

Abstract
The distribution and role of tumor‐infiltrating leucocytes in glioblastoma (GBM) remain largely unknown. Here, we investigated the cellular composition of 55 primary (adult) GBM samples by flow cytometry and correlated the tumor immune profile with patient features at diagnosis and outcome. GBM single‐cell suspensions were stained at diagnosis (n = 44) and recurrence following radiotherapy and chemotherapy (n = 11) with a panel of 8‐color monoclonal antibody combinations for the identification and enumeration of (GFAP+CD45−) tumor and normal astrocytic cells, infiltrating myeloid cells —i.e. microglial and blood‐derived tumor‐associated macrophages (TAM), M1‐like, and M2‐like TAM, neutrophils. and myeloid‐derived suppressor cells (MDSC)— and tumor‐infiltrating lymphocytes (TIL) —i.e. CD3+T‐cells and their TCD4+, TCD8+, TCD4−CD8−, and (CD25+CD127lo) regulatory (T‐regs) subsets, (CD19+CD20+) B‐cells, and (CD16+) NK‐cells—. Overall, GBM samples consisted of a major population (mean ± 1SD) of tumor and normal astrocytic cells (73% ± 16%) together with a significant but variable fraction of immune cells (24% ± 18%). Within myeloid cells, TAM predominated (13% ± 12%) including both microglial cells (10% ± 11%) and blood‐derived macrophages (3% ± 5%), in addition to a smaller proportion of neutrophils (5% ± 9%) and MDSC (4% ± 8%). Lymphocytes were less represented and mostly included TCD4+ (0.5% ± 0.7%) and TCD8+ cells (0.6% ± 0.7%), together with lower numbers of TCD4−CD8− T‐cells (0.2% ± 0.4%), T‐regs (0.1% ± 0.2%), B‐lymphocytes (0.1% ± 0.2%) and NK‐cells (0.05% ± 0.05%). Overall, three distinct immune profiles were identified: cases with a minor fraction of leucocytes, tumors with a predominance of TAM and neutrophils, and cases with mixed infiltration by TAM, neutrophils, and T‐lymphocytes. Untreated GBM patients with mixed myeloid and lymphoid immune infiltrates showed a significantly shorter patient overall survival versus the other two groups, in the absence of gains of the EGFR gene (p = 0.02). Here we show that immune cell infiltrates are systematically present in GBM, with highly variable levels and immune profiles. Patients with mixed myeloid and T‐lymphoid infiltrates showed a worse outcome.
Keywords: glioblastoma, immune cells, lymphocytes, microenvironment, microglia, myeloid cells
1. INTRODUCTION
Glioblastoma multiforme (GBM) is the most frequent and lethal malignant brain tumor type with a median overall survival rate of ≈1 year (y) (1). In past decades, detailed knowledge has been achieved about the (e.g., histopathological and genetic/molecular) characteristics of GBM cells (2). These studies also confirmed GBM displays highly heterogeneous molecular profiles (3). To a large extent, such heterogeneity is related to distinct tumor‐associated cytogenetic and genomic profiles (4), but also to highly variable interactions between the tumor cells and their microenvironment, particularly with tumor‐infiltrating leucocytes (5).
At present, it is well‐established that both the innate and adaptive immune systems play a critical role in immune surveillance (6). At the earlier phases of tumor development, neoplastic cells might trigger inflammatory responses with local infiltration by innate immune cells, such as macrophages and other antigen‐presenting cells (APC), as well as NK‐cells (7). Tumor antigens presented by local APC and the tumor cells themselves might further activate local tumor cell‐specific cytotoxic T‐cell and humoral B‐cell responses, aimed at controlling tumor development and growth (8). However, the GBM tumor microenvironment progressively becomes immunosuppressive, leading to immune tolerance, tumor growth, and progression (9, 10). Thus, GBM cells display impaired presentation of tumor antigens with decreased expression of HLA molecules, and they produce and release immunosuppressive and pro‐apoptotic signals to the infiltrating tumor‐specific immune cells (11, 12, 13), such as transforming growth factor beta (TGF‐β) and interleukin (IL)‐10 (14). Locally produced TGF‐β inhibits T‐cell activation, proliferation, and differentiation, while promoting regulatory T‐cells (T‐regs) and suppressing both NK‐cell and T‐cell cytotoxicity, as well as innate cell functions, thereby contributing also to immune escape and tumor growth (15). In turn, locally produced IL‐10 (16) exerts inhibitory effects on T‐helper (Th) cells, monocytes, macrophages, and dendritic cells (DC) (17). Altogether, this leads to an impaired balance between anti‐tumor immune responses and immune tolerance that progressively favors tumor development and growth (18), in which local infiltrating macrophages, T‐regs, and myeloid‐derived suppressor cells (MDSC) might play a critical role (3).
At present, it is well established that tumor‐infiltrating leucocytes consist of an admixture of several immune cells at variable proportions, such as tumor‐associated macrophages (TAM) and tumor‐infiltrating lymphocytes (TIL). TAM present in GBM might derive from two independent sources: brain‐resident microglia cells and bone marrow (BM) or blood‐derived monocytes/macrophages (5). Both types of TAM interact with tumor cells and might promote the growth and progression of GBM (19) due to an imbalanced ratio between pro‐inflammatory (M1) and anti‐inflammatory (M2) macrophages (20). In turn, locally increased T‐regs and MDSC have also been found in GBM in association with tumor progression, poor response to immunotherapy and more adverse patient outcome (21, 22, 23, 24). Of note, MDSC have been reported to be increased both inside the tumor and in blood of GBM patients (22).
Despite the potential relevance of immune surveillance versus immune tolerance in GBM, detailed information about the composition of immune cell infiltrates in GBM still remains relatively limited (3, 25, 26). This is partially due to technical limitations as regards the number of markers and cell populations that have been simultaneously detected (with both immunochemistry and flow cytometry) in otherwise limited (i.e., small) tumor samples (21, 27, 28, 29). Recent development and availability of flow cytometers capable of simultaneously evaluating higher numbers of markers and immune cell populations (30), paves the way for more detailed characterization of the distinct immune cell populations present in both tumor and blood specimens of GBM patients, at the expense of losing information about their spatial tissue location.
Here we used an 8‐color panel of antibody combinations for simultaneous (detailed) characterization of GBM tumor cells and tumor‐infiltrating immune cells by flow cytometry. Our goal was to gain insight into the cellular composition of the tumor microenvironment in GBM and to investigate in a pilot series of 55 tumor samples (44 primary diagnostic samples and 11 recurrent tumors studied after therapy) from 51 patients its potential association with disease features at diagnosis and patient outcome.
2. MATERIAL AND METHODS
2.1. Patients and samples
A total of 55 GBM (tumor) samples from 51 adult patients —22 females and 29 males; mean age of 60 years, ranging from 27 to 80 years— who underwent surgery at diagnosis (n = 43 patients; 44 samples) or at tumor progression/recurrence (n = 8 patients; 11 samples) at the Neurosurgery Service of the University Hospital of Salamanca (Salamanca, Spain), were studied (Table S1). In 3/51 patients, paired samples obtained at diagnosis, (including two samples from a bilateral tumor in which complete in block tumor resection was not achieved) and at tumor progression/recurrence were analyzed in parallel. Only those parts of the tumor showing both macroscopical and microscopical infiltration were used for immune cell analyses, avoiding necrotic areas and areas with the heterogeneous spatial distribution of cell populations. GBM diagnosis was made by an experienced pathologist based on WHO criteria, including immunohistochemical labeling for the IDH1 mutated protein, ATRX, GFAP, EGFR, TP53 or KI‐67 among other markers. In a subset of 10 GBM samples, further immunohistochemical analyses were performed in order to better identify and characterize residual non‐tumor cells and the immune cell infiltrates. All tissue samples used in this study were obtained after informed consent had been given by each individual patient, and the study was approved by the local ethics committee of the University Hospital of Salamanca. In 44 GBM patients (34 newly diagnosed patients and 10 recurrent tumors), peripheral blood samples were also obtained prior to surgery for the parallel evaluation of the distribution of the major subsets of leucocytes and lymphocytes.
2.2. Mutational analyses
Analysis of IDH1 and IDH2 gene mutations was based on DNA extracted from frozen tissue samples (n = 46) using organic solvents and subsequent digestion with Proteinase K, according to well‐established methods (31). Exon 4 DNA of both the IDH1 and the IDH2 genes was amplified by PCR and sequenced on a capillary automated sequencer (CEQ 8000; Beckman‐Coulter, Hialeah, FL); mutational analysis of the sequenced data was performed using the Sequencher, (version 4.7) software (Genes Codes, Ann Arbor, MI). Only 1/46 primary GBM cases analyzed showed IDH1 mutation (Table S2).
2.3. Immunophenotypic studies
Each tumor sample was stained with a 5‐tube 8‐color antibody panel that systematically contained the DRAQ5 live nucleated cell DNA dye (Cytognos, SL, Salamanca, Spain) for staining and identification of (all) nucleated cells and to evaluate their baseline autofluorescence levels (Table S3). Stainings were evaluated in either an LSRFortessa X20 or a FACSCanto II flow cytometer —Becton/Dickinson Biosciences (BD), San Jose, CA— for the following markers: CD45, CD3, CD4, CD8, CD11b, CD14, CD15, CD16, CD19, CD20, CD24, CD25, CD33, CD44, CD56, CD68, CD127, CD133, CD163, CD192, CD206, EGFR, GFAP, HLA‐DR, IL‐10, SOX2, and TGF‐β (Table S3).
Briefly, fresh GBM samples were placed in 1 ml of RPMI 1640 medium (Gibco™, Life Technologies, Grand Island, NY) and mechanically disaggregated with tweezers, as previously described (32). The resulting single‐cell suspension was placed in phosphate‐buffered saline (PBS) containing 2 mM EDTA (Merck, Darmstadt, Germany), 0.09% azide, 0.5% bovine serum albumin (BSA) (Sigma‐Aldrich, St. Louis, MO), and 10% fetal bovine serum (FBS) (Gibco™). After a short (5 min) sedimentation step, the supernatant was collected and the cell suspension passed through a 20‐25G gauge needle, centrifuged (10 min at 314 G), the supernatant removed and the cell pellet washed in PBS. Afterward, staining for cell surface membrane markers was performed by incubating ≥1 × 106 cells/aliquot with the corresponding monoclonal antibody combination for 30 min in the dark (room temperature, RT). In case of aliquots stained only for cell surface membrane markers, stained cells were subsequently washed in PBS, resuspended in 2ml of 1X FACS Lysing Solution —10X FACS Lysing Solution (BD Biosciences) diluted 1/10 (vol/vol) in distilled water—, incubated for another 15 min (RT) in the dark, washed in PBS and resuspended in 0.5 mL of PBS. In turn, for combined staining of cell surface plus cytoplasmic markers (i.e., GFAP, CD68, and SOX2), the Fix&Perm™ reagent (Nordic‐MUbio, Rangeerweg, The Netherlands) was used, following previously described protocols (33). Briefly, after staining for cell surface markers as described above, cells were sequentially washed in PBS, incubated for 15 min in the dark (RT) with 50 µl of solution A (fixative) of Fix&Perm™ and washed in PBS; monoclonal antibodies against cytoplasmic markers were then added together with 50 µl of solution B (permeability solution) of the Fix&Perm™ reagent, and stained cells were subsequently incubated for another 15 min in the dark (RT), washed, resuspended in 0.5 ml of PBS and measured in the flow cytometer, as described below. Immediately, prior to data acquisition, the DRAQ5 DNA dye was added, as described elsewhere (34).
Peripheral blood samples were stained with a 12‐marker, 8‐color, combination of monoclonal antibodies strictly following the EuroFlow standard operating procedures (SOPs) previously reported in detail (35) and that are available at Euroflow.org.
2.4. Detection of cytokine production by GBM tumor cells
Spontaneous production of both TGF‐β and IL‐10 was evaluated after short term (1 h) in vitro culture (37ºC) of tumor cells in RPMI 1640 medium to which 0.2% brefeldin A (Sigma‐Aldrich) per 100 µl volume had been added. Cultured cells were then stained for cell surface membrane markers (CD45, CD3, CD4, CD8, CD14, CD19, CD20, CD25, CD127, CD192, CD206, and HLA‐DR) and intracellular TGF‐β‐PECF594 and IL10‐PE, as described above (i.e., tubes 1 and 3 in Table S3). Prior to data acquisition in the flow cytometer, the DRAQ5 DNA dye was added to stain (all) nucleated cells.
2.5. Flow cytometry data acquisition and analysis
The FACSDiva™ software (BD Biosciences) was used for the acquisition of data on ≥106 events/tube. For data analysis, the Infinicyt software (Cytognos, SL) was employed. During data analysis, the percent distribution of each cell population in the sample together with its phenotype —percent of positive cells and mean fluorescence intensity (MFI) per marker— were recorded. The small fraction of residual normal astrocytic cells in the tumor tissue specimens (non‐tumoral non‐hematopoietic tissue cells) could not be clear cut identified and discriminated from the GBM cells coexisting in the same sample; thereby, they were systematically included within the percentage of non‐hematopoietic (tumor) cells.
2.6. Cell purification and characterization of FACS‐sorted cells
Purification of two different cell populations coexisting in all GBM samples was performed in a subset of 20 fresh tumor samples, using a 4‐way fluorescence‐activated cell sorter (FACSAria III; BD Biosciences) and the FACSDiva software. Prior to sorting, cells were resuspended in PBS containing 2 mM EDTA, 0.09% azide, 0.5% BSA, and 10% FBS, and stained with a 4‐color antibody combination (CD45‐PacB HLA‐DR‐PB CD11b‐FITC CD14‐PE) to which the DRAQ5 DNA dye was added, as previously described (36). The phenotype of the distinct purified (DRAQ5hi) nucleated cell populations was as follows: (i) CD45+ CD11b+ HLADR+ CD14+ macrophages and (ii) CD45− CD11b− HLADR− CD14− GBM cells, with a mean (±1SD) purity of 99% ± 0.4% and 99% ± 4%, respectively. A fraction of the sorted cells was concentrated and placed on a cytospin slide to assess their morphological appearance based on hematoxylin and eosin staining, analyzed in an Olympus BX5 microscope (Olympus, Melville, NY).
2.7. Interphase fluorescence in situ hybridization studies (iFISH)
iFISH studies were performed in whole GBM single‐cell suspensions of tumor samples obtained by mechanical disaggregation procedures as described above, after they had been fixed in methanol/acetic acid (3:1 v/v). For iFISH studies, the EGFR/CEP7 FISH probe Kit from Vysis Abbott Molecular Inc (Des Plaines, IL) was used. EGFR gene amplification was defined when ≥7 fluorescent signals (i.e., spots) were present for its specific probe; below this cut‐off, tumors with ≥3 copies of the EGFR gene (3–6 fluorescence signals) were considered to have genetic/chromosome gains.
2.8. Statistical analyses
For all statistical analyses, the SPSS software (SPSS 25.0, IBM SPSS, Armonk, NY) was used. The Student T, the Mann‐Whitney U, and the Chi‐square tests were used to compare different groups of patients for (parametric and non‐parametric) continuous parameters and for categorical variables, respectively. Unsupervised clustering based on t‐distributed stochastic neighbor embedding (t‐SNE) was used for the classification of GBM samples based on their immune cell composition (37). Survival curves were plotted according to the Kaplan and Meier method, and the (two‐sided) log‐rank test was used to assess the statistical significance of differences in overall survival (OS) between distinct groups of patients. For OS studies, only those GBM studied at diagnosis, who survived for >1 month after surgery and had a minimum follow‐up of 18 months (in case of patients remaining alive), were included in the analysis.
3. RESULTS
3.1. Identification of tumor cell and immune cell populations in GBM samples
Gating of flow cytometric data for accurate phenotypic identification of the distinct cell populations coexisting in GBM tumor samples was based on the phenotypic profiles described in Table 1, as illustrated in Figure 1. Briefly, nucleated cells were first gated based on their high DRAQ5‐associated fluorescence intensity, after excluding cell debris and doublets based in a forward high scatter‐area (FSC‐A) versus FSC‐Height (FSC‐H) dot plot histogram (Figure 1A). Subsequently, GBM tumor cells and normal residual astrocytic cells were identified as GFAP+ CD45− cells and subdivided into two SOX2− and SOX2+ cell populations (Figure 1A). In turn, tumor‐infiltrating leucocytes were defined as GFAP−CD45+ cells and microglial cells as CD45low HLADR+ CD14+ cells with high green autofluorescence levels (Figure 1A,B; Table 1). Blood‐derived tumor‐infiltrating monocytes/macrophages were CD45+GFAP− with low green autofluorescence (Figure 1B). In turn, TAM were discriminated from other immune infiltrating cells, particularly from other myeloid cells, based on their strong co‐expression of CD11b+and HLA‐DR+ and their positivity for CD14+ (Figure 1C; Table 1). TAM (both microglial cells and blood‐derived monocytes/macrophages) were further divided into HLA‐DR+CD14+CD192+ M1‐like versus HLA‐DR+CD14+CD163+and/or CD206+ M2‐like cells (Figure 1C and Table 1). Further identification of neutrophils (SSChi) among myeloid infiltrating cells was based on their unique CD15hi and CD16hi (or CD24hi) phenotype, in the absence of CD14 and HLA‐DR expression (Figure 1C and Table 1). MDSC were gated as CD11b+ HLA‐DR− CD33low CD45low GFAP− myeloid cells that showed variable levels of expression of both CD14 and CD15 (Figure 1C and Table 1). Finally, tumor‐infiltrating SSClo CD45hi lymphocytes were subdivided into CD3+ T‐cells, CD19+ CD20+ B‐cells and CD3−CD19−CD16+ NK‐cells (Figure 1D and Table 1); T‐cells were further subsetted into TCD4+ (CD3+CD4+CD8−), TCD8+ (CD3+CD4−CD8+) and T‐double negative (DN) (CD3+CD4−CD8−) T‐lymphocytes, in addition to CD4+CD25hiCD127low T‐regs (Figure 1D; Table 1).
TABLE 1.
Phenotypic features used for the identification of GBM tumor cells, microglial cells, and other infiltrating immune cell populations present in GBM tissue specimens (n = 55)
| Immunophenotypic marker | Cell population | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mature GBM tumor cells | Immature tumor cells | Immune cells | Myeloid cells | Microglial cells | Blood‐derived macrophages | M1‐like TAM | M2‐like TAM | MDSC | Neutrophils | Lymphocytes | T‐cells | CD4+CD8− T‐cells | CD4−CD8− T‐cells | CD4+ T‐regs | B‐cells | NK‐cells | |
| SSC | Het | Het | Het | Het | Het | Het | Het | Het | Het | High | Low | Low | Low | Low | Low | Low | Low |
| GAFL | − | − | −/+ | − | + | Low | −/+ | −/+ | − | − | − | − | − | − | − | − | − |
| CD45 | − | − | + | Low | Low | Low | Low | Low | Low | Low | High | High | High | High | High | High | High |
| GFAP | + | + | −/+ | −/+ | − | − | − | − | − | − | − | − | − | − | − | − | − |
| SOX2 | − | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CD133 | − | −/+ | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CD3 | − | − | −/+ | − | − | − | − | − | − | − | −/+ | + | + | + | + | − | − |
| CD4 | − | − | −/+ | −/+ | − | − | − | − | − | − | −/+ | + | + | − | + | − | − |
| CD8 | − | − | −/+ | − | − | − | − | − | − | − | −/+ | + | − | − | − | − | − |
| CD11b | − | − | −/+ | −/+ | + | + | + | + | + | + | − | − | − | − | − | − | − |
| CD14 | − | − | −/+ | −/+ | + | + | + | + | −/+ | − | − | − | − | − | − | − | − |
| CD15 | − | − | −/+ | −/+ | − | − | − | − | −/+ | + | − | − | − | − | − | − | − |
| CD16 | − | − | −/+ | −/+ | −/+ | −/+ | −/+ | −/+ | − | + | − | − | − | − | − | − | + |
| CD19‐CD20 | − | − | −/+ | − | − | − | − | − | − | − | −/+ | − | − | − | − | + | − |
| HLA‐DR | − | − | −/+ | −/+ | + | + | + | + | − | − | −/+ | − | − | − | − | + | − |
| CD25 | − | − | −/+ | − | − | − | − | − | − | − | −/+ | − | − | − | High | − | − |
| CD33 | − | − | −/+ | Low | Low | Low | Low | Low | Low | Low | − | − | − | − | − | − | − |
| CD127 | − | − | −/+ | − | − | − | − | − | − | − | −/+ | −/+ | − | − | Low | − | − |
| CD163 | − | − | −/+ | −/+ | −/+ | −/+ | − | + | − | − | − | − | − | − | − | − | − |
| CD192 | −/+ | − | −/+ | −/+ | −/+ | −/+ | + | − | − | − | −/+ | − | − | − | − | −/+ | − |
| CD206 | − | − | −/+ | −/+ | −/+ | −/+ | − | + | − | − | − | − | − | − | − | − | − |
Other markers evaluated in tumor cells were EGFR (growth factor receptor), CD24 and CD44 (adhesion markers), and CD192 (chemokine receptor). Some of these markers were also used for the identification and/or characterization of neutrophils (CD44, CD24), and lymphocytes (CD44), and CD68 was used to identify TAM.
Abbreviations: −, marker not expressed; +, marker expressed; −/+, marker expressed in a fraction of the cell population; GAFL: Green autofluorescence; TAM: Tumor‐associated macrophages.
FIGURE 1.
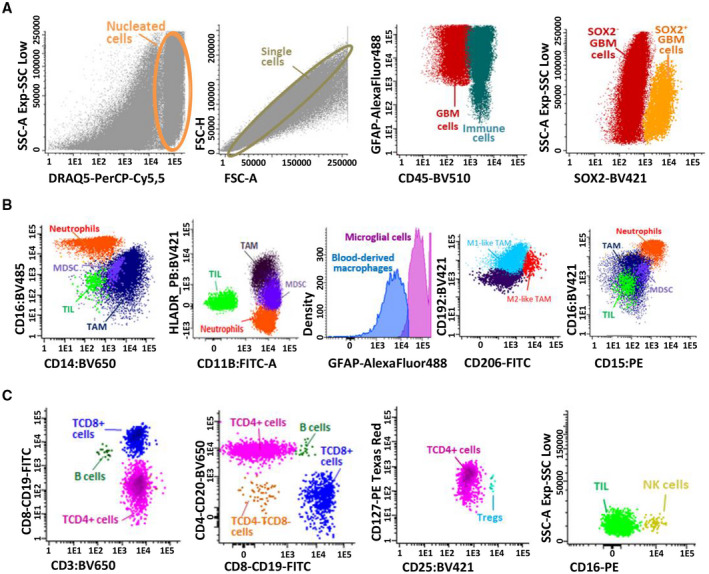
Immunophenotypic identification and characterization of distinct cell populations present in single‐cell suspensions derived from fresh GBM tumor samples identified based on 8‐color flow cytometry stainings. First, live cells stained with DRAQ5 were selected, doublets eliminated, and nucleated cells corresponding to immune and GBM tumor (and normal astrocytic) cells were evaluated according to CD45 and GFAP expression profile. GBM tumor and normal astrocytic cells stained with GFAP subdivided into mature and immature GBM cells based on the staining for SOX2 (panel A). Second, myeloid and lymphoid populations were distributed based on the stained for CD14, CD16, CD11b, and HLA‐DR markers (panel B). TAM population (CD11B+HLA‐DR+CD14+) was subsetted into microglial cells (CD45+ high green autofluorescence) and blood‐derived monocytes/macrophages (CD45+ low green autofluorescence). Two subsets of TAM were subsequently defined (M1 and M2‐like TAM) according to the pattern of expression of CD206 and CD192. Other tumor‐infiltrating leucocytes (CD45+ GFAP−CD14) were defined also as CD11b+ myeloid cells; neutrophils were also CD11b+ but in addition they showed a CD15hiCD16hiHLA‐DR− phenotype, while MDSC were HLA‐DR− CD16− with variable expression levels of CD14 and CD15 (Panel B). Among CD45hi lymphocytes, T‐cells were identified as CD3+ cells and they included TCD4 (CD4+CD8−), T CD8 (CD4−CD8+), T double‐negative (DN; CD4−CD8−) T‐cells, together with CD4+ T‐regs (CD25highCD127low); B‐cells were defined as CD20+CD19+CD3− lymphocytes and NK‐cells were identified based on high CD16+ expression in the absence of CD3 and CD19 (panel C)
FACS sorting of CD45+CD11b+HLA‐DR+CD14+ followed by hematoxylin and eosin staining confirmed the morphological characteristics of these cells were finally compatible with TAMs, while purified GBM tumor cells lacked positivity for CD45 by immunohistochemistry (Figure S1).
3.2. Distribution and phenotypic features of tumor cells and immune cells in GBM
Most cells in GBM samples corresponded to non‐hematopoietic tumor and normal residual astrocytic cells —mean ± 1 standard deviation (SD) of 73% ± 16%— while the remaining cells in the same tumor specimens were mostly immune cells (mean ± 1SD: 24% ± 18%) (Table 2). Coexistence of mature GFAP+CD45− GBM neoplastic cells (mean ± 1SD: 52% ± 25%) with a smaller fraction of (more immature) GFAP+SOX2+ GBM tumor cells (mean ± 1SD: 20% ± 26%) was systematically detected in every GBM sample analyzed (Table 2). However, highly variable percentages of each of these two tumor cell populations were found among the different tumors (Table 2). From the phenotypic point of view, GFAP+CD45− tumor cells displayed highly heterogeneous (low to intermediate) FSC and SSC features, together with variable percentages of cells (mean ± 1SD) positive for the EGFR tyrosine‐kinase growth factor receptor (14% ± 26%), the CD24 tetraspanin (24% ± 32%), the hyaluronic acid receptor CD44 (61% ± 34%) and the CD192 (CCR2) chemokine receptor (27% ± 26%). In turn, around half of all SOX2+ tumor cells also expressed the stem cell‐associated marker CD133 (mean ± 1SD: 48% ± 41%) for a total of (mean ± 1SD) 8% ± 10% of all cells in the tumor samples investigated (Table 2), thereby revealing the existence of a significant fraction of SOX2+CD133− cells.
TABLE 2.
Distribution of distinct populations of tumor cells, TAM (microglial cells and blood‐derived macrophages), and other tumor‐infiltrating immune cells in GBM samples (n = 55)
| Cell population and phenotype | Marker | % cells in the tumor sample | % cells within the parental population |
|---|---|---|---|
| Total GBM tumor cells (GFAP+) | 73% ± 16% | ||
| SOX2− GBM tumor cells (CD45− GFAP+ SOX2−) | 52% ± 25% | ||
| EGFR+ | 9% ± 13% | 14% ± 26% | |
| CD24+ | 16% ± 22% | 24% ± 32% | |
| CD44+ | 41% ± 30% | 61% ± 34% | |
| CD192+ | 13% ± 11% | 27% ± 26% | |
| SOX2+ GBM tumor cells (CD45−GFAP+SOX2+) | 20% ± 26% | ||
| CD133+ | 8% ± 10% | 48% ± 41% | |
| Immune cells (CD45+) | 24% ± 18% | ||
| Myeloid cells | 22% ± 17% | 90% ± 15% | |
| TAM (HLA‐DR+CD14+) | 13% ± 12% | 61% ± 28% | |
| M1‐like TAM (CD192+) | 4% ± 6% | 17% ± 18% | |
| M2‐like TAM (CD163+) | 3% ± 6% | 16% ± 20% | |
| CD206+ | 2% ± 3% | 12% ± 20% | |
| CD33+ | 13% ± 13% | 45% ± 29% | |
| CD16+ | 4% ± 8% | 16% ± 20% | |
| CD44+ | 8% ± 8% | 35% ± 30% | |
| CD68+ | 9% ± 9% | 42% ± 36% | |
| Microglial cells (HLA‐DR+CD14+ GAFL+) | 10% ± 11% | 44% ± 27% | |
| Blood‐derived macrophages (HLA‐DR+CD14+ GAFLlo−) | 3% ± 5% | 16% ± 20% | |
| Neutrophils (CD15hi/CD16hi HLA‐DR−) | 5% ± 9% | 20% ± 21% | |
| MDSC ( CD15lo CD16− HLA‐DR−) | 4% ± 8% | 15% ± 23% | |
| TIL | 2% ± 2% | 10% ± 15% | |
| T‐cells (CD3+) | 1.4% ± 2% | 8% ± 9% | |
| CD4+CD8− T‐cells | 0.5% ± 0.7% | 2% ± 4% | |
| CD8+CD4− T‐cells | 0.6% ± 0.7% | 5% ± 7% | |
| CD8−CD4− T‐cells | 0.2% ± 0.4% | 2% ± 4% | |
| CD4+CD25+CD127− T‐regs | 0.1% ± 0.2% | 0.4% ± 0.5% | |
| B‐cells (CD19+CD20+) | 0.1% ± 0.2% | 0.5% ± 1% | |
| NK‐cells (CD3− CD16+CD56+) | 0.05% ± 0.05% | 1% ± 5% | |
Results expressed as mean ± SD percentage of cells from the whole tumor sample cellularity and from the parental cell population (either tumor cells or all immune cells).
Abbreviations: GAFL, green autofluorescence; TAM, Tumor‐associated macrophages (i.e. microglial cells and blood‐derived macrophages); TIL, Tumor‐infiltrating lymphocytes.
CD45+ immune cells represented (mean ± 1SD) 24% ± 18% of the whole sample cellularity. Among them, myeloid cells clearly predominated (mean ± 1SD: 90% ± 15%) over lymphoid cells (mean ± 1SD: 10% ± 15%) (Table 2). Within myeloid cells, TAM was the most represented cell population (mean ± 1SD: 61% ± 28%) including similar frequencies of M1‐like (HLA‐DR+CD14+CD192+) and M2‐like (HLA‐DR+CD14+CD206+ and/or CD163+) TAM (Table 2). Overall, TAM showed variable levels of expression and heterogeneous percentages (mean ± 1SD) of CD33+ (45% ± 29%), CD16+ (16% ± 20%), CD44+ (35% ± 30%) and CD68+ (42% ± 36%) cells. Within TAM, microglial cells were almost three times more abundant than blood‐derived macrophages (mean percentage ± 1SD: 44% ± 27% vs. 16% ± 20%, respectively; p < 0.001). Apart from TAM, neutrophils were the second most frequent myeloid cell population and they accounted for a mean ± 1SD of 20% ± 21% of all immune cells, while MDSC were found at slightly lower frequencies (mean ± 1SD: 15% ± 23%) (Table 2). Within lymphocytes, T‐cells (mean ± 1SD: 8% ± 9%) predominated over both NK‐cells (mean ± 1SD: 1% ± 5%) and B‐cells (mean ± 1SD: 0.5% ± 1%), with progressively lower (mean ± 1SD) frequencies of CD8+CD4− T‐lymphocytes (5% ± 7%), CD4+CD8− (2% ± 4%) and CD4−CD8− double‐negative cytotoxic T‐lymphocytes (2% ± 4%), in addition to a minor population (0.4% ± 0.5%) of T‐regs (Table 2). Among all cells described above, only TAM showed baseline ex vivo production of TGF‐β (mean ± 1SD: 5% ± 9%) together with a minor proportion of IL‐10 producing cells (mean ± 1SD: 0.2% ± 0.5%).
Once GBM tissue specimens studied at diagnosis (n = 44) were compared with samples evaluated at tumor recurrence/progression (n = 11) after (median: 4 months) chemotherapy and radiotherapy, no significant differences (p > 0.05) in the relative distribution of the distinct tumor cell and immune cell subsets identified was observed between the two groups of samples (Table 3). Despite this, paired tumor samples from two GBM patients studied at diagnosis and at relapse showed a higher fraction of tumor‐infiltrating leucocytes at relapse versus diagnosis (24% vs. 70% and 12% vs. 20%, respectively) associated with either decreased or similar numbers of microglial cells (9% vs. 57% and 14% vs. 15%, respectively) and slightly increased neutrophil (5% vs. 1%, 2% vs. 0.6%, respectively) and MDSC (5% vs. 0.4% and 1% vs. 0.1%, respectively) counts. Different percentages of the later two myeloid cell populations were also found when paired samples from a bilateral tumor were compared with a unique sample at relapse (32% vs. 3% and 5% neutrophils and 22% vs. 17% MDSC in the right side with the exception of an increase of this population on the left side which was 50%, were found in the tumor at relapse versus right and left tumors at diagnosis, respectively) (Table S4).
TABLE 3.
Relative distribution of tumor cell and immune cell populations in fresh diagnostic versus recurrence GBM tumor tissue samples (n = 55)
| Cell population | Diagnostic tumor samples (n = 44 a ) | Recurrence tumor samples (n = 11) | p‐value |
|---|---|---|---|
| Total tumor cells | 71% (29‐97%) | 75% (26‐96%) | 0.53 |
| Stem cell‐like tumor (SOX2+) | 12% (0.8‐82%) | 1% (0.5‐19%) | 0.17 |
| Mature tumor cells (SOX2−) | 63% (11‐92%) | 76% (54‐78%) | 0.31 |
| Immune cells | 20% (0.2‐75%) | 18% (3‐65%) | 0.78 |
| Microglia | 8% (0.4‐57%) | 14% (7‐43%) | 0.09 |
| Blood‐derived macrophages | 2% (0.07‐34%) | 5% (0.2‐8%) | 0.86 |
| Neutrophils | 2% (<0.01‐45%) | 3% (0.02‐32%) | 0.68 |
| MDSC | 1% (<0.01‐51%) | 1% (0.3‐22%) | 0.38 |
| TIL | 1% (<0.01‐12%) | 1% (0.01‐6%) | 0.46 |
2/44 samples corresponded to paired samples from a patient with a bilateral tumor. Results expressed as median (range) percentage of cells from the whole tumor sample.
Immunohistochemical analyses performed in a subset of 10 GBM samples revealed a major fraction of GBM tumor cells (mean ± 1SD: 77% ± 20%) in addition to variable percentages of residual non‐tumor cells (mean ± 1SD: 16% ± 12%) along with a small percentage of TIL (mean ± 1SD: 2.5% ± 1.4%) in the absence of a significant correlation with the percentage of cells obtained by flow cytometry in the same tumor (Figure S2A). Similarly, no significant correlation was observed between the distribution of major peripheral blood leucocyte subsets in paired blood and tumor samples. (Figure S2B).
3.3. Association between the tumor immune profile and the clinical and genetic features of the disease
According to the relative cellular composition of tumor samples by flow cytometry, three different groups of GBM patients were identified by (multivariate) t‐SNE analysis (based on machine learning algorithms) among the 50 patients investigated (after excluding one outlier) (Figure 2A,B). These groups included: (1) cases with limited immune cell infiltrate (Group 1; n = 18); (2) tumors displaying an immune cell infiltrate with clear predominance of TAM and neutrophils together with residual percentages of lymphocytes, particularly T‐cells (Group 2; n = 26); and (3) tumors with an immune infiltrate consisting of a mixture of TAM, neutrophils, and lymphocytes, mostly TCD8+ and TCD4+ cells (Group 3; n = 6) (Table 4 and Figure 2A,B). No significant association was found between these three GBM immune profiles (Groups 1 to 3) and patient age, gender, Karnofsky index (KPS), and type of surgery performed at diagnosis (Table 5).
FIGURE 2.
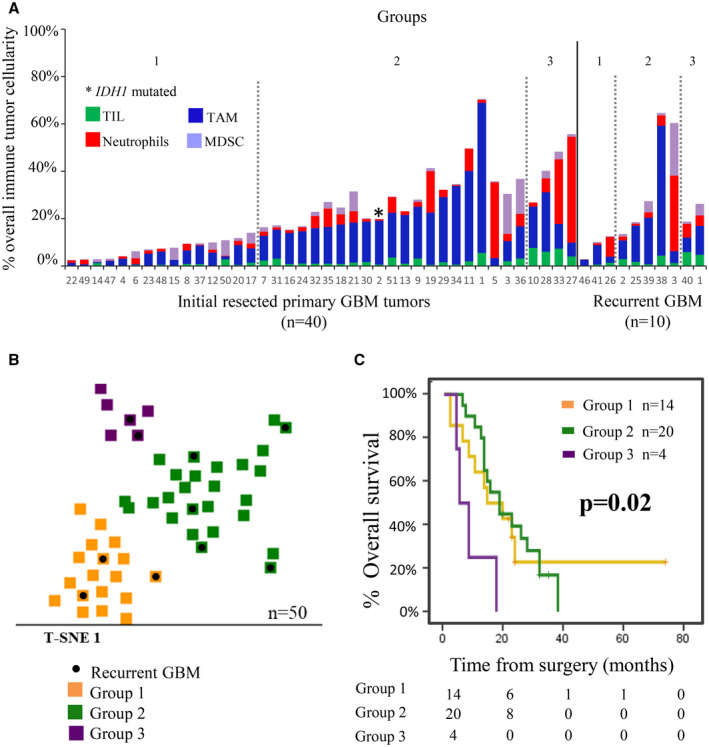
Immune cell composition of GBM samples classified according to their overall infiltrating immune profile and its impact on patient overall survival. Immune cell contents defined three subsets of primary diagnostic GBM samples (n = 40) including: (i) GBM with minor immune cell infiltrates (Group 1; n = 15); (ii) GBM with a predominance of TAM and neutrophils with minor percentages of lymphocytes (Group 2; n = 21); and (iii) GBM with immune cell infiltrates that consist of a mixture of TAM, neutrophils, and lymphocytes (Group 3; n = 4) (panel A). Based on the same criteria, recurrent GBM tumors (n = 10) were also classified into Group 1 (n = 3), Group 2 (n = 5), and Group 3 (n = 2) GBM. A 2‐dimension t‐SNE representation of the distribution of the 50 (color‐coded) tumors corresponding to each of the three groups identified—Group 1 (orange), Group 2 (green), and Group 3 (violet)—are shown in panel B, where recurrent GBM tumors are depicted in black. In panel C, the impact of the distinct immune cell profiles identified (labeled with the same color codes as in panel B) on overall survival of newly diagnostic patients after excluding those who were alive but had a follow‐up of less than 18 months and/or died within the 1st month after surgery (n = 38), is shown
TABLE 4.
Distribution of immune cell populations among the three groups of GBM patients showing distinct infiltrating immune profiles as identified by the t‐SNE machine learning algorithm (n = 50)
| GBM immune cell groups | ||||
|---|---|---|---|---|
| Group 1 (n = 18) | Group2 (n = 26) | Group 3 (n = 6) | p‐value | |
| % Immune cells | 8% ± 6% | 31% ± 15% | 35% ± 15% | <0.001*, ** |
| % TAM | 4% ± 3% | 20% ± 14% | 13% ± 7% | <0.001*, 0.002** |
| % Microglial cells | 2% ± 2% | 15% ± 13% | 9% ± 3% | <0.001*, 0.003** |
| % Blood‐derived macrophages | 1% ± 2% | 5% ± 6% | 3% ± 15% | 0.013* |
| % Neutrophils | 2% ± 2% | 6% ± 9% | 15% ± 17% | 0.003*, 0.001** |
| % MDSC | 2% ± 2% | 3% ± 6% | 2% ± 2% | NS |
| % TIL | 0.6% ± 0.7% | 2% ± 1% | 6% ± 1% | <0.001*, **, *** |
| % T‐cells | 0.5% 0.5% | 2% ± 1% | 6% ± 2% | 0.002*, 0.03**, *** |
| T CD4+CD8− | 0.2% ± 0.2% | 0.8% ± 0.8% | 3% ± 0.1% | 0.02*, 0.03** |
| T CD8+CD4− | 0.2% ± 0.2% | 0.6% ± 0.5% | 2% ± 1% | 0.006*, 0.026** |
| T CD8−CD4− | 0.06% ± 0.06% | 0.1% ± 0.1% | 1% ± 1% | 0.026** |
| T‐regs | 0.02% ± 0.02% | 0.2% ± 0.3% | 0.1% ± 0.07% | 0.004*, 0.048** |
| % B‐cells | 0.03% ± 0.04% | 0.1% ± 0.3% | 0.5% ± 0.4% | 0.026**, 0.049*** |
| % NK‐cells | 0.05 ± 0.05% | 0.03% ± 0.03% | 0.2% ± 0.03% | 0.026**, 0.015*** |
Results expressed as mean ± 1 SD percentage of cells from the whole sample cellularity.
Abbreviations: NS, no significant differences found (p > 0.05); t‐SNE: t‐distributed stochastic neighbor embedding.
Group 1 versus Group 2;
Group 1 versus Group 3;
Group 2 versus Group 3.
TABLE 5.
Association between clinical and genetic features of GBM and the frequency of different immune cells and immune profiles found in GBM samples
| Clinical and genetic variables | GBM Immune cell populations | GBM Immune cell profile | |||||
|---|---|---|---|---|---|---|---|
| TAM | Neutrophils | MDSC | TIL | Group 1 | Group 2 | Group 3 | |
| Karnofsky index | |||||||
| >70 | 15% ± 14% | 5% ± 8% | 3% ± 5% | 2% ± 2% | 70% | 63% | 60% |
| ≤70 | 12% ± 8% | 6% ± 12% | 2% ± 4% | 2% ± 2% | 30% | 37% | 40% |
| p‐value | NS | NS | NS | NS | NS | ||
| Age | |||||||
| 18‐45 years | 16% ± 22% | 2% ± 2% | 1% ± 1% | 1% ± 2% | 18% | 4% | – |
| 46‐65 years | 13% ± 12% | 4% ± 6% | 5% ± 11% | 2% ± 3% | 59% | 50% | 40% |
| 66‐85 years | 81% ± 11% | 8% ± 13% | 2% ± 3% | 2% ± 2% | 23% | 46% | 60% |
| p‐value | NS | NS | NS | NS | NS | ||
| Gender | |||||||
| Female | 12% ± 9% | 4% ± 6% | 2% ± 3% | 2% ± 2% | 50% | 42% | 40% |
| Male | 15% ± 14% | 7% ± 10% | 4% ± 10% | 2% ± 3% | 50% | 57% | 60% |
| p‐value | NS | NS | NS | NS | NS | ||
| Type of resection | |||||||
| Partial | 17% ± 12% | 4% ± 7% | 2% ± 4% | 2% ± 2% | 29% | 38% | 25% |
| Complete | 12% ± 13% | 6% ± 10% | 2% ± 4% | 2% ± 2% | 71% | 54% | 75% |
| p‐value | NS | NS | NS | NS | NS | ||
| EGFR profile b | |||||||
| No EGFR gains/AMP | 8% ± 9% | 3% ± 2% | 1% ± 2% | 5% ± 4% | 6% | – | 40% |
| EGFR gains | 18% ± 15% | 5% ± 9% | 1% ± 1% | 2% ± 2% | 39% | 65% | 40% |
| EGFR AMP | 9% ± 7% | 6% ± 9% | 3% ± 4% | 1% ± 2% | 56% | 35% | 20% |
| p‐value | 0.010 a | NS | NS | 0.040 a | 0.010 | ||
Results expressed as mean (±1SD) percentage of cells from the whole tumor sample (n = 50).
Abbreviations: AMP, Amplification; NS, no significant differences found (p > 0.05).
EGFR gains versus EGFR amplification.
IDH1 status evaluated by immunohistochemistry showed absence of labeling for IDH1 consistent with the wild‐type IDH1gene as confirmed by further molecular analyses in 45 of 46 samples.
From the genetic point of view, all patients investigated but one (45/46) showed a wild‐type IDH gene status (Table S1). In contrast, the great majority of the tumor samples analyzed (50/54; 93%) showed either the gain of EGFR (30/54; 55%) or EGFR amplification (20/54; 37%). EGFR protein expression levels by the flow cytometry were higher among GBM tumors that showed gain or amplification of EGFR (n = 50) versus no alteration (n = 4) of this gene —mean fluorescence intensity (MFI) of 2765 ± 4191 versus 387 ± 440, respectively (p = 0.001)— (Figure S1). Interestingly, patients that had gains of EGFR (n = 30) displayed a greater percentage of tumor infiltration by TAM (18% ± 15%; p = 0.010) and lymphocytes (2% ± 2%; p = 0.040) compared to those that displayed EGFR gene amplification (9% ± 7% and 1% ± 2%, respectively); in contrast, those few tumors (n = 4) that showed no gains or amplification of EGFR had lower percentages of TAM (8% ± 9%; p > 0.05) with greater levels of lymphocytes (5% ± 4%; p > 0.05). These differences translated into different EGFR genetic profiles among the three previously defined groups of GBM based on their tumor‐infiltrating immune cell profiles (Table 5). Thus, EGFR amplification predominated in Group 1 tumors (56% vs. 35% and 20% in groups 2 and 3, respectively), while EGFR gains were more frequently observed in Group 2 tumors (65% vs. 39% and 40% in groups 1 and 3, respectively) and most tumors with a diploid EGFR profile were concentrated in Group 3 (40% vs. 6% and 0% in groups 1 and 2, respectively) (p = 0.01) (Table 5).
From the prognostic point of view, the immune profile of newly diagnosed, untreated GBM showed a clear impact on patient survival (p = 0.02), cases with immune infiltrates consisting of a mixture of TAM, neutrophils, and T‐cells (Group 3) showing a significantly poorer outcome compared to the other two GBM patient groups (Groups 1 and 2): median overall survival of 6 months versus 16 and 18 months, respectively (Figure 2C).
4. DISCUSSION
In the present study, we used flow cytometry for the detailed phenotypic characterization of the different tumor cell and immune cell populations that coexist in GBM. In line with previous studies in other brain tumors (38) and also GBM, our results confirmed the systematic presence of immune cell infiltrates together with the GBM tumor cells, their relative proportion varying substantially among the individual tumors analyzed. Combined staining for GFAP and CD45, allowed clear cut discrimination between GFAP+CD45− GBM tumor and normal astrocytic cells and GFAP−CD45+ immune cells, in line with previous observations by others (3, 39). Despite the fact that GFAP is also expressed in normal astrocytes (40) and thereby, it is not a tumor‐specific marker, staining for GFAP together with CD45 contributed to the unequivocal identification of the tumor‐infiltrating immune cells in tissues that predominantly consisted of tumor cells. Among GBM tumor (and normal astrocytic) cells, two major cell subsets were further identified based on the presence versus absence of expression of SOX2: a major population of (phenotypically more mature) SOX2− tumor cells together with a smaller fraction of (phenotypically more immature) SOX2+ cells. A smaller fraction of the later tumor cells showed positivity for the CD133 cancer‐stem‐cell‐associated. Several studies have shown that CD133 is one of the most reliable markers for the identification of GBM cancer stem cells which also coexpress SOX2 (41, 42). Of note, most previous studies have specifically investigated SOX2 expression within the CD133+ population of GBM cells, while they do not provide information on the remaining CD133− tumor cells (43). In this regard, our results show coexpression of SOX2 on CD133+ GBM cells at the same time they reveal the existence of a significant fraction of SOX2+CD133− cells among GFAP+ GBM cells. Altogether, these findings would indicate that the minor subset of CD133+SOX2+ GBM tumor cells might contain GBM cancer stem cells which were represented at different percentages in each individual tumor sample. However, the biological and clinical significance of these CD133+SOX2+ tumor cells, and particularly of SOX2+CD133− cells, remains to be determined. Similar to CD133 and SOX2, variable percentages of tumor cells were also found to express other growth factor receptors (EGFR) and adhesion molecules (CD24, CD44) which are known to be positive in GBM tumor cells (44). These results further emphasize the existence of a highly heterogeneous tumor cell hierarchy in GBM with a clear association between specific genetic alterations and protein expression profiles (45), as confirmed here for the EGFR protein expression levels and the EGFR gene status. Despite this, no clear association between the protein expression profiles of GBM tumor cells for the different markers investigated here and neither patient features at diagnosis nor the patient outcome was observed (data not shown).
Regarding immune cells, highly variable levels of tumor‐infiltrating leucocytes consisting of different admixtures of immune cells in which myeloid cells predominated over lymphoid cells were also observed among the tumor samples analyzed herein, in line with previous observations (46). Among myeloid cells, microglial cells and blood‐derived TAM, neutrophils, and MDSC were found at progressively lower mean overall frequencies, in association with (much) smaller numbers of TIL (T‐lymphocytes and to a less extent also B‐cells and NK‐cells). To the best of our knowledge, this is the first study in which all above cell populations were simultaneously assessed in individual GBM tumor specimens from a relatively large series of human GBM samples based on a comprehensive multicolor antibody panel assessed by flow cytometry (47, 48). In previous studies in both murine and human models, microglial cells have been defined based on the expression of myeloid markers such as CD11b, and CD45 (49). However, in our study, these markers on their own were insufficient (39) for accurate identification of microglial cells and their discrimination from other myeloid cell populations of tumor‐infiltrating leucocytes. Thus, several other markers such as (CD14, HLADR, and CD15) were required —in addition to baseline (green) autofluorescence levels— to discriminate among the distinct populations of microglial and BM‐derived TAM, neutrophils, and MDSC. Simultaneous staining for CD15, CD16, and CD11b, in addition to HLA‐DR and CD14, provided unequivocal discrimination between CD14+ HLADR+ TAM (including microglial and blood‐derived TAM) and both CD15hi CD16hi CD11b+HLADR−CD14− neutrophils and CD15+CD16−CD11b+HLADR−/lo MDSC (46). Additional (conventional) (30, 50, 51) markers were used for the discrimination among the remaining CD45hi lymphoid cells including CD3+ T‐cells (and their subsets), CD4+ CD25hi T‐regs and both CD19+CD20+ B‐lymphocytes and SSCloCD16+ NK‐cells.
Overall, among tumor‐infiltrating leucocytes, TAM were the most represented cell population, including microglial cells and at lower percentages also blood‐derived macrophages. Previous studies suggest that predominance of M1‐like versus M2‐like TAM may be associated with a better outcome of GBM, while higher levels of M2‐like TAM could be related to tumor growth and progression (52). Here, we investigated the pattern of expression of three (CD192, CD206, and CD163) markers that have been related to these two different polarization states of TAM (52, 53). Despite variable and heterogeneous expression profiles were observed for each of the three markers, we could still discriminate between M1‐like and M2‐like TAM based on the expression of CD192 for the identification of M1‐like cells, and positivity for CD163 and/or CD206 for the definition of M2‐like cells. Based on these criteria, an overall similar proportion of M1‐like and M2‐like TAM was identified with no clearly defined tumor subgroups based on the predominance of M1‐like versus M2‐like TAM. In turn, neutrophils and MDSC were represented at almost similar, but lower and more variable proportions, whereas TIL were less represented (typically <10% of all tumor‐infiltrating leucocytes), also at variable percentages among different tumors. A more detailed analysis of the TIL showed clear predominance of T‐cells, particularly TCD8+ cells and to a less extent TCD4+and TCD4−CD8− T‐cells, with very minor numbers of T‐regs, B‐cells and NK‐cells. Overall, these results confirm and extend on previous observations in which only part of all above subsets of tumor‐infiltrating leucocytes had been assessed (26, 50). Despite all the above, no significant correlation was found between flow cytometry and immunohistochemical data evaluated in parallel for a few major cell populations, pointing out the need for more in‐depth studies to better define and understand the value of flow cytometric analyses of tumor‐infiltrating immune cells in GBM.
Interestingly, however, simultaneous assessment of all subsets of tumor cells and immune cells in individual tumors via multivariate analyses identified three clearly distinct immune cell profiles among our cases. These included: (i) a major group of GBM with limited immune cell infiltrates; (ii) tumors with the predominance of myeloid cells (TAM and neutrophils) together with residual percentages of lymphocytes, particularly T‐cells; and (iii) a smaller group of tumors with immune cell infiltrates that consisted of a mixture of TAM, neutrophils, and T‐lymphocytes, mostly TCD8+ and TCD4+ T‐cells. At present, there is only a limited number of studies in which various immune cell populations of tumor‐infiltrating leucocytes have been simultaneously assessed in large series of GBM (14, 26). Despite this, Zhang et al (54) have recently investigated immune profiles of GBM tumor based on gene expression profiling. Interestingly, they identified three different immune (microenvironment) profiles, among which the predominance of TAM‐related gene expression profiles was associated with a better patient outcome. Although the direct correlation between our data and the GEP defined by Zhang et al cannot be made, among our patients, those that had tumors with a predominance of TAM showed a comparable (better) outcome to tumors that displayed minimal immune cell infiltrates. In contrast, in our cohort, patients that showed mixed infiltrates of TAM, neutrophils, and lymphocytes, had a significantly poorer overall survival.
Previous studies in gliomas suggest that both the predominance of TAM over other immune cell populations (55), and the presence of tumor infiltrates enriched in neutrophils and MDSC are both associated with poorer prognostic disease features (24, 56), which may be due to the inhibitory effects of these myeloid cells on T‐cells, and their contribution to an immunosuppressive tumor microenvironment (57). Thus, in gliomas, the extent of neutrophil (58) and MDSC (57) infiltration has been positively correlated with tumor grade. In contrast, the presence of TIL, particularly of TCD8+ cells, has been associated in several studies with enhanced survival (50). However, controversial results have been reported in the literature in this regard and in some recent studies, higher levels of both TCD3+ and TCD8+ tumor‐infiltrating cells have been associated with shorter survival (26). Such apparent discrepancies may be due to the fact that patient outcome might not be directly related to the specific proportion of TAM, lymphocytes, TCD4+ or TCD8+ cells per se, but it is more likely associated with the overall tumor‐infiltrating leucocytes immune profile (25). In line with this hypothesis, here we did not find any significant association between the percentage of TAM, neutrophils, MDSC, and T‐cells on patient outcome. In contrast, a unique immune cell profile based on an admixture of TAM, neutrophils, and relatively high T‐lymphocyte counts, was associated with a significantly shorter OS. In contrast, despite the potential clinical relevance of NK‐cells and T‐regs, here they represented a very minor tumor cell compartment, their numbers showing no clear impact on disease outcome (59).
Previous studies have identified gender (26), age (50), KPS index (60), type of surgery (61), type of treatment (40), and tumor genetics (e.g., EGFR gene status (4, 62) and epigenetics (MGMT‐methylation) (61) to be associated with the outcome (i.e., OS) of GBM. In contrast, only a few studies have investigated the impact of the tumor microenvironment, particularly the immune microenvironment on patient OS. Our results showed no clear association between the tumor immune cell profile and the patient KPS, age, gender, and type of surgery. In contrast, a clear association between the cellular composition of the immune infiltrates the EGFR gene status was observed. EGFR has long been known to be altered in most GBM patients, and to play a key role in the oncogenesis and clinical behavior of GBM (62, 63). In this regard, a high percentage of chromosome 7 copy number alterations, (i.e., trisomy and polysomy associated or not with the amplification of the EGFR gene) have also been previously described in GBM in the literature, in which the percentage of EGFR gene amplification ranges from 30% to 70% of all tumors depending on the techniques used, at the same time numerical alterations of chromosome 7 would occur in up to 90% of EGFR non‐amplified cases (45, 64), in line with our results. Despite this, few studies have investigated the potential relationship between EGFR gene alterations and the immune profile in GBM. Thus, An et al (65) have shown that wild‐type EGFR and some EGFR mutants (e.g., EGFRvIII) cooperate to induce macrophage infiltration. In turn, Hao et al (66) reported higher levels of tumor infiltration by TCD4+ lymphocytes, neutrophils, and dendritic cells to be associated in low‐grade gliomas with shorter survival among EGFR‐mutated cases. Here, EGFR amplification was associated with minimal immune cell infiltrates, while EGFR gains predominated in cases with TAM plus neutrophil infiltrates and a diploid EGFR gene status was highly characteristic of tumors with mixed myeloid and T‐lymphoid tumor infiltration. These results are in line with previous studies in which an association between EGFR gene amplification and a better patient outcome has been reported (62) and point out a potential link between the tumor genetic and immune profiles.
In summary, here we show that immune cell infiltrates which consist of distinct immune profiles are systematically present at highly variable levels in GBM, the presence of mixed myeloid (TAM plus neutrophils) and T‐lymphoid infiltrates being associated with unique EGFR genetic features and a significantly poorer patient outcome.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
All authors have contributed significantly to this article. Study design: Alberto Orfao and María Dolores Tabernero. Samples and clinical data collection: Álvaro Otero, Daniel Arandia, Daniel Pascual, Laura Ruíz, Pablo Sousa, Andoni García, Juan Carlos Roa, Jorge Javier Villaseñor, Luis Torres, J Ortiz and Adelaida Nieto. Methodology and analysis: Maria do Rosario Almeida, María González‐Tablas and María Dolores Tabernero. Writing ‐ original draft preparation: María González‐Tablas, Alberto Orfao, and María Dolores Tabernero. All authors have read and approved the final version.
ETHICAL APPROVAL
All procedures performed in this study were in accordance with the 1964 Helsinki declaration. All enrolled patients provided written informed consent.
Supporting information
Fig S1
FIGURE S1 Immunohistochemical immunofluorescence and iFISH characterization of FACS‐isolated TAM and CD45‐ tumor and normal astrocytic cells. Immune cells were discriminated from tumor cells and other non‐hematopoietic cells based on CD45 positive expression and TAM were further identified based on the coexpression of CD14 and HLADR (panel A). Hematoxylin and eosin staining of FACS‐sorted CD45+HLADR+CD14+ cells showed a TAM compatible morphology (panel B) and positive expression for CD45 by immunofluorescence (IF), in contrast to FACS‐sorted tumor (and other non‐hematopoietic) cells that were CD45‐ (panel C). Further iFISH analyses revealed EGFR gene amplification associated with partial expression of EGFR by flow cytometry (panel D)
Fig S2
FIGURE S2 Correlation between the percentage of immune cell populations in tumor tissues as assessed by flow cytometry and histochemistry (panel A) and in peripheral blood (panels B and C). In panel A, the relationship between the number of leucocytes in peripheral blood and their percentage in the tumor (upper left panel) and between the percentage of neutrophils (upper right panel), monocytes/TAM (lower left panel), and lymphocytes (upper right panel) in blood versus the tumor, is shown. In panel B, the correlation between the percentage of tumor cells (left panel) and TIL (right panel) in paired tumor specimens evaluated by both flow cytometry and histochemistry is displayed
Table S1
TABLE S1 Overall features of fresh primary human GBM samples (n = 55) analyzed in this study derived from patients investigated at diagnosis (n = 43), after therapy (n = 8) and at both moments (n = 3)
Table S2
TABLE S2 Mutational status of IDH1 and IDH2 genes in 46 available GBM tumor samples
Table S3
TABLE S3 Antibody panels, markers and reagents (clones and manufacturers) used in this study for flow cytometry immunophenotypic analyses of primary GBM tumor specimens (n = 55)
Table S4
TABLE S4 Distribution of different subsets of tumor cells, microglial cells and infiltrating leucocytes in paired diagnostic vs recurrent (n = 2) or bilateral (n = 1) tumor samples from three GBM patients
ACKNOWLEDGEMENTS
This work was supported by CIBERONC (grant CB16/12/00400 CIBERONC, Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain) the ISCIII PI16/0476 grant (Instituto de Salud Carlos III), Fondos FEDER and GRS2049/A/19 grant (Consejería de Sanidad Junta de Castilla y León, Gerencia Regional de Salud, Spain).
Alberto Orfao and María Dolores Tabernero have equally contributed to this manuscript and both should be considered as the last author.
DATA AVAILABILITY STATEMENT
The clinical data associated with this paper are available in Table S1.
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 2. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129:829–48. [DOI] [PubMed] [Google Scholar]
- 3. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest. 2017;97:498–518. [DOI] [PubMed] [Google Scholar]
- 4. González‐Tablas M, Crespo I, Vital AL, Otero Á, Nieto AB, Sousa P, et al. Prognostic stratification of adult primary glioblastoma multiforme patients based on their tumor gene amplification profiles. Oncotarget. 2018;9:28083–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. 2018;9:1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eder K, Kalman B. The dynamics of interactions among immune and glioblastoma cells. NeuroMol Med. 2015;17:335–52. [DOI] [PubMed] [Google Scholar]
- 7. Domingues P, González‐Tablas M, Otero Á, Pascual D, Miranda D, Ruiz L, et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav Immun. 2016;53:1–15. [DOI] [PubMed] [Google Scholar]
- 8. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma‐infiltrating microglia/macrophages in mediating antitumor immune responses1. Neuro‐Oncology. 2006;8:261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer. 2018;119:1171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pellegatta S, Eoli M, Frigerio S, Antozzi C, Bruzzone MG, Cantini G, et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. OncoImmunology. 2013;2:e23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: parallels at non‐CNS sites. Front Oncol. 2015;5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang W, Li Y, Gao R, Xiu Z, Sun T. MHC class I dysfunction of glioma stem cells escapes from CTL‐mediated immune response via activation of Wnt/β‐catenin signaling pathway. Oncogene. 2020;39:1098–111. [DOI] [PubMed] [Google Scholar]
- 14. Jack AS, Lu J‐Q. Immune cell infiltrates in the central nervous system tumors. Austin Neurosurg Open Access. 2015;2(1):1024. [Google Scholar]
- 15. Kaminska B, Kocyk M, Kijewska M. TGF beta signaling and its role in glioma pathogenesis. Adv Exp Med Biol. 2013;986:171–87. [DOI] [PubMed] [Google Scholar]
- 16. Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol. 2004;22:1136–51. [DOI] [PubMed] [Google Scholar]
- 17. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin‐10 and the interleukin‐10 receptor. Annu Rev Immunol. 2001;19:683–765. [DOI] [PubMed] [Google Scholar]
- 18. Gousias K, von Ruecker A, Voulgari P, Simon M. Phenotypical analysis, relation to malignancy and prognostic relevance of ICOS+T regulatory and dendritic cells in patients with gliomas. J Neuroimmunol. 2013;264:84–90. [DOI] [PubMed] [Google Scholar]
- 19. Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget. 2015;6:15077–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. [DOI] [PubMed] [Google Scholar]
- 21. Alban TJ, Alvarado AG, Sorensen MD, Bayik D, Volovetz J, Serbinowski E, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune‐suppression signatures associated with prognosis. JCI Insight. 2018;3:e122264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gielen PR, Schulte BM, Kers‐Rebel ED, Verrijp K, Petersen‐Baltussen HMJM, Ter Laan M, et al. Increase in both CD14‐positive and CD15‐positive myeloid‐derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15‐positive myeloid‐derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol. 2015;74:390–400. [DOI] [PubMed] [Google Scholar]
- 23. Jacobs JFM, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJM, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. 2010;225:195–9. [DOI] [PubMed] [Google Scholar]
- 24. Raychaudhuri B, Rayman P, Huang P, Grabowski M, Hambardzumyan D, Finke JH, et al. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol. 2015;122:293–301. [DOI] [PubMed] [Google Scholar]
- 25. Farmer J‐P, Antel JP, Freedman M, Cashman NR, Rode H, Villemure J‐G. Characterization of lymphoid cells isolated from human gliomas. J Neurosurg. 1989;71:528–33. [DOI] [PubMed] [Google Scholar]
- 26. Orrego E, Castaneda CA, Castillo M, Bernabe LA, Casavilca S, Chakravarti A, et al. Distribution of tumor‐infiltrating immune cells in glioblastoma. CNS . Oncology. 2018;7:CNS21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bregy A, Wong TM, Shah AH, Goldberg JM, Komotar RJ. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat Rev. 2013;39:891–907. [DOI] [PubMed] [Google Scholar]
- 28. Buerki RA, Chheda ZS, Okada H. Immunotherapy of primary brain tumors: facts and hopes. Clin Cancer Res. 2018;24:5198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang B, Zhang H, Gu L, Ye B, Jian Z, Stary C, et al. Advances in immunotherapy for glioblastoma multiforme. J Immunol Res. 2017;2017:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Han S, Ma E, Wang X, Yu C, Dong T, Zhan W, et al. Rescuing defective tumor‐infiltrating T‐cell proliferation in glioblastoma patients. Oncology Letters. 2016;12:2924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cengage G, ed. DNA Isolation Methods. World of Forensic Science; 2006. [Google Scholar]
- 32. Paz‐Bouza JI, Orfao A, Abad M, Ciudad J, Garcia MC, Lopez A, et al. Transrectal fine needle aspiration biopsy of the prostate combining cytomorphologic, DNA ploidy status and cell cycle distribution studies. Pathol Res Pract. 1994;190:682–9. [DOI] [PubMed] [Google Scholar]
- 33. Groeneveld K, te Marvelde JG, van den Beemd MW, Hooijkaas H, van Dongen JJ. Flow cytometric detection of intracellular antigens for immunophenotyping of normal and malignant leukocytes. Leukemia. 1996;10:1383–9. [PubMed] [Google Scholar]
- 34. Smith PJ, Wiltshire M, Davies S, Patterson LH, Hoy T. A novel cell permeant and far red‐fluorescing DNA probe, DRAQ5, for blood cell discrimination by flow cytometry. J Immunol Methods. 1999;229:131–9. [DOI] [PubMed] [Google Scholar]
- 35. Kalina T, Flores‐Montero J, van der Velden VHJ, Martin‐Ayuso M, Böttcher S, Ritgen M, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26:1986–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matarraz S, Fernandez C, Albors M, Teodosio C, López A, Jara‐Acevedo M, et al. Cell‐cycle distribution of different cell compartments in normal versus reactive bone marrow: a frame of reference for the study of dysplastic hematopoiesis. Cytometry B Clin Cytom. 2011;80B:354–61. [DOI] [PubMed] [Google Scholar]
- 37. Van der Maaten L, Hinton G. Visualizing data using t‐SNE. J Mach Learn Res. 2008;9:2579–605. [Google Scholar]
- 38. Domingues PH, Teodósio C, Ortiz J, Sousa P, Otero Á, Maillo A, et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am J Pathol. 2012;181:1749–61. [DOI] [PubMed] [Google Scholar]
- 39. Balik V, Mirossay P, Bohus P, Sulla I, Mirossay L, Sarissky M. Flow cytometry analysis of neural differentiation markers expression in human glioblastomas may predict their response to chemotherapy. Cell Mol Neurobiol. 2009;29:845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang J, Hu G, Quan X. Analysis of the factors affecting the prognosis of glioma patients. Open Medicine. 2019;14:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Glumac PM, LeBeau AM. The role of CD133 in cancer: a concise review. Clin Transl Med. 2018;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. [DOI] [PubMed] [Google Scholar]
- 43. Song W‐S, Yang Y‐P, Huang C‐S, Lu K‐H, Liu W‐H, Wu W‐W, et al. Sox2, a stemness gene, regulates tumor‐initiating and drug‐resistant properties in CD133‐positive glioblastoma stem cells. J Chin Med Assoc. 2016;79:538–45. [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y, Li B, Zhang X, Sonpavde GP, Jiao K, Zhang A, et al. CD24 is a genetic modifier for risk and progression of prostate cancer. Mol Carcinog. 2017;56:641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. González‐Tablas M, Arandia D, Jara‐Acevedo M, Otero Á, Vital A‐L, Prieto C, et al. Heterogeneous EGFR, CDK4, MDM4, and PDGFRA gene expression profiles in primary GBM: no association with patient survival. Cancers. 2020;12:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169–80. [DOI] [PubMed] [Google Scholar]
- 47. Kumar R, de Mooij T, Peterson TE, Kaptzan T, Johnson AJ, Daniels DJ, et al. Modulating glioma‐mediated myeloid‐derived suppressor cell development with sulforaphane. PLoS One. 2017;12:e0179012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31:326–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma‐infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1:e85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kmiecik J, Poli A, Brons NHC, Waha A, Eide GE, Enger PØ, et al. Elevated CD3+ and CD8+ tumor‐infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71–83. [DOI] [PubMed] [Google Scholar]
- 51. Kmiecik J, Zimmer J, Chekenya M. Natural killer cells in intracranial neoplasms: Presence and therapeutic efficacy against brain tumours. J Neurooncol. 2014;116:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma‐associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hao NB, Lü MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang B, Shen R, Cheng S, Feng L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 2019;8:2897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7:e43339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khan S, Mittal S, McGee K, Alfaro‐Munoz KD, Majd N, Balasubramaniyan V, et al. Role of neutrophils and myeloid‐derived suppressor cells in glioma progression and treatment resistance. Int J Mol Sci. 2020;21:1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guadagno E, Presta I, Maisano D, Donato A, Pirrone CK, Cardillo G, et al. Role of macrophages in brain tumor growth and progression. Int J Mol Sci. 2018;19(4):1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. 2014;20:187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alizadeh D, Zhang L, Brown CE, Farrukh O, Jensen MC, Badie B. Induction of anti‐glioma natural killer cell response following multiple low‐dose intracerebral CpG therapy. Clin Cancer Res. 2010;16:3399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liang J, Lv X, Lu C, Ye X, Chen X, Fu J, et al. Prognostic factors of patients with gliomas – an analysis on 335 patients with glioblastoma and other forms of gliomas. BMC Cancer. 2020;20:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar N, Kumar P, Angurana SL, Khosla D, Mukherjee KK, Aggarwal R, et al. Evaluation of outcome and prognostic factors in patients of glioblastoma multiforme: a single institution experience. J Neurosci Rural Pract. 2013;4:S46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–56. [DOI] [PubMed] [Google Scholar]
- 63. Felsberg J, Hentschel B, Kaulich K, Gramatzki D, Zacher A, Malzkorn B, et al. Epidermal growth factor receptor variant III (EGFRvIII) positivity in EGFR‐amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res. 2017;23:6846–55. [DOI] [PubMed] [Google Scholar]
- 64. Lopez‐Gines C, Gil‐Benso R, Ferrer‐Luna R, Benito R, Serna E, Gonzalez‐Darder J, et al. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol. 2010;23(6):856–65. [DOI] [PubMed] [Google Scholar]
- 65. An Z, Knobbe‐Thomsen CB, Wan X, Fan QW, Reifenberger G, Weiss WA. EGFR cooperates with EGFRvIII to recruit macrophages in glioblastoma. Can Res. 2018;78:6785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hao Z, Guo D. EGFR mutation: novel prognostic factor associated with immune infiltration in lower‐grade glioma; an exploratory study. BMC Cancer. 2019;19:1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
FIGURE S1 Immunohistochemical immunofluorescence and iFISH characterization of FACS‐isolated TAM and CD45‐ tumor and normal astrocytic cells. Immune cells were discriminated from tumor cells and other non‐hematopoietic cells based on CD45 positive expression and TAM were further identified based on the coexpression of CD14 and HLADR (panel A). Hematoxylin and eosin staining of FACS‐sorted CD45+HLADR+CD14+ cells showed a TAM compatible morphology (panel B) and positive expression for CD45 by immunofluorescence (IF), in contrast to FACS‐sorted tumor (and other non‐hematopoietic) cells that were CD45‐ (panel C). Further iFISH analyses revealed EGFR gene amplification associated with partial expression of EGFR by flow cytometry (panel D)
Fig S2
FIGURE S2 Correlation between the percentage of immune cell populations in tumor tissues as assessed by flow cytometry and histochemistry (panel A) and in peripheral blood (panels B and C). In panel A, the relationship between the number of leucocytes in peripheral blood and their percentage in the tumor (upper left panel) and between the percentage of neutrophils (upper right panel), monocytes/TAM (lower left panel), and lymphocytes (upper right panel) in blood versus the tumor, is shown. In panel B, the correlation between the percentage of tumor cells (left panel) and TIL (right panel) in paired tumor specimens evaluated by both flow cytometry and histochemistry is displayed
Table S1
TABLE S1 Overall features of fresh primary human GBM samples (n = 55) analyzed in this study derived from patients investigated at diagnosis (n = 43), after therapy (n = 8) and at both moments (n = 3)
Table S2
TABLE S2 Mutational status of IDH1 and IDH2 genes in 46 available GBM tumor samples
Table S3
TABLE S3 Antibody panels, markers and reagents (clones and manufacturers) used in this study for flow cytometry immunophenotypic analyses of primary GBM tumor specimens (n = 55)
Table S4
TABLE S4 Distribution of different subsets of tumor cells, microglial cells and infiltrating leucocytes in paired diagnostic vs recurrent (n = 2) or bilateral (n = 1) tumor samples from three GBM patients
Data Availability Statement
The clinical data associated with this paper are available in Table S1.