

Abstract
Meningiomas are common in adults (~35% of brain tumors) but rare in children, where they exhibit unique clinical, pathological and molecular features compared to adult counterparts. Thus, data generated from adult cohorts may be imperfectly suited to guiding diagnostic, prognostic and treatment decisions for children. We studied 50 meningioma patients ≤18 years with available clinical and pathological data to address the need for data obtained in the pediatric setting. As previously described, we noted a slight bias toward male patients and a higher proportion of spinal tumors compared to adults. Thirty‐eight of 50 specimens were further analyzed by next generation sequencing. Loss‐of‐function mutations in NF2 and chromosome 22 losses were common, but pathogenic variants in other genes (SMARCB1, FUBP1, BRAF, TERT promoter, CHEK2, SMAD and GATA3) were identified in a minority of cases. Copy number variants outside of chromosomes 22 and 1 were infrequent. H3K27 hypomethylation, a useful biomarker in adult tumors, was not found in our cohort. In exploring the correlation between mitotic count and recurrence‐free survival, we found a threshold of six mitoses per 10 high powered fields as the optimal cutoff in predicting recurrence‐free survival. If independently validated in larger studies, adjusted grading thresholds could enhance the clinical management of pediatric meningiomas.
Keywords: clinicopathological, meningioma, molecular, NF2, pediatric, sporadic, WHO grade
Introduction
Meningiomas comprise approximately 35% of all primary intracranial neoplasms in adults (41). In contrast, they are relatively rare in children, accounting for just 1%–2% of primary brain tumors in persons <21 years of age (33). Studies have shown that pediatric meningioma have unique features compared to their adult counterparts. For instance, they are associated with an increased frequency of higher‐grade (WHO Grades II and III) tumors, aggressive histological types (eg, papillary, clear cell, etc.), higher mitotic and proliferation indices, more frequent brain invasion, higher recurrence rates and a predilection for atypical sites (particularly the spinal cord) (13, 15, 17, 19, 28, 34, 38, 44). They also lack the female sex bias seen in adult tumors (17), are more often associated with inherited genetic syndromes (eg, Neurofibromatosis type II (5, 27, 34, 43, 50) and Gorlin's syndrome (3, 31)), have increased sensitivity to environmental risk factors (such as exposure to ionizing radiation) (18, 20, 21, 46) and show a unique mutational profile (7). The dissimilarities in tumor characteristics and behavior may reflect distinct molecular and cytogenetic mechanisms in adult vs. pediatric meningiomas (44).
Though many studies have explored the molecular landscape and progression of meningiomas, the vast majority have exclusively or predominantly focused on adult patients (8, 9, 10, 23, 39, 40). In part due to their rarity, there is a paucity of molecular data describing meningiomas in the pediatric setting, so management strategies for pediatric patients are largely inferred from adult cohorts. Given the significant differences between adult and pediatric tumors, it is not surprising that WHO grading is less predictive of outcome in children (43). Novel strategies are needed to optimize stratification and clinical management of children with meningiomas. These should, ideally, be based on data obtained from pediatric rather than adult tumors.
Methods
Case selection
Fifty cases were collected from a retrospective search of patients aged ≤18 years in the Washington University School of Medicine, University of California San Francisco and Nationwide Children's Hospital electronic medical records between 1989 and 2017. Review of medical records and specimen collection was approved by the Institutional Review Boards of the individual institutions involved in the study.
Clinical review of patient data
Patients' charts and pathology reports were reviewed for the following data: age at diagnosis, sex, familial syndrome(s), WHO grade, tumor location, treatment, recurrence, death and any additional diagnostic workup (immunohistochemistry [IHC] or fluorescence in situ hybridization [FISH] testing). NF2 status was inferred based on the modified Baser criteria (6) in instances where it was not explicitly stated in the patient's chart.
Pathology review of tumor specimens
Stored slides were retrieved and reviewed for 46 (of 50) individuals included in the study cohort. Per the 2016 WHO Classification of Tumors of the Central Nervous System (4), tumors were designated as grade II, atypical if they demonstrated ≥4 mitoses per 10 high power fields (400×, HPF), brain invasion and/or three of the following five “soft” criteria: (i) sheeting architecture, (ii) small cell formation, (iii) prominent or macronucleoli, (iv) hypercellularity, (v) spontaneous necrosis. Tumors harboring >50% chordoid or clear cell histology were also designated as grade II. Tumors were designated as grade III (anaplastic) if they demonstrated >50% rhabdoid or papillary histology, ≥20 mitoses per 10 HPF or frank anaplasia (ie, sarcoma‐, carcinoma‐ or melanoma‐like appearance). Detailed data on ancillary studies (ie, progesterone receptor status, epithelial membrane antigen staining, Ki‐67 index, FISH studies and NF2 gene testing) was reviewed and recorded where possible. In four cases lacking available slides and/or blocks, the original grade assignment was maintained.
Next‐generation sequencing
A targeted, hybridization capture assay was used to support high‐depth sequencing of genes with known relevance in cancer, specifically tumors of the CNS. DNA was prepared and sequenced as previously described (14, 39). Briefly, cores were punched from selected areas of formalin‐fixed, paraffin‐embedded (FFPE) tumor blocks. Genomic DNA was isolated from tissue cores using the QIAamp DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). NGS libraries were prepared using the KAPA Hyper Prep Kit (KAPA Biosystems, Wilmington, MA) according to the manufacturer's suggested protocol. Five hundred nanograms of each sequencing library was hybridized to a collection of custom designed, single‐stranded cDNA capture probes to enrich for targeted regions (Table S1). Enriched libraries were sequenced on a NovaSeq6000 (Illumina, San Diego, CA) to generate 2 × 150 bp reads.
Base call (bcl) files from the NovaSeq6000 were demultiplexed and converted to fastq format using bcl2fastq2 (Illumina). Bioinformatic analyses were carried out using open source software implemented in bcbio‐nextgen v1.0.8 (https://github.com/bcbio/bcbio‐nextgen) to predict variants that fall within the coding exons of 98 genes (Table S1). Briefly, paired‐end reads were aligned to the human reference assembly (GRCh37) using bwa‐mem (37) (coverage metrics reported in Table S2). Single nucleotide variants (SNVs) and small insertions and deletions (indels) were predicted using VarScan2 (32), Freebayes (22) and VarDict (35). Copy number variants were inferred from alignments using CNVkit (49). Ten non‐cancer FFPE specimens were used to generate the normal depth profile for the assay; control and test cases were processed according to CNVkit's standard batch pipeline.
The clinical relevance of predicted variants was determined and reported as previously described (36). Benign polymorphisms represented in gnomAD v2.0 (https://gnomad.broadinstitute.org/) were discarded from this analysis. Exonic variants (including chromosomal syntax, protein syntax and clinical classifications) are reported in Table S3.
Immunohistochemistry (IHC)
FFPE tissue blocks were sectioned to 5 µm and mounted on poly‐L‐lysine coated slides. Slides were stained using a monoclonal rabbit antibody against human trimethylated H3K27 (clone C36B11, 1:50 dilution, Cell Signaling Technology, Danvers, MA) according to standard histological technique on a Leica Bond 3 automated immunostainer (Leica, Buffalo Grove, IL). Appropriate positive and negative controls were used, and endothelial and inflammatory cells were evaluated as internal positive controls. H3K27me3 status was categorized as "0" denoting lost (<5% cells staining) vs. retained (>5% cells staining) expression indicated as “1.”
Similarly, FFPE slides were stained using a 1:100 dilution of a monoclonal mouse antibody against human BRAF p.V600E (Spring Bioscience, Pleasanton, CA) with the assistance of a Benchmark Ultra IHC instrument (Ventana Medical Systems, Inc., Tucson, AZ). Again, endothelial and inflammatory cells served as internal negative controls.
Statistical analysis
The distributions of recurrence‐free survival (RFS) and overall survival (OS) were described using the Kaplan–Meier product‐limit method and compared by logrank test. The optimal threshold of mitotic counts was searched using software X‐Tile (12). All other analyses were performed using SAS 9.4 (SAS Institutes, Cary, NC). The overall predictive ability of RFS curves for the stated mitotic count thresholds was also summarized using Harrell's Concordance Statistics (C‐Statistics, (24)) which ranged from 0.5 to 1.0, with higher values indicating stronger association.
Results
Clinical and histopathological characteristics
A search of medical records from three institutions yielded 50 cases of pediatric meningioma with available clinicopathologic data (Table S3). The 22 females and 28 males included in this cohort ranged in age from 2 to 18 years (average 12.76 ± 4.17 years). Thirty‐eight of the described specimens were taken following the initial diagnosis while 12 represented recurrent tumors in cases where primary tumor specimens were not available. Nine of the 12 recurrent tumors were taken from male patients. Excluding the recurrent tumors, the average age at initial diagnosis was somewhat younger for males compared to females (11.4 ± 4.09 and 14.16 ± 3.40 years, respectively; P = 0.031 for two‐tailed t test with equal variance). The median time to follow‐up was 60 months (range: 2–282 months) in the 47 patients with accessible data. Seven deaths and 13 recurrences of the primary tumor were documented. Nineteen patients were disease‐free at the time of data collection.
Clinical data from 14 individuals (seven females, seven males) described a diagnosis of Neurofibromatosis type 2 (NF2); a 15 patient's was classified as positive for NF2 based on clinical imaging data using the modified Baser criteria (6) (Table S3). All seven of the individuals in this cohort with multiple primary tumors had a clinical NF2 diagnosis. Of the 12 specimens taken in the recurrent setting, only three were from individuals with NF2.
Tumor grade
The present cohort contained 24 WHO grade I, 20 WHO grade II and 6 WHO grade III meningiomas. Approximately 30% of the tumors in this cohort had a high mitotic index (n = 15 with ≥4 mitoses per 10 HPF). Of the 20 grade II tumors, 11 were graded on ≥4 mitoses per 10 HPF, five on specific cytology (two chordoid and three clear cell) and two on CNS parenchymal invasion; two were graded solely on “soft” criteria (see methods). The sex ratio was balanced for grades I and III, but a skew toward males (13 males, 7 females) was noted among the grade II tumors.
Tumor location
Information on the specific anatomical location of the tumor was available for 47 of 50 cases. The predominant location was convexity (n = 21), followed by skull base (n = 16), and spine (n = 6). One tumor each localized to the ventricular wall, the orbit and the subcutis of the scalp. A skew in favor of skull base tumors was noted among NF2 patients (9 of 15 patients) and females (10 of 19 patients).
Loss of function variants in NF2
Additional material was available to facilitate targeted NGS on DNA isolated from 38 of the 50 tumors. Sequence variants were identified from the coding exons of 98 genes relevant to meningioma and other tumors of the central nervous system (Figure 1, Table S3). NGS data were also used to infer copy number data across the genome (Figure 2). As expected, the majority of the pathogenic and likely pathogenic (P/LP) variants identified in this cohort were inactivators of NF2. Eighteen cases (47.4%) had SNVs or indels in NF2; all of these were previously characterized as deleterious (Figure 3). The tumor‐only NGS assay employed in this study cannot confidently discriminate germline from somatic mutations, but the NF2 variants tended to have high variant allelic fraction compared to other likely‐somatic variants identified in this cohort (range 27.63%–75.79% and average 56.36 ± 16.20% for patients without NF2 diagnosis compare to range 59.09%–83.93% and average 75.00 ± 10.34% for patients with NF2 diagnosis). All but one of the loss‐of‐function sequence variants in NF2 were present in combination with chromosome 22 loss (Figure 1).
Figure 1.
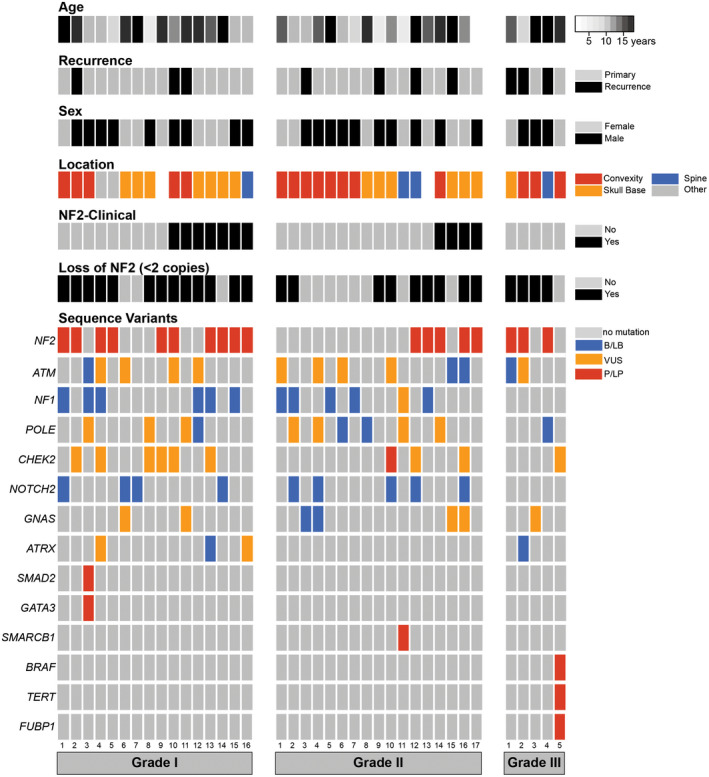
Copy number and sequence variants identified in pediatric meningiomas. Discrete copy number and non‐synonymous sequence variants are shown for the 16 Grade I, 17 Grade II and 5 Grade III tumors that were sequenced for this study. Ninety‐eight genes specifically relevant to CNS tumors were interrogated (Table S1); all mutations present in this cohort are described in Table S3; only the most frequently mutated genes are described in the figure. Abbreviations are as follows: neurofibromatosis type 2, NF2; benign or likely benign, B/LB; uncertain clinical significance, VUS; pathogenic or likely pathogenic, P/LP.
Figure 2.
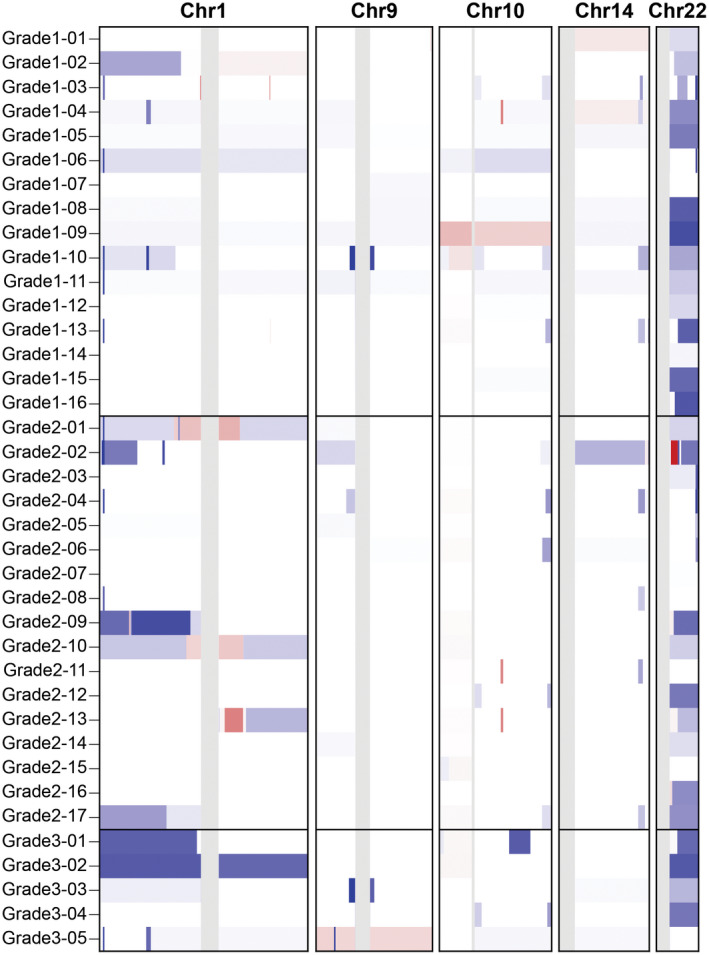
Copy number variants in pediatric meningioma. Copy number losses (blue) and gains (red) are plotted across the chromosomes 1, 9, 10, 14 and 22. The most frequent alteration noted in this cohort was the loss of 22q. Losses on chromosome 1 were noted in 11 cases. No recurrent variants were detected involving chromosomes 9, 10 or 14.
Figure 3.
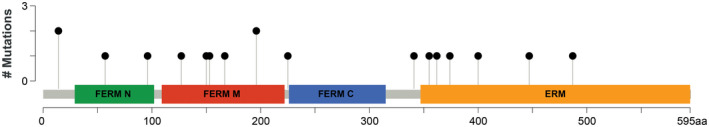
Pathogenic mutations in the NF2 gene. Pathogenic, loss‐of‐function mutations were identified along the length of the NF2 gene, including 3 of 4 functional protein domains.
Eight of the 11 sequenced patients with a clinical NF2 diagnosis had pathogenic sequence variants in the NF2 gene and another showed NGS‐based evidence of chromosome 22 loss (Figure 1). Two NF2 patients had no molecular data to corroborate their assigned status. It is possible that their disease is driven by a large indel or another variant that is not reliably detected using short‐read NGS technology, as these variant types are well‐known in the setting of NF2 disease (26, 42, 51).
Other pathogenic sequence variants
While a number of benign variants and variants of uncertain significance were identified (Figure 1), P/LP variants outside NF2 were rare in this cohort. Only four cases contained P/LP point mutations outside NF2 (Figure 1, Table 1); none of these cases had concomitant NF2 mutation or clinical NF2 diagnosis. One case of rhabdoid meningioma (Figure S1) had a p.V600E mutation in BRAF alongside an activating mutation in the TERT promoter. Expression of the BRAF p.V600E variant was confirmed via IHC (Figure 4). Another case of clear cell meningioma (Figure S1) had a P/LP variant in SMARCB1. These variants are unusual in the context of meningioma, but they are well‐known pathogenic variants in other cancers (16, 29). Besides NF2, the most frequently mutated genes in this cohort were ATM (13 cases), NF1 (12 cases), POLE (11 cases) and CHEK2 (10 cases); however, almost all these mutations were classified as likely benign or having uncertain clinical significance.
Table 1.
Pathogenic mutations outside NF2.
| Patient | Gene | Variant genomic syntax (protein syntax) | Variant allelic fraction (%) | Consequence |
|---|---|---|---|---|
| Grade 1–03 | SMAD2 | 18:45395609C > CCTACCTGGTGTCTCAACTCTCTGATAGTGGTAAAAAAGGGAAAAAGCCCCAATTTTT (splice region) | 6.84 | Splice |
| Grade 1–03 | GATA3 | 10:8115779C > A (p.Cys376Ter) | 3.29 | Missense |
| Grade 2–10 | CHEK2 | 22:29121087A > G (p.Ile200Thr) | 55.84 | Missense |
| Grade 2–11 | SMARCB1 | 22:24143269G > A (splice donor) | 70.79 | Nonsense |
| Grade 3–05 | BRAF | 7:140453136A > T (p.Val600Glu) | 56.05 | Missense |
| Grade 3–05 | TERT | 5:1295228G > A (promoter) | 29.95 | Promoter |
| Grade 3–05 | FUBP1 | 1:78414837T > C (splice) | 41.21 | Splice |
Figure 4.
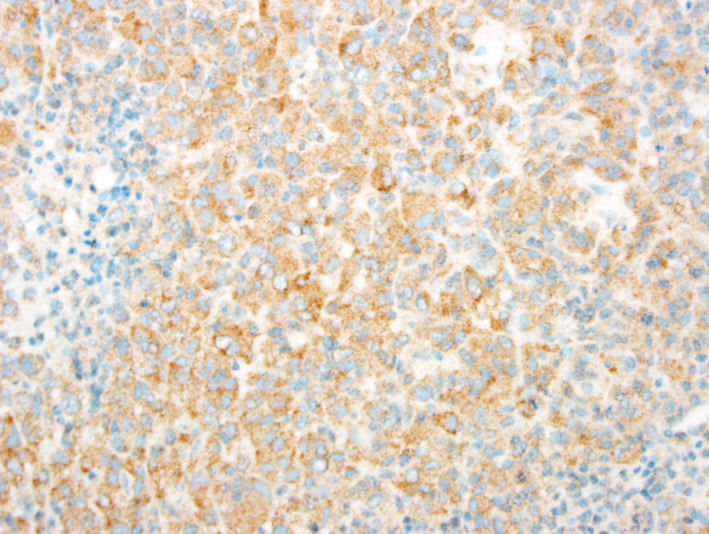
Immunohistochemical staining of BRAF p.V600E. Immunohistochemical staining using an antibody against human BRAF p.V600E indicated strong staining (brown) in a specimen found to harbor the mutation by next‐generation sequencing (see specimen Grade 3–05 in Table S3).
Copy number variation (CNV)
Loss of chromosome 22 was prevalent among tumors of all grades, occurring in 26 of 38 (68%) of sequenced specimens (Figures 1 and 2). Copy number losses on chromosome 1 were noted in 11 cases (29%), 9 of which also showed loss of chromosome 22 (Figure 2). No losses were detected on chromosome 10 or chromosomes 1p/14q.
H3K27me3 protein expression
The expression of trimethylated H3K27 was assessed by immunohistochemistry in 46 tumor specimens with available material. Staining was successful in 45 of 46 specimens; none of these demonstrated loss of H3K27me3 expression (Figure 5).
Figure 5.
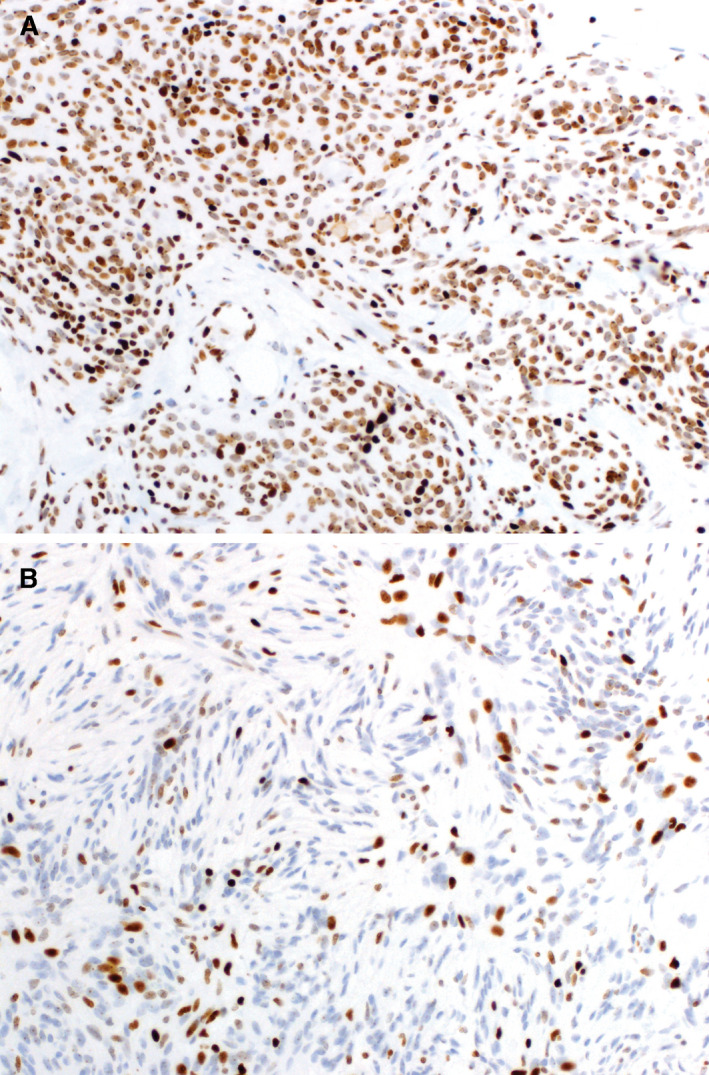
Immunohistochemical staining of H3K27me3. Immunohistochemical staining using an antibody against human trimethylated H3K27 showed strong (A) or patchy (B) staining of tumor nuclei, demonstrating the expression of H3K27me3. Complete loss of staining (defined as staining of <5% of nuclei tumor, per Katz et al (30)) was not observed in this cohort.
Recurrence‐free and overall survival
Considering all patients together, grade III tumors had a significantly worse recurrence‐free survival (RFS) and overall survival (OS) compared to grades I and II (P = 0.02 and P = 0.001, respectively, Figure 6A,B), but there was no difference in RFS or overall OS between grades I and II. Tumor location did not affect RFS or OS in uni‐ or multivariate analyses in this cohort, nor did it specifically correlate with the histological grade. Likewise, none of the molecular features identified in this study were shown to correlate with differences in patient outcome.
Figure 6.
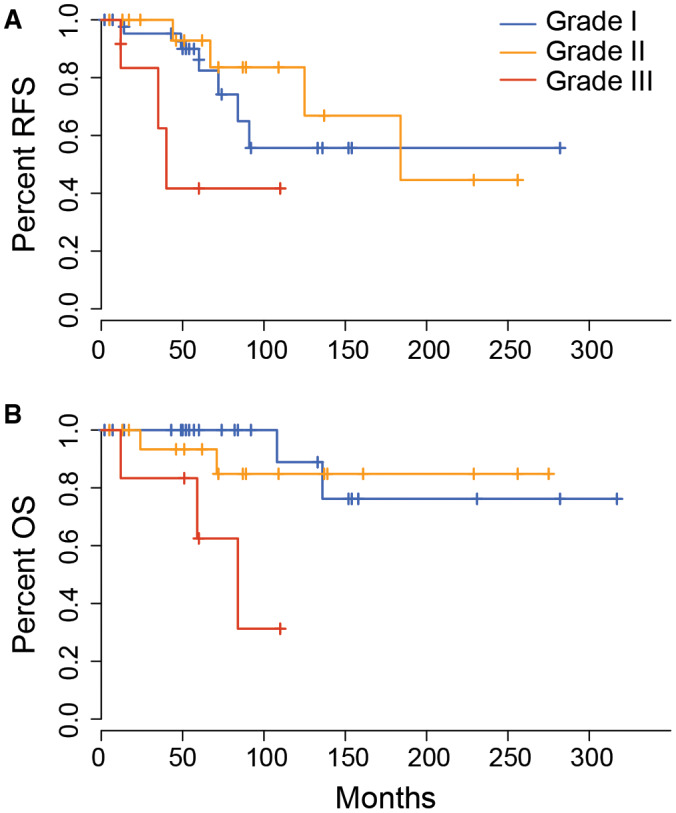
The impact of grade on recurrence‐free and overall survival in pediatric meningioma. Kaplan–Meier plots indicate a difference in recurrence‐free survival (A) and overall survival (B) between patients with grade III tumors compared to those with grade I and grade II (log rank test P = 0.046 and P = 0.000181, respectively).
An exploratory analysis was performed to specifically assess the impact of mitotic count on patient outcomes. Grades I and II are typically separated based on a count of ≥4 mitoses per 10 HPF. There was no significant difference in RFS in all patients separated based on presence/absence of at least four mitoses per 10 HPF (Figure 7A, P = 0.383). This was also true when considering NF2 and non‐NF2 patients separately. In our cohort, a mitotic count of ≥6 per 10 HPF was identified as the optimal cutoff in predicting RFS (Figure 7B, P = 0.002, C‐statistics = 0.69). In contrast, the standard threshold (≥4 mitoses) showed a relatively weak predictive accuracy on RFS (C‐statistics = 0.63).
Figure 7.
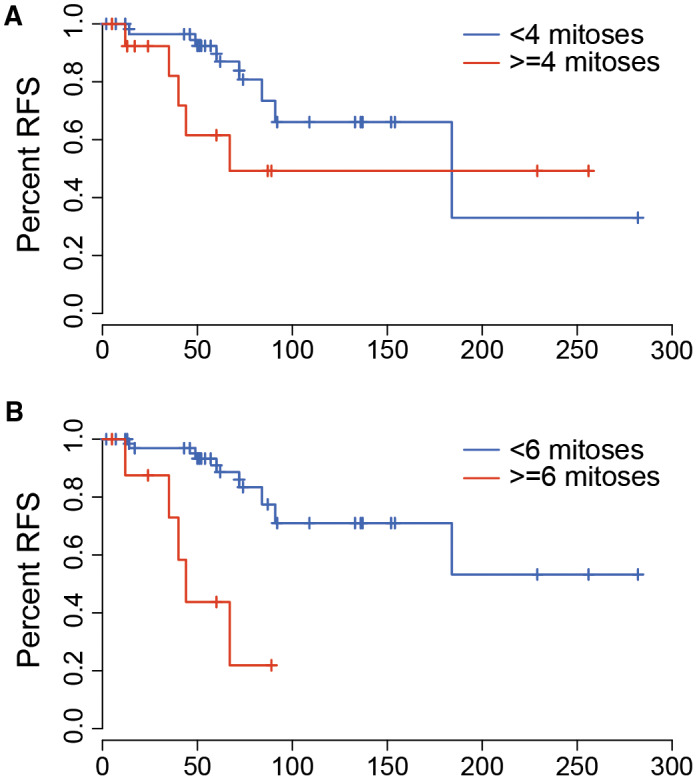
The impact of mitotic count on recurrence‐free survival in pediatric meningioma. Kaplan–Meier plots indicate no difference in recurrence‐free survival in patients using a cutoff of four mitoses per 10 high powered (40×) fields (log rank test P = 0.383, C‐statistics = 0.63). However, significant differences in RFS were seen when using a cutoff of 6 (P = 0.002, C‐statistics = 0.69).
Discussion
In this study, we collected data from 50 pediatric meningioma patients, including all three WHO grades, NF2‐related and sporadic tumors, and various histological subtypes. Our study confirmed many of the clinical features that have been previously reported for pediatric meningioma. For example, we noted a slight skew toward male patients (22 females vs. 28 males), a higher frequency of tumors in uncommon sites (n = 7 in the spine) compared to adult populations, a higher prevalence of skull base tumors in both female patients and patients with clinical NF2 diagnoses, and an increased frequency of tumors with high mitotic counts.
Thirty‐eight of 50 tumors were sequenced to explore their mutational profile. The NGS‐based characterization further emphasized the distinction between adult and pediatric meningioma. P/LP mutations outside NF2 were rare, but some were found in genes that were not previously described in adult or pediatric meningioma populations (eg, SMAD2, GATA3, CHEK2).
Copy number variants were less frequent in our cohort compared to adult meningioma (39) and previous pediatric studies (7). We did not see any loss of chromosome 10, which is a notable feature of high‐grade meningioma in adults (39). Likewise, we did not identify any chromosome 1p and 14q codeletion events, which were reported in 28.5% of high grade tumors in previous study (total n = 7 pediatric tumors graded as II and III) (7). This discrepancy could be due to methodological differences or due to differences in the patient population (eg, prevalence of NF2 patients in the current study as compared to the predominance of sporadic meningiomas in the previous study).
H3K27me3 suppresses transcription of nearby genes that are typically turned on during development; conversely, hypomethylation can allow unchecked cell division. Hypomethylation of H3K27, which serves as a predictor of poor prognosis in adult meningioma (30), was not noted in any of our patients, as trimethylation was retained in 100% of specimens (n = 45). This is notably interesting given the study by Katz et al (30), where hypomethylation of H3K27 was seen in 25 (10.8%) of 232 adult meningiomas. Of note, Katz et al reported that 26% (9 of 34) of NF2 mutant tumors were associated with hypomethylation compared to none of the 50 NF2 wild‐type tumors (30). If there is a causal link between NF2 mutation status and hypomethylation of H3K27, our study argues this may be less common in the pediatric setting.
A recent study highlighted the potential role of YAP1 fusions in pediatric meningiomas without NF2 inactivation (47). Unfortunately, the NGS assay selected for this study does not support the identification of mutations or fusions in this gene. This remains a limitation of our study, and the role of gene fusions, particularly in patients without other well‐characterized pathogenic mutations, should be assessed in future work. Other notable features, such as methylation or other epigenetic modifications should also be considered.
Forty percent of the specimens included in this study were classified as grade II (n = 20). Grade II tumors are particularly challenging in the clinical setting, as evidenced by their increased risk for recurrence (45). A cutoff of four mitoses per 10 high powered microscopic fields (HPF; one HPF equal to 0.20 mm2) is typically used to distinguish WHO grade I from WHO grade II tumors. In our cohort, a cutoff of four mitoses per 10 HPF did not have a significant correlation with RFS; a cutoff of six was identified as the optimal threshold in predicting RFS. Lack of predictability in tumor behavior can hamper optimal treatment, including over‐treating tumors which will not likely recur (1, 2, 43, 44, 48). Though delivery and dosage of radiation therapy to intracranial neoplasms is becoming more sophisticated with better targeting and newer modalities (11), there is still concern for adverse effects, particularly to the developing brain (25). Due to limited sample size, our study lacks adequate power to support the adjustment of clinical guidelines at this time. However, if larger multi‐institution studies independently validate our conclusions, revised classification criteria, specifically mitotic count threshold, may aid in risk stratifying pediatric patients and directing therapeutic decisions.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. Histopathological characterization of two cases with unusual variant profiles. Histopathology of a rhabdoid meningioma harboring BRAF p.V600E and TERT promoter variants (see Grade 3–05 in Table S3) demonstrated areas of necrosis, rhabdoid appearance of tumor cells with moderate to abundant cytoplasm, and frequent mitoses (A, hematoxylin and eosin stain shown at 400× magnification) along‐with patchy epithelial membrane antigen immunoreactivity (B, epithelial membrane antigen stain shown at 400× magnification). The tumor was negative for glial (GFAP, OLIG2), myogenic (Myf4, myogenin, MyoD1), and/or melanocytic (S100, HMB45, melan‐A), differentiation and INI‐1 was retained. A clear cell meningioma example (see Grade 2–11 in Table S3) harboring SMARCB1 mutation demonstrated clear cells with conspicuous collagen deposition in the stroma (C, hematoxylin and eosin stain shown at 400× magnification) along‐with patchy, membranous, EMA reactivity (D, epithelial membrane antigen stain shown at 400× magnification). There was also strong expression of vimentin, and non‐reactivity for glial (GFAP) and neuronal (SYN and NFP) markers.
Table S1. Genes enriched for seuqencing and subjected to variant calling.
Table S2. Sequencing metrics for 38 specimens of pediatric meningioma.
Table S3. All described features of 50 pediatric meningioma specimens.
Acknowledgments
We would like to thank the Washington University Division of Neuropathology, Department of Pathology and Immunology for funding this study and the Genome Technology Access Center at Washington University for assistance with next‐generation sequencing. We would also like to thank Jingqin Luo, PhD and Feng Gao, PhD of the Washington University Department of Public Health for their assistance with statistical analyses.
Presented in part at the International Congress of Neuropathology meeting in Tokyo, Japan, September 17‐23, 2018.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Aghi MK, Carter BS, Cosgrove GR, Ojemann RG, Amin‐Hanjani S, Martuza RL et al (2009) Long‐term recurrence rates of atypical meningiomas after gross total resection with or without postoperative adjuvant radiation. Neurosurgery 64:56–60. [DOI] [PubMed] [Google Scholar]
- 2. Aizer AA, Arvold ND, Catalano P, Claus EB, Golby AJ, Johnson MD et al (2014) Adjuvant radiation therapy, local recurrence, and the need for salvage therapy in atypical meningioma. Neuro Oncol 16:1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albrecht S, Goodman JC, Rajagopolan S, Levy M, Cech DA, Cooley LD (1994) Malignant meningioma in Gorlin's syndrome: cytogenetic and p53 gene analysis. Case report. J Neurosurg 81:466–471. [DOI] [PubMed] [Google Scholar]
- 4. Ansari J, Shackelford RE, El‐Osta H (2016) Epigenetics in non‐small cell lung cancer: from basics to therapeutics. Transl Lung Cancer Res 5:155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arivazhagan A, Devi BI, Kolluri SVR, Abraham RG, Sampath S, Chandramouli BA (2008) Pediatric intracranial meningiomas – do they differ from their counterparts in adults? Pediatr Neurosurg 44:43–48. [DOI] [PubMed] [Google Scholar]
- 6. Baser ME, Friedman JM, Joe H, Shenton A, Wallace AJ, Ramsden RT, Evans DGR (2011) Empirical development of improved diagnostic criteria for neurofibromatosis 2. Genet Med 13:576–581. [DOI] [PubMed] [Google Scholar]
- 7. Battu S, Kumar A, Pathak P, Purkait S, Dhawan L, Sharma MC et al (2018) Clinicopathological and molecular characteristics of pediatric meningiomas. Neuropathology 38:22–33. [DOI] [PubMed] [Google Scholar]
- 8. Bi WL, Abedalthagafi M, Horowitz P, Agarwalla PK, Mei Y, Aizer AA et al (2016) Genomic landscape of intracranial meningiomas. J Neurosurg 125:525–535. [DOI] [PubMed] [Google Scholar]
- 9. Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK et al (2017) Genomic landscape of high‐grade meningiomas. NPJ Genom Med 2:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bi WL, Mei Y, Agarwalla PK, Beroukhim R, Dunn IF (2016) Genomic and epigenomic landscape in meningioma. Neurosurg Clin N Am 27:167–179. [DOI] [PubMed] [Google Scholar]
- 11. Bindra RS, Wolden SL (2015) Advances in radiation therapy in pediatric neuro‐oncology. J Child Neurol 31:506–516. [DOI] [PubMed] [Google Scholar]
- 12. Camp RL, Dolled‐Filhart M, Rimm DL (2004) X‐tile: a new bio‐informatics tool for biomarker assessment and outcome‐based cut‐point optimization. Clin Cancer Res 10:7252–7259. [DOI] [PubMed] [Google Scholar]
- 13. Caroli E, Russillo M, Ferrante L (2006) Intracranial meningiomas in children: report of 27 new cases and critical analysis of 440 cases reported in the literature. J Child Neurol 21:31–36. [DOI] [PubMed] [Google Scholar]
- 14. Cottrell CE, Al‐Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ et al (2014) Validation of a next‐generation sequencing assay for clinical molecular oncology. J Mol Diagn 16:89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dash C, Pasricha R, Gurjar H, Singh PK, Sharma BS (2016) Pediatric intraventricular meningioma: a series of six cases. J Pediatr Neurosci 11:193–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. [DOI] [PubMed] [Google Scholar]
- 17. Dudley RWR, Torok MR, Randall S, Beland B, Handler MH, Mulcahy‐Levy JM et al (2018) Pediatric versus adult meningioma: comparison of epidemiology, treatments, and outcomes using the Surveillance, Epidemiology, and End Results database. J Neurooncol 137:621–629. [DOI] [PubMed] [Google Scholar]
- 18. Elbabaa SK, Gokden M, Crawford JR, Kesari S, Saad AG (2012) Radiation‐associated meningiomas in children: clinical, pathological, and cytogenetic characteristics with a critical review of the literature. J Neurosurg‐Pediatr 10:281–290. [DOI] [PubMed] [Google Scholar]
- 19. Erdinçler P, Lena G, Sarioğlu AÇ, Kuday C, Choux M (1998) Intracranial meningiomas in children: review of 29 cases. Surg Neurol 49:136–140. [DOI] [PubMed] [Google Scholar]
- 20. Felicetti F, Fortunati N, Garbossa D, Biasin E, Rudà R, Daniele D et al (2015) Meningiomas after cranial radiotherapy for childhood cancer: a single institution experience. J Cancer Res Clin Oncol 141:1277–1282. [DOI] [PubMed] [Google Scholar]
- 21. Galloway TJ, Indelicato DJ, Amdur RJ, Swanson EL, Morris CG, Marcus RB (2011) Favorable outcomes of pediatric patients treated with radiotherapy to the central nervous system who develop radiation‐induced meningiomas. Int J Radiat Oncol Biol Phys 79:117–120. [DOI] [PubMed] [Google Scholar]
- 22. Garrison E, Marth G (2012) Haplotype‐based variant detection from short‐read sequencing. arXiv. (arXiv:1207.3907). [Google Scholar]
- 23. Harmanci AS, Youngblood MW, Clark VE, Coskun S, Henegariu O, Duran D et al (2017) Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun 8:14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrell FE Jr, Lee KL, Califf RM, Pryor DB, Rosati RA (1984) Regression modelling strategies for improved prognostic prediction. Stat Med 3:143–152. [DOI] [PubMed] [Google Scholar]
- 25. Hoffman KE, Yock TI (2009) Radiation therapy for pediatric central nervous system tumors. J Child Neurol 24:1387–1396. [DOI] [PubMed] [Google Scholar]
- 26. Honda M, Arai E, Sawada S, Ohta A, Niimura M (1995) Neurofibromatosis 2 and neurilemmomatosis gene are identical. J Invest Dermatol 104:74–77. [DOI] [PubMed] [Google Scholar]
- 27. Hui M, Uppin MS, Saradhi MV, Sahu BP, Purohit AK, Sundaram C (2015) Pediatric meningiomas an aggressive subset: a clinicopathological and immunohistochemical study. J Postgrad Med 61:32–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huntoon K, Pluto CP, Ruess L, Boué DR, Pierson CR, Rusin JA, Leonard J (2017) Sporadic pediatric meningiomas: a neuroradiological and neuropathological study of 15 cases. J Neurosurg Pediatr 20:141–148. [DOI] [PubMed] [Google Scholar]
- 29. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katz LM, Hielscher T, Liechty B, Silverman J, Zagzag D, Sen R et al (2018) Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol 135:955–963. [DOI] [PubMed] [Google Scholar]
- 31. Kimonis VE, Mehta SG, Digiovanna JJ, Bale SJ, Pastakia B (2004) Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet Med 6:495–502. [DOI] [PubMed] [Google Scholar]
- 32. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L et al (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kotecha RS, Junckerstorff RC, Lee S, Cole CH, Gottardo NG (2011) Pediatric meningioma: current approaches and future direction. J Neuro‐Oncol 104:1–10. [DOI] [PubMed] [Google Scholar]
- 34. Kotecha RS, Pascoe EM, Rushing EJ, Rorke‐Adams LB, Zwerdling T, Gao X et al (2011) Meningiomas in children and adolescents: a meta‐analysis of individual patient data. Lancet Oncol 12:1229–1239. [DOI] [PubMed] [Google Scholar]
- 35. Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R et al (2016) VarDict: a novel and versatile variant caller for next‐generation sequencing in cancer research. Nucleic Acids Res 44:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S et al (2017) Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19:4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Z, Li H, Jiao Y, Ma J, Wang S, Zhao J, Cao Y (2017) A comparison of clinicopathological features and surgical outcomes between pediatric skull base and non‐skull base meningiomas. Child Nerv Syst 33:595–600. [DOI] [PubMed] [Google Scholar]
- 39. McNulty SN, Schwetye K, Goldstein M, Carter J, Schmidt RE, Ansstas G et al (2018) Analysis of point mutations and copy number variation in Grade II and III meningioma. Exp Mol Pathol 105:328–333. [DOI] [PubMed] [Google Scholar]
- 40. Olar A, Wani KM, Wilson CD, Zadeh G, DeMonte F, Jones DTW et al (2017) Global epigenetic profiling identifies methylation subgroups associated with recurrence‐free survival in meningioma. Acta Neuropathol 133:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ostrom QT, Gittleman H, Liao P, Vecchione‐Koval T, Wolinsky Y, Kruchko C, Barnholtz‐Sloan JS (2017) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro‐Oncol 19(suppl_5):v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parry DM, MacCollin MM, Kaiser‐Kupfer MI, Pulaski K, Nicholson HS, Bolesta M et al (1996) Germ‐line mutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet 59:529–539. [PMC free article] [PubMed] [Google Scholar]
- 43. Perry A, Dehner LP (2003) Meningeal tumors of childhood and infancy. An update and literature review. Brain Pathol 13:386–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perry A, Giannini C, Raghavan R, Scheithauer BW, Banerjee R, Margraf L et al (2001) Aggressive phenotypic and genotypic features in pediatric and NF2‐associated meningiomas: a clinicopathologic study of 53 cases. J Neuropathol Exp Neurol 60:994–1003. [DOI] [PubMed] [Google Scholar]
- 45. Rogers L, Barani I, Chamberlain M, Kaley T, McDermott M, Raizer J et al (2015) Meningiomas: knowledge base, treatment outcomes, and uncertainties. A RANO review. J Neurosurg 122:4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sahm F, Toprak UH, Hübschmann D, Kleinheinz K, Buchhalter I, Sill M et al (2017) Meningiomas induced by low‐dose radiation carry structural variants of NF2 and a distinct mutational signature. Acta Neuropathol 134:155–158. [DOI] [PubMed] [Google Scholar]
- 47. Sievers P, Chiang J, Schrimpf D, Stichel D, Paramasivam N, Sill M et al (2019) YAP1‐fusions in pediatric NF2‐wildtype meningioma. Acta Neuropathol 139:215–218. [DOI] [PubMed] [Google Scholar]
- 48. Sun SQ, Hawasli AH, Huang J, Chicoine MR, Kim AH (2015) An evidence‐based treatment algorithm for the management of WHO Grade II and III meningiomas. Neurosurg Focus 38:E3. [DOI] [PubMed] [Google Scholar]
- 49. Talevich E, Shain AH, Botton T, Bastian BC (2016) CNVkit: genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thuijs NB, Uitdehaag BMJ, Van Ouwerkerk WJR, van der Valk P, Vandertop WP, Peerdeman SM (2012) Pediatric meningiomas in the Netherlands 1974–2010: a descriptive epidemiological case study. Child Nerv Syst 28:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zucman‐Rossi J, Legoix P, Der Sarkissian H, Cheret G, Sor F, Bernardi A et al (1998) NF2 gene in neurofibromatosis type 2 patients. Hum Mol Genet 7:2095–2101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Histopathological characterization of two cases with unusual variant profiles. Histopathology of a rhabdoid meningioma harboring BRAF p.V600E and TERT promoter variants (see Grade 3–05 in Table S3) demonstrated areas of necrosis, rhabdoid appearance of tumor cells with moderate to abundant cytoplasm, and frequent mitoses (A, hematoxylin and eosin stain shown at 400× magnification) along‐with patchy epithelial membrane antigen immunoreactivity (B, epithelial membrane antigen stain shown at 400× magnification). The tumor was negative for glial (GFAP, OLIG2), myogenic (Myf4, myogenin, MyoD1), and/or melanocytic (S100, HMB45, melan‐A), differentiation and INI‐1 was retained. A clear cell meningioma example (see Grade 2–11 in Table S3) harboring SMARCB1 mutation demonstrated clear cells with conspicuous collagen deposition in the stroma (C, hematoxylin and eosin stain shown at 400× magnification) along‐with patchy, membranous, EMA reactivity (D, epithelial membrane antigen stain shown at 400× magnification). There was also strong expression of vimentin, and non‐reactivity for glial (GFAP) and neuronal (SYN and NFP) markers.
Table S1. Genes enriched for seuqencing and subjected to variant calling.
Table S2. Sequencing metrics for 38 specimens of pediatric meningioma.
Table S3. All described features of 50 pediatric meningioma specimens.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.