3A mutation or HIST1H3B mutation (72% vs. 83.3%. respectively, P = 1).
ACVR1 mutations were significantly associated with HIST1H3B mutation (57.1% vs. 0%, P = 0.001), whereas p53 pathway alterations were significantly associated with H3F3A mutation (75% vs. 28.6%, P = 0.033).
Within the H3F3A‐mutant subgroup, PDGFRA amplification was found in 7 patients (25.9%), loss of 17p in 8 patients (29.6%) and a complex chromosomal profile in 20 patients (74.1%). Within the HIST1H3B‐mutant subgroup, no PDGFRA amplification was identified (0%, P = 0.3), and loss of 17p and a complex chromosomal profile were identified in one patient (14.3%, P = 0.6) and four patients (57.1%, P = 0.4), respectively.
Histone wild‐type subgroup
In this subgroup of nine patients, the median age at diagnosis was 11 years (1–19) and the median overall survival was 27.3 months (IQR 13.2; −). Two tumors were initially diagnosed as diffuse glioma grade IV (22.2%), five as grade III (55.6%) and two as grade II (22.2%).
Loss of H3K27me3 was seen in five patients (55.6%); three of them had ACVR1 mutation and the two others had TP53 mutation (see Figure 2). The median overall survival for these five patients was 13.2 months (IQR 10.3; 15.1). We did not find any additional mutation for the four patients with retained H3K27me3.
We identified homozygous deletion of CDKN2A in only one patient who had additional loss of H3K27me3. Loss of 17p was only found in one patient, and no PDGFRA amplification was identified. Statistical comparisons performed for each chromosomal alteration showed that ‐13 was the only alteration significantly associated with H3K27M mutation (41.2% vs. 0%, P = 0.020). Histone wild‐type tumors appeared to have less chromosomal alterations than H3K27M‐mutant tumors (see Figure 2). However, with a threshold of at least five altered chromosomes, this finding was not statistically significant (H3K27M‐mutant 70.6% vs. histone wild‐type 33.3%, P = 0.058).
Discussion
Diffuse pontine and midline gliomas are devastating pediatric diseases. Despite recent advances in the knowledge of their biology, survival remains poor. Pretreatment samples are rare and provide a fascinating opportunity to identify additional molecular and chromosomal alterations as well as new prognostic markers. Histone mutations are the most adverse prognostic markers in pediatric diffuse midline gliomas, regardless of tumor location 22. Diffuse midline gliomas, H3K27M‐mutant, as defined by the 2016 WHO classification 41, share the same type of histone mutations (K27M in H3F3A, HIST1H3B/C and HIST2H3A/C), which are the hallmark of this entity. These tumors are located throughout the midline and can be highly infiltrative 2, with up to 25% of DIPG involving the thalamus or upper cervical cord 6.
However, although H3K27M‐mutant tumors are all classified as grade IV according to the latest WHO classification of CNS tumors, the clinical course of these tumors remains heterogeneous 10, 11. Notably, HIST1H3B‐mutant tumors are less aggressive and have a better response to treatments than H3F3A‐mutant tumors 10. These differences in clinical behavior can be explained by significant molecular and chromosomal abnormalities. This was first suggested by Puget et al. in 2012, which was before the identification of recurrent H3K27M mutations within diffuse midline gliomas 34. This study showed the existence of two subgroups among DIPG with gene expression profiling, including the oligodendroglial subtype, which is driven by PDGFRA and is associated with worse outcomes. Later, additional molecular alterations were found to be more likely associated with H3F3A mutation (ie, TP53 and PDGFRA) or with HIST1H3B mutation (ie, ACVR1 and BCOR) 10, 27, 38. In this context, it seems particularly interesting to focus on H3K27M‐mutant tumors to try to identify new prognostic markers. This approach could help refine more precisely their prognosis and optimize the clinical management of patients.
Here, we aimed to describe the molecular and, especially, the chromosomal landscape of a series of non‐pretreated diffuse midline gliomas with full clinical follow‐up, using immunohistochemistry, next‐generation sequencing and aCGH. We identified new features associated with H3K27M‐mutant tumors, including three prognostic markers within this specific subgroup: loss of 17p, PDGFRA amplification and, more interestingly, a complex chromosomal profile.
Characterization of diffuse midline gliomas H3K27M‐mutant
We identified that approximately 80% of tumors had H3F3A or HIST1H3B mutation. This rate is consistent with previous studies 25. No HIST1H3C mutation was found, likely reflecting its rarity 27. The median overall survival of patients with H3K27M‐mutant tumors was 7.95 months, which is in line with previous studies 23. As expected, within histone mutant tumors, p53 pathway alteration was the most frequent finding (65.71%), followed by PIK3CA/PIK3R1 mutations (14.29%) 25, 29. ACVR1 mutations were identified in 11.76% of H3K27M‐mutant tumors. The identification of these mutations confirmed the association between histone mutations and obligate partners (TP53 or PPM1D, ACVR1 and PIK3R1) essential for tumorigenesis as previously highlighted 28. The analysis of only biopsy or small resection samples here gives more credit to the absolute necessity of molecular analyses in the characterization of these lesions; molecular alterations are far more homogeneous than histological findings 28.
In our study, BRAFV600E mutations were found in two thalamic gliomas and co‐occurred with the H3F3A K27M mutation. This finding has already been described in diffuse midline gliomas 4, 43, especially in thalamic gliomas 35, 37. We also detected two FGFR1 mutations and point activating mutations in EGFR, KIT and MET, which were already described in previous studies 27, 35, 43.
The MAX mutation R60Q was identified in one sample. MAX is a transcription factor which can form homodimers and heterodimers with the oncogenic protein MYC 16. R60Q seems to be a hotspot mutation since it has been described in numerous types of cancer, including diffuse intrinsic pontine gliomas 24, 38. One study highlighted the importance of arginine 60 in the homodimerization of MAX and its interaction with DNA in silico 16. However, the effect of R60Q substitution on MAX heterodimerization with MYC is still unknown.
We also identified the MED12 L36R mutation in the same sample, co‐occurring with the MAX R60Q mutation; this mutation has never been described in diffuse midline gliomas. MED12 (Mediator of RNA polymerase II transcription subunit 12 homologue) encodes a subunit of the multiprotein complex Mediator. Mediator interacts with RNA polymerase II and acts as both an activator and repressor of gene transcription 17. MED12 is considered a cancer driver gene, and its mutations have been described in different human tumors, such as uterine leiomyomas, breast fibroepithelial tumors, prostate cancers and thyroid cancers 13. The MED12 L36R mutation is a hotspot mutation affecting a highly conserved amino acid 21. More data are needed to assess its role in oncogenesis.
The most frequent large‐scale chromosomal alterations found in the H3K27M‐mutant group were ‐14, +1q, ‐10q, ‐13, ‐16q, ‐5q, ‐11q and ‐17p. These alterations have already been described in high‐grade pediatric gliomas 27, 30, including diffuse midline gliomas and diffuse intrinsic pontine gliomas 10, 40, 44. In our study, except for + 1q, these abnormalities were observed more frequently in the H3K27M‐mutant subgroup than in the histone wild‐type subgroup (Figure 2). Notably, ‐13 was significantly associated with H3K27M‐mutant gliomas rather than histone wild‐type gliomas. As this combination of chromosomal abnormalities has already been described together in pediatric high grade‐gliomas in previous studies regardless of histone mutation status, it seems interesting to study its potential association with histone mutations. This association could represent a potential chromosomal signature for H3K27M‐mutant gliomas and should be further validated in studies with larger sample size.
Prognostic markers in diffuse midline gliomas H3K27M‐mutant
With aCGH array, we identified three interesting prognostic markers within histone mutant tumors.
First, loss of 17p was found in 26.47% of H3K27M‐mutant tumors. In our study, this deletion was strongly associated with a worse outcome in H3K27M‐mutant tumors (P = 0.008). This prognostic marker has already been described by others in pediatric high‐grade gliomas and diffuse midline gliomas 27, 34 but never in the specific subgroup of H3K27M‐mutant tumors. Interestingly, TP53 is located at the locus 17p13.1. In our cohort, all samples with loss of 17p were also TP53 mutant. This configuration is commonly found in human solid and blood cancers 26. TP53 deleterious mutations were more frequent than loss of 17p (57.1%) and had no prognostic value in the H3K27M‐mutant tumors. Hence, this finding suggests that the combination of both loss of 17p and TP53 deleterious mutation could explain the worse prognosis associated to loss of 17p. In multiple myeloma, loss of 17p has been suggested to precede TP53 mutation; the association of the two alterations conferred a poorer prognosis 12, as well as in acute myeloid leukemia 26. Another hypothesis was raised by Liu et al who provided evidence that the clinical impact of loss of 17p could be independent of TP53 locus loss alone 26. Indeed, the size of the 17p deletion is variable and may involve different genes, which could play a role in the biological behavior of the tumor.
Secondarily, we found recurrent focal gains for different receptor tyrosine kinase (RTKs) genes, the most frequent being PDGFRA amplification (20.59%). Interestingly, this alteration was exclusively detected in gliomas with H3F3A mutation, as previously described by others 8, 10. PDGFRA amplification has been associated with worse outcomes and more aggressive tumors in pediatric high‐grade gliomas and DIPG 27, 34. Our findings are similar within the H3K27M‐mutant subgroup (P = 0.01). Moreover, it has been shown that the PDGFRA pathway is often activated in DIPG 19, 39. In this context, PDGFRA appears as a relevant therapeutic target 34, 44. A previous study demonstrated the in vitro efficacy of dasatinib in human DIPG cell lines 39 with promising results. Although two phase I clinical trials reported significant side effects of dasatinib combined with crizotinib 5 and imatinib 32, a phase II trial is currently ongoing in Europe.
A complex chromosomal profile was defined as ≥5 chromosomes with copy number alterations. This threshold has already been used by others in adult glioblastomas 15 and is easy to apply to aCGH reports. Before histone mutation identification in 2012, several teams described many copy number alterations in DIPG 30, 44 and pediatric high‐grade gliomas 3. Interestingly, Warren et al found that the tumors with more aggressive morphology are also those with the highest number of chromosomal alterations 40. More recently, in the study of Buczkowicz et al H3K27M‐mutant tumors were found to have highly unstable genomes with more copy number alterations than histone wild‐type tumors 8. This finding is consistent with our results. Moreover, in our study, a complex chromosomal profile was associated with a worse outcome in the H3K27M‐mutant subgroup. To our knowledge, this finding has never been described in previous studies.
Due to a relatively small number of patients that reflects the epidemiology of the disease and the rarity of tumor samples, prognostic markers could not be evaluated in a multivariate analysis; however, the three prognostic markers identified are robust features in univariate analysis. For the same reasons, we did not find any significant differences in survival between the H3F3A mutant and HIST1H3B mutant subgroups, unlike other studies 10, 27. We only noted that the median overall survival in the H3F3A mutant subgroup is slightly shorter than the median overall survival in the HIST1H3B mutant subgroup. Interestingly, when focusing on the three prognostic markers identified within histone mutant patients, PDGFRA amplification, loss of 17p and a complex chromosomal profile were more frequently identified within the H3F3A mutant subgroup. This finding may partly explain the difference in survival between the H3F3A and HIST1H3B mutant subgroups, as described in the literature.
Histone wild‐type subgroup and loss of H3K27me3
Within the histone wild‐type subgroup, five samples showed loss of H3K27me3 in IHC. Three patients had ACVR1 mutation, and the two others had TP53 mutation. Given that ACVR1 and TP53 have been shown to be obligate partners of H3F3A and HIST1H3B mutations in diffuse midline gliomas tumorigenesis 28, we hypothesize that we were not able to identify histone gene mutation for these five tumors. Indeed, our next‐generation panel only targeted H3F3A, HIST1H3B and HIST1H3C genes and not rarer histone genes, such as HIST2H3C. Interestingly, the median overall survival in this group of five patients was 13.2 months, which was not far from the median overall survival of 7.9 months in the histone mutant subgroup. This gives value to the use of H3K27me3 immunostaining which can help to identify tumors probably displaying the same clinical behavior as molecularly proven H3K27M‐mutant tumors.
In conclusion, this study provides a description of a rare cohort of untreated diffuse midline gliomas, especially H3K27M‐mutant tumors. Interestingly, within the H3K27M‐mutant tumors, we identified three chromosomal prognostic markers: loss of 17p, PDGFRA amplification and a complex chromosomal profile. These markers can be screened using either copy‐number profiling (ie, array CGH or SNP array) or using more simple and targeted methods (ie, loss of heterozygosity with microsatellite markers or fluorescent in situ hybridization (FISH)). Considering that H3K27M‐mutant tumors display a certain degree of heterogeneity, these markers may be relevant to refine the prognosis of this entity.
Conflict of Interest
None to declare.
Supporting information
Figure S1 . Representative morphology and immunohistochemistry of H3K27M‐mutant tumors included in the study. All are diffuse midline gliomas, H3K27M‐mutant, WHO grade IV, using the 2016 WHO Classification: (A) Diffuse glioma with moderate cellularity and nuclear atypia without appreciable mitotic activity, necrosis or microvascular proliferation. Thus, this tumor has grade II histologic features of a diffuse astrocytoma, but the presence of the H3K27M mutation equals an integrated diagnosis of diffuse midline glioma, H3K27M‐mutant, WHO grade IV (H&E staining, 200x); (B) Diffuse glioma with high cellularity, nuclear atypia, and frequent mitoses but no necrosis or microvascular proliferation. Thus, this tumor has grade III histologic features of an anaplastic astrocytoma, but the presence of H3K27M mutation equals an integrated diagnosis of diffuse midline glioma, H3K27M‐mutant, WHO grade IV (H&E staining, 200x); (C) Diffuse glioma with high cellularity, marked nuclear atypia, frequent mitoses, microvascular proliferation and palisading necrosis. Thus, this tumor has grade IV histologic features of a glioblastoma, but the presence of H3K27M mutation equals an integrated diagnosis of diffuse midline glioma, H3K27M‐mutant, WHO grade IV (H&E staining, 200x); (D) p53 overexpression by immunohistochemistry (IHC, 200x), (E) loss of H3K27me3 in tumor cells (black arrow), expression is retained in lymphocytes (IHC, 200x), (F) for comparison, conservation of H3K27me3 in histone wild‐type tumor (IHC, 200x), (G) c‐MET overexpression (IHC, 200x) score 200 (100% of positive tumor cells, staining intensity 2+), (H) EGFR overexpression (IHC, 200x) score 300 (100% of positive tumor cells, staining intensity 3+).
Table S1. NGS Custom‐Made Panel.
Table S2. Survival data. Blue boxes indicate patients alive at the end of data collection.
Table S3. NGS details. *Analysis was performed in another center for this patient, these data are not available. **For patients 41–44, we did not identify any mutation with our NGS assay. ***For patients 45–48, NGS data were uninterpretable.
Acknowledgements
FR acknowledges funding from Lille University Hospital Programme “Fonds Hospitalier d'aide à l’émergence”, 2016. CD acknowledges funding from the French Society of Pathology (Resident bursary 2017). All the authors gratefully acknowledge financial support from the Siric ONCOLille and the “Ligue contre le cancer.” The authors would like to thank the Tumor Bank of the Lille University Hospital and the technicians and engineers of the Departments of Pathology and Molecular Oncology of the Lille University Hospital for their technical assistance.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Azizi AA, Paur S, Kaider A, Dieckmann K, Peyrl A, Chocholous M et al (2018) Does the interval from tumour surgery to radiotherapy influence survival in paediatric high grade glioma? Strahlenther Onkol 194:552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barrow J, Adamowicz‐Brice M, Cartmill M, MacArthur D, Lowe J, Robson K et al (2011) Homozygous loss of ADAM3A revealed by genome‐wide analysis of pediatric high‐grade glioma and diffuse intrinsic pontine gliomas. Neuro‐Oncology 13:212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bax DA, Mackay A, Little SE, Carvalho D, Viana‐Pereira M, Tamber N et al (2010) A distinct spectrum of copy number aberrations in pediatric high‐grade gliomas. Clin Cancer Res 16:3368–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bozkurt SU, Dagcinar A, Tanrikulu B, Comunoglu N, Meydan BC, Ozek M, Oz B (2018) Significance of H3K27M mutation with specific histomorphological features and associated molecular alterations in pediatric high‐grade glial tumors. Childs Nerv Syst 34:107–116. [DOI] [PubMed] [Google Scholar]
- 5. Broniscer A, Jia S, Mandrell B, Hamideh D, Huang J, Onar‐Thomas A et al (2018) Phase 1 trial, pharmacokinetics, and pharmacodynamics of dasatinib combined with crizotinib in children with recurrent or progressive high‐grade and diffuse intrinsic pontine glioma. Pediatr Blood Cancer 65:e27035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C (2014) Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol 128:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buczkowicz P, Hawkins C (2015) Pathology, molecular genetics, and epigenetics of diffuse intrinsic pontine glioma. Front Oncol 5:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M et al (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calculating H‐Score – The ASCO Post [Internet]. Available at: http://www.ascopost.com/issues/april-10-2015/calculating-h-score/. (accessed 22 May 2018).
- 10. Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N et al (2015) Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castel D, Philippe C, Kergrohen T, Sill M, Merlevede J, Barret E et al (2018) Transcriptomic and epigenetic profiling of “diffuse midline gliomas, H3 K27M‐mutant” discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol Commun 6:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chin M, Sive JI, Allen C, Roddie C, Chavda SJ, Smith D et al (2017) Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J 7:e610–e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark AD, Oldenbroek M, Boyer TG (2015) Mediator kinase module and human tumorigenesis. Crit Rev Biochem Mol Biol 50:393–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cohen KJ, Jabado N, Grill J (2017) Diffuse intrinsic pontine gliomas‐current management and new biologic insights. Is there a glimmer of hope? Neuro‐Oncology 19:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dahlback H‐SS, Brandal P, Meling TR, Gorunova L, Scheie D, Heim S (2009) Genomic aberrations in 80 cases of primary glioblastoma multiforme: pathogenetic heterogeneity and putative cytogenetic pathways. Genes Chromosomes Cancer 48:908–924. [DOI] [PubMed] [Google Scholar]
- 16. Dela Cruz FS, Diolaiti D, Turk AT, Rainey AR, Ambesi‐Impiombato A, Andrews SJ et al (2016) A case study of an integrative genomic and experimental therapeutic approach for rare tumors: identification of vulnerabilities in a pediatric poorly differentiated carcinoma. Genome Med 8:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding N, Zhou H, Esteve P‐O, Chin HG, Kim S, Xu X et al (2008) Mediator links epigenetic silencing of neuronal gene expression with x‐linked mental retardation. Mol Cell 31:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gatta G, Peris‐Bonet R, Visser O, Stiller C, Marcos‐Gragera R, Sánchez M‐J et al (2017) Geographical variability in survival of European children with central nervous system tumours. Eur J Cancer 82:137–148. [DOI] [PubMed] [Google Scholar]
- 19. Hoeman C, Shen C, Becher OJ (2018) CDK4/6 and PDGFRA signaling as therapeutic targets in diffuse intrinsic pontine glioma. Front Oncol 8:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M et al (2018) Clinical, radiologic, pathologic, and molecular characteristics of long‐term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the international and European society for pediatric oncology DIPG registries. J Clin Oncol 36:1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ibrahimpasic T, Xu B, Landa I, Dogan S, Middha S, Seshan V et al (2017) Genomic alterations in fatal forms of non‐anaplastic thyroid cancer: identification of MED12 and RBM10 as novel thyroid cancer genes associated with tumor virulence. Clin Cancer Res 23:5970–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karremann M, Gielen GH, Hoffmann M, Wiese M, Colditz N, Warmuth‐Metz M et al (2018) Diffuse high‐grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro‐Oncology 20:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khuong‐Quang D‐A, Buczkowicz P, Rakopoulos P, Liu X‐Y, Fontebasso AM, Bouffet E et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koschmann C, Farooqui Z, Kasaian K, Cao X, Zamler D, Stallard S et al (2017) Multi‐focal sequencing of a diffuse intrinsic pontine glioma establishes PTEN loss as an early event. NPJ Precis Oncol 1:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lapin DH, Tsoli M, Ziegler DS (2017) Genomic insights into diffuse intrinsic pontine glioma. Front Oncol 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J et al (2016) Deletions linked to TP53 loss drive cancer through p53‐independent mechanisms. Nature 531:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal‐Salom J, Taylor KR et al (2017) Integrated molecular meta‐analysis of 1,000 pediatric high‐grade and diffuse intrinsic pontine glioma. Cancer Cell 32:520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M et al (2016) Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7:11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Panditharatna E, Yaeger K, Kilburn LB, Packer RJ, Nazarian J (2015) Clinicopathology of diffuse intrinsic pontine glioma and its redefined genomic and epigenomic landscape. Cancer Genet 208:367–373. [DOI] [PubMed] [Google Scholar]
- 30. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz‐Brice M, Zhang J et al (2010) Integrated molecular genetic profiling of pediatric high‐grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Plessier A, Le Dret L, Varlet P, Beccaria K, Lacombe J, Mériaux S et al (2017) New in vivo avatars of diffuse intrinsic pontine gliomas (DIPG) from stereotactic biopsies performed at diagnosis. Oncotarget 8:52543–52559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pollack IF, Jakacki RI, Blaney SM, Hancock ML, Kieran MW, Phillips P et al (2007) Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro‐Oncology 9:145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Puget S, Beccaria K, Blauwblomme T, Roujeau T, James S, Grill J et al (2015) Biopsy in a series of 130 pediatric diffuse intrinsic pontine gliomas. Childs Nerv Syst 31:1773–1780. [DOI] [PubMed] [Google Scholar]
- 34. Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier M‐P et al (2012) Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS ONE 7:e30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ryall S, Krishnatry R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R et al (2016) Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun 4:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Siegel RL, Miller KD, Jemal A (2017) Cancer statistics, 2017. CA Cancer J Clin 67:7–30. [DOI] [PubMed] [Google Scholar]
- 37. Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJJ, Perry A (2016) Diffuse midline gliomas with histone H3–K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol 26:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C et al (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Truffaux N, Philippe C, Paulsson J, Andreiuolo F, Guerrini‐Rousseau L, Cornilleau G et al (2015) Preclinical evaluation of dasatinib alone and in combination with cabozantinib for the treatment of diffuse intrinsic pontine glioma. Neuro‐Oncology 17:953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warren KE, Killian K, Suuriniemi M, Wang Y, Quezado M, Meltzer PS (2012) Genomic aberrations in pediatric diffuse intrinsic pontine gliomas. Neuro‐Oncology 14:326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weltgesundheitsorganisation . (2016) WHO Classification of Tumours of the Central Nervous System. Revised 4th edn. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds). 408 p. International Agency for Research on Cancer: Lyon. (World Health Organization classification of tumours). [Google Scholar]
- 42. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A et al (2010) Whole‐genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet‐derived growth factor receptor alpha and poly (ADP‐ribose) polymerase as potential therapeutic targets. J Clin Oncol 28:1337–1344. [DOI] [PubMed] [Google Scholar]
- 45. Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J et al (2014) Exome sequencing identifies somatic gain‐of‐function PPM1D mutations in brainstem gliomas. Nat Genet 46:726–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 . Representative morphology and immunohistochemistry of H3K27M‐mutant tumors included in the study. All are diffuse midline gliomas, H3K27M‐mutant, WHO grade IV, using the 2016 WHO Classification: (A) Diffuse glioma with moderate cellularity and nuclear atypia without appreciable mitotic activity, necrosis or microvascular proliferation. Thus, this tumor has grade II histologic features of a diffuse astrocytoma, but the presence of the H3K27M mutation equals an integrated diagnosis of diffuse midline glioma, H3K27M‐mutant, WHO grade IV (H&E staining, 200x); (B) Diffuse glioma with high cellularity, nuclear atypia, and frequent mitoses but no necrosis or microvascular proliferation. Thus, this tumor has grade III histologic features of an anaplastic astrocytoma, but the presence of H3K27M mutation equals an integrated diagnosis of diffuse midline glioma, H3K27M‐mutant, WHO grade IV (H&E staining, 200x); (C) Diffuse glioma with high cellularity, marked nuclear atypia, frequent mitoses, microvascular proliferation and palisading necrosis. Thus, this tumor has grade IV histologic features of a glioblastoma, but the presence of H3K27M mutation equals an integrated diagnosis of diffuse midline glioma, H3K27M‐mutant, WHO grade IV (H&E staining, 200x); (D) p53 overexpression by immunohistochemistry (IHC, 200x), (E) loss of H3K27me3 in tumor cells (black arrow), expression is retained in lymphocytes (IHC, 200x), (F) for comparison, conservation of H3K27me3 in histone wild‐type tumor (IHC, 200x), (G) c‐MET overexpression (IHC, 200x) score 200 (100% of positive tumor cells, staining intensity 2+), (H) EGFR overexpression (IHC, 200x) score 300 (100% of positive tumor cells, staining intensity 3+).
Table S1. NGS Custom‐Made Panel.
Table S2. Survival data. Blue boxes indicate patients alive at the end of data collection.
Table S3. NGS details. *Analysis was performed in another center for this patient, these data are not available. **For patients 41–44, we did not identify any mutation with our NGS assay. ***For patients 45–48, NGS data were uninterpretable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.