

Abstract
Medulloblastoma (MB) represents a spectrum of biologically and clinically distinct entities. Initially described histopathologically as a small, round blue cell tumor arising in the cerebellum, MB has emerged as a paradigm for molecular classification in cancer. Recent advances in genomic, transcriptomic and epigenomic profiling of MB have further refined molecular classification and complemented conventional histopathological diagnosis. Herein, we review the main clinical and molecular features of the four consensus subgroups of MB (WNT, SHH, Group 3 and Group 4). We also highlight hereditary predisposition syndromes associated with increased risk of MB. Finally, we explore advances in the classification of the consensus molecular groups while also presenting cutting‐edge frontiers in identifying intratumoral heterogeneity and cellular origins of MB.
Keywords: medulloblastoma, genomics, pediatrics, molecular pathology
Introduction
Medulloblastoma (MB) is the most common malignant pediatric brain tumor 70. Arising from the cerebellum, the median age of diagnosis is approximately 6 years of age. Clinical manifestations of MB, often due to raised intracranial pressure secondary to hydrocephalus, may be non‐specific and include headaches, nausea, early morning emesis and poor academic performance. More specific symptoms, such as ataxia, cranial nerve palsies and visual problems, may be attributable to direct mass effect and/or raised intracranial pressure. Diagnostic work‐up includes magnetic resonance imaging and cytologic examination of the cerebrospinal fluid. Clinical risk stratification integrates the aforementioned work‐up in the Chang staging system 10. Treatment involves neurosurgical resection, irradiation (usually restricted to patients older than 3 years of age) and cytotoxic chemotherapy. The 5‐year overall survival of MB hovers around 75%, but many survivors face serious cognitive, endocrine and psychosocial sequalae 29, 43, 72. Outcomes of treatment, as implemented according to conventional clinicopathologic risk stratification, varies according to risk group, with average‐risk disease defined in patients older than 3 years with gross total resection and no metastasis at diagnosis (5‐year overall survival > 80%) and high‐risk disease defined in patients younger than 3 years with subtotal resection at metastasis at diagnosis (5‐year overall survival < 70%) 29, 30, 43, 71, 72.
As an embryonal tumor of the cerebellum, MB exhibits an undifferentiated cytology, being composed of small, round uniform cells with hyperchromatic nuclei 94. Such cytology suggests the emergence of these tumors from various stem/progenitor populations during early neurodevelopment. In the histopathologic evaluation of MB, other undifferentiated tumors occurring in the posterior fossa, such as atypical teratoid/rhaboid tumor and the embryonal tumor with multilayered rosettes must be considered in the differential diagnosis 25, 36. Despite early controversy surrounding the classification of MB and other embryonal tumors, such as atypical teratoid/rhabdoid tumor and the now obsolete CNS primitive neuroectodermal tumor (PNET), transcriptomic profiling has definitively separated MB from other intracranial embryonal tumors 45, 81, 94, 103. Additionally, these and other early studies (discussed below) paved the way for a molecular classification of MB that stratifies patients more robustly than clinical parameters alone 81. The consensus molecular subgroups of MB (WNT, SHH, Group 3 and Group 4) were incorporated into the 2016 WHO update on CNS tumor classification 58.
History
Termed by neurosurgeons Cushing and Bailey in 1925, “medulloblastoma” was initially described as a cerebellar tumor in children 2. The embryonal derivation of MB was reflected in its nomenclature despite no “medulloblast” ever being definitively identified 78. Nonetheless, defining clinical characteristics, including a cerebellar localization, metastatic tendency and male preponderance, were appreciated in early descriptions 16. Furthermore, the extent of neurosurgical resection was identified as a key determinant of survival, as patients undergoing biopsy alone fared poorly compared to those undergoing more complete resection 2.
Though roentgen therapy was used to treat MB postoperatively in Cushing’s era, outcomes did not improve significantly until almost 1950, when craniospinal irradiation (CSI) of the entire neuraxis emerged 76. The importance of such therapy highlights previous observations that MB could spread along the leptomeninges, seed the cerebrospinal fluid with abnormal cells, and recur in distant sites along the neuraxis 42. Despite favorable survival outcomes, exposure of younger patients to CSI often led to neurocognitive and neuroendocrine deficits 38, 52. Furthermore, the risk of secondary malignancies, particularly high‐grade gliomas, also increased as a result of CSI 12.
The efficacy of adjuvant chemotherapy in treating MB was initially demonstrated in the late 1970s. These early studies utilized vincristine and lomustine after surgery and radiotherapy. Despite some limitations, these early studies strongly suggested a survival benefit conferred by chemotherapy for patients with more extensive disease at diagnosis 105. Subsequent addition of platinum‐based agents formed the backbone of the modern chemotherapeutic regimens used today 73, 74. Given the adverse sequelae associated with CSI, risk stratification for multimodal management hinged, until the molecular era, on age, extent of resection and presence or absence of metastasis.
Molecular Subgroups of MB: Overview
MB represents a biologically and clinically heterogenous disease that can vary widely with respect to demographic biases, phenotypic presentation and clinical outcomes. Resolving the varied clinical behavior of some tumors by molecular biomarkers has long represented a key hurdle in dissecting the intertumoral heterogeneity within MB. For instance, nuclear β‐catenin expression in the tumors of patients with favorable outcome foreshadowed the eventual identification of the concordant WNT group of MB26, 29. Similarly, histopathologic correlates of patient outcome were also identified. For example, desmoplastic morphology and a relatively favorable prognosis in pediatric MB patients recapitulates a subset of SHH group tumors, while a large cell/anaplastic (LCA) morphology is associated with poor prognosis and MYC amplification, eventually being codified into Group 3 tumors 1, 3, 31, 39, 64, 79, 81. Such diversity in clinical behavior and molecular phenotype strongly motivated the consideration of MB as a composite of distinct entities.
Early transcriptomic studies by microarrays identified between four and six transcriptomic groups of clinical relevance, dependent on cohort size and clustering approaches 11, 50, 65, 108. From these efforts, the four consensus molecular groups (WNT, SHH, Group 3 and Group 4) emerged 107. These molecular groups have distinct demographic features, genetic lesions and gene expression patterns, forming a new framework for studying the disease in the laboratory and the clinic. Additionally, these molecular groups exhibit drastically divergent responses to conventional therapy, enabling molecularly guided risk stratification with the potential to abrogate some of the treatment‐related sequelae that impact quality of life in survivors.
With the emergence of high‐density DNA methylation arrays to query the epigenomes of many MBs, additional heterogeneity and subtypes have been defined within molecular subgroups 8, 63, 98 (Figure 1A). Though consensus on the definition, nomenclature and methodology required to define these subtypes is ongoing, such additional granularity will likely aid detailed exploration of MB tumor biology while informing clinical assessment. Furthermore, such heterogeneity among epigenetically defined subtypes highlights a potential avenue into understanding chromatin and epigenetic dysregulation as common themes in MB biology (Figure 1B).
Figure 1.
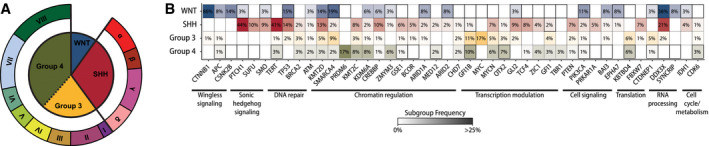
Molecular classification of medulloblastoma. A. Consensus molecular groups of MB are depicted according to relative frequency of incidence while emerging molecular subtypes of SHH (α, β, γ, δ) and Groups 3/4 (I–VIII) tumors are shown on the outer ring. B. Mutational landscape of the molecular groups of medulloblastoma is shown in a heatmap with genes arranged according to functional classes.
WNT‐activated MBs
Though only accounting for approximately 10% of MB diagnoses, WNT‐activated MBs carry the best prognosis with over 95% of children surviving this disease after 5 years 8, 29, 108. Tumors usually occur in older children (>4 years) with equal incidence between males and females 107. These tumors are also rarely metastatic at diagnosis (Figure 2A). Of note, adults with WNT tumors tend not to have the favorable outcome seen in childhood disease 13, 89.
Figure 2.
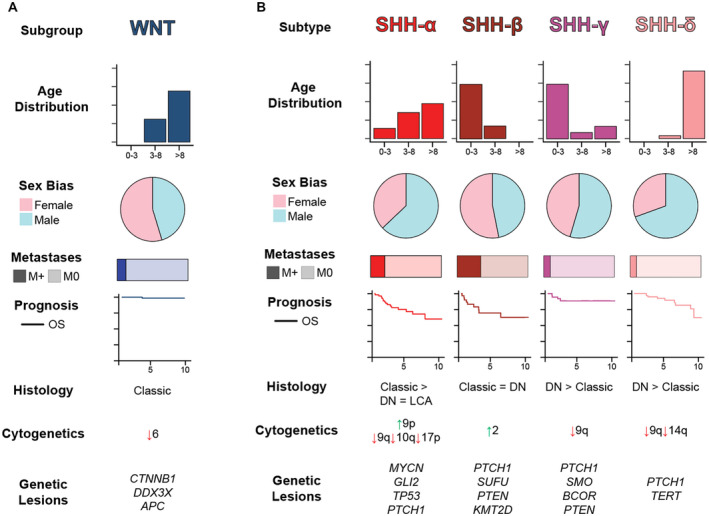
Summary of WNT and SHH medulloblastomas. A. WNT MBs are summarized by key clinicodemographic and molecular features. B. SHH MBs are summarized according to molecular subtypes proposed by Cavalli et al.
Readily identifiable by a WNT gene expression signature, these tumors are so named due to activation of the canonical Wingless (WNT) signal transduction pathway 14, 26, 117. Nearly 90% of WNT MBs harbor somatic activating mutations in exon 3 of CTTNB1 63. These mutations stabilize the gene product, β‐catenin, preventing its degradation by a cytoplasmic destruction complex (containing APC) and facilitating its unfettered translocation to the nucleus to act as a transcriptional co‐activator of TCF/LEF family transcription factors 24, 37. Most patients whose tumors lack somatic CTTNB1 mutations carry pathogenic germline APC variants, necessitating genetic testing for Turcot syndrome in these patients 113. Therefore, constitutive activation of the WNT pathway leading to cellular growth and proliferation in these tumors may emerge as a result of increased stability of β‐catenin itself or hampered degradation.
Additional recurrently mutated genes in WNT tumors include DDX3X, SMARCA4, TP53, CSNK2B, PIK3CA and EPHA7 63. While SMARCA4, PIK3CA and TP53 are commonly mutated across a variety of human cancers, mutations in CSNK2B highlight the essential role of WNT signaling in these tumors as it encodes the β‐subunit of casein kinase II, a positive regulator of WNT signaling 21, 46, 61, 84, 115. DDX3X encodes an RNA helicase that may promote WNT tumorigenesis by expanding the population of lower rhombic lip progenitors, the putative cell of origin for these tumors 32, 92. EPHA7 also plays a critical role in developmental patterning of neuronal populations, yet the exact molecular pathogenesis underlying such mutations remains unknown 17, 82. Given the implication of chromatin dysregulation across molecular subgroups of MB, mutations in SMARCA4 and other members of the SWI‐SNF chromatin remodeling complexes in WNT tumors underscores subgroup‐specific dependencies and cellular vulnerabilities underlying tumorigenesis.
The genomes of WNT MBs tend to be cytogenetically balanced, with the exception of monosomy 6 occurring in up to 85% of tumors 67. Despite the high frequency of hallmark genetic events in WNT tumors, utilizing only monosomy 6 and CTTNB1 mutations as WNT tumor biomarkers will fail to identify up to 15% of WNT MBs. WNT subgroup heterogeneity manifests in two age‐related subtypes, stratifying children and adults.8 Of note, the frequency of chromosome 6 euploidy is much higher in adults. As outcomes are generally favorable for children with WNT MBs, current efforts in this patient population are geared toward therapy de‐escalation and minimizing toxicities 51.
SHH‐activated MBs
SHH‐activated MBs account for approximately two‐thirds of MBs in infants (<3 years) and adults (>16 years), while only accounting for around 15% of MBs in children (ages 3–16) 49. Unlike the largely homogeneous WNT MBs, SHH MBs exhibit a striking degree of biological, pathological, and clinical heterogeneity 8, 98 (Figure 2B). The prognosis of these tumors is largely dictated by genetic factors in addition to clinicopathologic parameters such as patient age, metastatic status and morphology 101.
Named for activation of the sonic hedgehog (SHH) signaling pathway, SHH MBs possess alterations in genes leading to ligand‐independent activation of this canonical signaling pathway, facilitating uncontrolled cellular growth and proliferation 18, 33, 111. Such alterations include loss‐of‐function mutations or deletions in PTCH1 and SUFU, activating mutations of SMO and amplifications of GLI1, GLI2 and/or MYCN 63. In canonical SHH signaling, soluble SHH ligand binds to PTCH1 on the cell surface, thereby de‐repressing SMO. Activated SMO can then transduce the SHH signal intracellularly by releasing SUFU‐mediated repression of GLI1/2, allowing these transcription factors to translocate to the nucleus and to elicit expression of target genes, such as MYC family proto‐oncogenes, growth‐promoting cyclins and PTCH1 itself (to effect pathway feedback inhibition) 23, 69. A molecular understanding of SHH MBs has facilitated the implementation of molecularly targeted therapies with SMO inhibitors in skeletally mature patients 48, 90, 91.
The hallmark cytogenetic events in SHH MBs include losses of chromosomes 9q and 10q, potentiating loss of heterozygosity for PTCH1 (located on 9q) and SUFU (located on 10q) 48, 63, 101, 107. Haploinsufficiency of these crucial negative regulators of SHH signaling underscores the role of PTCH1 and SUFU as tumor suppressors 106. Additionally, loss of function mutations, either germline or somatic/mosaic, in TP53 can lead to defects in DNA repair and may contribute to clustered chromosomal rearrangements, known as chromothripsis, which are observed in tumors with coincident oncogene amplifications 88. TP53 mutations in children and adolescents with SHH MBs portend an abysmal prognosis, in stark contrast to the prognosis for WNT tumors with somatic TP53 mutations 116. As such, the SHH MB with TP53 mutation is listed separately in the WHO classification and is considered very high risk clinically 58. Such age‐dependent segregation of genetic lesions is also highlighted by the predominance of somatic TERT promoter mutations in nearly all adults with SHH MBs while only 10%–20% of tumors in pediatric patients harbor such events 48. In addition to WNT tumors, DDX3X is also mutated in SHH MBs, albeit at a slightly lower frequency of about 20% 63.
Other genes recurrently altered in SHH tumors include classes of chromatin modifiers, transcriptional regulators and signal transduction components. KMT2C and KMT2D, mutated in 7% and 13% of SHH MBs, respectively, are members of the MLL/COMPASS family and methylate H3K4 to regulate promoters and/or enhancers 83. Hotspot mutations in IDH1, occurring in 4% of SHH MBs, mirror the hypermethylation phenotype observed in adult gliomas 9, 63, 68. Mutations in PTEN, a negative regulator of the pro‐proliferative PI3K/AKT pathway, occur in 7% of SHH MBs and may also confer intrinsic or acquired resistance to molecular therapies against SHH pathway activation 60. Despite poorly understood molecular mechanisms of the underlying epigenetic or signaling cascade vulnerability, constitutive activation of SHH likely synergizes with dysregulation of chromatin and canonical signal transduction pathways to promote tumorigenesis in vulnerable cell populations 55, 97, 100.
Intertumoral heterogeneity among SHH MBs has been substantiated at both clinical and molecular levels 8, 98. Four distinct subtypes, termed α, β, γ and δ, with various demographic compositions and molecular landscapes have been identified 8. Among patients younger than 5 years, two comparable subtypes, iSHH‐I/II (corresponding to SHH‐β/γ), have been identified with 5‐year progression‐free survivals of approximately 25% and 75%, respectively 93. Additionally, comparison of single‐cell RNA sequencing of SHH MBs and cerebellar developmental atlases has identified age‐related developmental trajectories that seem to recapitulate varying degrees of neuronal differentiation between SHH tumors in infants and older children 40. Taken together, these new SHH MB data highlight advances in linking tumor biology to clinical behavior.
Group 3 MBs
Occurring commonly during infancy and early childhood, Group 3 tumors comprise approximately a quarter of MBs. These tumors are also twice as common in males and are noted for a high incidence of metastasis at the time of presentation 107. Considered the most aggressive MB subgroup, Group 3 tumors confer a 5‐year overall survival of <60% 11, 65. Certain hallmark genomic features, such as MYC amplification, represent extremely high‐risk disease 63, 79, 80. Aneuploidy, particularly isochromosome 17q, gains of chromosomes 1q and 7 and losses of chromosome 8, 10q and 16q, is notable among Group 3 tumors 67. The naming of Group 3 tumors highlights the current lack of framework for unifying molecular derangements to underlying tumor biology in this molecular subgroup.
Nearly 20% of Group 3 tumors are characterized by high‐level MYC amplification 63, 67. Aberrant activation of MYC functions as a central node in a myriad of pro‐tumorigenic cellular pathways, including mRNA processing and protein translation 19. Additional gene level amplifications include MYCN (5%) and OTX2 (3%). The contribution of MYCN to underlying MB tumor biology is well appreciated with roles in tumor initiation, maintenance and progression 47, 77. As a master transcriptional regulator in neurodevelopment, OTX2 is thought to confer a stem‐like state to MB cells 4, 5. Additionally, MYC and OTX2 may function synergistically to promote Group 3 tumorigenesis through transcriptional cross‐regulation 6.
In addition to overexpression of the aforementioned genes as a result of gene amplification and other genetic events, GFI1 and GFI1B are upregulated in ~ 15% of Group 3 MBs, in a mutually exclusive manner 63. These transcriptional repressors are key dictators of developmental cell fate decisions 22, 53. The mechanism governing overexpression of these genes occurs through enhancer hijacking, whereby structural variants juxtapose normally distant and often unrelated gene regulatory elements with the promoters of oncogenes (Figure 3). In Group 3 MBs, highly active enhancers overlapping DDX31 are repositioned proximal to GFI1B, leading to its overexpression 66. Such striking genetic–epigenetic interplay highlights the necessity to contextualize genomic events with annotations of gene regulatory elements.
Figure 3.
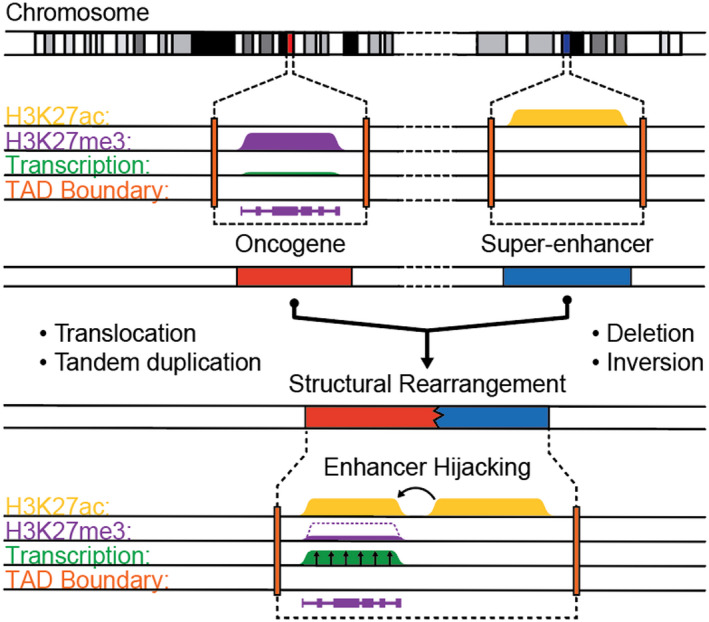
Schematic mechanism of enhancer hijacking in medulloblastoma. Juxtaposition of transcriptionally repressed oncogene locus with active gene regulatory elements (eg, super‐enhancers) can occur by various structural rearrangements. Such alterations to genomic architecture lead to reorganization of topologically associated domains (TADs, depicted by dashed boxes) by disruption of native boundary elements (orange). As a result, active gene regulatory elements (marked by H3K27ac, yellow) can spread to the normally repressed proto‐oncogene promoters (marked by H3K27me3, purple), leading to oncogene transcription (green).
The landscape of somatically altered genes in Group 3 MBs is relatively sparse with only SMARCA4, KBTBD4, CTDNEP1 and KMT2D recurrently mutated in greater than 5% of tumors 44, 63, 83. KBTBD4 is a poorly characterized member of the Kelch–BTB–BACK family, which consists of proteins involved in the ubiquitin–proteasome pathway 7. Somatic in‐frame insertions in KBTBD4 are clustered in the conserved Kelch domain, potentially leading to dysregulation of substrate recognition 63. CTDNEP1 functions as a protein phosphatase, with substrates including BMP receptors 95, 96. The molecular pathogenesis of CTDNEP1 mutations, which can occur as hotspot frameshifts in the phosphatase domain, are poorly understood 44. Whether the contribution of SMARCA4 and KMT2D mutations in Group 3 tumors is distinct from that seen in WNT and SHH MBs, respectively, remains unknown.
Group 4 MBs
As the most common molecular subgroup of MB, Group 4 tumors account for up to 40% of all cases and tend to occur in older children 107. Group 4 tumors have a gender bias toward males of nearly 3:1. Despite being considered intermediate in terms of survival, approximately 33% of patients have metastatic disease at presentation, and relapse latency is longer for Group 4 tumors compared to other MBs 8, 86. Like their Group 3 counterparts, Group 4 tumors remain poorly characterized in terms of underlying tumor biology.
Though Group 4 MBs fail to disclose driver genes recurrently mutated at frequencies above 10%, the class of somatically altered genes highlight the likely role of chromatin modifier dysregulation in the molecular pathogenesis of this molecular subgroup 63, 75. Mutually exclusive loss of function mutations in the chromatin modifiers KDM6A, ZMYM3 and KMT2C suggest possible convergent dysregulation of the epigenome in tumorigenesis 41, 63, 92. KDM6A functions as a demethylase of H3K27 and associates with the MLL complexes to effect changes in H3K4 methylation 112. KMT2C, a member of the MLL family and COMPASS complex member, can modulate activity of gene regulatory elements through its H3K4 methyltransferase action at promoters and enhancers 87. ZMYM3 has been described in association other chromatin modifiers and implicated in the DNA damage response 54. Disruptions of these well‐described chromatin modifiers may perturb the chromatin and transcriptional regulatory landscape at key developmental gene promoters and/or enhancers to promote Group 4 tumorigenesis. Additionally, disruption of these chromatin modifiers may alter cellular responses to DNA damage.
The most prevalent putative driver event in Group 4 MBs involves the overexpression of PRDM6 via enhancer hijacking, in which tandem duplications and other structural variants at the SNCAIP locus juxtapose a highly active super‐enhancer with promoter elements of PRDM6 63. PRDM6 has been described as a chromatin modifier and transcriptional regulator in the developing cardiovascular system, though functional validation of its role in MB is lacking 20, 114. CDK6, also amplified exclusively in Group 4 tumors, encodes a key cell cycle regulator of the G1‐S transition 59, 85. Hotspot mutations in the T‐box domain of TBR1, a neurodevelopmental transcriptional factor, occur exclusively in Group 4 tumors 28, 44.
Similar to their Group 3 counterparts, Group 4 MBs harbor gene‐level amplifications of MYCN as well as somatic mutations in KBTBD4 63. As amplifications of OTX2 also occur in Group 4 tumors, the overlapping spectrum of altered genes between Group 3 and Group 4 MBs suggests a possible continuum in terms of tumor biology. At the cytogenetic level, Group 4 tumors are characterized by high rates of isochromosome 17q, losses of chromosomes 8 and 11 and gains of chromosomes 7 63, 67. Certain cytogenetic events, namely chromosome 11 loss and chromosome 17 gain, have been associated with favorable prognosis in Group 4 MB patients 101.
Molecular Subtypes in Group 3/4 MBs
Representing nearly two‐thirds of all MBs, Group 3 and Group 4 tumors exhibit a spectrum of clinical behaviors with many patients relapsing despite having average‐risk disease 34. Furthermore, molecular heterogeneity within Group 3 and Group 4 MBs has been recognized, with identification of varying numbers of Group 3/4 subtypes using different methods 8, 11, 50, 63, 67, 98. However, the extent to which these clinical and molecular subtypes can ultimately be reconciled remains in question (Figure 4).
Figure 4.
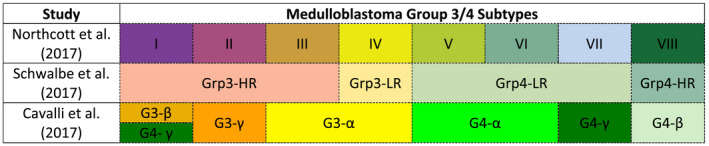
Overview of proposed Group 3/4 MB subtypes. Three independent studies have described four to eight molecular subtypes of Group 3 and Group 4 medulloblastoma. While concordance between subtypes across different studies is not definitive, an overlay of these subtypes according to study are depicted above according to nomenclature adopted within each study. LR = low‐risk; HR = high‐risk.
Because of differing analytical approaches and cohort composition biases in prior studies, distinct numbers of Group 3/4 subtypes were identified. Northcott et al. utilized t‐distributed stochastic neighbor embedding dimensionality reduction and density‐based clustering on a cohort of 740 Group 3/4 tumors profiled by DNA methylation array 63. Schwalbe et al. also utilized DNA methylation arrays but implemented non‐negative matrix factorization on 243 Group 3/4 tumors 98. Cavalli et al. integrated gene expression and DNA methylation arrays using similarity network fusion on 470 Group 3/4 samples 8. An overview of the molecular subtypes identified in these studies is presented in Figure 4. In attempting to unify these large‐scale analyses, Sharma et al. recently performed a meta‐analysis based on the aforementioned studies to summarize the second‐generation subtypeing of Group 3/4 MBs and developed a random forest classifier for eight molecular subtypes of Group 3/4 MBs based on DNA methylation array 99 (Figure 5).
Figure 5.
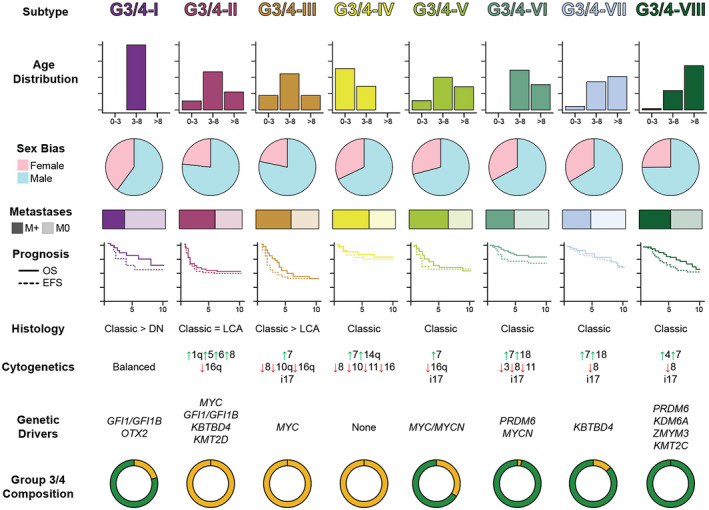
Summary of Group 3/4 medulloblastoma subtypes. Clinicopathologic and molecular summary of eight molecular subtypes of Group 3 and Group 4 medulloblastomas according to Northcott et al. (2017) and Sharma et al. (2019). The composition of each subtype according to consensus Group 3 or Group 4 profiling is shown at the bottom of the panel. LCA = large cell anaplastic; DN = desmoplastic/nodular.
Germline Predispositions
MB can be associated with rare, hereditary tumor predisposition syndromes. Gorlin syndrome, characterized by damaging mutations in SUFU or PTCH1, has been associated with increased risk of MB .102. Other syndromes associated with aberrant SHH signaling, such as Curry–Jones syndrome (mosaic SMO mutations) or Greig cephalopolysyndactyly syndrome (GLI3 mutations), underscores the dysregulation of SHH pathway as a unifying pathophysiologic mechanism for SHH‐activated MB 27, 35, 109. Familial adenomatous polyposis (Turcot) syndrome, characterized by mutations in APC, predisposes to WNT MB 15. In addition to developmental signaling axes, other molecular processes commonly affected in germline predisposition syndromes with increased risk of MB include germline defects in DNA damage response/repair machinery, such is in Li–Fraumeni syndrome (TP53 mutations) and constitutional mismatch repair (mutations in MLH1, MSH2, MSH6 or PMS2) 48, 56, 57, 62, 88, 104.
Recent studies have identified damaging germline mutations in approximately 10% of all patients with MB 113. The most commonly altered predisposition genes include APC, BRCA2, PALB2, PTCH1, SUFU and TP53, accounting for 6% of MBs. Of note, the burden and distribution of germline predisposition to MB is not distributed uniformly across molecular groups. While 20% of patients with SHH MBs harbor predisposing germline variants, such predisposition based on known hereditary tumor genes is much rarer in Group 3 and Group 4 tumors. Furthermore, the specific mutations associated with germline predisposition dictate risk for MB and thereby influence the age at diagnosis as well as association with molecular group. Clinical manifestations and syndromic symptomatology vary according to specific genetic predisposition (Figure 6). Coupled with the incidence of cancers in the family history, the various altered predisposition genes in MB underscore the differing necessities for genetic testing, family counseling and surveillance monitoring.
Figure 6.
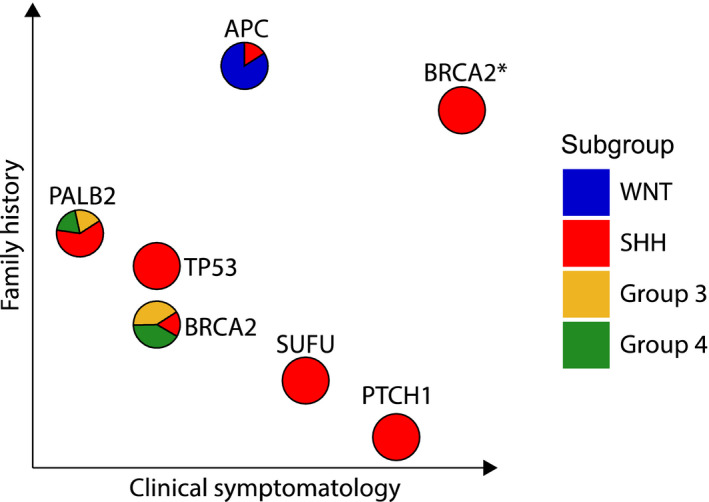
Germline predisposition genes in medulloblastoma. Six most commonly mutated genes (BRCA2* = compound heterozygous mutations) predisposing to medulloblastoma are depicted. Molecular subgroups of tumors associated with alterations in respective germline predisposition genes are plotted as pie charts. Distance along each axis represents the proportion of patients with positive family history or recognizable clinical signs of germline predisposition syndromes.
Outlook
The modern era of MB genomics has afforded tremendous insights into basic tumor biology, refined molecular diagnostic approaches and informed clinical management. Nonetheless, considerable effort is needed to tailor treatment strategies to risk stratification guided by conventional histopathologic and clinical features and adapted to the context of MB molecular subgroups and subtypes. Furthermore, molecular risk features identified through large‐scale genomics of retrospective trials must be validated and contextualized in prospective studies. Implementation of molecularly guided therapies requires not only careful consideration of rational drug targets based on tumor biology but also the rigorous evaluation of these agents in appropriate preclinical models. Furthermore, tumor evolution and the nature of relapsed disease must be explored comprehensively to guide efforts to salvage patients who fail first‐line therapies. Likewise, systematic characterization of patient germlines must be conducted beyond known cancer predisposition genes to uncover previously unknown genetic risks that will guide long‐term surveillance and genetic counseling. Finally, despite incremental advances in survival for certain patients, improvements in overall survival must also be considered in terms of quality of life with effort geared toward therapeutic de‐escalation in patients with favorable outcome.
While appreciation of intertumoral heterogeneity among MBs has facilitated biologically and clinically relevant molecular classification, deeper characterization of these tumors using single‐cell sequencing techniques has uncovered additional layers of intratumoral heterogeneity. Furthermore, such approaches can be utilized to unravel normal developmental cascades of cellular differentiation to correlate the transcriptomic signatures of individual tumor cells to specific cellular populations within the developing cerebellum. Two recent single‐cell transcriptomic studies have leveraged the developing mouse cerebellum as a reference atlas for uncovering cellular hierarchies and transcriptional programs underlying the various molecular subgroups of MB 40, 110. In addition to recapitulating granule neuron progenitors as the cellular origins for SHH MBs, both subgroups identified unipolar brush cells are the putative cellular origins of Group 4 MBs. Additionally, age‐associated developmental hierarchies were uncovered for SHH MBs, wherein the differentiation state of cells within a tumor was inversely correlated with age of the patient at diagnosis. Furthermore, the cellular compositions of Group 3/4 MBs was shown to vary according to proportion of differentiated vs. undifferentiated cells, with Group 3 tumors largely comprised of the latter. These novel insights may suggest the necessity to reconsider Group 3/4 MBs as a continuum, particularly for those subtypes intermediate between Group 3 and Group 4.
While these studies implicate specific cellular and mechanistic susceptibilities to MB tumorigenesis in the context of normal cerebellar development, additional characterization of MB at the epigenomic and chromatin level will be necessary to deconvolute the respective contributions of cellular origins and transcriptional aberrancies imposed by mutations in epigenetic machinery to the overall signature of a given tumor. With the emergence of single‐cell approaches to profile the chromatin landscape and transcriptional factor binding, refinement of the molecular structure of MB will hopefully continue to motivate a more profound understanding of this devastating disease.
Conclusions
The modern molecular era of MB genomics has facilitated the classification of the disease into WNT, SHH, Group 3 and Group 4 tumors. These molecular subgroups recapitulate distinct tumor biology as evidenced by demographic biases, distinct genetic lesions and clinical behavior. Further subtyping of the MBs has unveiled additional granularity in SHH and Group 3/4 tumors. While continued efforts are required to reconcile the various proposed MB subtypes, such information will provide a molecular framework for the design of rational and targeted clinical trials in attempts to improve survival for high‐risk patients while mitigating long‐term sequelae in lower risk patients by de‐escalation of therapy. Continued exploration of tumor biology underlying MB will afford crucial insights into novel oncogenic mechanisms and uncover additional therapeutic vulnerabilities. As such, MB represents a paradigm of integrated clinicomolecular study of pediatric cancers.
References
- 1. Badiali M, Pession A, Basso G, Andreini L, Rigobello L, Galassi E et al (1991) N‐myc and c‐myc oncogenes amplification in medulloblastomas. Evidence of particularly aggressive behavior of a tumor with c‐myc amplification. Tumori J 77:118–121. [DOI] [PubMed] [Google Scholar]
- 2. Bailey P, Cushing H (1925) Medulloblastoma cerebelli: a common type of midcerebellar glioma of childhood. Arch Neurol Psychiatry 14:192–224. [Google Scholar]
- 3. Bigner SH, Friedman HS, Vogelstein B, Oakes WJ, Bigner DD (1990) Amplification of the c‐myc gene in human medulloblastoma cell lines and xenografts. Cancer Res 50:2347–2350. [PubMed] [Google Scholar]
- 4. Boulay G, Awad ME, Riggi N, Archer TC, Iyer S, Boonseng WE et al (2017) OTX2 Activity at distal regulatory elements shapes the chromatin landscape of group 3 medulloblastoma. Cancer Discov 7:288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bunt J, Hasselt NE, Zwijnenburg DA, Hamdi M, Koster J, Versteeg R et al (2012) OTX2 directly activates cell cycle genes and inhibits differentiation in medulloblastoma cells. Int J Cancer 131:E21–E32. [DOI] [PubMed] [Google Scholar]
- 6. Bunt J, Hasselt NE, Zwijnenburg DA, Koster J, Versteeg R, Kool M (2011) Joint binding of OTX2 and MYC in promotor regions is associated with high gene expression in medulloblastoma. PLoS ONE 6:e26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A et al (2013) Structural basis for Cul3 protein assembly with the BTB‐Kelch family of E3 ubiquitin ligases. J Biol Chem 288:7803–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B et al (2017) Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31:737–754 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang CH, Housepian EM, Herbert C Jr (1969) An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 93:1351–1359. [DOI] [PubMed] [Google Scholar]
- 11. Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H et al (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christopherson KM, Rotondo RL, Bradley JA, Pincus DW, Wynn TT, Fort JA et al (2014) Late toxicity following craniospinal radiation for early‐stage medulloblastoma. Acta Oncol 53:471–480. [DOI] [PubMed] [Google Scholar]
- 13. Clifford SC, Lannering B, Schwalbe EC, Hicks D, O'Toole K, Nicholson SL et al (2015) Biomarker‐driven stratification of disease‐risk in non‐metastatic medulloblastoma: Results from the multi‐center HIT‐SIOP‐PNET4 clinical trial. Oncotarget 6:38827–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D et al (2006) Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub‐group of medulloblastomas associated with a favorable prognosis. Cell Cycle 5:2666–70. [DOI] [PubMed] [Google Scholar]
- 15. Cohen SB (1982) Familial polyposis coli and its extracolonic manifestations. J Med Genet 19:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cushing H (1930) Experiences with the cerebellar medulloblastomas. Acta Pathol Microbiol Scand 7:1–86. [Google Scholar]
- 17. Cwiek P, Leni Z, Salm F, Dimitrova V, Styp‐Rekowska B, Chiriano G et al (2015) RNA interference screening identifies a novel role for PCTK1/CDK16 in medulloblastoma with c‐Myc amplification. Oncotarget 6:116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dahmane N, Ruiz i Altaba A. (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–3100. [DOI] [PubMed] [Google Scholar]
- 19. Dang CV (2012) MYC on the path to cancer. Cell 149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis CA, Haberland M, Arnold MA, Sutherland LB, McDonald OG, Richardson JA et al (2006) PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol 26:2626–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M et al (2019) Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep 28:1370–1384 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duan Z, Horwitz M (2003) Targets of the transcriptional repressor oncoprotein Gfi‐1. Proc Natl Acad Sci U S A 100:5932–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duman‐Scheel M, Weng L, Xin S, Du W (2002) Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 417:299–304. [DOI] [PubMed] [Google Scholar]
- 24. Eberhart CG, Tihan T, Burger PC (2000) Nuclear localization and mutation of beta‐catenin in medulloblastomas. J Neuropathol Exp Neurol 59:333–337. [DOI] [PubMed] [Google Scholar]
- 25. Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G et al (2011) Medulloblastoma: clinicopathological correlates of SHH, WNT, and non‐SHH/WNT molecular subgroups. Acta Neuropathol 121:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE et al (2005) beta‐Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23:7951–7957. [DOI] [PubMed] [Google Scholar]
- 27. Erez A, Ilan T, Amariglio N, Muler I, Brok‐Simoni F, Rechavi G et al (2002) GLI3 is not mutated commonly in sporadic medulloblastomas. Cancer 95:28–31. [DOI] [PubMed] [Google Scholar]
- 28. Fink AJ, Englund C, Daza RA, Pham D, Lau C, Nivison M et al (2006) Development of the deep cerebellar nuclei: transcription factors and cell migration from the rhombic lip. J Neurosci 26:3066–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE et al (2006) Risk‐adapted craniospinal radiotherapy followed by high‐dose chemotherapy and stem‐cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma‐96): long‐term results from a prospective, multicentre trial. Lancet Oncol 7:813–820. [DOI] [PubMed] [Google Scholar]
- 30. Gandola L, Massimino M, Cefalo G, Solero C, Spreafico F, Pecori E et al (2009) Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J Clin Oncol 27:566–571. [DOI] [PubMed] [Google Scholar]
- 31. Giangaspero F, Rigobello L, Badiali M, Loda M, Andreini L, Basso G et al (1992) Large‐cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol 16:687–693. [PubMed] [Google Scholar]
- 32. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277:1109–1113. [DOI] [PubMed] [Google Scholar]
- 34. Goschzik T, Schwalbe EC, Hicks D, Smith A, Zur Muehlen A, Figarella‐Branger D et al (2018) Prognostic effect of whole chromosomal aberration signatures in standard‐risk, non‐WNT/non‐SHH medulloblastoma: a retrospective, molecular analysis of the HIT‐SIOP PNET 4 trial. Lancet Oncol 19:1602–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grange DK, Clericuzio CL, Bayliss SJ, Berk DR, Heideman RL, Higginson JK et al (2008) Two new patients with Curry‐Jones syndrome with trichoblastoma and medulloblastoma suggest an etiologic role of the sonic hedgehog‐patched‐GLI pathway. Am J Med Genet A 146A:2589–2597. [DOI] [PubMed] [Google Scholar]
- 36. de Haas T, Oussoren E, Grajkowska W, Perek‐Polnik M, Popovic M, Zadravec‐Zaletel L et al (2006) OTX1 and OTX2 expression correlates with the clinicopathologic classification of medulloblastomas. J Neuropathol Exp Neurol 65:176–186. [DOI] [PubMed] [Google Scholar]
- 37. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT et al (1998) Identification of c‐MYC as a target of the APC pathway. Science 281:1509–1512. [DOI] [PubMed] [Google Scholar]
- 38. Heitzer AM, Ashford JM, Harel BT, Schembri A, Swain MA, Wallace J et al (2019) Computerized assessment of cognitive impairment among children undergoing radiation therapy for medulloblastoma. J Neurooncol 141:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. von Hoff K, Hartmann W, von Bueren AO, Gerber NU, Grotzer MA, Pietsch T et al (2010) Large cell/anaplastic medulloblastoma: outcome according to myc status, histopathological, and clinical risk factors. Pediatr Blood Cancer 54:369–376. [DOI] [PubMed] [Google Scholar]
- 40. Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw ML, Baumgartner A et al (2019) Resolving medulloblastoma cellular architecture by single‐cell genomics. Nature 572:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huether R, Dong L, Chen X, Wu G, Parker M, Wei L et al (2014) The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun 5:3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ingraham FD, Bailey OT, Barker WF (1948) Medulloblastoma cerebelli; diagnosis, treatment and survivals, with a report of 56 cases. N Engl J Med 238:171–174. [DOI] [PubMed] [Google Scholar]
- 43. Jakacki RI, Burger PC, Zhou T, Holmes EJ, Kocak M, Onar A et al (2012) Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children's Oncology Group Phase I/II study. J Clin Oncol 30:2648–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M et al (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kadin ME, Rubinstein LJ, Nelson JS (1970) Neonatal cerebellar medulloblastoma originating from the fetal external granular layer. J Neuropathol Exp Neurol 29:583–600. [DOI] [PubMed] [Google Scholar]
- 46. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J et al (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C et al (2012) A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21:168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V et al (2014) Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell 25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA et al (2012) Molecular subgroups of medulloblastoma: an international meta‐analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P et al (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 3:e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Korshunov A, Sahm F, Zheludkova O, Golanov A, Stichel D, Schrimpf D et al (2019) DNA methylation profiling is a method of choice for molecular verification of pediatric WNT‐activated medulloblastomas. Neuro Oncol 21:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lafay‐Cousin L, Bouffet E, Hawkins C, Amid A, Huang A, Mabbott DJ (2009) Impact of radiation avoidance on survival and neurocognitive outcome in infant medulloblastoma. Curr Oncol 16:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee C, Rudneva VA, Erkek S, Zapatka M, Chau LQ, Tacheva‐Grigorova SK et al (2019) Lsd1 as a therapeutic target in Gfi1‐activated medulloblastoma. Nat Commun 10:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Leung JW, Makharashvili N, Agarwal P, Chiu LY, Pourpre R, Cammarata MB et al (2017) ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev 31:260–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M et al (2016) Active medulloblastoma enhancers reveal subgroup‐specific cellular origins. Nature 530:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lindsay H, Jubran RF, Wang L, Kipp BR, May WA (2013) Simultaneous colonic adenocarcinoma and medulloblastoma in a 12‐year‐old with biallelic deletions in PMS2. J Pediatr 163:601–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ling C, Yang W, Sun H, Ge M, Ji Y, Han S et al (2018) Rare compound heterozygous mutations in gene MSH6 cause constitutive mismatch repair deficiency syndrome. Clin Case Rep 6:1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. [DOI] [PubMed] [Google Scholar]
- 59. Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE et al (2005) Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol 23:8853–8862. [DOI] [PubMed] [Google Scholar]
- 60. Metcalfe C, Alicke B, Crow A, Lamoureux M, Dijkgraaf GJ, Peale F et al (2013) PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer Res 73:7034–7042. [DOI] [PubMed] [Google Scholar]
- 61. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R (2016) Landscape of phosphatidylinositol‐3‐kinase pathway alterations across 19784 diverse solid tumors. JAMA Oncol 2:1565–1573. [DOI] [PubMed] [Google Scholar]
- 62. Nguyen A, Bougeard G, Koob M, Chenard MP, Schneider A, Maugard C et al (2016) MSI detection and its pitfalls in CMMRD syndrome in a family with a bi‐allelic MLH1 mutation. Fam Cancer 15:571–577. [DOI] [PubMed] [Google Scholar]
- 63. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al (2017) The whole‐genome landscape of medulloblastoma subtypes. Nature 547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M et al (2011) Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol 122:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D et al (2014) Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T et al (2012) Subgroup‐specific structural variation across 1,000 medulloblastoma genomes. Nature 488:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM et al (2003) Transcriptional profiling of the Sonic hedgehog response: a critical role for N‐myc in proliferation of neuronal precursors. Proc Natl Acad Sci U S A 100:7331–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz‐Sloan JSCBTRUS (2018) Statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol 20(Suppl. 4):iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Oyharcabal‐Bourden V, Kalifa C, Gentet JC, Frappaz D, Edan C, Chastagner P et al (2005) Standard‐risk medulloblastoma treated by adjuvant chemotherapy followed by reduced‐dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23:4726–4734. [DOI] [PubMed] [Google Scholar]
- 72. Packer RJ, Gajjar A, Vezina G, Rorke‐Adams L, Burger PC, Robertson PL et al (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average‐risk medulloblastoma. J Clin Oncol 24:4202–4208. [DOI] [PubMed] [Google Scholar]
- 73. Packer RJ, Siegel KR, Sutton LN, Evans AE, D'Angio G, Rorke LB et al (1988) Efficacy of adjuvant chemotherapy for patients with poor‐risk medulloblastoma: a preliminary report. Ann Neurol 24:503–508. [DOI] [PubMed] [Google Scholar]
- 74. Packer RJ, Sutton LN, Goldwein JW, Perilongo G, Bunin G, Ryan J et al (1991) Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg 74:433–440. [DOI] [PubMed] [Google Scholar]
- 75. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC et al (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Paterson E, Farr RF (1953) Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol 39:323–336. [DOI] [PubMed] [Google Scholar]
- 77. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ et al (2012) An animal model of MYC‐driven medulloblastoma. Cancer Cell 21:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Penfield W (1931) The classification of gliomas and neuroglia cell types. Arch Neurol Psychiatry 26:745–753. [Google Scholar]
- 79. Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J et al (2009) Outcome prediction in pediatric medulloblastoma based on DNA copy‐number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27:1627–1636. [DOI] [PubMed] [Google Scholar]
- 80. Pietsch T, Schmidt R, Remke M, Korshunov A, Hovestadt V, Jones DT et al (2014) Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol 128:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME et al (2002) Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415:436–442. [DOI] [PubMed] [Google Scholar]
- 82. Prost G, Braun S, Hertwig F, Winkler M, Jagemann L, Nolbrant S et al (2016) The putative tumor suppressor gene EphA7 is a novel BMI‐1 target. Oncotarget 7:58203–58217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J et al (2012) Medulloblastoma exome sequencing uncovers subtype‐specific somatic mutations. Nature 488:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rabalski AJ, Gyenis L, Litchfield DW (2016) Molecular pathways: Emergence of Protein Kinase CK2 (CSNK2) as a potential target to inhibit survival and DNA damage response and repair pathways in cancer cells. Clin Cancer Res 22:2840–2847. [DOI] [PubMed] [Google Scholar]
- 85. Raleigh DR, Choksi PK, Krup AL, Mayer W, Santos N, Reiter JF (2018) Hedgehog signaling drives medulloblastoma growth via CDK6. J Clin Invest 128:120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ et al (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rampias T, Karagiannis D, Avgeris M, Polyzos A, Kokkalis A, Kanaki Z et al (2019) The lysine‐specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep 20:e46821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J et al (2012) Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Remke M, Hielscher T, Northcott PA, Witt H, Ryzhova M, Wittmann A et al (2011) Adult medulloblastoma comprises three major molecular variants. J Clin Oncol 29:2717–2723. [DOI] [PubMed] [Google Scholar]
- 90. Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A et al (2017) Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8:69295–69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I et al (2015) Vismodegib exerts targeted efficacy against recurrent sonic hedgehog‐subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC‐025B and PBTC‐032. J Clin Oncol 33:2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L et al (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KS et al (2018) Risk‐adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19:768–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rorke LB (1983) The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol 42:1–15. [PubMed] [Google Scholar]
- 95. Satow R, Chan TC, Asashima M (2002) Molecular cloning and characterization of dullard: a novel gene required for neural development. Biochem Biophys Res Commun 295:85–91. [DOI] [PubMed] [Google Scholar]
- 96. Satow R, Kurisaki A, Chan TC, Hamazaki TS, Asashima M (2006) Dullard promotes degradation and dephosphorylation of BMP receptors and is required for neural induction. Dev Cell 11:763–774. [DOI] [PubMed] [Google Scholar]
- 97. Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG et al (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh‐induced medulloblastoma. Cancer Cell 14:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D et al (2017) Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18:958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sharma T, Schwalbe EC, Williamson D, Sill M, Hovestadt V, Mynarek M et al (2019) Second‐generation molecular subgrouping of medulloblastoma: an international meta‐analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138:309–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shi X, Wang Q, Gu J, Xuan Z, Wu JI (2016) SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying Shh‐type medulloblastoma development. Oncogene 35:5746–5758. [DOI] [PubMed] [Google Scholar]
- 101. Shih DJ, Northcott PA, Remke M, Korshunov A, Ramaswamy V, Kool M et al (2014) Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32:886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Smith MJ, Beetz C, Williams SG, Bhaskar SS, O'Sullivan J, Anderson B et al (2014) Germline mutations in SUFU cause Gorlin syndrome‐associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32:4155–4161. [DOI] [PubMed] [Google Scholar]
- 103. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 164:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Taeubner J, Wimmer K, Muleris M, Lascols O, Colas C, Fauth C et al (2018) Diagnostic challenges in a child with early onset desmoplastic medulloblastoma and homozygous variants in MSH2 and MSH6. Eur J Hum Genet 26:440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tait DM, Thornton‐Jones H, Bloom HJ, Lemerle J, Morris‐Jones P (1990) Adjuvant chemotherapy for medulloblastoma: the first multi‐centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer 26:464–9. [PubMed] [Google Scholar]
- 106. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X et al (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310. [DOI] [PubMed] [Google Scholar]
- 107. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC et al (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC et al (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931. [DOI] [PubMed] [Google Scholar]
- 109. Twigg SRF, Hufnagel RB, Miller KA, Zhou Y, McGowan SJ, Taylor J et al (2016) A recurrent mosaic mutation in SMO, encoding the hedgehog signal transducer smoothened, is the major cause of curry‐jones syndrome. Am J Hum Genet 98:1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vladoiu MC, El‐Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y et al (2019) Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wallace VA (1999) Purkinje‐cell‐derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9:445–448. [DOI] [PubMed] [Google Scholar]
- 112. Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH et al (2017) A UTX‐MLL4‐p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol Cell 67:308–321 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S et al (2018) Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 19:785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wu Y, Ferguson JE 3rd, Wang H, Kelley R, Ren R, McDonough H et al (2008) PRDM6 is enriched in vascular precursors during development and inhibits endothelial cell proliferation, survival, and differentiation. J Mol Cell Cardiol 44:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang Y, Kwok‐Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH et al (2017) A Pan‐Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 31:820–832 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC et al (2013) Subgroup‐specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31:2927–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zurawel RH, Chiappa SA, Allen C, Raffel C (1998) Sporadic medulloblastomas contain oncogenic beta‐catenin mutations. Cancer Res 58:896–899. [PubMed] [Google Scholar]