

Abstract
Pericytes are vascular mural cells that surround capillaries of the central nervous system (CNS). They are crucial for brain development and contribute to CNS homeostasis by regulating blood–brain barrier function and cerebral blood flow. It has been suggested that pericytes are lost in Alzheimer's disease (AD), implicating this cell type in disease pathology. Here, we have employed state‐of‐the‐art stereological morphometry techniques as well as tissue clearing and two‐photon imaging to assess the distribution of pericytes in two independent cohorts of AD (n = 16 and 13) and non‐demented controls (n = 16 and 4). Stereological quantification revealed increased capillary density with a normal pericyte population in the frontal cortex of AD brains, a region with early amyloid β deposition. Two‐photon analysis of cleared frontal cortex tissue confirmed the preservation of pericytes in AD cases. These results suggest that pericyte demise is not a general hallmark of AD pathology.
Keywords: Alzheimer's disease (AD), angiogenesis, blood–brain barrier (BBB), capillaries, clarity, pericytes, stereology, two‐photon microscopy
Introduction
Pericytes are paramount for the development and function of the capillary network (1). Pericytes are contractile under certain conditions, although it is still debated if this is a ubiquitous ability, and to which extent it contributes to the brain's functional blood flow regulation (16, 20, 24). In the CNS, vessels establish a tight barrier to blood constituents that might interfere with proper functioning of neural tissue—the blood–brain barrier (BBB). Pericyte coverage correlates with the tightness of the endothelial cell lining in different organs of the body, and is exceptionally high in the CNS (51). The investment of endothelium by pericytes is dependent upon platelet‐derived growth factor (PDGFR)β signaling, and experimental models that exploit this dependence collectively revealed the importance of pericytes in the development and regulation of the BBB. Mice lacking PDGFRβ show diminished coverage of endothelial cells by pericytes as well as structurally aberrant vessels and increased BBB permeability during early development, resulting in neonatal lethality (31). Genetically engineered mice with incomplete deficiency of PDGFRβ signaling exhibit reduced pericyte coverage in adults as well as increased BBB permeability (2, 5, 14, 38). Recently, targeted diphtheria‐toxin (DT)‐mediated depletion of pericytes was shown to result in the rapid collapse of brain perfusion, and BBB disruption (40). Some murine models of impaired PDGFRβ‐signaling, as well as mice heterozygous for the PDGFRB gene or with two hypomorphous PDGFRB alleles show a progressive reduction of cortical pericyte coverage accompanied by increased BBB permeability, preceding neurodegeneration and behavioral alterations (5, 38). However, other mouse models of PDGFRβ signaling deficiency do not present with an age‐dependent loss of pericyte coverage in the brain (61), suggesting that the observed functional differences may not necessarily be linked to pericyte demise.
Alzheimer's disease (AD) is a neurodegenerative disorder, whose hallmarks are the deposition of amyloid β (Aβ) in the brain parenchyma, the presence of hyperphosphorylated tau filaments in neurons, and reactive gliosis (43). In a large fraction of subjects with AD pathology, Aβ is deposited in the vascular walls of leptomeningeal and parenchymal arteries and arterioles (cerebral amyloid angiopathy, CAA), and these neuropathological changes are associated with accelerated cognitive decline (54). Ultrastructurally, Aβ is deposited around the basement membrane, thus, in direct, intimate contact to smooth muscle cells and pericytes of small arterioles and capillaries (63). In culture, Aβ is toxic to pericytes; this effect is partly dependent of apolipoprotein E (APOE) 4 genotype, the strongest genetic risk factor for the development of late‐onset AD (9, 57, 62, 64). Pericytes may participate in Aβ clearance (32) and pericyte loss leads to increased deposition of Aβ in vessel walls and brain parenchyma in AD transgenic mice (47). Recent data show that soluble Aβ might lead to constriction of pericytes in microvessels (41). Altogether, the data suggest a complex interaction between the trafficking of Aβ and mural cell function in AD.
Post‐mortem studies of AD brain tissue have detected diminished absolute pericyte numbers and area coverage of capillaries in the frontal cortex and hippocampus, correlating with BBB leakage (50). This effect was increased in carriers of the APOE4 genotype (21). Others have shown decreased levels of PDGFRβ along with increased BBB permeability in post‐mortem tissue obtained from AD cases (35) and recently, another study correlated BBB disruption with increased levels of PDGFRβ in the cerebrospinal fluid (CSF), as well as neurocognitive decline independent of Aβ or tau pathology in APOE4 carriers (37). In these studies, the alterations of the levels of PDGFRβ in the brain tissue or in the CSF were considered a surrogate marker of pericyte damage. Due to the well documented impact of pericyte function on the BBB, these results collectively suggest that pericyte dysfunction or loss could represent a cause of BBB breakdown, leading ultimately to neurodegeneration in AD [as well as in amyotrophic lateral sclerosis, Ref. (65)]. However, earlier ultrastructural studies did not show increased pericyte degeneration in AD (15, 52).
To reassess the impact of AD pathology on the pericyte population, we have used stereological techniques, which are considered the gold standard in morphometric studies, as well as clearing and two‐photon imaging of post‐mortem samples to measure capillary and pericyte density in two independent AD cohorts. We have focused on the frontal cortex, a region with early Aβ deposition (55). Our results indicate that pericyte loss is not a general feature of AD tissue. Instead, we find a denser capillary network with a normal pericyte population in the frontal cortex of AD subjects.
Materials and Methods
Samples
For the stereological study, we obtained a total of 16 frozen samples from patients with a clinical and neuropathologically confirmed diagnosis of AD as well as 16 age‐ and sex‐matched control cases without history of dementia, from the Neurological Foundation Human Brain Bank in New Zealand. In controls, the lack of history of dementia was confirmed by the general practitioner. For AD cases, neuropathological scores (four‐tier neurofibrillary tangle (NFT) score, CERAD score) were provided by the brain bank. For the analysis of cleared tissue, which precluded the use of archival frozen or formalin‐fixed materials, we obtained freshly fixed tissue from 14 cases with neuropathologically confirmed AD, as well as four control cases without history of dementia deceased during the same time period from the Netherlands Brain Bank. Neuropathological data (Thal, Braak, CERAD and CAA scores) were made available for both controls and AD cases by the brain bank. One case was excluded from the analysis due to incomplete tissue clearing. Human brain tissue was obtained in compliance with ethical and legal guidelines and the studies were approved by the institutional review boards. The available demographic and clinical data for all included cases are presented in Tables 1 and 2.
Table 1.
Clinical and neuropathological data of subjects in the stereological study.
| Case | Brain bank ID | Group | Age (y) | Sex | PMT (h) | APOE genotype | Atrophy | Duration of dementia (y) | NFT score | Neuritic plaque score (CERAD) (36) | Capillary CAA (54) | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H122 | CTRL | 72 | F | 9 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Emphysema |
| 2 | H109 | CTRL | 81 | M | 7 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Coronary atherosclerosis |
| 3 | H110 | CTRL | 83 | F | 14 | E2/E2 | n.d. | 0 | n.d. | n.d. | Neg. | Ruptured aortic aneurysm |
| 4 | H137 | CTRL | 77 | F | 12 | E3/E4 | n.d. | 0 | n.d. | n.d. | Neg. | Coronary atherosclerosis |
| 5 | H131 | CTRL | 73 | F | 13 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Ischemic heart disease |
| 6 | H150 | CTRL | 78 | M | 12 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Ruptured myocardial infarct |
| 7 | H123 | CTRL | 78 | M | 7.5 | E2/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Ruptured aortic aneurysm |
| 8 | H103 | CTRL | 70 | M | 5 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Myocardial infarct |
| 9 | H79 | CTRL | 75 | M | 11 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Myocardial infarct |
| 10 | H77 | CTRL | 75 | M | 3.4 | E3/E4 | n.d. | 0 | n.d. | n.d. | Neg. | Aortic aneurysm |
| 11 | H241 | CTRL | 76 | F | 12 | E3/E4 | n.d. | 0 | n.d. | n.d. | Neg. | Metastatic bowel cancer |
| 12 | H190 | CTRL | 72 | F | 19 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Ruptured myocardial infarct |
| 13 | H181 | CTRL | 78 | F | 20 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Aortic aneurysm |
| 14 | H238 | CTRL | 63 | F | 16 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Dissecting aortic aneurysm |
| 15 | H164 | CTRL | 73 | M | 13 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Ischemic heart disease |
| 16 | H152 | CTRL | 79 | M | 18 | E3/E3 | n.d. | 0 | n.d. | n.d. | Neg. | Congestive heart failure |
| 17 | AZ46 | AD | 82 | F | 22 | E4/E4 | 1 | n.d. | 2 | 2 | Neg. | Coronary artery disease |
| 18 | AZ37 | AD | 83 | M | 4 | E3/E4 | 3 | n.d. | 1 | 2 | Pos. | Bronchopneumonia |
| 19 | AZ33 | AD | 65 | M | 20 | E3/E3 | 1 | 7 | 1 | 2 | Neg. | Hypostatic pneumonia |
| 20 | AZ22 | AD | 81 | F | 2 | E2/E3 | 1 | 10 | 2 | 2 | Pos. | Bronchopneumonia |
| 21 | AZ20 | AD | 72 | F | 3 | n.d. | 3 | 7 | 2 | 3 | Neg. | Cachexia |
| 22 | AZ34 | AD | 74 | F | 18 | n.d. | 2 | 5 | 2 | 2 | Neg. | Bronchopneumonia, breast carcinoma |
| 23 | AZ29 | AD | 76 | F | 20 | n.d. | 2 | n.d. | 2 | 2 | Pos. | pneumonia |
| 24 | AZ32 | AD | 75 | F | 3 | n.d. | 2 | n.d. | 1 | 2 | Neg. | Bronchostatic pneumonia |
| 25 | AZ28 | AD | 75 | F | 8 | n.d. | 1 | 4 | 2 | 2 | Neg. | Pneumonia |
| 26 | AZ23 | AD | 71 | F | 10 | E3/E4 | 1 | 2 | 2 | 2 | Neg. | Bronchopneumonia |
| 27 | AZ38 | AD | 80 | M | 5.5 | E3/E3 | 2 | 17.5 | 2 | 3 | Neg. | Respiratory arrest/ pulmonary edema |
| 28 | AZ39 | AD | 74 | M | 12 | E3/E3 | 1 | 20 | 2 | 2 | Neg. | Sepsis |
| 29 | AZ80 | AD | 77 | M | 4.5 | E3/E4 | 3 | 13 | 3 | 3 | Neg. | Myocardial infarct |
| 30 | AZ90 | AD | 73 | M | 4 | E3/E3 | 3 | n.d. | 3 | 3 | Neg. | Gastrointestinal hemorrhage |
| 31 | AZ43 | AD | 80 | M | 21 | n.d. | 1 | n.d. | 2 | 2 | Neg. | Bronchopneumonia |
| 32 | AZ88 | AD | 83 | M | 21 | E3/E3 | 2 | n.d. | 3 | 3 | Neg. | Pneumonia |
PMT = post‐mortem time; NFT = neurofibrillary tangles; Atrophy and NFT stage = 1, mild; 2, moderate; 3, severe; Neuritic plaque score (CERAD) = 0, absent; 1, sparse; 2, moderate; 3, frequent; Capillary CAA was determined in one slide of the frontal cortex. n.d. = not determined due to technical reasons.
Table 2.
Clinical and neuropathological data of subjects in the cleared tissue study.
| Case | Group | Age (y) | Sex | PMT (h) | Brain weight (g) | APOE Genotype | Aβ plaque score (55) | NFT stage (7) | Neuritic plaque score (CERAD (36) | ABC score (26) | CAA (54) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | Type | Stage | ||||||||||
| 2016‐137 | CTRL | 77 | M | 12.8 | 1581 | E3/E3 | 2 | III | 0 | 1 | 2 | 0 | n.d. | n.d. |
| 2017‐003 | CTRL | 96 | F | 6.25 | 1135 | E3/E3 | 3 | III | 1 | 2 | 1 | 1 | 2 | 1 |
| 2017‐005 | CTRL | 60 | F | 5.5 | 1215 | E3/E3 | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 1 |
| 2017‐043 | CTRL | 80 | M | 9.5 | 1460 | E2/E3 | 0 | II | 0 | 0 | 1 | 0 | n.d. | n.d. |
| 2016‐120 | AD | 88 | F | 6.67 | 905 | E2/E2 | 5 | V | 3 | 3 | 3 | 3 | 1 | 2 |
| 2016‐129 | AD | 90 | F | 9 | 1275 | E3/E4 | 4 | IV | 1 | 3 | 2 | 1 | 1 | 1 |
| 2016‐130 | AD | 73 | M | 5.83 | 1300 | E3/E3 | 5 | VI | 3 | 3 | 3 | 3 | 2 | 1 |
| 2016‐132 | AD | 66 | F | 8 | 1115 | E3/E3 | 5 | VI | 3 | 3 | 3 | 3 | 2 | 1 |
| 2017‐018 | AD | 65 | M | 7.83 | 1305 | E3/E3 | 4 | V | 3 | 2 | 3 | 3 | 1 | 2 |
| 2017‐024 | AD | 59 | M | 6.5 | 1305 | E4/E4 | 5 | V | 3 | 3 | 3 | 3 | 1 | 3 |
| 2017‐030 | AD | 80 | M | 10.8 | 1455 | E3/E3 | 5 | VI | 3 | 3 | 3 | 3 | 1 | 2 |
| 2017‐035 | AD | 95 | F | 6.58 | 890 | E3/E4 | 5 | VI | 3 | 3 | 3 | 3 | 1 | 1 |
| 2017‐039 | AD | 88 | M | 8.58 | 1445 | E3/E3 | 5 | V | 3 | 3 | 3 | 3 | 1 | 1 |
| 2017‐045 | AD | 61 | F | 6.83 | 920 | E2/E4 | 5 | VI | 3 | 3 | 3 | 3 | 1 | 1 |
| 2017‐049 | AD | 96 | M | 4.42 | 1095 | E3/E3 | 5 | V | 3 | 3 | 3 | 3 | 2 | 1 |
| 2017‐051 | AD | 94 | M | 6.75 | 1107 | E4/E4 | 5 | V | 3 | 3 | 3 | 3 | 2 | 1 |
| 2017‐054 | AD | 69 | F | 6.17 | 1065 | E3/E3 | 5 | VI | 3 | 3 | 3 | 3 | 1 | 1 |
PMT = post‐mortem time; NFT = neurofibrillary tangles; CAA = cerebral amyloid angiopathy; Neuritic plaque score (CERAD) = 0, absent; 1, sparse; 2, moderate; 3, severe; n.d. = not determined.
Preparation of samples for stereology
Fresh frozen (−80°C) blocks were cut orthogonally to the middle frontal gyrus at a thickness of 60 µm. We fixed the mounted slices for 10 minutes in 100% of methanol at −20°C, except for slices stained for the proliferation marker Ki67, which were fixed in ice‐cold 4% of paraformaldehyde (PFA). All primary antibodies were incubated in 1% of normal donkey serum (NDS), 0.3% of Triton X‐100 and Tris‐buffered saline (TBS) overnight at 4°C. For the stereological measurement of pericyte linear density and proliferation, the following primary antibodies were employed: laminin (mouse anti‐human, γ1, clone 2E8, Millipore, 1:800); PDGFRβ (rabbit anti‐human, Y92, GeneTex, 1:100); Ki67 (Abcam, 1:1000). To reveal endothelia, mouse anti‐CD31 (Clone JC70A, DAKO, 1:100) was employed. Binding of the primary antibodies was detected by incubation with the secondary antibodies Alexa‐488 donkey anti‐mouse and Alexa 568 donkey anti‐rabbit (Invitrogen, both 1:1000) in 5% of NDS, 0.3% of Triton X‐100 in TBS. 4′,6‐diamidino‐2‐phenylindole (DAPI) was used to stain nuclei.
For the stereological quantification of total and vascular Aβ, fresh frozen mounted slices were fixed in methanol. Incubation with 0.3% of H2O2 was used to inactivate endogenous peroxidase. Aβ was stained with a primary antibody against Aβ (clone DE2B4, Santa Cruz, 1:500) revealed by a biotinylated secondary horse anti‐mouse antibody (Vector Laboratories, 1:200). We applied the avidin‐biotin complex (ABC) kit (Vector Laboratories) according to the manufacturer's instructions and nuclei were stained with Harris' hematoxylin.
Stereological analysis
The stereological approach for the measurement of pericyte and vascular densities is described in the Supporting Figure S1. We counted pericyte bodies, defined as PDGFRβ‐immunoreactive cells with round or oval nuclei, whose cell bodies were embedded in the laminin‐immunoreactive basement membrane (1). To identify cells, we used a LEICA DMIL microscope equipped with a 40× (NA 1.25–0.75) oil‐immersion objective. Pericyte density and linear density was quantified by stereological sampling over three second‐consecutive slices per subject. The optical fractionator was implemented as detailed previously (27). Briefly, the dedicated stereological software (Stereo investigator 10, MBF Bioscience) drove an x/y stage to access sampling sites in a random and systematic manner from the entire cortical tissue represented in the probe. At those places, a counting frame was projected over the live images acquired with a CCD camera. Pericytes were then identified and counted online. The size of the counting frame area in each sampling site (14 400 µm2) was chosen so as to contain about two pericytes. Typically, we counted about 450 pericytes per subject from more than 200 sampling sites encompassing the entire cortical tissue presented in the sections. Capillaries were defined as laminin‐immunoreactive tubular vascular structures with a diameter ≤9 µm (29). We implemented the space balls method to analyze capillary density (39), counting every intersection between the projected hemisphere and the central axis of the capillary. Pericyte linear density was calculated as the pericyte number divided by the total capillary length for a given subject. In addition, we estimated the density of intermediate (diameter >9 and ≤50 µm) and large vessels (diameter >50 µm).
For pericyte coverage analysis, images of the laminin‐immunoreactive vasculature and PDGFRβ‐immunoreactive structures were obtained with a 100 × oil‐immersion objective (NA 1.4) at random systematically placed locations selected with the stereological optical fractionator. A total of ~10 intersections were sampled per subject, obtained from one of the three consecutive sections included in the stereological pericyte and vascular density analysis. After manual thresholding in ImageJ (v1.51s), the fraction of area of the laminin‐immunoreactive vessels covered by PDGFRβ‐immunoreactive signal was determined.
Aβ analysis was performed on a single slice per subject and the Aβ signal was divided into two fractions: “vascular” and “parenchymal.” We used the stereological area fraction fractionator as previously described, projecting ~200 counting grids into the counting space at each crossing of the lattice per subject (25). This analysis yielded the Aβ‐immunoreactive area fraction as a percent of the total gray matter area. The presence or absence of capillary CAA (Table 1) was determined in the same frontal sample.
For illustration purposes, we acquired confocal images using a Leica SP5 confocal microscope.
Clearing
Cortical tissue from the middle frontal gyrus obtained from the Netherlands Brain Bank was dissected and fixated in 4% of PFA for 4 hours before being transferred into PBS containing 0.035% of sodium azide. Clearing was performed using the original Clarity protocol with slight modifications (12, 58). These were directed to minimize the duration of the incubation with the PFA fixative. The cortical tissue was cut to obtain a cuboid of approximately 1 × 1 × 3–5 mm entailing all cortical layers and a small fraction of subcortical tissue. The tissue was incubated in the polyacrylamide solution overnight, and thereafter in 4% of PFA‐containing polyacrylamide solution for 4 h. For cross‐linking of proteins with the polyacrylamide scaffold, the tissue was degassed and incubated at 37°C for another 2 h. After cross‐linking, the excess acrylamide was scraped off the tissue. The cross‐linked tissue was then incubated at 37°C in 4% of SDS‐containing PBS for 7–10 days, after which an optimum of clearing was typically reached. SDS was then removed by repeated washes in PBS with 0.5% of Triton‐X before immunostaining. In order to reveal the pericyte‐containing vascular network as well as Aβ plaques, we incubated with rabbit primary antibodies against the pericyte marker PDGFRβ (clone Y92, GeneTex) and against Aβ peptide (Clone DE2B4, Santa Cruz, both at 1:100) for 72 h at 37°C. After washing off the primary antibodies with PBS containing 0.5% of Triton‐X, incubation with secondary Alexa‐488 anti‐mouse and Alexa‐638 anti‐rat antibodies (1:100) was performed for another 72 h at 37°C.
Two‐photon imaging of cleared tissue
Prior to imaging, the samples were incubated in 30% of 2,20‐Thiodiethanol (TDE) to match the refractive index of water (13), and then, placed in a custom‐built chamber. Imaging was performed on a Nikon A1R MP multiphoton microscope equipped with a 25x N.A. 1.1 objective, Nikon) and a Ti:Sa laser (Spectra Physics, Insight DeepSee) at the AMBIO Life Cell Imaging Core Facility (http://AMBIO.charite.de). For each fragment, we produced 15–20 stacks arranged as tiles with 10% overlap covering the entire fragment. At each of the position of the tile, the tissue was subsequently scanned with the IR laser tuned for excitation at 980 nm for Alexa‐488 imaging or 1200 for Alexa‐647 imaging. The fluorescence of the Alexa dyes was recorded with 525/50 or 575/25 nm bandpass filters, respectively. A third image was acquired with excitation at 980 nm and the emission bandpass filter at 575/25 to record the tissue autofluorescence. The size of a single tile was typically with a voxel size of about 1 µm × 1 µm × 4 µm. Using this approach, the typical imaging time for a whole tissue fragment lasted for about 3 h.
Image processing
We stitched the tiles using the Nikon software. We applied a Gaussian filter for noise reduction and resampled the images to a half of the original resolution to make further analysis feasible using ImageJ. The contrast of the PDGFRβ‐immunoreactive signal was enhanced using the CLAHE ImageJ plugin. In order to increase the specificity of the detection of the PDGFRβ‐immunoreactive and Aβ‐immunoreactive signal, we employed a color extraction procedure in the imaging processing program Imaris 8.3.1 (Bitplane). This enhanced the fluorescent signals over tissue autofluorescence. Using Imaris, both the PDGFRβ‐immunoreactive and Aβ‐immunoreactive signals were segmented. The rendered volume of the PDGFRβ‐immunoreactive vascular network was skeletonized using a custom‐written plugin based on the Skel2Graph3D MATLAB function (28); MATLAB version R2015a, MathWorks). This procedure provided a network of segments, connected by nodes, from which several metrics were calculated: length of the vascular network, tortuosity (the quotient of the vascular segment length and the distance between their endpoints) as well as, for every voxel, the distance to the nearest vessel. In addition, the volume occupied by Aβ positive immunostaining was measured. A scheme of the image analysis is presented in the Supporting Figure S2.
APOE genotyping
For APOE genotyping, we extracted genomic DNA either from unfixed frozen or fixed brain tissue. We employed a semi‐nested PCR assay based on (18). Outer primers for amplification of exon 4 of the APOE gene were 5′‐TACAAATCGGAACTGGAGGAAC‐3′ (forward) and 5′‐CCTGCTCCTTCACCTCGTC‐3′(reverse); inner primers were 5′‐GGCACGGCTGTCCAAGGAGC‐3′(forward) and 5′‐ACGCGGCCCTGTTCCACCAG‐3′ (reverse). The product of the nested PCR was digested with the HhaI restriction enzyme to yield bands specific for the APOE2, APOE3 and APOE4 genotypes (18). For statistical analysis, subjects presenting any APOE4‐allele, either homo‐ or heterozygotes, were pooled in the “APOE4” group. Subjects with any combination of the APOE2 and APOE3 alleles were pooled into the “APOE2‐3” group.
Statistics
The effect of diagnosis (AD vs. control) on pericyte density, pericyte linear density, capillary density, intermediate vessel density and large vessel density was determined by a multivariate analysis (one‐way MANOVA). Univariate analysis (one‐way ANOVA) was used in case of significant multivariate analysis (P < 0.05). We carried out a Mann–Whitney U Test to investigate the effect of diagnosis (AD vs. control) or genotype on the non‐normally distributed variables. Comparison between more than two groups was performed with the Kruskal–Wallis nonparametric test followed by the Dunn‐Šidák correction for multiple comparisons. Pericyte and capillary densities were correlated with total Aβ and vascular Aβ (Spearman's rank‐order correlation). Data are presented as means ± standard deviation or the median for non‐normally distributed variables. In boxplots, box and whiskers indicate median and 25th and 75th percentiles and whiskers represent 5th and 95th percentiles. SPSS (version 23, IBM) and MATLAB were used for statistical analysis.
RESULTS
Demographic, clinical and neuropathological data
Demographic and neuropathological characteristics of all included cases are shown in Tables 1 and 2. Matched samples of 16 control and 16 AD subjects were included in the stereological study. Median ages were 75.5 years for both the control and AD groups, and the sex distribution was matched to eight males and eight females (Table 1). Median post‐mortem times were 12 and 9 h in controls and AD subjects, respectively. In AD subjects, the CERAD score of neuritic plaques (36) ranged from moderate (11/16) to frequent (5/16). In a semiquantitative four‐tier classification of the presence of NFTs, the scores of AD subjects varied from mild (3/16) to moderate (10/16) and severe (3/16). AD subjects showed mild (7/16) to moderate (5/16) and severe (4/16) degrees of atrophy. We did not find any statistically significant differences for any morphometric parameter among the different stages of pathology, and therefore, pooled all data.
Tissue employed for clearing comprised 13 AD cases as well as four control cases. The necessity of using freshly fixed tissue for clearing and staining for PDGFRβ accounted for the skewed group sizes. All demographic and neuropathological data of subjects included in the cleared tissue study are provided in the Table 2. Mean ages were 78.3 ± 14.8 and 78.8 ± 13.8 years and sex distribution (f:m) was 2:2 and 6:7 in the control and AD groups, respectively. Aβ plaque, Braak and CERAD scores were known for all subjects and differed significantly between the control and AD groups [Mann–Whitney U test, Supporting Figure S3; Refs. (7, 36, 55)]. Mean brain weight for controls and AD subjects were 1348 ± 208 g and 1168 ± 195 g, respectively, not reaching a statistically significant difference (Supporting Figure S3). Out of all AD cases, four cases displayed grade 2 or 3 type 1 (capillary) CAA. We did not find any statistically significant difference for any metric between cases with grade 2 or 3 CAA and the cases without CAA, and therefore, we pooled all cases in one single AD group.
Stereological analysis
We identified pericytes as PDGFRβ‐immunoreactive cells with cellular bodies and projections embedded in the laminin‐immunoreactive vascular basement membrane (Figure 1A). As PDGFRβ‐immunoreactivity surrounded the CD31‐immunoreactive endothelium, we confirmed the specific staining of mural cells by the anti‐PDGFRβ antibody. (Supporting Figure S4). Pericyte density was increased by 28% in AD cases compared to controls (controls: 2,412 ± 376 cells/mm3 vs. AD: 3,081 ± 584 cells/mm3; P = 0.0006; one‐way‐ANOVA; Figure 1B). The increase in pericyte density was paralleled by a similar increase in the capillary density (see below), so that the linear pericyte density remained unchanged (controls: 8.3 ± 0.7 vs. AD: 8.5 ± 1 cells/mm; Figure 1C). Pericyte coverage (the fraction of laminin‐immunoreactive area occupied by PDGFRβ immunoreactivity) was not different between AD cases and controls (control: 75.6 %, AD: 75.8 %; Figure 1D). The percentage of brain tissue immunoreactive for Aβ (Aβ burden) was increased in AD cases compared to non‐demented controls, although a fraction of the latter also presented with some Aβ immunoreactivity (median 0.18% in control vs. 0.91% in AD cases, P < 0.001, Mann–Whitney U Test, Figure 2A,E. The vascular fraction of Aβ also differed between controls (0%) and AD cases (0.07%) (P < 0.0001, Mann–Whitney U Test, Figure 2F). The increase in pericyte density correlated with total Aβ burden (r s = 0.54, P < 0.01; Spearman's rank‐order correlation; Figure 2G). Among AD cases, neither pericyte density nor pericyte coverage varied significantly between different stages of the four‐tier NFT or neuritic plaque (CERAD) classifications (Supporting Figure S5). In summary, total pericyte numbers increased in AD subjects, while no statistically significant changes in pericyte linear density (i.e. corrected for vascular density) or endothelial area coverage were detected in AD cases. These findings show that pericyte loss was not present in the frontal cortex of AD cases.
Figure 1.
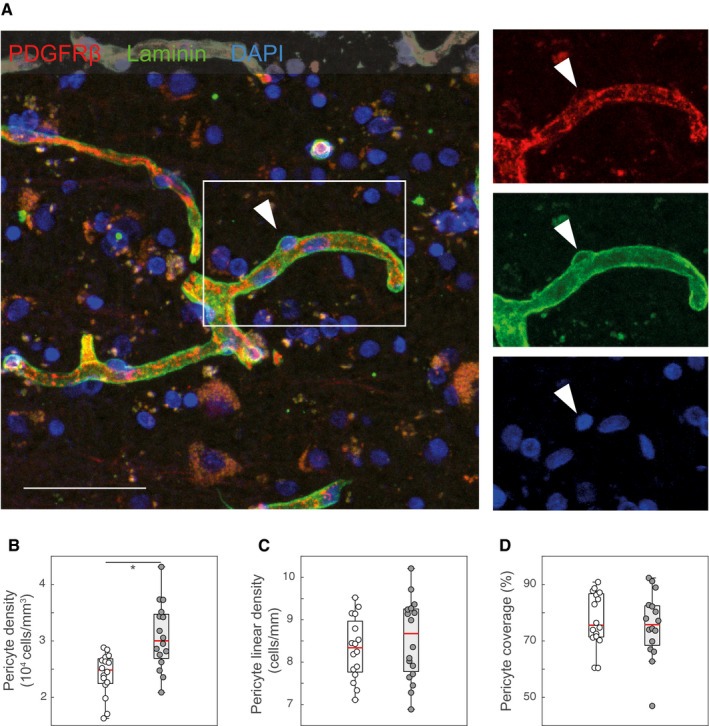
Pericyte density changes in AD. A. Representative confocal image showing PDGFRβ‐immunoreactive pericytes and the laminin‐immunoreactive vascular basement membrane in the frontal cortex of an AD case. The arrowhead demonstrates a PDGFRβ‐immunoreactive pericyte body contained within the laminin‐immunoreactive basement membrane. Those were counted with the stereological fractionator method (see Supporting Figure S1 for details). B. The density of pericytes (number of pericytes in a given tissue volume) is increased in AD cases (one‐way ANOVA; *P = 0.00058). C and D. The linear density of pericytes (pericyte number in a given capillary segment; C; one‐way ANOVA; P = 0.46) as well as the vascular coverage (D; Mann–Whitney U test; P = 0.78) did not change in AD cases compared to controls. Scale bar: 50 µm. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
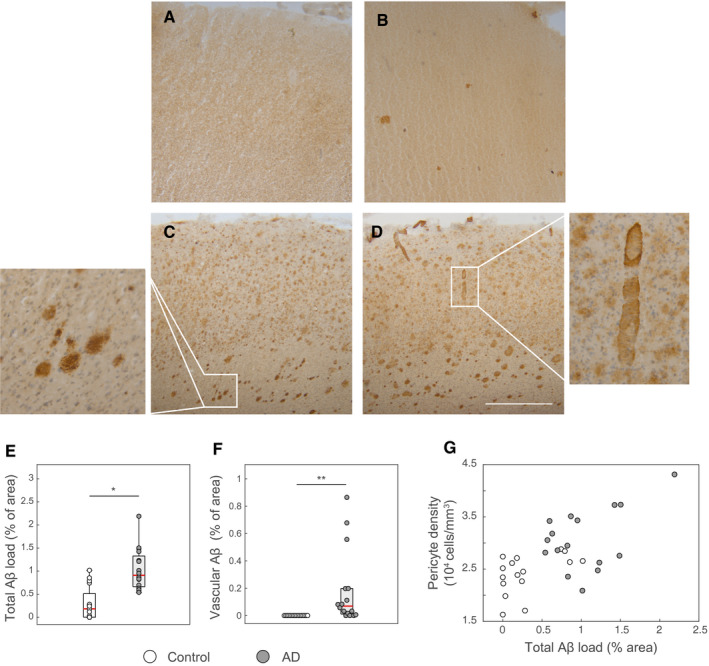
Correlation of pericyte density with AD pathology. A‐D. Representative images showing immunoreactivity for β‐amyloid in two non‐demented cases (A and B) and two AD cases (C and D). Non‐demented controls showed variable degree of β‐amyloid deposition. The insets in C and D demonstrate plaques and parenchymal vascular amyloid deposition, respectively. E and F. Boxplots showing the fraction of the area occupied by total Aβ immunoreactivity (E) or vessel‐associated Aβ immunoreactivity (F) in non‐demented control and AD cases. *P = 0.00015, **P = 0.000016; Mann–Whitney U Test. G. Pericyte density correlated with the total Aβ load (r s = 0.54, P = 0.0012, Spearman's rank‐order correlation. Scale bar: 100 µm.
We stained for the basement marker laminin to measure vascular density (Figure 3A,B). The microvascular network of AD subjects displayed a 24% increase in capillary density compared to controls (controls: 291 ± 39 mm/mm3 vs. AD: 361 ± 48 mm/mm3; P = 0.000079; one‐way‐ANOVA; Figure 3C). In contrast, no significant differences between AD cases and controls was detected for the densities of intermediate sized vessels (controls: 72 ± 23 vs. AD: 73 ± 27 mm/mm3; Figure 3C) or large vessels (controls: 0.55 ± 0.25 vs. AD: 0.43 ± 0.30 mm/mm3; P = 0.24; one‐way‐ANOVA; Figure 3C). The capillary density correlated with the total Aβ burden (r s = 0.6419, P < 0.0001; Spearman's rank‐order correlation; Figure 3D). In AD subjects, vascular density of intermediate vessels was significantly higher in subjects with a score of 3 when compared with those with a score of 2 (median of 47.6 vs. 87.9, P < 0.005, Mann–Whitney U Test). Otherwise, the density of vessels did not vary significantly between different stages of the four‐tier NFT or neuritic plaque (CERAD) classifications (Supporting Figures S6 and S7).
Figure 3.
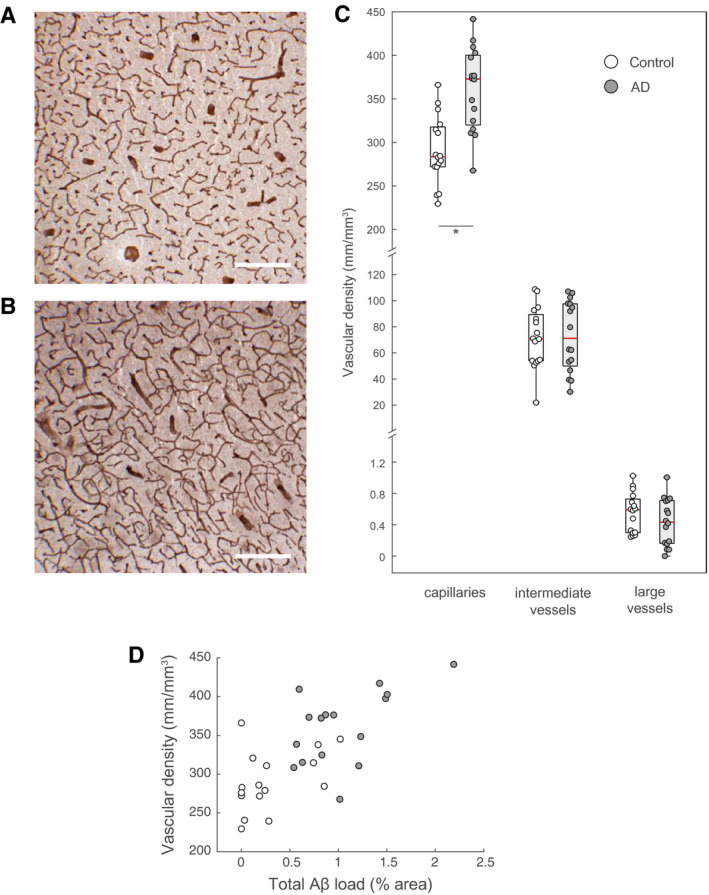
Vascular density changes in AD. A and B. Representative immunohistochemical stains for laminin showing changes in capillary density in a non‐demented control (A) versus an AD case (B). The discontinuous pattern of staining is owed to the sectioning of fresh‐frozen samples. C. The density of capillaries (defined as vessels with diameters ≤9 µm) is significantly increased in AD cases compared to controls (*P = 0.000079, one‐way ANOVA). The density of intermediate and large vessels (including arterioles and veins) remains constant (P = 0.9 and 0.24, respectively; one‐way ANOVA). D. Vascular density correlated with the total Aβ load (r s = 0.6419, P = 0.000075, Spearman's rank‐order correlation). Scale bars: 250 µm.
Some of the non‐demented control cases showed Aβ‐immunoreactive plaques (Figure 2G). We separated the control cases into subjects with negligible Aβ‐immunoreactivity (< 0.1% area fraction; n = 5), and non‐demented subjects with positive Aβ immunoreactivity (Aβ‐immunoreactive plaques in> 0.1%; n = 9). The analysis of pericyte and capillary densities showed significant differences between these two groups and the AD group, while no difference was found between control and non‐demented subjects with AD‐like pathology for any other metric studied (Supporting Figure S8). No differences were found for pericyte linear density, intermediate vascular or large vascular densities (Supporting Figure S8). These results suggest that clinically patent and neuropathologically confirmed AD, and not merely the presence of Aβ‐immunoreactive plaques, is associated with the observed increases in pericyte and capillary density. The APOE genotype did not influence Aβ plaque load, pericyte or vascular density in neither the control nor the AD groups, although a trend to a higher amount of Aβ vascular deposition was found in carriers of the APOE4 allele (Supporting Figures S9 and S10).
Analysis of cleared samples
Upon clearing, double‐labeling and two‐photon imaging of brain tissue samples revealed Aβ deposits as well as the PDGFRβ‐immunoreactive vascular network (Figure 4A,B). Pericyte‐containing vessels were detected in both controls and in AD cases along all cortical layers, often in close apposition to Aβ plaques (Figure 4B). The PDGFRβ‐immunoreactive mural cell lining of arteries, capillaries and veins was clearly rendered (Figure 4C,E). The Supporting Videos S1 and S2 demonstrate the staining and imaging of whole tissue fragments. The volume occupied by Aβ deposition was significantly higher in AD cases than controls, although we encountered some degree of Aβ immunoreactivity in one control case (median 0% in control vs. 0.6% in AD cases, P < 0.005, Mann–Whitney U Test, Supporting Figure S11A). Aβ deposition did not vary significantly in dependence of the APOE genotype (Supporting Figure S11B).
Figure 4.
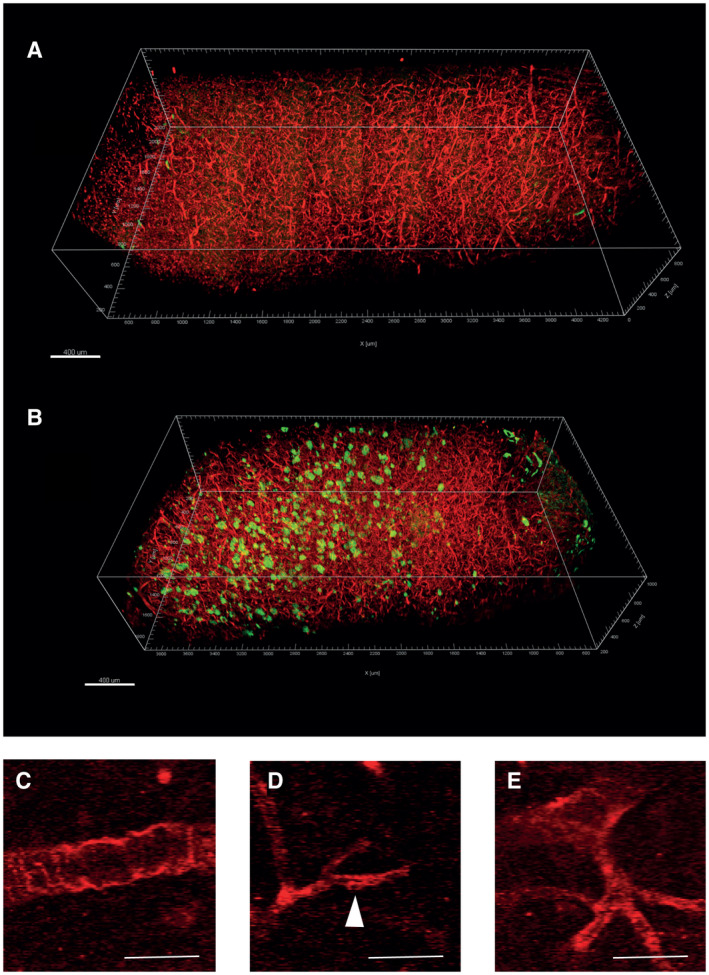
Pericyte vascular network in cleared brain samples. A and B. 3D‐rendering of stitched and processed two‐photon images from two cleared specimens, one non‐demented control case (A) and one AD case (B) showing PDGFRβ‐immunoreactive (red) and Aβ‐immunoreactive (green) signals. The spurious green signal in the surface of A represent contaminating particles of the tissue piece. The density of PDGFRβ‐immunoreactive structures is similar between the control and AD tissue. C–E. raw scanning images of the signal corresponding to PDGFRβ‐immunoreactivity. Under the imaging protocol employed for the sampling of large tissue volumes, two‐photon imaging did not resolve the fine cellular structures of pericytes. However, the staining revealed unambiguously the lining of PDGFRβ‐immunoreactive mural cells in arterioles, capillaries and venules. Scale bars: 400 µm.
For the morphometric analysis, we first produced a skeleton of the PDGFRβ‐immunoreactive vascular network, which comprised pericyte‐containing capillaries in addition to mural cell‐containing larger arterioles and venules. (Figure 5A,D). The linear density of PDGFRβ‐immunoreactive vessels as well as the density of junctions were calculated as independent morphometric endpoints. Further, the mean parenchymal distance to the next vessel and the tortuosity were calculated. The values of the linear vascular density matched with previously published results (29) and the stereological measurements of laminin‐immunoreactive capillaries reported above (Figure 3). We did not find statistically significant differences of PDGFRβ‐immunoreactive vascular density, mean parenchymal distance to the next vessel or tortuosity between AD cases and controls (Figure 5E,G). Also, the PDGFRβ‐immunoreactive vascular density did not change significantly among APOE genotypes, Aβ plaque scores, NFT (Braak) stages or neuritic plaque (CERAD) scores (Supporting Figure S12).
Figure 5.
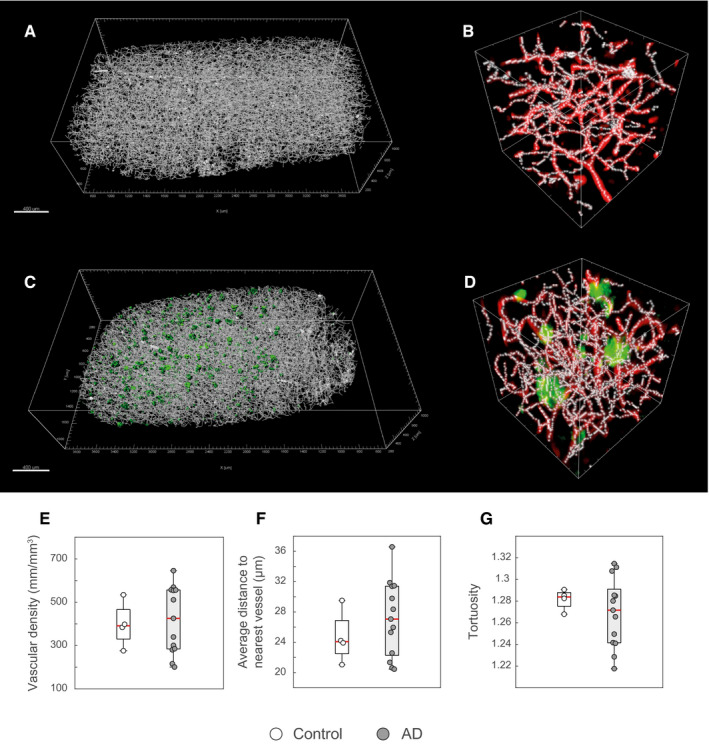
Morphometric analysis of the pericyte vascular network in cleared samples. A–D. 3D‐rendering of the skeletonized PDGFRβ‐immunoreactive vascular network of a non‐demented control (A and B, same as in Figure 4A) and an AD case (C,D, same as in Figure 4 B). The green surfaces in C represent thresholded Aβ‐immunoreactive plaques. The boxes in B and D display the skeleton along the PDGFRβ‐ or Aβ‐signals (box comprises 75 µm in each dimension). E–G. No differences between AD cases and non‐demented controls were found for neither PDGFRβ‐immunoreactive vascular network (E; Mann–Whitney U test; P = 1) nor the average distance in the parenchyma to the nearest vessel (F; Mann–Whitney U test; P = 0.83) or the tortuosity of the vascular structures (G; Mann–Whitney U test; P = 0.50). Scale bars: 400 µm.
Collectively, our results show a preservation of capillary pericytes in the frontal cortex of AD.
Discussion
Located in capillaries throughout the CNS at the interface between the neural tissue and the circulatory system, pericytes are involved in the development of the brain's vascular system and the regulation of the BBB (1). Pericytes appear to be contractile under certain conditions, and a population of pericyte‐like mural cells in the arteriolar vasculature contributes to the control of blood flow in the CNS (16, 20, 24, 45). Pericyte function thus determines the maintenance of a homeostatic environment in the CNS. An adequate blood supply and the isolation of the neural tissue from blood electrolytes, proteins and inflammatory mediators are necessary conditions to ensure proper brain function. Experimental conditions which target the brain's pericyte population, either by interfering with PDGFRβ signaling (2, 5, 14, 31) or by iDTR‐mediated ablation of pericytes (40), result in CNS pathology (disturbances of the BBB and CNS blood flow, inflammation and neurodegeneration) and are in some cases associated with cognitive dysfunction or even death.
The effect of CNS pathology on the pericyte population is not fully known. Ischemia leads to a rapid demise of pericytes (17, 20). Loss of pericytes has also been described in neurodegenerative disorders, particularly in AD (21, 50). Some ultrastructural studies showed increased signs of pericyte degeneration in post‐mortem AD brains, such as the accumulation of intracellular lipofuscin‐like material and damaged mitochondria (3). However, other studies did not confirm pericyte degeneration (15, 52). Aβ is internalized by pericytes and has toxic effects on cultured pericytes (9, 57, 62, 64). These observations have led to the hypothesis that pericyte loss might be a major factor in the cascade leading to neurodegeneration in AD (21, 50).
Here, we used state‐of‐the‐art techniques, such as stereology which is considered the gold standard in morphometric studies, as well as novel tissue clearing and two‐photon imaging techniques to characterize the presence of pericytes in two independent samples of AD brains and controls. Our results clearly demonstrate that pericytes are not diminished in the frontal cortex of individuals with clinically diagnosed AD. These findings are in line with previous ultrastructural studies of pericytes in the AD brain (15, 52). However, our results contradict more recent reports suggesting that capillary pericytes are lost in AD, in particular in dependence of the APOE genotype (21, 50).
This discrepancy could be explained by the different techniques employed. We did not produce measurements of the area covered by pericyte or endothelial markers above a certain intensity threshold, as in (21, 50), because this measure is influenced by the amount of expressed proteins, which is subject to regulation in disease. For instance, the Ulex europaeus agglutinin I used to reveal endothelia in (21, 50) binds to fucosylated glycoproteins, which are effectors of leukocyte adhesion and could be upregulated during endothelial activation (30). It is not well established how AD affects the expression levels of pericyte markers, but in vitro studies show that pericytes shed PDGFRβ into the medium upon hypoxia or exposure to β‐amyloid, which may explain why lower levels of PDGFRβ protein are found in AD brains (35, 47), even in the absence of pericyte degeneration. In our stereological study, pericyte numbers were assessed visually by an investigator blinded to the groups, who identified pericytes and vessels independently of the intensity of the immunofluorescence signal. Similarly, in the cleared tissue analysis, skeletonization of the PDGFRβ‐immunoreactive signal yields measurements that are largely independent of the cross‐sectional area of positive immunoreactivity.
In line with previous results (4, 22, 34, 44), our stereological analysis demonstrates an increased capillary density in the AD cortex. Increased capillary density has also been described in the brains of different AD mouse models by chronic intravital imaging (6). Since the cortex is subject to atrophy in AD, the increase in capillary density could be explained by the persistence of the vascular network in a context of parenchymal volume shrinking. However, we did not detect increased densities of intermediate sized and large vessels, which is hard to reconcile with a passive effect of atrophy. The increased density of capillaries is more likely to result from angiogenesis, since angiogenic genes are upregulated in AD (6, 42). Upregulation of ANGPTL4 expression has been associated to angiogenesis and increased capillary density in in patients with capillary CAA (11). The intrathecal concentration of vascular endothelial growth factor (VEGF), an angiogenic molecule, is increased in patients with AD and might serve as a biomarker (53, 56). The rise in angiogenesis and increase in capillary density could reflect a regulatory response to reduced cerebral blood perfusion or an effect of brain inflammation, among other potential mechanisms (59).
One limitation of our study is the focus on only one brain region, namely the frontal cortex. Although amyloid deposits are found early on in the frontal cortex of AD cases (55), neurofibrillary tangles and cortical atrophy occur first and predominantly in limbic structures, such as the hippocampus or the entorhinal cortex (8, 48). More advanced AD pathology, including atrophy, might be associated with a different vascular phenotype. However, our study recapitulates the increase in capillary density found by others in the hippocampus or temporal cortex (4, 10, 44), showing that vascular remodeling is a common feature of AD among different brain regions, including the frontal cortex.
It is important to note that our results do not imply that pericyte function is intact in AD. CAA affects parenchymal vessels in about one‐third of AD cases (23) and contributes to microhemorrhages and infarcts, having thus a direct impact on vascular function (19, 33). Aβ oligomers constrict pericytes in capillaries (41). Vascular amyloid deposition may lead to immune reaction and vasculitis (49), and pericytes actively participate in neuroinflammatory responses through the production of a plethora of chemokines and cytokines (46).
In conclusion, our study demonstrates that pericyte loss is not a general feature of AD. Methods to isolate pericytes, endothelia, and other vascular cells in the neurovascular niche are rapidly evolving (60) and will shed light on functional changes of pericytes and their contribution to the cascade of neurodegeneration in AD.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Fig S1
Figure S1. Methods for stereological counting of pericyte density. Frozen samples of the middle frontal gyrus were provided by the Neurological Foundation Douglas Human Brain Bank in New Zealand. About 60 µm thick slides were stained for PDGFRβ and laminin expression. For assessment of the pericyte density (pericytes in a given tissue volume), we counted pericyte bodies with the optical fractionator. The space ball method was employed for measuring the vascular density (length of vascular segments in a given volume). The pericyte linear density (number of pericytes in a given vascular segment) was inferred from the above measurements.
Fig S2
Figure S2. Image processing and analysis of the cleared brain tissue samples. The cleared samples were imaged with the two‐photon microscope to obtain three different images, corresponding to the Alexa‐647 signal (PDGFRβ), the Alexa‐488 signal (Aβ) or tissue autofluorescence, largely due to lipofuscin deposits in the aged brain tissue. Using Imaris “Channel Extraction” color deconvolution algorithm, the autofluorescence) was subtracted from the Aβ‐immunoreactive image. For PDGFRβ detection, a far‐red fluorochrome was employed, whose emission spectrum did not overlap with the autofluorescence. The PDGFRβ‐immunoreactive signal was enhanced using a rolling contrast enhancing algorithm (CLAHE) using ImageJ. Both PDGFRβ‐immunoreactive and Aβ‐immunoreactive signals were segmented. For the analysis of the vascular network, the segmented images were skeletonized using a custom MATLAB Imaris plugin based on the Skel2Graph3D MATLAB function (28).
Fig S3
Figure S3. Neuropathology scores of donors of cleared brain tissue samples. Boxplots showing A the Aβ plaque scores (*P = 0.0008, Mann–Whitney U test), B NFT (Braak) stages (*P = 0.0008, Mann–Whitney U test), C neuritic plaque (CERAD) scores (*P = 0.0017, Mann–Whitney U test) and D the brain weight (P = 0.12, Mann–Whitney U test) of donors of cleared brain tissue.
Fig S4
Figure S4. PDGFRβ‐CD31 double staining. Maximum projection (A) as well as single confocal slice and orthogonal views (B) of a confocal stack of frontal brain tissue double stained for PDGFRβ (pericytes) and CD31 (endothelial cells). The images demonstrate the distinct PDGFRβ‐positive immunoreactivity of pericytes lining endothelia in capillaries. Scalebar: 50 µm.
Fig S5
Figure S5. Effect of NFT and CERAD scores on pericyte density in AD subjects. A,B Boxplots represent pericyte density, pericyte linear density, pericyte area coverage of AD subjects stratified along the different scores of the four‐tier NFT classification (A) or the neurofibrillary plaque (CERAD) score (B).The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons. n.p. = not present (no matching cases).
Fig S6
Figure S6. Effect of NFT score on vascular density in AD subjects. Boxplots represent vascular densities of capillaries, intermediate or large vessels of AD subjects stratified along the different scores of the four‐tier NFT classification. The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons. n.p. = not present (no matching cases).
Fig S7
Figure S7. Effect of CERAD score on vascular density in AD subjects. Boxplots represent vascular densities of capillaries, intermediate or large vessels of AD subjects stratified along the different CERAD scores. The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons. Intermediate vessels display a higher density in AD subjects with CERAD score of 3 compared to 2. n.p. = not present (no matching cases).
Fig S8
Figure S8. Analysis of non‐demented subjects with AD‐like pathology. Boxplots display pericyte density (A), pericyte linear density (B), pericyte area coverage (C) and vascular density (D) in control subjects with negligible Aβ immunoreactivity, non‐demented subjects with Aβ immunoreactivity and AD cases. The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons.
Fig S9
Figure S9. Effect of APOE genotype on total and vascular Aβ load. Boxplots display total (A) or vascular Aβ load (B) of subjects stratified along the APOE genotype for the control and AD groups. Numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons (A) or of the Mann–Whitney U Test (B).
Fig S10
Figure S10. Effect of APOE genotype on pericyte and vascular densities. Boxplots display pericyte density (A), pericyte linear density (B), pericyte area coverage (C) and vascular density (D) of subjects stratified along the APOE genotype for the control and AD groups. Numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons.
Fig S11
Figure S11. Aβ load in cleared brain tissue samples. A Boxplots display the fraction of tissue volume occupied by Aβ immunoreactivity (Aβ load) in control and AD subjects. *P = 0.0017, Mann–Whitney U Test. B Aβ load in subjects stratified by clinical diagnosis and APOE genotype. The number represents the P‐values of the of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons.
Fig S12
Figure S12. PDGFRβ‐immunoreactive vascular density contrasted by APOE Genotype and neuropathological scores. Boxplots display the density of PDGFRβ‐immunoreactive vessels in control and AD subjects stratified by APOE genotype (A), Aβ plaque score (B), NFT stage (Braak) stage (C) or neuritic plaque (CERAD) score (D). No significant differences were found (A: P = 0.58, P = 0.67, P = 0.51, P = 0.50, respectively; Kruskal–Wallis test). n.p. = not present (no matching cases).
Supplementary Material
Movie S1. 3D‐reconstruction of the cortical volume of a non‐demented control, showing the orthogonal slicing of the sample. PDGFRβ‐immunoreactivity is displayed in red pseudocolor. Green‐pseudocolored signal at the surface of the piece of tissue represents autofluorescent debris. Scale bar: 500 µm.
Supplementary Material
Movie S2. 3D‐reconstruction of the cortical volume of an AD case, showing the orthogonal slicing of the sample. PDGFRβ‐immunoreactivity is displayed in red, Aβ plaques in green pseudocolor. Green‐pseudocolored signal at the surface of the piece of tissue represents autofluorescent debris. Scale bar: 500 µm.
Acknowledgments
This work was supported by grants from the German Research Foundation (SFB/TRR 167). Open access funding enabled and organized by Projekt DEAL.
[Correction added on October 1, 2020, after first online publication: Projekt Deal funding statement has been added.]
Contributor Information
Francisco Fernandez‐Klett, Email: francisco.fernandez@uk-halle.de.
Josef Priller, Email: josef.priller@charite.de.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding authors. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Armulik A, Genové G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215. [DOI] [PubMed] [Google Scholar]
- 2. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C et al (2010) Pericytes regulate the blood‐brain barrier. Nature 468:557–561. [DOI] [PubMed] [Google Scholar]
- 3. Baloyannis SJ, Baloyannis IS (2012) The vascular factor in Alzheimer's disease: a study in Golgi technique and electron microscopy. J Neurol Sci 322:117–121. [DOI] [PubMed] [Google Scholar]
- 4. Bell MA, Ball MJ (1981) Morphometric comparison of hippocampal microvasculature in ageing and demented people: diameters and densities. Acta Neuropathol 53:299–318. [DOI] [PubMed] [Google Scholar]
- 5. Bell RD, Winkler EA, Sagare AP, Singh I, Larue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark Tv 2018) Tau induces blood vessel abnormalities and angiogenesis‐related gene expression in P301L transgenic mice and human Alzheimer's disease. Proc Natl Acad Sci U S A 115:E1289–E1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathologica 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 9. Bruinsma IB, Wilhelmus MMM, Kox M, Veerhuis R, de Waal RMW, Verbeek MM (2010) Apolipoprotein E protects cultured pericytes and astrocytes from D‐Abeta(1–40)‐mediated cell death. Brain Res 1315:169–180. [DOI] [PubMed] [Google Scholar]
- 10. Burke MJC, Nelson L, Slade JY, Oakley AE, Khundakar AA, Kalaria RN (2014) Morphometry of the hippocampal microvasculature in post‐stroke and age‐related dementias. Neuropathol Appl Neurobiol 40:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chakraborty A, Kamermans A, van het Hof B, Castricum K, Aanhane E, van Horssen J et al (2018) Angiopoietin like‐4 as a novel vascular mediator in capillary cerebral amyloid angiopathy. Brain 141:3377–3388. [DOI] [PubMed] [Google Scholar]
- 12. Chung K, Wallace J, Kim S‐Y, Kalyanasundaram S, Andalman AS, Davidson TJ et al (2013) Structural and molecular interrogation of intact biological systems. Nature 497:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Costantini I, Ghobril J‐P, Di Giovanna AP, Mascaro ALA, Silvestri L, Müllenbroich MC et al (2015) A versatile clearing agent for multi‐modal brain imaging. Sci Rep 5:168–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood‐brain barrier integrity during embryogenesis. Nature 468:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farkas E, De Jong GI, de Vos RA, Jansen Steur EN, Luiten PG (2000) Pathological features of cerebral cortical capillaries are doubled in Alzheimer's disease and Parkinson's disease. Acta Neuropathol 100:395–402. [DOI] [PubMed] [Google Scholar]
- 16. Fernández‐Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U (2010) Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci U S A 107:22290–22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernández‐Klett F, Potas JRJR, Hilpert D, Blazej K, Radke J, Huck J et al (2013) Early loss of pericytes and perivascular stromal cell‐induced scar formation after stroke. J Cereb Blood Flow Metab 33:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gioia L, Vogt LJ, Freeman WM, Flood A, Vogt BA, Vrana KE (1998) PCR‐based apolipoprotein E genotype analysis from archival fixed brain. J Neurosci Methods 80:209–214. [DOI] [PubMed] [Google Scholar]
- 19. Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al‐Shahi Salman R, Warach S et al (2009) Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA et al (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV (2015) Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J Cereb Blood Flow Metab 36:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hase Y, Ding R, Harrison G, Hawthorne E, King A, Gettings S et al (2019) White matter capillaries in vascular and neurodegenerative dementias. Acta Neuropathol Com 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hecht M, Krämer LM, von Arnim CAF, Otto M, Thal DR (2018) Capillary cerebral amyloid angiopathy in Alzheimer's disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol 135:681–694. [DOI] [PubMed] [Google Scholar]
- 24. Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, Grutzendler J (2015) Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87:95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Howard V, Reed M (2004) Unbiased Stereology. Oxon, UK: Garland Science/BIOS Scientific. [Google Scholar]
- 26. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al (2012) National Institute on Aging–Alzheimer“s Association guidelines for the neuropathologic assessment of Alzheimer”s disease. Alzheimer's & Dementia 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keuker JI, Vollmann‐Honsdorf GK, Fuchs E (2001) How to use the optical fractionator: an example based on the estimation of neurons in the hippocampal CA1 and CA3 regions of tree shrews. Brain Res Protoc 7:211–221. [DOI] [PubMed] [Google Scholar]
- 28. Kollmannsberger P, Kerschnitzki M, Repp F, Wagermaier W, Weinkamer R, Fratzl P (2017) The small world of osteocytes: connectomics of the lacuno‐canalicular network in bone. New J Phys 19:073019. [Google Scholar]
- 29. Kreczmanski P, Schmidt‐Kastner R, Heinsen H, Steinbusch HWM, Hof PR, Schmitz C (2005) Stereological studies of capillary length density in the frontal cortex of schizophrenics. Acta Neuropathol 109:510–518. [DOI] [PubMed] [Google Scholar]
- 30. Li J, Hsu H‐C, Mountz JD, Allen JG (2018) Unmasking Fucosylation: from Cell Adhesion to Immune System Regulation and Diseases. Cell Chemical Biology 25:499–512. [DOI] [PubMed] [Google Scholar]
- 31. Lindahl P, Johansson BR, Levéen PL, Betsholtz C. (1997) Pericyte loss and microaneurism formation in PDGF‐B-deficient mice. Science 277:242–245. [DOI] [PubMed] [Google Scholar]
- 32. Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC et al (2018) Blood‐brain barrier‐associated pericytes internalize and clear aggregated amyloid‐β42 by LRP1‐dependent apolipoprotein E isoform‐specific mechanism. Mol Neurodegener 13:57–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mandybur TI (1986) Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol 45:79–90. [PubMed] [Google Scholar]
- 34. Meier‐Ruge W, Ulrich J, Stähelin HB (1985) Morphometric investigation of nerve cells, neuropil and senile plaques in senile dementia of the Alzheimer type. Arch Gerontol Geriatr 4:219–229. [DOI] [PubMed] [Google Scholar]
- 35. Miners JS, Schulz I, Love S (2017) Differing associations between Aβ accumulation, hypoperfusion, blood–brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer's disease. J Cereb Blood Flow Metab 38:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer“s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer”s disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 37. Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A et al (2020) APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D et al (2018) Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med 24:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39. Mouton PR, Gokhale AM, Ward NL, West MJ (2002) Stereological length estimation using spherical probes. J Microsc 206:54–64. [DOI] [PubMed] [Google Scholar]
- 40. Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT et al (2019) Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci 22:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z et al (2019) Amyloid β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science 365:eaav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pogue AI, Lukiw WJ (2004) Angiogenic signaling in Alzheimer's disease. NeuroReport 15:1507–1510. [DOI] [PubMed] [Google Scholar]
- 43. Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362:329–344. [DOI] [PubMed] [Google Scholar]
- 44. Richard E, van Gool WA, Hoozemans JJM, van Haastert ES, Eikelenboom P, Rozemuller AJM, van de Berg WDJ (2010) Morphometric changes in the cortical microvascular network in Alzheimer's disease. J Alzheimers Dis 22:811–818. [DOI] [PubMed] [Google Scholar]
- 45. Rungta RL, Chaigneau E, Osmanski B‐F, Charpak S (2018) Vascular compartmentalization of functional hyperemia from the synapse to the pia. Neuron 99:362–375.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rustenhoven J, Jansson D, Smyth LC, Dragunow M (2017) Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci 38:291–304. [DOI] [PubMed] [Google Scholar]
- 47. Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV (2013) Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun 4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC (2002) Mapping the evolution of regional atrophy in Alzheimer's disease: Unbiased analysis of fluid‐registered serial MRI. Proc Natl Acad Sci U S A 99:4703–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J et al (2005) Abeta‐related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128:500–515. [DOI] [PubMed] [Google Scholar]
- 50. Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV (2012) Deficiency in mural vascular cells coincides with blood‐brain barrier disruption in Alzheimer's disease. Brain Pathol 23:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sims DE (1986) The pericyte—a review. Tissue Cell 18:153–174. [DOI] [PubMed] [Google Scholar]
- 52. Stewart PA, Hayakawa K, Akers MA, Vinters HV (1992) A morphometric study of the blood‐brain barrier in Alzheimer's disease. Lab Invest 67:734–742. [PubMed] [Google Scholar]
- 53. Tarkowski E, Issa R, Sjögren M, Wallin A, Blennow K, Tarkowski A, Kumar P (2002) Increased intrathecal levels of the angiogenic factors VEGF and TGF‐β in Alzheimer's disease and vascular dementia. Neurobiol Aging 23:237–243. [DOI] [PubMed] [Google Scholar]
- 54. Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–1301. [DOI] [PubMed] [Google Scholar]
- 55. Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 56. Thirumangalakudi L, Samany PG, Owoso A, Wiskar B, Grammas P (2006) Angiogenic proteins are expressed by brain blood vessels in Alzheimer's disease. J Alzheimers Dis 10:111–118. [DOI] [PubMed] [Google Scholar]
- 57. Timmer NM, Schirris TJJ, Bruinsma IB, Otte‐Höller I, van Kuppevelt TH, de Waal RMW, Verbeek MM (2010) Aggregation and cytotoxic properties towards cultured cerebrovascular cells of Dutch‐mutated Abeta40 (DAbeta(1–40)) are modulated by sulfate moieties of heparin. Neurosci Res 66:380–389. [DOI] [PubMed] [Google Scholar]
- 58. Tomer R, Ye L, Hsueh B, Deisseroth K (2014) Advanced CLARITY for rapid and high‐resolution imaging of intact tissues. Nat Protoc 9:1682–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vagnucci AH Jr, Li WW (2003) Alzheimer's disease and angiogenesis. Lancet 361:605–608. [DOI] [PubMed] [Google Scholar]
- 60. Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F et al (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480. [DOI] [PubMed] [Google Scholar]
- 61. Vanlandewijck M, Lebouvier T, Andaloussi Mäe M, Nahar K, Hornemann S, Kenkel D et al (2015) Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS One 10:e0143407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Verbeek MM, Van Nostrand WE, Otte‐Holler I, Wesseling P, de Waal RM (2000) Amyloid‐beta-induced degeneration of human brain pericytes is dependent on the apolipoprotein E genotype. Ann N Y Acad Sci 903:187–199. [DOI] [PubMed] [Google Scholar]
- 63. Vinters HV, Secor DL, Read SL, Frazee JG, Tomiyasu U, Stanley TM et al (2009) Microvasculature in brain biopsy specimens from patients with Alzheimer's disease: an immunohistochemical and ultrastructural study. Ultrastruct Pathol 18:333–348. [DOI] [PubMed] [Google Scholar]
- 64. Wilhelmus MMM, Otte‐Höller I, Davis J, van Nostrand WE, de Waal RMW, Verbeek MM (2005) Apolipoprotein E genotype regulates amyloid‐beta cytotoxicity. J Neurosci 25:3621–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV (2012) Blood–spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol 125:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Figure S1. Methods for stereological counting of pericyte density. Frozen samples of the middle frontal gyrus were provided by the Neurological Foundation Douglas Human Brain Bank in New Zealand. About 60 µm thick slides were stained for PDGFRβ and laminin expression. For assessment of the pericyte density (pericytes in a given tissue volume), we counted pericyte bodies with the optical fractionator. The space ball method was employed for measuring the vascular density (length of vascular segments in a given volume). The pericyte linear density (number of pericytes in a given vascular segment) was inferred from the above measurements.
Fig S2
Figure S2. Image processing and analysis of the cleared brain tissue samples. The cleared samples were imaged with the two‐photon microscope to obtain three different images, corresponding to the Alexa‐647 signal (PDGFRβ), the Alexa‐488 signal (Aβ) or tissue autofluorescence, largely due to lipofuscin deposits in the aged brain tissue. Using Imaris “Channel Extraction” color deconvolution algorithm, the autofluorescence) was subtracted from the Aβ‐immunoreactive image. For PDGFRβ detection, a far‐red fluorochrome was employed, whose emission spectrum did not overlap with the autofluorescence. The PDGFRβ‐immunoreactive signal was enhanced using a rolling contrast enhancing algorithm (CLAHE) using ImageJ. Both PDGFRβ‐immunoreactive and Aβ‐immunoreactive signals were segmented. For the analysis of the vascular network, the segmented images were skeletonized using a custom MATLAB Imaris plugin based on the Skel2Graph3D MATLAB function (28).
Fig S3
Figure S3. Neuropathology scores of donors of cleared brain tissue samples. Boxplots showing A the Aβ plaque scores (*P = 0.0008, Mann–Whitney U test), B NFT (Braak) stages (*P = 0.0008, Mann–Whitney U test), C neuritic plaque (CERAD) scores (*P = 0.0017, Mann–Whitney U test) and D the brain weight (P = 0.12, Mann–Whitney U test) of donors of cleared brain tissue.
Fig S4
Figure S4. PDGFRβ‐CD31 double staining. Maximum projection (A) as well as single confocal slice and orthogonal views (B) of a confocal stack of frontal brain tissue double stained for PDGFRβ (pericytes) and CD31 (endothelial cells). The images demonstrate the distinct PDGFRβ‐positive immunoreactivity of pericytes lining endothelia in capillaries. Scalebar: 50 µm.
Fig S5
Figure S5. Effect of NFT and CERAD scores on pericyte density in AD subjects. A,B Boxplots represent pericyte density, pericyte linear density, pericyte area coverage of AD subjects stratified along the different scores of the four‐tier NFT classification (A) or the neurofibrillary plaque (CERAD) score (B).The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons. n.p. = not present (no matching cases).
Fig S6
Figure S6. Effect of NFT score on vascular density in AD subjects. Boxplots represent vascular densities of capillaries, intermediate or large vessels of AD subjects stratified along the different scores of the four‐tier NFT classification. The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons. n.p. = not present (no matching cases).
Fig S7
Figure S7. Effect of CERAD score on vascular density in AD subjects. Boxplots represent vascular densities of capillaries, intermediate or large vessels of AD subjects stratified along the different CERAD scores. The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons. Intermediate vessels display a higher density in AD subjects with CERAD score of 3 compared to 2. n.p. = not present (no matching cases).
Fig S8
Figure S8. Analysis of non‐demented subjects with AD‐like pathology. Boxplots display pericyte density (A), pericyte linear density (B), pericyte area coverage (C) and vascular density (D) in control subjects with negligible Aβ immunoreactivity, non‐demented subjects with Aβ immunoreactivity and AD cases. The numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons.
Fig S9
Figure S9. Effect of APOE genotype on total and vascular Aβ load. Boxplots display total (A) or vascular Aβ load (B) of subjects stratified along the APOE genotype for the control and AD groups. Numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons (A) or of the Mann–Whitney U Test (B).
Fig S10
Figure S10. Effect of APOE genotype on pericyte and vascular densities. Boxplots display pericyte density (A), pericyte linear density (B), pericyte area coverage (C) and vascular density (D) of subjects stratified along the APOE genotype for the control and AD groups. Numbers represent the P‐values of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons.
Fig S11
Figure S11. Aβ load in cleared brain tissue samples. A Boxplots display the fraction of tissue volume occupied by Aβ immunoreactivity (Aβ load) in control and AD subjects. *P = 0.0017, Mann–Whitney U Test. B Aβ load in subjects stratified by clinical diagnosis and APOE genotype. The number represents the P‐values of the of the Kruskal–Wallis test followed by post hoc Dunn‐Šidák correction for multiple comparisons.
Fig S12
Figure S12. PDGFRβ‐immunoreactive vascular density contrasted by APOE Genotype and neuropathological scores. Boxplots display the density of PDGFRβ‐immunoreactive vessels in control and AD subjects stratified by APOE genotype (A), Aβ plaque score (B), NFT stage (Braak) stage (C) or neuritic plaque (CERAD) score (D). No significant differences were found (A: P = 0.58, P = 0.67, P = 0.51, P = 0.50, respectively; Kruskal–Wallis test). n.p. = not present (no matching cases).
Supplementary Material
Movie S1. 3D‐reconstruction of the cortical volume of a non‐demented control, showing the orthogonal slicing of the sample. PDGFRβ‐immunoreactivity is displayed in red pseudocolor. Green‐pseudocolored signal at the surface of the piece of tissue represents autofluorescent debris. Scale bar: 500 µm.
Supplementary Material
Movie S2. 3D‐reconstruction of the cortical volume of an AD case, showing the orthogonal slicing of the sample. PDGFRβ‐immunoreactivity is displayed in red, Aβ plaques in green pseudocolor. Green‐pseudocolored signal at the surface of the piece of tissue represents autofluorescent debris. Scale bar: 500 µm.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding authors. The data are not publicly available due to privacy or ethical restrictions.