

Abstract
Temporal lobe epilepsy (TLE) is defined as the sporadic occurrence of spontaneous recurrent seizures, and its pathogenesis is complex. SHP‐2 (Src homology 2‐containing protein tyrosine phosphatase 2) is a widely expressed cytosolic tyrosine phosphatase protein that participates in the regulation of inflammation, angiogenesis, gliosis, neurogenesis and apoptosis, suggesting a potential role of SHP‐2 in TLE. Therefore, we investigated the expression patterns of SHP‐2 in the epileptogenic brain tissue of intractable TLE patients and the various effects of treatment with the SHP‐2‐specific inhibitor SHP099 on a pilocarpine model. Western blotting and immunohistochemistry results confirmed that SHP‐2 expression was upregulated in the temporal neocortex of patients with TLE. Double‐labeling experiments revealed that SHP‐2 was highly expressed in neurons, astrocytes, microglia and vascular endothelial cells in the epileptic foci of TLE patients. In the pilocarpine‐induced C57BL/6 mouse model, SHP‐2 upregulation in the hippocampus began one day after status epilepticus, reached a peak at 21 days and then maintained a significantly high level until day 60. Similarly, we found a remarkable increase in SHP‐2 expression at 1, 7, 21 and 60 days post‐SE in the temporal neocortex. In addition, we also showed that SHP099 increased reactive gliosis, the release of IL‐1β, neuronal apoptosis and neuronal loss, while reduced neurogenesis and albumin leakage. Taken together, the increased expression of SHP‐2 in the epileptic zone may be involved in the process of TLE.
Keywords: apoptosis, blood–brain barrier, gliosis, hippocampus, neurogenesis, SHP‐2, temporal lobe epilepsy
Introduction
TLE is one of the most commonly acquired seizure disorders and is often resistant to antiepileptic drug therapy 20. The unpredictable and recurrent nature of seizures associated with TLE is the most disabling feature of this devastating disease. Epilepsy involves several processes, such as neuronal loss, neurogenesis, gliosis, gene regulation, axonal sprouting, synaptic plasticity and inflammation 36, 43. However, to date, the molecular and biochemical mechanisms of TLE are still incompletely understood. Over the past few decades, much attention has been shifted toward ion channels, neuroinflammation, neurotransmitter receptors and extracellular proteinases 36 regulation in epileptogenesis and seizure propagation; however, little research has focused on the function of intracellular enzymes.
SHP‐2 is a classical nonreceptor protein tyrosine phosphatase with two src homology 2 domains (N‐terminal SH2, C‐terminal SH2) 4. Mammalian SHP‐2 was previously identified by several groups and is variably named SH‐PTP2, SH‐PTP3, PTP2C, PTP1D, PTPN11 and Syp 40, 51. Molecular biology and genetic studies have shown that SHP‐2 regulates signaling events downstream of several growth factor/cytokine receptors in various cell types 6. SHP‐2 is highly expressed in neural precursor cells, neurons, astrocytes and oligodendrocytes 26, 38. The significance of SHP‐2 in the mammalian central nervous system cannot be ignored. For instance, deletion of SHP‐2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality 23. Its role in the focal adhesion, differentiation and migration of neural stem cells was demonstrated by a variety of experimental data 19. A previous study suggests that SHP‐2 is a critical signal transducer downstream of SDF‐1α/CXCR4 in guiding granule cell migration during cerebellar development 16. It has been widely accepted that the development of epilepsy involves the abnormal proliferation and migration of neurons in both human and animal epileptogenic zones 14. Additionally, SHP‐2 is also highly expressed in mature neurons, and may contribute to synaptic transmission, short‐term synaptic plasticity and memory formation in the mature brain under pathologic status 27. Furthermore, SHP‐2 is an appealing molecule in the context of the regulation of neurogenesis and gliogenesis 7, 23. Recently, SHP‐2 was found to participate in many important physiological and pathological processes, including the regulation of synaptic plasticity 27, 30, neurite outgrowth 11, 41, neuronal excitotoxicity 42, neuronal death 13 and oxidative stress 21. Notably, these processes are also critical for the occurrence and development of temporal lobe epilepsy. However, little is known about the role of SHP‐2 in TLE.
In this study, we utilized western blotting to measure SHP‐2 expression in the epileptogenic foci of TLE patients. Immunohistochemistry and double‐immunofluorescence labeling were used to examine the cellular distributions of SHP‐2 in the TLE patients. We also investigated the expression of SHP‐2 in the temporal neocortex and hippocampus of a pilocarpine‐induced mouse model. Furthermore, we utilized a potent, selective inhibitor, SHP099, to directly inhibit SHP‐2 activity 3, 12, 18, 29 in mice following pilocarpine‐induced SE. Next, we detected the effects of SHP099 on glial proliferation, neuronal apoptosis, neuronal loss, neurogenesis and albumin leakage after an episode of pilocarpine‐induced SE.
Materials and Methods
Subjects and clinical data
A total of 24 temporal neocortex specimens from patients with medically intractable TLE were examined in the present study. The diagnosis of each drug resistant epilepsy patient was based on the criteria established by the International League Against Epilepsy (ILAE) 28. No other nervous system diseases (glioma, cavernous hemangioma, cyst, cortical dysplasia, etc.) were detected in these patients by brain computerized tomography (CT), magnetic resonance imaging (MRI), interictal and ictal electroencephalogram studies, neuropsychological tests and pathological examinations. The clinical characteristics derived from the patient's medical records are summarized in (Table 1). For control experiments, we used presumably normal temporal neocortices from 10 patients being treated for increased intracranial pressure due to severe traumatic brain injury. In particular, we selected tissues from areas distant from the direct injury area and obtained <4 h after the brain injury 46, 50. None of the controls had a history of neurological or psychiatric disorders, and all temporal neocortex specimens were normal, as confirmed by neuropathological examination. Relevant clinical data for the controls are summarized in (Table 2).
Table 1.
Clinical characteristics of the patients with TLE. AED, antiepileptic drugs; CBZ, carbamazepine; CLB, clonazepam; F, female; G, gliosis; GBP, gabapentin; IHC, Immunohistochemistry; LH, left hippocampus; LTG, lamotrigine; LTN, left temporal neocortex; M, male; NL, neuron loss. PO, postoperative outcome (Engel class); OXC, oxcarbazepine; PB, phenobarbital; PHT, phenytoin; RH, right hippocampus; RTN, right temporal neocortex; TPM, topiramate; VPA, valproic acid; WB, western blotting.
| Case no. | Gender | Age (year) | Duration (year) | AEDs before surgery | Resected tissue | Pathology | PO | Application in present study |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 22 | 8 | CBZ, PB | RTN, RH | G | I | WB |
| 2 | M | 22 | 7 | OXC,VPA, PHT | LTN, LH | G | II | WB, IHC |
| 3 | F | 17 | 3 | CBZ, VPA | LTN | NL | I | WB, IHC |
| 4 | M | 21 | 5 | CBZ, VPA | RTN | G, NL | I | WB, IHC |
| 5 | F | 29 | 11 | CBZ, VPA, CBL | LTN, LH | G, NL | III | WB, IHC |
| 6 | F | 32 | 11 | OXC, CBL, VPA | LTN | G, NL | III | WB, IHC |
| 7 | M | 17 | 3 | OXC, VPA, GBP | LTN | G | I | WB, IHC |
| 8 | F | 18 | 4 | CBZ, VPA | RTN, RH | G, NL | II | WB |
| 9 | M | 33 | 10 | CBZ, VPA, TPM | LTN | G, NL | II | WB, IHC |
| 10 | F | 28 | 12 | VPA, CBZ, PHT | RTN | G | I | WB, IHC |
| 11 | M | 21 | 4 | OXC, VPA | LTN | G, NL | I | WB, IHC |
| 12 | M | 35 | 9 | CBZ, VPA, CBL | RTN | G, NL | III | WB, IHC |
| 13 | F | 34 | 13 | CBZ, VPA, TPM | LTN, LH | G | II | WB, IHC |
| 14 | F | 24 | 8 | OXC, VPA, PHT | RTN | G, NL | I | WB, IHC |
| 15 | M | 27 | 9 | CBZ, PHT, VPA | LTN | G | II | WB, IHC |
| 16 | F | 19 | 5 | OXC, PHT | RTN, RH | G, NL | I | IHC |
| 17 | F | 20 | 4 | OXC, VPA | RTN, RH | G, NL | II | IHC |
| 18 | M | 41 | 22 | CBZ, PHT, TMP | RTN | G, NL | II | IHC |
| 19 | F | 33 | 16 | CBZ, PHT, LTG | RTN | NL | I | IHC |
| 20 | M | 20 | 8 | CBZ, PHT | LTN, LH | G, NL | III | IHC |
| 21 | M | 15 | 3 | VPA, PHT | RTN, RH | G | II | IHC |
| 22 | F | 22 | 6 | CBZ, LTG | LTN | G, NL | II | IHC |
| 23 | F | 37 | 9 | OXC, CBL, VPA | LTN, LH | G, NL | III | IHC |
| 24 | F | 26 | 5 | OXC, VPA | LTN, LH | G, NL | I | IHC |
Table 2.
Clinical data from 10 control subjects. F, female; IHC, Immunohistochemistry; LTN, left temporal neocortex; M, male; RTN, right temporal neocortex; TBI, Traumatic Brain Injury; WB, western blotting.
| Case no. | Gender | Age (year) | Etiologic factor | Resected tissue | Pathology | Seizure | Application in present study |
|---|---|---|---|---|---|---|---|
| 1 | M | 26 | TBI | LTN | Normal | None | WB, IHC |
| 2 | M | 28 | TBI | RTN | Normal | None | WB, IHC |
| 3 | M | 23 | TBI | RTN | Normal | None | WB, IHC |
| 4 | F | 31 | TBI | LTN | Normal | None | WB, IHC |
| 5 | F | 19 | TBI | RTN | Normal | None | WB, IHC |
| 6 | M | 17 | TBI | LTN | Normal | None | WB, IHC |
| 7 | F | 33 | TBI | LTN | Normal | None | WB, IHC |
| 8 | F | 22 | TBI | RTN | Normal | None | WB, IHC |
| 9 | M | 21 | TBI | RTN | Normal | None | WB, IHC |
| 10 | F | 20 | TBI | LTN | Normal | None | IHC |
Animal preparation
Adult male C57BL/6 mice (21–24 g) were purchased from the Daping Hospital Animal Center of the Army Medical University. All mice were housed under climate‐controlled conditions in a 12 h light/dark cycle (lights on 8:00 am to 8:00 pm) and provided with standard food and water.
Mouse model of temporal lobe epilepsy
Mice were injected with scopolamine methyl nitrate (5 mg/kg, i.p., Sigma‐Aldrich, USA) to limit the peripheral effects of the convulsant 22. Thirty minutes later, pilocarpine (280–300 mg/kg, i.p., Sigma‐Aldrich, USA) was administered to induce SE 9. After pilocarpine treatment, the mice were observed for the development of continuous seizure activity. Seizure severity was rated using a modified version of the Racine scale 45: Grades 1 and 2 (ie, facial automatisms, tail stiffening and wet‐dog shakes), Grade 3 (ie, low‐intensity tonic–clonic seizures marked by unilateral forelimb myoclonus in addition to the symptoms above), grade 4 (ie, the addition of bilateral forelimb myoclonus and rearing) and grade 5 (ie, bilateral fore‐ and hindlimb myoclonus and transient loss of postural control). Only those mice that attained grades 4–5 were used for our further study. SE was allowed to continue for up to 2 h, at which point seizure activity was stopped with 1 mg/ml diazepam (4 mg/kg, i.p., Sigma‐Aldrich, USA), and repeated as needed to suppress convulsions. The post‐SE supportive care of the animals included body temperature maintenance and rehydration therapy to increase the number of survivors. Injections (i.p.) of sterile 0.9% saline were administered to replenish fluids. Sliced peeled apples were placed in all mouse cages in addition to rodent chow. The mortality rate of the mice in the first 24 h after SE was 30%. Mice were sacrificed at the following time points after SE: 6 h, 1, 7, 21 and 60 days. Age‐matched control mice received the same treatment with scopolamine methyl nitrate and diazepam, but we used saline instead of pilocarpine.
SHP099 treatment
SHP099 was obtained from MedChem Express, USA (HY‐100388). SHP099 was dissolved in sterile saline and administered by oral gavage at 100 mg/kg daily 12, 18. SHP099 concurrently binds to the interface of the N‐terminal SH2, C‐terminal SH2 and protein tyrosine phosphatase domains, thus inhibiting SHP‐2 activity through an allosteric mechanism 3, 12. SHP099 or vehicle treatment lasted from the 1st day to the 21st day post‐SE. Age‐ and weight‐matched control mice were administered orally, one time per day, for 21 days with vehicle or SHP099 at the same dose. We chose to administer the two substances continuously for 21 days since in our western blotting results, we found peak expression of SHP‐2 on the 21st day in model mice. The final treatment groups were as follows: 1 vehicle only, 2 SHP099 only, 4 pilocarpine + vehicle and 3 pilocarpine + SHP099. Mice were sacrificed after 21 days of treatment. Brains were extracted for, use in an immunohistochemistry assay and western blotting analysis.
Tissue preparation
Tissues from patients were immediately divided into two parts at the time of surgery. One set of tissue was immediately frozen in liquid nitrogen and stored at −80°C for western blotting analysis. The other samples were fixed in 4% phosphate‐buffered paraformaldehyde for 48 h, then embedded in paraffin and sectioned at 6 μm thickness for immunohistochemistry and 10 μm for immunofluorescence staining. Bilateral temporal neocortices and hippocampi from the mice were dissected for protein extraction. For immunohistochemistry observation, the mouse brains were obtained after systemic saline and 4% phosphate‐buffered paraformaldehyde infusion, then further processed as described above for the human tissue.
Western blotting
Total proteins were extracted from tissues using a whole protein extraction kit (Beyotime Institute of Biotechnology, Jiangsu, China). Total tissue lysates were centrifuged at 14 000 × g for 10 min at 4°C, and the protein concentration in the supernatant was determined using a bicinchoninic acid (BCA) protein assay (Bio‐Rad, Hercules, CA, USA). Equal amounts of protein (60 μg/lane) were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) (5% spacer gel, 80 V,25 min; 10% separating gel, 120 V, 60 min) and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Temecula, CA, USA) using a semidry electroblotting system (Transblot SD; Bio‐Rad) (80 min at 300 mA). Next, the membranes were incubated at room temperature for 2 h in 5% nonfat dry milk to block nonspecific binding. Then, the membranes were incubated overnight at 4°C with relevant primary antibodies: anti‐SHP‐2 (ab131541, rabbit polyclonal, 1/1000; Abcam, UK); anti‐SHP‐1 (sc‐287, rabbit polyclonal, 1/500; Santa Cruz Biotechnology, USA); anti‐Albumin (ab19194, goat polyclonal, 1/5000; Abcam, UK); anti‐glial fibrillary acidic protein (GFAP, SAB5201113; mouse monoclonal, 1/500; Sigma); anti‐IL‐1β (ab9722, rabbit polyclonal, 1/1000; Abcam, UK); anti‐Iba1 (016‐20001, Rabbit, 1/800; Wako); anti‐cleaved caspase3 (9664, rabbit polyclonal, 1/500; Cell Signaling Technology; Boston, MA); anti‐Doublecortin (DCX, ab18723; rabbit polyclonal, 1/1000; Abcam, UK) and anti‐GAPDH (ab181602, rabbit monoclonal, 1/10000; Abcam, UK). After several washes in TBST (20 mmol/l Tris‐HCl, pH 8.0, 150 mmol/l NaCl, 0.5% Tween‐20), the samples were treated with horseradish peroxidase‐conjugated goat anti‐rabbit, goat anti‐mouse or rabbit anti‐goat secondary antibody (ZB‐2301, ZB‐2305, ZB‐2306, respectively, 1/1000; Zhongshan Golden Bridge Biotechnology, China) for 1 h in a 37°C incubator. The immunoreactive bands were visualized using enhanced chemiluminescence and were scanned and analyzed with Quantity One software (Bio‐Rad Laboratories, Hercules, CA, USA). The optical densities (ODs) of each protein band were calculated relative to the OD of the reference protein, GAPDH.
Immunohistochemistry
Paraffin‐embedded sections (6 μm) were mounted on polylysine‐coated slides and used for immunohistochemistry (IHC). Paraffin sections were deparaffinized in xylene and rehydrated through a graduated alcohol series. Endogenous peroxidase activity was blocked with 3% H2O2 in methyl alcohol. All of the samples were placed into phosphate‐buffered saline (0.01 M, pH 7.3) and heated for 20 min in a microwave oven for antigen retrieval. Sections were then blocked in bovine serum (Boster Biological Technology, Wuhan, China) for 60 min at room temperature. After removal of excess serum, sections were incubated with primary antibody overnight at 4°C. The following primary antibodies were used: anti‐SHP‐2 (ab131541, rabbit polyclonal, 1/100; Abcam, UK), and anti‐Doublecortin (DCX, ab18723; rabbit polyclonal, 1/1000; Abcam, UK). Anti‐SHP‐2 antibody was used to detect the expression of SHP‐2 in the brain tissue of the patients. The expression of DCX in the hippocampus of mice was detected by anti‐DCX antibody. Then, the sections were incubated with goat anti‐rabbit immunoglobulin conjugated to peroxidase‐labeled dextran polymer (EnVision + System‐HRP; Boster, China) for 1 h at 37°C and subsequently incubated in 3,3‐diaminobenzidine (DAB, Boster) for the appropriate time. The sections were counterstained with hematoxylin, dehydrated and coverslipped. No immunoreactive cells were observed in negative control experiments, which included omission of the primary antibody, preabsorption with a tenfold excess of specific blocking antigen, and incubation with an isotype‐matched rabbit polyclonal antibody. Images of SHP‐2 immunostaining were obtained with a 40X objective for each clinical surgical specimen section (Olympus microscope digital camera system; DP80, Tokyo, Japan). The clinical surgical specimens from 22 TLE patients and 10 controls were used in this study, and three sections were randomly selected from each specimen. Ten nonoverlapping cortical visual fields from each section were randomly chosen for further semiquantitative analysis of SHP‐2 expression (Image‐Pro plus 6.0 software; Media Cybermetrics Inc., USA). For each field, areas of interest were selected for the measurement of integrated optical density (OD) and area. Moreover, each integrated OD/area value was calculated by subtracting the background integrated OD/area value from the directly measured integrated OD/area value. Then, the mean value of these 30 fields was used as the expression level of SHP‐2 for that TLE patient or control. These analyses were conducted in a blinded fashion.
Three consecutive coronal sections from each mouse were selected. The coordinates according to the atlas of Paxinos and Franklin (−1.94 mm caudal to bregma). Images of DCX immunoreactivity were captured using a 40X objective in an Olympus microscope (DP80; Olympus, Tokyo, Japan) and Image‐Pro plus 6.0 software. All of the mouse section samples used for the analysis were viewed in a blinded manner. We randomly selected four fields of view of the dentate gyrus (DG) to obtain the DCX positive immature neuron number of the DG for each mouse. We averaged these results for each group.
Immunofluorescence staining
For double‐immunofluorescence staining , patients' paraffin sections were incubated at 4°C overnight with primary antibody anti‐SHP‐2 (ab131541, rabbit polyclonal, 1/100; Abcam, UK) combined with anti‐NF200 (BM0100, mouse monoclonal, 1/200; Boster, China), anti‐GFAP (SAB5201113, mouse monoclonal, 1/500; Sigma), anti‐human leukocyte antigen‐DR (HLA‐DR, M077501‐2; mouse monoclonal, 1/300, Dako, Denmark) and anti‐CD31 (ab9498, mouse monoclonal, 1/200; Abcam, UK). After rinsing 3 times in PBS, the sections were incubated with a mixture of FITC (fluorescein isothiocyanate)‐conjugated goat anti‐rabbit IgG (ZF‐0311, 1/300; Zhongshan Goldenbridge Biotechnology Co.) and AlexaFluor 594 goat anti‐mouse IgG (R37121, 1/300; Invitrogen, Carlsbad, CA) for 1 h at 37°C. Next, 4′,6‐diamidino‐2‐phenylindole (DAPI, 10 μg/ml, Beyotime, China) was used to counterstain the cell nuclei. Fluorescence label staining for NeuN in mouse model sections was performed with anti‐NeuN (MAB377, mouse monoclonal, 1/200, Millipore, USA) and AlexaFluor 594 goat anti‐mouse IgG (R37121, 1/300; Invitrogen, Carlsbad, CA) as the primary and secondary antibodies, respectively. The fluorescent signals were acquired using a confocal fluorescence microscope (TCS‐TIV; Leica, Nussloch, Germany). Based on the literature 1, 35, we evaluated the density of neurons in the CA1 and CA3 regions of the hippocampus using a modified method. Three areas from each section's CA1 or CA3 region were randomly selected, three consecutive coronal sections (coordinate position: 1.94 mm posterior to bregma) from each mouse and six mice were measured for each group. The number of NeuN positive neurons was counted using ImageJ software (version 1.42 V, NIH, USA). These analyses were conducted in a blinded fashion. The results are expressed as the mean ± SD number of cells per mm2.
Determination of albumin concentrations in the brain
The mice were anesthetized with 10% chloral hydrate and perfused with cold and sterile PBS over a period of 10 min to eliminate proteins from the blood circulation. Then, the concentration of albumin was measured in brain homogenates by western blotting.
Statistical analysis
The SPSS Statistical 16 package (SPSS Inc., Chicago, IL, USA) was used for statistical analyses. Data were expressed as the mean ± standard deviation (S.D.), and the analysis was carried out using Student's t‐test between the TLE and control groups. The chi‐square test was used to compare the gender differences between the groups of TLE patients and controls. One‐way analysis of variance (ANOVA) followed by a post hoc Bonferroni's test was used to determine the differences among multigroup comparisons. Following the statistical tests, P < 0.05 was considered statistically significant.
Results
Clinical characteristics
An examination of sex (P = 0.825) and age (P = 0.549) revealed no difference in the subjects between the control and epileptic groups. The control group contained five female and five male individuals with a mean age of 24.00 ± 5.312 years (range: 17–33 years).The mean age of the intractable TLE patients was 25.54 ± 7.235 years (range: 15–41 years), with 11 males and 13 females. All patients had taken two or more AEDs and had at least a 3‐year history of seizure recurrence.
The expression of SHP‐1/2 in the temporal neocortex of intractable TLE patients
Another important nonreceptor protein tyrosine phosphatase, SHP‐1, has a similar structure to SHP‐2. SHP‐1 and SHP‐2 can employ similar or parallel cellular pathways to participate in cell growth, inflammation and injury 6. Therefore, we first examined surgical samples from intractable TLE patients (n = 15) and controls (n = 9) for alterations in the protein expression levels of SHP‐1 and SHP‐2. Immunoreactive proteins were detected by immunoblotting at the predicted size of 68 kDa for SHP‐1 and SHP‐2. As shown in Figure 1A, SHP‐2 bands in the temporal neocortex from TLE patients were denser than those from the cortex of control (CTX) tissues; however, SHP‐1 bands were similar in the epileptic and control groups. GAPDH (37 kDa) was used as an internal control and confirmed equal protein loading in each lane (Figure 1A). No immunoreactive bands were detected when the primary antibody was omitted (data not shown), indicating the specificity of the staining. SHP‐1 and SHP‐2 expressions were normalized by calculating the OD ratio of the immunoreactive bands corresponding to GAPDH. The analysis demonstrated that the protein levels of SHP‐2 (Figure 1C), but not SHP‐1 (Figure 1B), were significantly increased in the epileptic temporal neocortex (P < 0.05).
Figure 1.
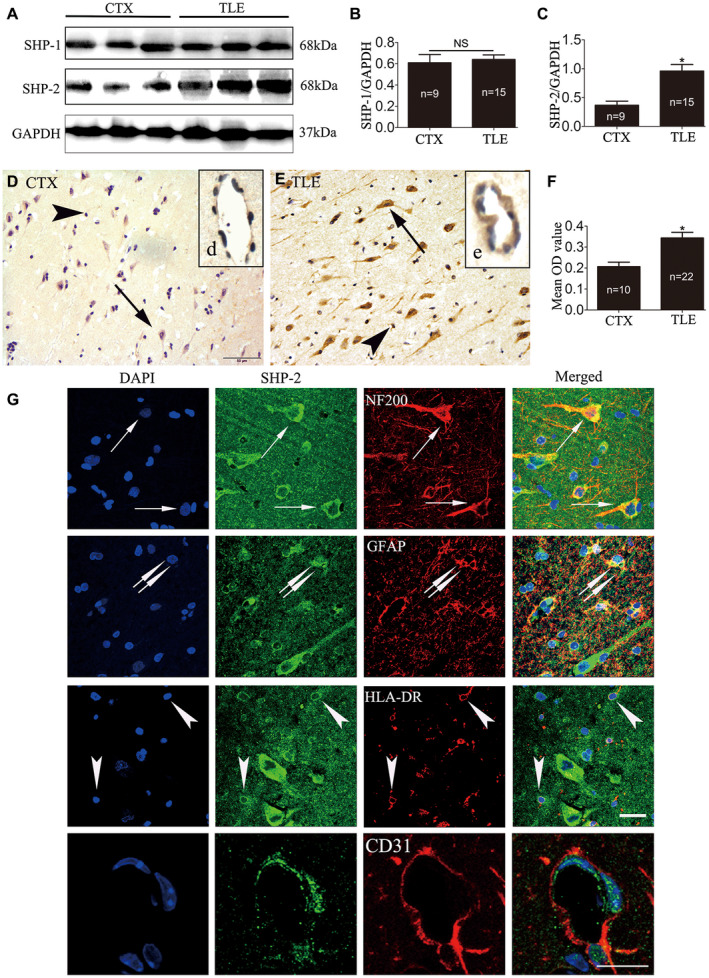
The expression pattern of SHP‐1/2 in the temporal neocortex of patients with TLE and in the CTX group. The levels of SHP‐1/2 protein were measured by western blotting in the temporal neocortex of intractable TLE patients and in that of the controls (A–C). Densitometric analysis showed that immunoreactive protein levels for SHP‐2 (C), but not SHP‐1 (B), were significantly increased in TLE patients compared with control subjects. Faint SHP‐2 immunoreactivity in neurons (arrows in D), glial cells (arrowheads in D) and vascular endothelial cells (d, insert in D) were observed in the temporal neocortex of CTX (D). The relatively strong immunoreactivity of SHP‐2 in neurons (arrows in E), glial cells (arrowheads in E) and in vascular endothelial cells (e, insert in E) was visualized in the TLE patients (E). F. The mean OD value of SHP‐2 is significantly greater in the TLE group (n = 22) compared with the control group (n = 10). G. Double‐labeled immunofluorescence staining shows SHP‐2 was coexpressed with NF200 in neurons (arrows). Colocalization of SHP‐2 with GFAP in astrocytes (doublearrows). SHP‐2 and HLA‐DR were also colocalized (arrows). Colocalization of SHP‐2 with CD31 in vascular endothelial cells. Data are expressed as the means ± SD. NS, not significant. *P < 0.05. Scale bar 50 μm for (D,E), 25 μm for (G).
To further confirm the changes in SHP‐2 expression in human epileptogenic tissue, we examined the expression of SHP‐2 using IHC. In CTX (n = 10), SHP‐2 displayed weak to moderate IR (Figure 1D, d). However, SHP‐2 showed strong staining in the temporal neocortex of intractable TLE patients (n = 22), and the immunostaining was presumably from neurons (Figure 1E), glial cells (Figure 1E) and vascular endothelial cells (Figure 1e). Compared to the CTX, the mean OD value of the SHP‐2 protein was significantly higher for patients with TLE (Figure 1F; P < 0.05). Immunohistochemical analysis of SHP‐1 showed no difference between the epilepsy group and the control group (data not shown).
Location of SHP‐2 in the temporal neocortex of intractable TLE patients
Subsequently, double‐labeled immunofluorescence was used to locate the distribution of SHP‐2 in the epileptic temporal neocortex. Double‐labeling experiments (Figure 1G) demonstrated that SHP‐2 colocalized with the neuronal marker NF200 and the astrocyte marker GFAP. Moreover, SHP‐2 IR was detectable in HLA‐DR‐positive microglia and CD31‐positive vascular endothelial cells.
Dynamic expression of SHP‐2 in the hippocampus and temporal neocortex in the pilocarpine mouse model
To validate our findings from TLE patients and to clarify whether AEDs may have an effect on SHP‐2 expression, we used an animal model of TLE generated by an intraperitoneal injection of pilocarpine. The changes in SHP‐2 protein levels in both the hippocampus (Figure 2A, left panel) and temporal neocortex (Figure 2A, right panel) at different time points post‐SE were investigated by western blotting (n = 6 for each group). SHP‐2‐immunoreactive bands were present at approximately 68 kDa, whereas the positive control band GAPDH was detected at 37 kDa (Figure 2A). SHP‐2 relative to GAPDH protein ratios between control and model mice 6 h–60 days post‐SE is presented (Figure 2B). As shown in Figure 2B, SHP‐2 expression in the hippocampus of the epileptic mice began to increase from 1 day (acute stage) (P < 0.05) to 7 days (latent stage) (P < 0.05) after SE, reached a peak at 21 days (P < 0.01), and was maintained at a relatively high level during the chronic stage (60 days; P < 0.05). We observed a remarkable increase in SHP‐2 expression at 1 day (P < 0.05), 7 days (P < 0.01), 21 days (P < 0.05) and 60 days (P < 0.05) after SE in the temporal neocortex.
Figure 2.
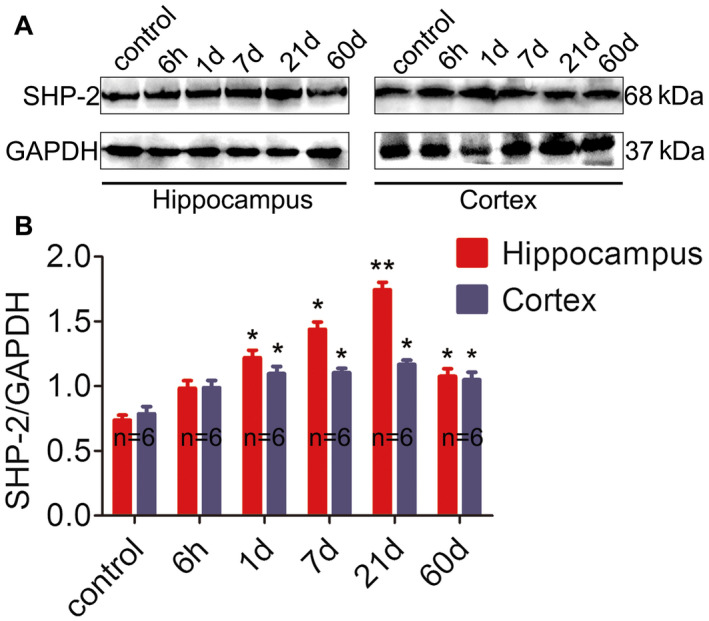
Western blotting analysis of SHP‐2 in the pilocarpine‐induced model. A. Representative western blot image of SHP‐2 (65 kDa) and corresponding GAPDH (37 kDa) expression in the hippocampus (left panel) and cortex (right panel). Lane 1 represents the normal mouse sample, and lanes 2–6 represent different time points (6 h, 1 day, 7 days, 21 days and 60 days) post‐SE. A distinctly stronger immunoblot was observed in experimental mice at each time point compared to the controls. B. Comparison of the intensity ratio of SHP‐2 expression between the control and epileptic mice at different time points post‐SE in the hippocampus and cortex. n = 6 for each group. *P < 0.05, **P < 0.01.
Different glial responses upon SHP099 treatment in control and post‐SE conditions
Findings from some previous studies point to a pivotal role of SHP‐2 in the negative regulation of gliogenesis 7, leading us to ask whether SHP099 affected the activation state of astrocytes and microglia following SE. As a test of astrocyte and microglia activation, we determined the protein levels of GFAP (50 kDa), IL‐1β (17 kDa) and Iba1 (17 kDa) in the hippocampus of the four groups (n = 6 for each group) by western blotting (Figure 3A). As shown in Figure 3B‐D, vehicle‐treated pilocarpine mice and SHP099‐treated pilocarpine mice exhibited higher protein levels of GFAP, IL‐1β and Iba1 than vehicle‐treated control mice and SHP099‐treated control mice, respectively (P < 0.05 or P < 0.01). The protein levels of GFAP and IL‐1β, but not Iba1 were significantly increased in SHP099‐treated pilocarpine mice compared with vehicle‐treated pilocarpine mice (P < 0.05). SHP099 treatment, however, did not significantly change the protein levels of GFAP, IL‐1β, and Iba1 in control mice (vehicle‐treated control mice vs. SHP099‐treated control mice; P > 0.05).
Figure 3.
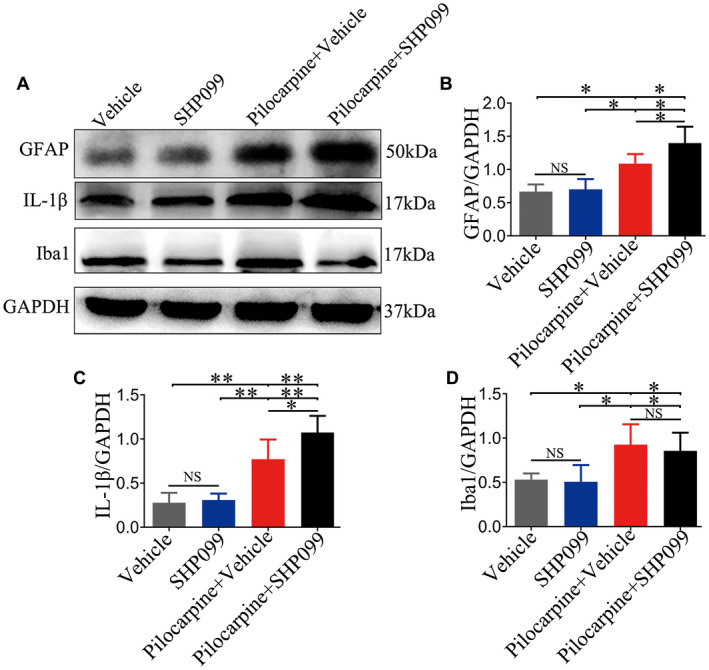
Effect of SHP099 on the activation of astrocytes and microglia in control and post‐SE conditions. Representative western blotting image of GFAP (50 kDa), IL‐1β (17 kDa), Iba1 (17 kDa) and the corresponding GAPDH (37 kDa) expression in the hippocampus (A). Densitometric analysis results of GFAP (B), IL‐1β (C) and Iba1 (D) in the four groups. n = 6 for each group. Data are presented as the mean ± SD. NS, not significant, *P < 0.05, **P < 0.01.
SHP099 treatment affects neuronal apoptosis and loss in the hippocampus
SHP‐2 can determine the fate of the neuron under pathological and physiological conditions. Thus, we detected the expression of cleaved caspase3 in the hippocampus to determine the apoptotic status of the four groups (n = 6 for each group). Representative immunoblotting bands are presented in Figure 4A. There was a large increase in cleaved caspase3 protein levels in the vehicle‐treated pilocarpine mice, and SHP099‐treated pilocarpine mice versus the vehicle‐treated control mice and SHP099‐treated control mice, respectively (P < 0.01). In addition, the protein levels of cleaved caspase3 were obviously elevated in SHP099‐treated pilocarpine mice compared to vehicle‐treated pilocarpine mice (P < 0.05). We also confirmed that the protein levels of cleaved caspase3 were significantly increased in vehicle‐treated pilocarpine mice compared to SHP099‐treated control mice (P < 0.05). No difference in cleaved caspase3 protein levels between the vehicle‐treated control mice and SHP099‐treated control mice was detected (P > 0.05). All of the above analysis results are shown in Figure 4B. Furthermore, we examined whether SHP099 treatment affected neuronal loss in the hippocampal CA1 and CA3 regions (n = 6 for each group; Figure 4C‐E). Our data showed that there was no significant difference between vehicle‐treated control mice and SHP099‐treated control mice in both regions of the hippocampus (P > 0.05). A significant reduction in number of NeuN‐expressing cells in CA1 and CA3 was found in vehicle‐treated pilocarpine mice and SHP099‐treated pilocarpine mice compared to vehicle‐treated control mice and SHP099‐treated control mice, respectively (P < 0.001). Additionally, the level of NeuN‐positive cells was significantly decreased in SHP099‐treated pilocarpine mice compared to vehicle‐treated pilocarpine mice in the CA1 (P < 0.01) and CA3 (P < 0.001) regions.
Figure 4.
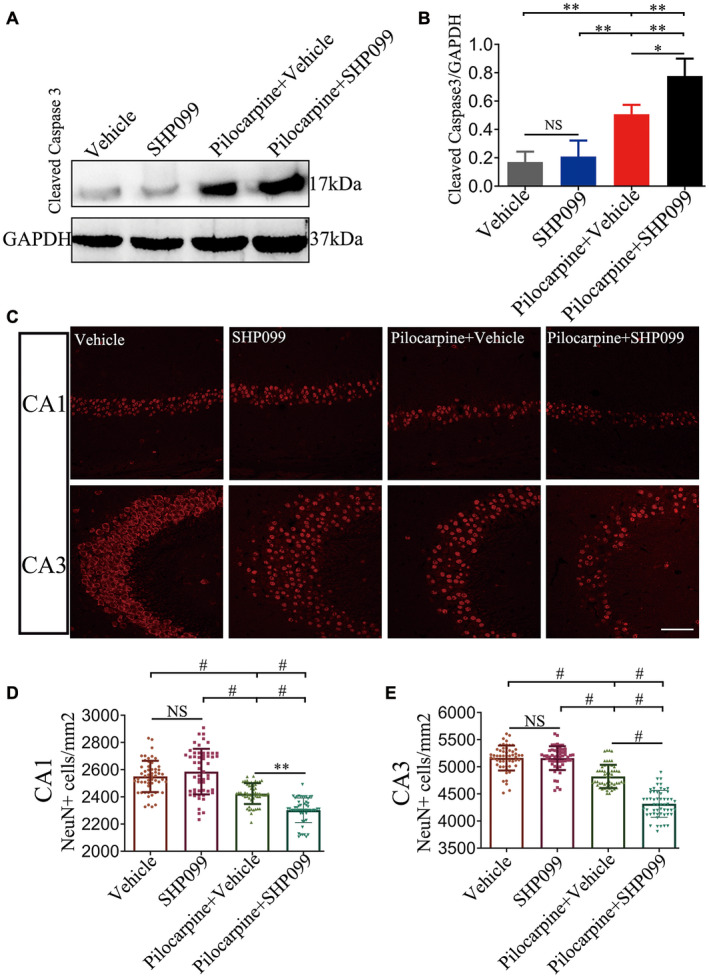
SHP099 treatment affects neuronal apoptosis and loss in the hippocampus. Representative immunoblotting showing immunoreactive protein expression of cleaved caspase 3 (17 kDa) in the hippocampus of the four groups (A). Densitometric analysis results of cleaved caspase 3 in the four groups (B). NeuN fluorescence staining in CA1 and CA3 regions of the hippocampus (C). The number of NeuN‐positive cells in the CA1 (D) and CA3 (E) regions of the hippocampus in the four groups (per mm2). n = 6 for each group. Data are presented as the mean ± SD. NS, not significant, *P < 0.05, **P < 0.01, # P < 0.001. Scale bar = 50 μm for (C).
Effect of SHP099 treatment on neurogenesis in the hippocampus
Neurogenesis occurs in the hippocampal DG at early and developmental stages of TLE 52, 53. DCX is a specific marker of neurogenesis for representing a snapshot of cells undergoing neuronal maturation 37. Therefore, we tested DCX‐labeled positive cells in the DG region of four groups (n = 6 for each group) by IHC. As shown in Figure 5A–E, pilocarpine treatment significantly increased the number of DCX‐positive cells in the DG of the hippocampus (SHP099‐treated pilocarpine mice or vehicle‐treated pilocarpine mice vs. SHP099‐treated control mice or vehicle‐treated control mice, respectively; P < 0.01 or P < 0.05). The number of DCX‐positive cells in the SHP099‐treated control mice was approximately equal to that of vehicle‐treated control mice (P > 0.05). Compared with vehicle‐treated pilocarpine mice, there was a remarkable reduction in DCX‐positive cells in the SHP099‐treated pilocarpine mice (P < 0.01). In addition, we detected the expression of DCX protein (45 kDa) in the hippocampus by western blotting (n = 6 for each; Figure 3F‐G). First, SHP099 did not affect the expression of DCX in the hippocampus under normal conditions (P > 0.05). Second, the expression of DCX protein in the hippocampus of vehicle‐treated pilocarpine mice and SHP099‐treated pilocarpine mice was higher than that of the control groups (SHP099‐treated control mice and vehicle‐treated control mice, P < 0.01). It is worth noting that the expression of DCX was lower in SHP099‐treated pilocarpine mice compared to vehicle‐treated pilocarpine mice (P < 0.05).
Figure 5.
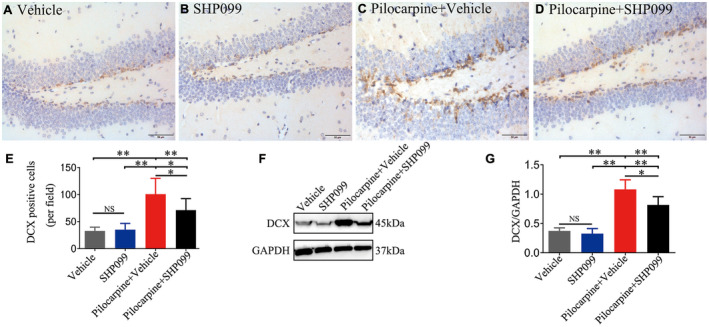
SHP099 treatment influences neurogenesis. Representative DCX immunostaining (brown) is shown for the DG of the hippocampus in vehicle‐treated control mice (A), SHP099‐treated control mice (B), vehicle‐treated pilocarpine mice (C) and SHP099‐treated pilocarpine mice (D). (E) The number of DCX‐positive cells in the above four groups in the DG (per field). F. Representational immunoblotting bands for DCX (45 kDa) and GAPDH (37 kDa). G. Densitometric analysis results of DCX in the four groups. n = 6 for each group. Data are presented as the mean ± SD. NS, not significant, *P < 0.05, **P < 0.01. Scale bar = 50 μm (A–D).
SHP099 treatment influences the permeability of BBB
Albumin, as a serum protein, is normally excluded from brain tissue by an intact BBB. We confirmed that the protein levels of albumin were observably increased in vehicle treated‐pilocarpine mice compared to vehicle‐treated control mice, SHP099‐treated control mice and SHP099‐treated pilocarpine mice (n = 6 for each group; Figure 6A,B; P < 0.01 or P < 0.05). In addition, the protein levels of albumin were significantly enhanced in SHP099‐treated pilocarpine mice compared to vehicle‐treated control mice and SHP099‐treated control mice (Figure 6A,B; P < 0.01 or P < 0.05, respectively). However, no difference in albumin concentrations between the vehicle‐treated control mice and SHP099‐treated control mice was observed (Figure 6A,B; P > 0.05).
Figure 6.
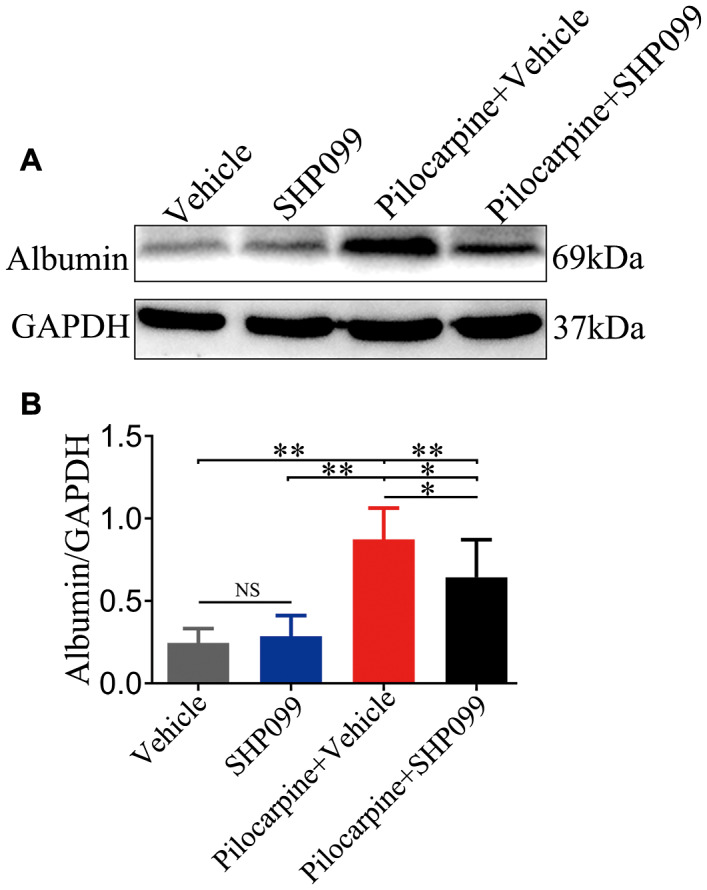
SHP099 treatment influences BBB permeability. A. The levels of albumin (69 kDa) in brain parenchyma were measured by western blotting. B. Statistical analysis results of albumin concentration in the four groups. n = 6 for each group. Data are presented as the mean ± SD. NS, not significant, *P < 0.05, **P < 0.01.
Discussion
In the current study, we noted that the protein levels of SHP‐2, but not SHP‐1, in the temporal neocortex of patients with TLE were markedly increased. Next, immunofluorescence labeling assay revealed that SHP‐2 was mainly localized within neurons, reactive astrocytes, microglia and vascular endothelial cells. In addition, consistent with the observations from clinical surgical samples, both the hippocampus and cortex exhibited upregulation of SHP‐2 in a mouse model of epilepsy. Furthermore, we found that SHP099 can alter glial proliferation, neuronal apoptosis, neuronal loss, neurogenesis and albumin leakage following SE. Based on these results, we deduce that SHP‐2 may be involved in the process of TLE.
Our previous study 46 and those from other groups 10, 54 have provided evidence indicating that glial cell activation has been associated with increased expression of inflammatory cytokines, which may contribute to disease pathology and progression in epileptic patients. SHP‐2 negatively regulated TLR4‐ and TLR3‐activated IFN‐β productions have been demonstrated in 2. Moreover, SHP‐2 also inhibited TLR3‐activated and TLR9‐activated proinflammatory cytokine IL‐6 and TNF‐a production. SHP‐2 participates in constitutive MHC‐I molecules negatively regulating TLR‐triggered inflammatory responses. All the above data suggest a critical role of SHP‐2 in the cytokine circuit for inflammatory and immune responses 49. Shifting our focus to the function of SHP‐2 in glial cells, previous research indicates that elevated chemokine CXCL8 expression in astrocytes can be caused by SHP‐2 overexpression 32. In vitro, SHP‐2, as a negative regulator of Janus kinase (JAK) activity, participates in curcumin‐mediated anti‐inflammatory effects in the brain‐activated microglia 24. In our study, immunohistochemical assays clarified a characteristically high level of SHP‐2 expression in glial cells. Moreover, double‐labeling experiments showed that SHP‐2 colocalized with GFAP and HLA‐DR in reactive astrocytes and microglia, respectively. Accordingly, it is reasonable to presume that the local high concentration of glia‐derived SHP‐2 may participate in TLE by modifying neuroinflammation.
BBB damage is one of the earliest characteristic pathophysiological disturbances during SE and therefore may contribute to the progression and development of epilepsy or aggravate seizure activity 15. It is well recognized that pathologic angiogenesis is a significant causal factor of BBB damage in TLE 15. Coincidentally, many studies have expatiated that SHP‐2 is closely related to angiogenesis. For instance, 33 reported that inhibition of SHP‐2 suppresses angiogenesis (impairs capillary‐like structure formation and new vessel growth) and increases endothelial apoptosis in vitro and in vivo. Additionally, in brain vascular endothelial cells, SHP‐2 interacts with platelet endothelial cell adhesion molecule (PECAM‐1) or VE‐cadherin, thereby regulating endothelial cell migration and leukocyte infiltration 8. On the other hand, previous studies have suggested that albumin leakage occurs due to BBB damage induced by epileptic seizures 31, 39. Our immunoblotting experiments revealed that SHP‐2 inhibition decreased the albumin concentration in brain parenchyma after an episode of pilocarpine‐induced SE (vehicle‐treated pilocarpine mice vs. SHP099‐treated pilocarpine mice). Thus, we speculate that the upregulation of SHP‐2 may participate in the occurrence and development of TLE by facilitating BBB disruption.
The role of gliosis in epileptogenesis remains largely unknown. Previous studies have indicated that SHP‐2 inhibits gliogenesis in vitro and in vivo 7, 23. Our present findings showed that SHP‐2 inhibition using SHP099 increased reactive astrogliosis (indicated by the protein levels of GFAP and IL‐1β) post‐SE in pilocarpine mice compared to vehicle‐treated control mice, vehicle‐treated pilocarpine mice and SHP099‐treated control mice. However, SHP099 treatment had no effect on reactive astrogliosis under control conditions. These data suggest that the increased expression of SHP‐2 in the epileptic zone might play a role by repressing astrogliogenesis in TLE.
Apoptosis has been implicated in seizure‐induced neuronal death and the pathogenesis of TLE 44. Activation of caspase3 is a significant surrogate measure of apoptotic neurodegeneration, as it is recognized to be a remarkable cell executioner 17, 47. Our western blotting results presented that SHP‐2 inhibition using SHP099 increased the protein levels of cleaved caspase3 21 days post‐SE in the pilocarpine mice compared to the vehicle‐treated control mice, the SHP099‐treated control mice and the vehicle‐treated pilocarpine mice. The protein levels of cleaved caspase3 were also significantly increased in vehicle‐treated pilocarpine mice compared to vehicle‐treated control mice. In addition, we can conclude that apoptosis was not affected by SHP099 treatment in control mice by comparing the protein levels of cleaved caspase3 between vehicle‐treated control mice and SHP099‐treated control mice. To further verify that inhibiting SHP2 can aggravate the apoptosis of hippocampal neurons 21 days after status epilepticus, we found that compared with vehicle‐treated pilocarpine mice, the number of hippocampal CA1 and CA3 neurons in SHP099‐treated pilocarpine mice decreased significantly. Until now, it has been widely accepted that SHP‐2 functions as an intracellular inhibitor of apoptosis 34, 48. Earlier studies support a neuroprotective role of SHP‐2 in response to ischemic brain injury 13. The apoptosis of primary cultured neurons exposed to NO (nitric oxide) was increased after the inhibition of SHP‐2 5. Thus, combined with our findings mentioned above, we can infer that the overexpression of SHP‐2 in TLE may alleviate apoptosis and the loss of neurons after epileptic seizure.
The relationship between neurogenesis and TLE epileptogenesis remains controversial. Hippocampal DG neurogenesis may be induced by brain damage after seizures 25. There is a growing body of evidence indicating that the seizure‐induced upregulation of VEGF, BDNF and FGF‐2 is capable of enhancing neurogenesis 52. Interestingly, SHP‐2 is an important regulator of the above molecules. Findings from recent studies point to a pivotal role of SHP‐2 in the positive regulation of neurogenesis 7, 23. Our data demonstrated that SHP‐2 inhibition decreased both the number of DCX‐positive cells and the DCX protein levels in pilocarpine mice (vehicle‐treated pilocarpine mice vs. SHP099‐treated pilocarpine mice). Based on this fact, we speculate that the upregulation of SHP‐2 in the DG of the hippocampus can increase neurogenesis and therefore participate in the pathophysiology of TLE.
Our study also has some limitations. Most importantly, hippocampal lesion is a characteristic pathological change of TLE patients. Unfortunately, as a result of ethical requirements, we could not obtain normal hippocampal samples during our study period. In addition, to avoid the influence of traumatic factors on SHP‐2 expression, we also examined the expression of SHP‐2 in direct traumatic tissues and distant tissues within 4 h after traumatic brain injury, and found no difference between them, which further verified the rationality of our control samples (data not shown). In summary, to our knowledge, this study is the first to indicate the increased expression of SHP‐2 in TLE, which may be related to inflammation, gliosis, neuronal apoptosis, neuronal loss, neurogenesis and BBB breakdown. Following the present descriptive study, additional studies are required to confirm that SHP‐2 should be considered a possible target for intervention in the epileptic process.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Ethics Statement
This study and all its procedures were approved by the ethics committee of the Army Medical University, China. And the human brain specimens were used in a manner compliant with the Declaration of Helsinki. All patients signed informed consent for using the biologic material. All animal experimental procedures were reviewed and approved by the Internal Animal Care and Use Committee of the Army Medical University. Appropriate measures were taken during experimental procedures to minimize animal suffering and reduce the number of animals used.
Acknowledgements
This work was supported by National Natural Science Foundation of China (NO.81771217, NO.81771394). The authors sincerely thank the patients and their families for their participation in this work.
Contributor Information
Chunqing Zhang, Email: cqzhang@tmmu.edu.cn.
Shiyong Liu, Email: liushi2425@163.com.
Hui Yang, Email: huiyangtg2018@aliyun.com.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Ahmadian SR, Ghasemi‐Kasman M, Pouramir M, Sadeghi F (2019) Arbutin attenuates cognitive impairment and inflammatory response in pentylenetetrazol‐induced kindling model of epilepsy. Neuropharmacology 146:117–127. [DOI] [PubMed] [Google Scholar]
- 2. An H, Zhao W, Hou J, Zhang Y, Xie Y, Zheng Y, et al (2006) SHP‐2 phosphatase negatively regulates the TRIF adaptor protein‐dependent type I interferon and proinflammatory cytokine production. Immunity 25:919–928. [DOI] [PubMed] [Google Scholar]
- 3. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia‐Fortanet J, Acker MG, et al (2016) Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535:148–152. [DOI] [PubMed] [Google Scholar]
- 4. Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, et al (2006) Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol 70:562–570. [DOI] [PubMed] [Google Scholar]
- 5. Chong ZZ, Lin S‐H, Kang J‐Q, Maiese K (2003) The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell Mol Neurobiol 23:561–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chong ZZ, Maiese K (2007) The Src homology 2 domain tyrosine phosphatases SHP‐1 and SHP‐2 diversified control of cell growth, inflammation, and injury. Histol Histopathol 22:1251–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coskun V, Zhao J, Sun YE (2007) Neurons or glia? Can SHP2 know it all? Sci STKE 2007:pe58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Couty JP, Rampon C, Leveque M, Laran‐Chich MP, Bourdoulous S, Greenwood J, Couraud PO (2007) PECAM‐1 engagement counteracts ICAM‐1‐induced signaling in brain vascular endothelial cells. J Neurochem 103:793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Curia G, Longo D, Biagini G, Jones RS, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172:143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das A, Wallace GCt, Holmes C, McDowell ML, Smith JA, Marshall JD, et al (2012) Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience. 220:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fuchikawa T, Nakamura F, Fukuda N, Takei K, Goshima Y (2009) Protein tyrosine phosphatase SHP2 is involved in Semaphorin 4D‐induced axon repulsion. Biochem Biophys Res Comm 385:6–10. [DOI] [PubMed] [Google Scholar]
- 12. Garcia Fortanet J, Chen CH‐T, Chen Y‐NP, Chen Z, Deng Z, Firestone B, et al (2016) Allosteric inhibition of SHP2: identification of a potent, selective, and orally efficacious phosphatase inhibitor. J Med Chem 59:7773–7782. [DOI] [PubMed] [Google Scholar]
- 13. Gee CE, Mansuy IM (2005) Protein phosphatases and their potential implications in neuroprotective processes. Cell Mol Life Sci 62:1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goldberg EM, Coulter DA (2013) Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci 14:337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gorter JA, van Vliet EA, Aronica E (2015) Status epilepticus, blood‐brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav 49:13–16. [DOI] [PubMed] [Google Scholar]
- 16. Hagihara K, Zhang EE, Ke YH, Liu G, Liu JJ, Rao Y, Feng GS (2009) Shp2 acts downstream of SDF‐1alpha/CXCR16 in guiding granule cell migration during cerebellar development. Dev Biol 334:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hannan JL, Matsui H, Sopko NA, Liu X, Weyne E, Albersen M, et al (2016) Caspase‐3 dependent nitrergic neuronal apoptosis following cavernous nerve injury is mediated via RhoA and ROCK activation in major pelvic ganglion. Sci Rep 6:29416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hill KS, Roberts ER, Wang X, Marin E, Park TD, Son S, et al (2019) PTPN11 plays oncogenic roles and is a therapeutic target for BRAF wild‐type melanomas. Mol Cancer Res 17:583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang YS, Cheng CY, Chueh SH, Hueng DY, Huang YF, Chu CM, et al (2012) Involvement of SHP2 in focal adhesion, migration and differentiation of neural stem cells. Brain Develop 34:674–684. [DOI] [PubMed] [Google Scholar]
- 20. Jerome Engel J (2001) Mesial temporal lobe epilepsy: what have we learned? Neuroscientist 7:340–352. [DOI] [PubMed] [Google Scholar]
- 21. Jo A, Park H, Lee SH, Ahn SH, Kim HJ, Park EM, Choi YH (2014) SHP‐2 binds to caveolin‐1 and regulates Src activity via competitive inhibition of CSK in response to H2O2 in astrocytes. PLoS ONE 9:e91582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karlocai MR, Toth K, Watanabe M, Ledent C, Juhasz G, Freund TF, Magloczky Z (2011) Redistribution of CB1 cannabinoid receptors in the acute and chronic phases of pilocarpine‐induced epilepsy. PLoS ONE 6:e27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, Klein R, et al (2007) Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol 27:6706–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim HY, Park EJ, Joe Eh, Jou I (2003) Curcumin Suppresses Janus Kinase‐STAT Inflammatory Signaling through Activation of Src Homology 2 Domain‐Containing Tyrosine Phosphatase 2 in Brain Microglia. J Immunol 171:6072–6079. [DOI] [PubMed] [Google Scholar]
- 25. Korn MJ, Mandle QJ, Parent JM (2016) Conditional Disabled‐1 Deletion in Mice Alters Hippocampal Neurogenesis and Reduces Seizure Threshold. Front Neurosci 10:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuo E, Park DK, Tzvetanova ID, Leiton CV, Cho BS, Colognato H (2010) Tyrosine phosphatases Shp1 and Shp2 have unique and opposing roles in oligodendrocyte development. J Neurochem 113:200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kusakari S, Saitow F, Ago Y, Shibasaki K, Sato‐Hashimoto M, Matsuzaki Y, et al (2015) Shp2 in forebrain neurons regulates synaptic plasticity, locomotion, and memory formation in mice. Mol Cell Biol 35:1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al (2010) Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51:1069–1077. [DOI] [PubMed] [Google Scholar]
- 29. LaRochelle JR, Fodor M, Vemulapalli V, Mohseni M, Wang P, Stams T, et al (2018) Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat Commun 9:4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee YS, Ehninger D, Zhou M, Oh JY, Kang M, Kwak C, et al (2014) Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat Neurosci 17:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Z, Liu J, Wang S, Liu S, Zhao Y (2016) Neuronal uptake of serum albumin is associated with neuron damage during the development of epilepsy. Exp Ther Med 12:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mamik MK, Ghorpade A (2012) Src homology‐2 domain‐containing protein tyrosine phosphatase (SHP) 2 and p38 regulate the expression of chemokine CXCL8 in human astrocytes. PLoS ONE 7:e45596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mannell H, Hellwig N, Gloe T, Plank C, Sohn HY, Groesser L, et al (2008) Inhibition of the tyrosine phosphatase SHP‐2 suppresses angiogenesis in vitro and in vivo. J Vasc Res 45:153–163. [DOI] [PubMed] [Google Scholar]
- 34. Morales LD, Casillas Pavon EA, Shin JW, Garcia A, Capetillo M, Kim DJ, Lieman JH (2014) Protein tyrosine phosphatases PTP‐1B, SHP‐2, and PTEN facilitate Rb/E2F‐associated apoptotic signaling. PLoS ONE 9:e97104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Naeimi R, Safarpour F, Hashemian M, Tashakorian H, Ahmadian SR, Ashrafpour M, Ghasemi‐Kasman M (2018) Curcumin‐loaded nanoparticles ameliorate glial activation and improve myelin repair in lyolecithin‐induced focal demyelination model of rat corpus callosum. Neurosci Lett 674:1–10. [DOI] [PubMed] [Google Scholar]
- 36. O'Dell CM, Das A, Gt Wallace, Ray SK, Banik NL (2012) Understanding the basic mechanisms underlying seizures in mesial temporal lobe epilepsy and possible therapeutic targets: a review. J Neurosci Res 90:913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Leary JD, Hoban AE, Murphy A, O'Leary OF, Cryan JF, Nolan YM (2018) Differential effects of adolescent and adult‐initiated exercise on cognition and hippocampal neurogenesis. Hippocampus 29:352–365. [DOI] [PubMed] [Google Scholar]
- 38. Paul S, Lombroso PJ (2003) Receptor and nonreceptor protein tyrosine phosphatases in the nervous system. Cell Mol Life Sci 60:2465–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ralay Ranaivo H, Hodge JN, Choi N, Wainwright MS (2012) Albumin induces upregulation of matrix metalloproteinase‐9 in astrocytes via MAPK and reactive oxygen species‐dependent pathways. J Neuroinflammation 9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robert M, Freeman J, Plutzky J, Neel BG (1992) Identification of a human src homology 2‐containing proteintyrosine‐phosphatase: A putative homolog of Drosophila corkscrew. Proc Natl Acad Sci 89:11239–11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rosario M, Franke R, Bednarski C, Birchmeier W (2007) The neurite outgrowth multiadaptor RhoGAP, NOMA‐GAP, regulates neurite extension through SHP2 and Cdc42. J Cell Biol 178:503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rusanescu G, Yang W, Bai A, Nee BG, Feig LA (2005) Tyrosine phosphatase SHP‐2 is a mediator of activity‐dependent neuronal excitotoxicity. EMBO J 24:305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ryan K, Liang LP, Rivard C, Patel M (2014) Temporal and spatial increase of reactive nitrogen species in the kainate model of temporal lobe epilepsy. Neurobiol Dis 64:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schindler CK, Pearson EG, Bonner HP, So NK, Simon RP, Prehn JH, Henshall DC (2006) Caspase‐3 cleavage and nuclear localization of caspase‐activated DNase in human temporal lobe epilepsy. J Cerebr Blood F MET 26:583–589. [DOI] [PubMed] [Google Scholar]
- 45. Shibley H, Smith BN (2002) Pilocarpine‐induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD‐1 mice. Epilepsy Res 49:109–120. [DOI] [PubMed] [Google Scholar]
- 46. Shu H‐F, Zhang C‐Q, Yin Q, An N, Liu S‐Y, Yang H (2010) Expression of the interleukin 6 system in cortical lesions from patients with tuberous sclerosis complex and focal cortical dysplasia type IIb. J Neuropathol Exp Neurol 69:838–849. [DOI] [PubMed] [Google Scholar]
- 47. Smart AD, Pache RA, Thomsen ND, Kortemme T, Davis GW, Wells JA (2017) Engineering a light‐activated caspase‐3 for precise ablation of neurons in vivo. Proc Natl Acad Sci U S A 114:E8174–E8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang G, Jin C, Hou Y, Zhang L, Li S, Zhang L, et al (2012) Overexpression of Shp‐2 attenuates apoptosis in neonatal rat cardiac myocytes through the ERK pathway. Exp Mol Pathol 93:50–55. [DOI] [PubMed] [Google Scholar]
- 49. Xu S, Liu X, Bao Y, Zhu X, Han C, Zhang P, et al (2012) Constitutive MHC class I molecules negatively regulate TLR‐triggered inflammatory responses via the Fps‐SHP‐2 pathway. Nat Immunol 13:551–559. [DOI] [PubMed] [Google Scholar]
- 50. Yue J, Li W, Liang C, Chen B, Chen X, Wang L, et al (2016) Activation of LILRB2 signal pathway in temporal lobe epilepsy patients and in a pilocarpine induced epilepsy model. Exp Neurol 285(Pt A):51–60. [DOI] [PubMed] [Google Scholar]
- 51. Zheng J, Huang S, Huang Y, Song L, Yin Y, Kong W, et al (2016) Expression and prognosis value of SHP2 in patients with pancreatic ductal adenocarcinoma. Tumour Biol 37:7853–7859. [DOI] [PubMed] [Google Scholar]
- 52. Zhong Q, Ren BX, Tang FR (2016) Neurogenesis in the Hippocampus of Patients with Temporal Lobe Epilepsy. Curr Neurol Neurosci Rep 16:20. [DOI] [PubMed] [Google Scholar]
- 53. Zhu X, Dong J, Shen K, Bai Y, Chao J, Yao H (2016) Neuronal nitric oxide synthase contributes to pentylenetetrazole‐kindling‐induced hippocampal neurogenesis. Brain Res Bull 121:138–147. [DOI] [PubMed] [Google Scholar]
- 54. Zurolo E, Iyer A, Maroso M, Carbonell C, Anink JJ, Ravizza T, et al (2011) Activation of Toll‐like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain 134(Pt 4):1015–1032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.