

Abstract
Stroke, in association with its complications, is one of the leading causes of mortality and morbidity worldwide. Cerebral ischemia triggers an inflammatory response in the brain that is controlled by the activation of resident microglia as well as the infiltration of peripheral myeloid and lymphoid cells into the brain parenchyma. This inflammation has been shown to have both beneficial and detrimental effects on stroke outcome. The focus of this review lies on the functions of myeloid cells and their interaction with infiltrating lymphocytes in different phases of stroke. A detailed and time‐specific understanding of the contribution of different immune cell subsets during the course of cerebral ischemia is crucial to specifically promote beneficial and inhibit detrimental effects of inflammation on stroke outcome.
Keywords: lymphocytes, myeloid cells, neuroinflammation, stroke
Stroke is one of the leading causes of mortality and morbidity worldwide. Brain inflammation, mediated by myeloid and lymphoid immune cells, is an inherent component of stroke pathology and contributes to the functional outcome after ischemia.

Introduction
Stroke is one of the leading causes of death worldwide. In 2016 alone, approximately 5.6 million people worldwide died of stroke. Furthermore, the incidence of stroke is steadily increasing and prognoses predict 12 million deaths in 2030 (36, 117). One of the main complications following stroke are stroke‐associated infections, notably pneumonia. Stroke‐associated infections occur in approximately 30% of patients and pneumonia in up to 10% of stroke cases (15, 49, 69, 116). The increased risk of pneumonia is attributed to a general immunosuppression caused by an activation of the sympathetic nervous system and the hypothalamus‐pituitary‐adrenal gland axis causing lymphocyte apoptosis and dysfunction as well as parasympathetic, cholinergic pathways inhibiting innate immune cells occurring after central nervous system (CNS) injury and notably stroke (11, 33, 89). Another integral part of stroke pathology is the local inflammation in the affected brain area itself. It is well established that this local immune reaction contributes to ischemic pathology, influencing the death/survival of neurons and contributing to the functional outcome (15, 51). Besides the acute ischemic damage, research in recent years also showed that a chronic adaptive immune response in the ischemic hemisphere might contribute to the occurrence of poststroke dementia, which is another common complication after stroke and affects up to 30% of stroke survivors (28, 29, 86). The peripheral immune reaction dominated by stroke‐associated immunosuppression and infections and the central inflammatory reaction are closely linked. Stroke‐associated immunosuppression is thought to be a protective mechanism to limit brain inflammation, and thus, further damage (94). On the contrary, the increased occurrence of infections has been shown to function as a booster of CNS inflammation, as patients with poststroke infections show an increased CNS autoreactivity in the blood compared to stroke patients without infections, correlating with a worse clinical outcome (7, 8).
In this review, we will focus on the local CNS inflammation associated with stroke with a special focus on resident and infiltrating myeloid cells as well as their interaction with adaptive lymphocytes. Most of the data presented comes from animal models of cerebral ischemia, which only partly have been confirmed in human stroke patients so far. For the sake of clarity, the most important phenomena are summarized in Table 1, highlighting evidence from animal models and human data.
Table 1.
Contribution of myeloid cells to brain inflammation after ischemia in animal models and human stroke.
| Cell type | Animal studies | Human studies |
|---|---|---|
| Neutrophils |
|
|
| Microglia |
|
|
| Monocytes/macrophages |
|
|
Table 1 summarizes key features of myeloid cell function after stroke, comparing data from animal models to results from human studies.
Microglia and Infiltrating Myeloid Cells in Cerebral Ischemia
As CNS‐resident cells, microglia are the first immune cells to respond to acute ischemic damage in the brain. Microglia morphology is a good indicator of the cell’s activation status. Under homeostatic conditions, microglia have long and ramified processes. Upon activation, ramification is decreased and the cells adopt an amoeboid appearance. These different microglia morphologies are also observed in the ischemic hemisphere: in the lesion core, the number and length of microglia processes are significantly reduced as early as 8 hours after stroke. In the periphery of the lesion, microglia still show a ramified morphology, although with shorter processes than in the resting state (75). The majority of microglia accumulate in the periphery of the ischemic lesion, whereas the infarct core is mainly occupied by infiltrating myeloid cells in the first days poststroke. This activation of microglia primarily aims at clearing cellular debris that accumulates through neuronal cell death upon ischemia (9, 75, 81). In fact, mice deficient in triggering receptor expressed on myeloid cells 2 (TREM2) show impaired phagocytosis, increased infarct size, and worse functional outcome after stroke (58). During resolution of the acute inflammation, microglia produce anti‐inflammatory cytokines (IL‐10, TGFβ), growth factors (e.g., IGF‐1), and remove cellular debris by phagocytosis thus promoting angiogenesis and tissue reorganization (51). Following the same line of evidence, depletion of microglia in mouse models of cerebral ischemia has been associated with increased infarct sizes, aggravated neurological deficits, and enhanced brain inflammation accompanied by a stronger infiltration of leukocytes (neutrophils, T cells, natural killer cells, and monocytes/macrophages) (55). This effect was found to be mediated by a cross talk between microglia and astrocytes, as microglia depletion dramatically increased the production of pro‐inflammatory cytokines by astrocytes after stroke which directly induce neuronal apoptosis (55). In vitro and in vivo studies demonstrate that microglia can phagocytose infiltrating neutrophils, hence, reducing their cell number and contribution to the ischemic injury (25, 78, 81). Furthermore, microglia depletion alters neuronal function in the hours after ischemia. In vivo calcium imaging revealed that loss of microglia leads to the rapid development of neuronal calcium oscillations shortly after the induction of ischemia (105). Interestingly, the neurons showing these oscillations mostly died within 24 hours, and infarct sizes were increased when compared to mice without microglia depletion (105). The data presented so far highlights anti‐inflammatory effects of microglia in the early phase after stroke. Nevertheless, microglia also produce large amounts of pro‐inflammatory cytokines, reactive oxygen species, and other pro‐inflammatory mediators, thus, contributing to poststroke inflammation (51).
Activation of microglia after stroke is controlled by two mechanisms: 1) inhibitory interactions with neurons or other CNS cells are lost as well as 2) release of damage‐associated molecules upon ischemia. In the healthy brain, the interaction between neurons and microglia promotes a homeostatic microglia phenotype. For instance, the interaction between CD200 on neurons and its receptor, CD200R1 on microglia helps to keep microglia in a resting state. CD200 expression is known to decrease during tissue damage and aging, causing a reduced restraint on microglia activation (82). This could also occur during stroke when neuronal cells are lost because of the ischemic damage. In fact, CD200 knockout mice present a stronger neuroinflammatory response after stroke with increased immune cell infiltration and microgliosis. This was associated with a worsened functional outcome at 7 days after stroke (93).
Another important signal regulating microglia activation is provided by the fractalkine receptor CX3CR1 also expressed on microglia and its ligand CX3CL1 that is primarily produced by neurons. This is also an inhibitory signal, which is often lost in pathological conditions as neuronal expression of CX3CL1 is decreased or cells are lost. However, both detrimental and beneficial effects have been attributed to CX3CR1/CX3CL1 signaling in stroke. CX3CR1 or CX3CL1 knockout mice developed smaller lesions 1 day after cerebral ischemia. On the contrary, exogenous administration of CX3CL1 has also been shown to be protective during stroke in mice (82). It is hypothesized that depending on the context (ie, homeostatic brain or inflamed CNS containing infiltrating myeloid cells possibly also expressing CX3CR1), the CX3CR1‐CX3CL1 axis could have different effects on microglia/macrophage activation and brain inflammation (12).
Upon ischemia, neuronal function is greatly disturbed in the affected area. Stressed and dying cells start releasing danger molecules that directly act on microglia. Prominent molecules belonging to this category of danger‐associated signals are the nucleotides adenosine and uridine triphosphate (ATP, UTP), heat‐shock proteins (HSPs), and high mobility group box 1 (HMGB1). The released ATP acts on microglial P2X7 receptors and induces the release of pro‐inflammatory factors. Nucleotides also control microglia migration and phagocytosis (45), which can promote engulfment of still viable neurons, and therefore, contribute to brain damage upon stroke (9). On the contrary, damage‐associated molecules also bind to toll‐like receptors (TLRs) and scavenger receptors, equally inducing a pro‐inflammatory phenotype in microglia. Upon this activation, microglia rapidly release cytokines (IL‐1β, tumor necrosis factor α (TNFα), IL‐6), chemokines (CCL2, CCL3, CXCL2/3, CXCL8), and reactive oxygen species (51).
Following the activation of microglia, neutrophils are the first blood‐borne cells found in the ischemic hemisphere after stroke. In fact, neutrophil adhesion to the activated endothelium and rolling can be observed as early as 1 hour after experimental stroke (52). Whether the adhering neutrophils actually infiltrate into the ischemic hemisphere or remain trapped in perivascular spaces is controversial (34). Irrespective of this controversy, neutrophils have been shown to release neurotoxic substances such as reactive oxygen and nitrogen species, matrix metalloproteases (i.e., MMP9), and neutrophil elastase and contribute to blood–brain barrier disruption and ischemic damage (40, 100, 103). In fact, a higher neutrophil‐to‐lymphocyte ratio indicating increased numbers of neutrophils is associated with worse outcome and hemorrhagic transformation in human stroke patients (46, 65, 101). In addition, neutrophils release neutrophil extracellular traps consisting of DNA, histones, and granule proteins that contribute to thrombus formation and resistance to recanalization by thrombolysis (30, 62, 85). On the contrary, neutrophils are nowadays also considered to be heterogeneous cells and so‐called N2 or anti‐inflammatory neutrophils have also been shown to exert protective effects during cerebral ischemia (22, 41).
With regard to infiltrating monocytes, experimental studies also provide evidence for both detrimental and protective roles of invading monocytes/macrophages in brain ischemia. CCR2+Ly6Chi monocytes are the main population of infiltrating monocytes in mouse studies of stroke (39, 72). Once in the ischemic hemisphere, they spread within the ischemic core and penumbra and differentiate into CX3CR1+ macrophages that can remain in the brain for extended periods of time (39). This spreading and differentiation is most likely controlled by chemokines and cytokines produced by local brain cells. Early after ischemia (i.e., 3 days), CCR2+ cells are evenly distributed in the damaged tissue. However, at 14 and 28 days after stroke, CCR2+ cells are mainly found in the lesion core, which is surrounded by CX3CR1+ monocytes/macrophages as shown by the transfer of CX3CR1GFP/+CCR2RFP/+ bone marrow into wild‐type mice. The number of CCR2+ monocytes peaks around day 3 post‐ischemia, whereas CX3CR1+ cells reach their peak much later, around 14–28 days after induction of ischemia (39).
The functional consequence of this monocyte/macrophage infiltration is controversial as different lines of evidence suggest protective as well as deleterious features. It was shown that selective deficiency of CCR2 in monocytes leads to a reduced infiltration after experimental stroke. This was associated with a worse functional outcome, a decrease of angiogenesis‐associated genes, and a reduced vessel density in the ischemic brain region (83). Furthermore, prevention of monocyte infiltration by clodronate‐liposomes and diphtheria toxin‐mediated monocyte depletion in CD11b‐DTR (diphtheria toxin receptor) mice increased the risk of hemorrhagic transformation of the infarct in the first week after ischemia, further highlighting the importance of monocytes/macrophages in angiogenesis and vessel integrity after stroke (43). In an additional study, circulating CCR2+ monocytes were depleted with an anti‐CCR2 monoclonal antibody during the first week after stroke, which caused a reduced recovery of motor function as late as 11 weeks post‐ischemia (113). However, deficiency of CCR2 or its ligand CCL2, and thus, reduced monocyte infiltration after stroke has also been associated with smaller infarct sizes, a reduction in pro‐inflammatory cytokine expression, and thus, a better functional outcome (27, 50). Additional studies could not find any effect of infiltrating monocytes on the outcome after ischemia. However, these studies did not assess cytokine production, angiogenesis, or vessel integrity (72, 96).
In addition to microglia and infiltrating monocytes/macrophages, the CNS harbors also resident macrophage populations called border‐associated macrophages (BAMs) that reside around blood vessels, in leptomeningeal spaces and the choroid plexus. In a rat model of ischemic stroke, it was shown that these cells rapidly respond to ischemic damage and contribute to the recruitment of granulocytes, increase vascular permeability, and participate in neurological dysfunction. This contribution of BAMs to the ischemic injury was studied in the very early phase of stroke (16 hours after induction) where peripheral immune cell infiltration is still negligible (84).
Microglia–T Cell Interactions
Several experimental studies have shown that T lymphocytes modify stroke outcome, although the chronological sequence and types of T cell‐mediated effects have been controversial. Certain animal studies of stroke report T cell infiltration as early as 24 hours after ischemia, whereas in other cases, significant T cell infiltration is only observed in the chronic inflammatory phase from day 7 post‐ischemia onward (42, 104). Irrespective of the exact timing, T lymphocytes need signals to be attracted to the brain. Microglia may provide these signals through direct release of pro‐inflammatory molecules or by the chemoattraction of peripheral myeloid cells, which then in turn secrete the T cell‐attracting molecules.
The rapid release of TNFα, IL‐6, IL‐1β, and interferon γ (IFNγ) by microglia upon ischemia induces the upregulation of adhesion molecules such P‐Selectin, E‐Selectin, intercellular adhesion molecule (ICAM), and vascular cell adhesion molecule (VCAM) by endothelial cells (111). This prepares the ground for T cell adhesion and successful transmigration into the CNS. Through the release of chemokines such as CXCL2, CCL2, CXCL8, CXCL9, and CXCL10, microglia can directly attract T cells that express the corresponding receptors (51, 111).
It is now well established that T lymphocytes can polarize into different effector cells. T helper cells type 1 (Th1) and type 17 (Th17) promote neuroinflammation, whereas T helper cells type 2 (Th2) and regulatory T cells (Treg) rather dampen the inflammatory response in the brain (3, 4, 38, 64, 92). The generation of Th1 and Th17 cells requires certain cytokine profiles (IL‐12, IFNγ for Th1 and IL‐6, TGFβ, IL‐23 for Th17 cells), which can be provided by activated microglia and/or infiltrating myeloid cells (51, 67, 110). Once in the brain, the effector functions of T lymphocytes will be influenced by the local cytokine microenvironment, which is partly controlled by activated myeloid cells.
As mentioned above, after the first acute inflammatory phase, microglia is considered to adopt a rather anti‐inflammatory phenotype. Increased phagocytosis as well as the production of anti‐inflammatory cytokines such as IL‐4, IL‐10, and TGFβ generate a cytokine milieu favoring the generation of regulatory T cells that inhibit inflammation. In fact, Tregs infiltrate into the ischemic hemisphere and accumulate for as long as 30 days poststroke. Treg depletion markedly increases infarct volumes after middle cerebral artery occlusion (MCAO). Interestingly, depletion of Tregs also resulted in an increase of microglia cell numbers at day 3 after MCAO, most of which produced TNFα, highlighting the bidirectional communication between microglia and T cells (64). However, another study investigating depletion of Tregs in experimental stroke did not find any effects on functional outcome or immune cell numbers (104). The differences between these two studies might be in part explained by the use of different stroke models (transient vs. permanent ischemia) and the timing of Treg depletion (3 and 14 days post‐MCAO vs. 2 days before MCAO) (63).
On the contrary, infiltrating T cells produce cytokines that influence microglia function. Th1 cells are major producers of IFNγ, whereas Th17 cells produce mainly IL‐17. Microglia express receptors for both cytokines and can thus be influenced by their presence. Treatment of primary mouse microglia cultures with IFNγ induces the expression of IL‐1β, IL‐6, and TNFα (112). Coculture of primary human microglia with activated T cells also induces the expression of the pro‐inflammatory cytokines TNFα and IL‐12 (14).
Beyond CD4+ helper T cells, CD4−CD8− γδ T cells were shown to infiltrate as early as 12–24 hours after ischemia and contribute significantly to tissue damage (42, 71). Infiltrating γδ T cells are major producers of the neurotoxic cytokine IL‐17. In vitro studies showed that TLR‐stimulated primary microglia are able to induce IL‐17 production in γδ T cells which in turn cause neuronal apoptosis (26). As TLR ligands, such as HSPs, HMGB1, and ATP, are abundantly present in the ischemic brain (73), similar mechanisms could take place in vivo after stroke. Furthermore, IL‐23 produced in the ischemic brain by microglia and macrophages further amplifies IL‐17 production by infiltrating γδ T cells (51, 99). Coculture of primary microglia with double‐negative T cells isolated from mouse spleens 3 days after MCAO was shown to increase the expression of the pro‐inflammatory molecules CD86 and CD16 while reducing the expression of the anti‐inflammatory marker CD206 in microglia (71).
The abovementioned studies mostly focus on the early phase after ischemia, meaning the first hours to 1 week post‐surgery. However, in recent years it has become increasingly obvious that stroke also generates long‐lasting, chronic immune responses in the affected brain regions (29, 98, 104, 118). Myeloid cells and relatively small numbers of pro‐inflammatory Th1 and anti‐inflammatory Treg cells mainly dominate the inflammatory response in the first week after stroke. However, stroke also generates antigen‐specific CD4 T cell responses (54). This delayed process involves antigen‐presenting cells, which phagocytose CNS proteins and present epitopes of them to CD4 T lymphocytes in the context of major histocompatibility complex (MHC) class II surface proteins. As activated microglia upregulate MHC class II, these cells could be in a unique position to prime or reactive infiltrating autoreactive T lymphocytes. In addition, microglia also express co‐stimulatory molecules such as CD80 and CD86 that are essential for a successful antigen‐dependent T cell activation (119). Antigen‐specific T cells occur with an increased frequency in the blood of stroke patients and have also been shown to infiltrate the ischemic brain in rodent models (60, 94). Through the transfer of myelin oligodendrocyte protein (MOG)35‐55‐specific T cells into Rag2−/− mice, it was shown that these cells are reactivated in the ischemic brain and proliferate. In addition, cells with other specificities (i.e., to the peptides MOG91‐108 and MOG103‐125) were also locally expanded. This reactivation and clonal expansion was at least in part controlled by antigen presentation by microglia, as depletion of microglia reduced this phenomenon. Furthermore, these antigen‐specific T cells clearly play a deleterious role, as transfer of WT or ovalbumin‐specific T cells into Rag2‐/‐ mice generated smaller ischemic lesions compared to the transfer of brain antigen‐specific T cells (53).
Nevertheless, it is not definitely clear whether microglia play a similar role in WT mice in the presence of a polyclonal T cell pool. In the case of experimental autoimmune encephalitis (EAE), a rodent model of multiple sclerosis, it was shown that microglia, although the most abundant myeloid cells in the CNS, are not the main antigen‐presenting cells (APC) presenting myelin antigens to autoreactive T cells (44). Furthermore, several studies showed that CNS‐antigens are found in draining lymph nodes after stroke in patients and rodent models (88, 122). Therefore, it is possible that T cell activation through antigen presentation occurs in the periphery, before the autoreactive T cells infiltrate into the ischemic brain. Furthermore, dendritic cells are also observed in the ischemic hemisphere in rodent models and could present CNS antigens to infiltrating T cells (37).
Antigen presentation by microglia has also been shown to play a role in the induction of regulatory T cells. In vitro stimulation of microglia cultures with high‐dose IFNγ and MOG peptide was shown to increase proliferation of MOG‐specific effector T cells, whereas low‐dose IFNγ and MOG peptide preferentially led to the de novo generation and proliferation of MOG‐specific regulatory T cells. These microglia‐induced regulatory T cells significantly reduced disease severity when transferred to mice suffering from EAE. As speculated by the authors, this could present a mechanism for limiting inflammation and autoreactive responses after a first pro‐inflammatory phase likely also occurring in ischemic brain injury (32).
Whether antigen presentation occurs in the ischemic CNS and which myeloid cell type contributes to this phenomenon remains to be further studied.
Microglia and B Lymphocytes
The role of B lymphocytes in cerebral ischemia has been neglected for a long time. First experiments carried out in µMT−/− mice, which lack mature B cells revealed larger infarct volumes in the absence of B cells. The lack of B cells was accompanied by increased numbers of neutrophils, T cells, microglia, and infiltrating myeloid cells in the ischemic hemisphere (91). This effect is probably because of the release of the anti‐inflammatory cytokine IL‐10 by early infiltrating B cells, as adoptive transfer of IL‐10 producing B cells led to a reduction in infarct sizes, whereas IL‐10‐deficient B cells did not have an effect on acute infarct size (16, 91). However, other studies found no significant B cell infiltration into the ischemic brain or at least no clinical relevance for the infiltrating cells in the first week after stroke (42, 97).
RAG−/− mice, lacking mature B and T cells were shown to have smaller infarcts compared to WT mice. Transfer of T cells abolished this phenomenon, whereas B cell transfer did not have this effect. This could either support the idea that B cell infiltration does not play a critical role early after ischemia or that a cross talk between B and T cells is needed to exert their effect on stroke‐associated inflammation (61).
The studies mentioned so far only analyzed the effect of B cells in the acute and subacute phase of experimental stroke. Here, B cells function mainly as cytokine producers. However, recent studies revealed that B lymphocytes might play an important role in the chronic phase occurring weeks after the onset of ischemia. Experimental studies demonstrated that B cells infiltrate in a delayed manner into the ischemic mouse brain and contribute to the occurrence of poststroke cognitive decline. This is thought to be mediated by the production of brain‐reactive antibodies that interfere with neuronal function (28, 29), as has been shown to occur in human stroke patients (23, 57, 79, 114). The occurrence of local, intrathecal antibody production after stroke has been observed for a long time (90, 107). B lymphocytes are present in the brains of stroke patients, and their numbers are significantly higher in those with poststroke dementia (28, 29).
Interestingly, the abovementioned studies as well as our own results show that the infiltrating B cells are not randomly spread but cluster within the ischemic lesion (10, 29). This organization is reminiscent of ectopic lymphoid structures (ELS) that occur in different chronic inflammatory and autoimmune diseases. In these disease settings, B cells differentiate into antibody‐secreting and memory cells in these ELS and contribute to the local clearance of pathogens or the propagation of autoimmune inflammation (2, 21, 87). In murine stroke models, CD138+ plasmablasts or plasma cells can be readily detected in the ischemic hemisphere (10, 29). Little is known about how B lymphocytes are attracted to the ischemic brain and how this organization is controlled. The formation of ELS closely mirrors the development of secondary lymphoid organs during which the expression of lymphoid chemokines (CXCL12, CXCL13, CCL19, and CCL21) by stromal cells, so‐called lymphoid tissue organizer cells, control the positioning of lymphocytes within the forming follicles in spleen and lymph nodes. Expression of these chemokines also occurs in ELS. However, in the adult organism during the formation of ELS, these chemokines are rather expressed by activated myeloid cells or lymphocytes. In the case of stroke, the situation is less clear. CXCL12 and its receptor CXCR4 are expressed constitutively in the brain but are upregulated after ischemia. In animal models, CXCL12 expression after ischemia is mainly attributed to endothelial cells and astrocytes and, besides regulating leukocyte chemotaxis, also contributes to angiogenesis and recruitment of neuronal progenitor cells. However, the main B cell‐attracting chemokine is CXCL13 (2, 20). It has been shown that Cxcl13 mRNA is upregulated as early as 1 day after stroke and that protein production mainly occurs in the ischemic hemisphere by endothelial cells (74). However, this early after stroke, endothelial‐derived CXCL13 may not be responsible for the delayed infiltration of ELS‐forming B cells. A different experimental study analyzing the transcriptome of the ischemic penumbra found that Cxcl13 mRNA is still upregulated as late as 8 weeks after stroke. In addition, such a delayed upregulation was also observed for Cxcl12 and Ccl19 mRNAs (17). Nevertheless, this study did not determine which cell types produce these chemokines or how their expression is regulated. Research on other inflammatory conditions of the CNS shows that microglia could potentially contribute to B cell infiltration through CXCL13 production. Cxcl13 transcripts are increasing with age in retinal microglia (66). In addition, microglia have also been shown to produce CXCL13 during viral encephalitis (35).
Several reports show that CXCL13 can also be expressed by monocytes/macrophages in autoimmune diseases that are accompanied by the generation of ELS. This occurs for instance in the joints of rheumatoid arthritis patients and in the salivary glands of patients with Sjögren’s Syndrome (2, 6, 13, 20). In addition, stimulation with TNFα, IL‐10, or TLR7 ligands of human monocytes induces the secretion of CXCL13 (19). In the EAE mouse model, Cxcl13 expression was shown to increase during the disease progression and was detected in CD11c+ dendritic cells in the spinal cord (5). Furthermore, a population of murine peritoneal macrophages constitutively expresses CXCL13 (1). Whether infiltrating monocytes/macrophages or dendritic cells also express CXCL13 and contribute to B cell infiltration in stroke remains to be determined.
Other studies show that B lymphocytes also infiltrate in remote areas that are not directly affected by the ischemic damage. Here, B cells have been shown to promote motor recovery and neurogenesis (80). The exact mechanism of this phenomenon is not clear yet. However, as the antibodies produced by infiltrating B cells likely bind CNS structures, it is possible that they also enhance phagocytosis through opsonization of cell debris, and thus, promote wound healing in the ischemic core. A similar phenomenon has been described in a study of peripheral nerve injuries. Here, natural antibodies accumulate in the damaged nerve tissue and promote clearance of myelin debris (109). As myelin and other cellular debris are also abundant in the ischemic brain, similar mechanisms could take place here, where the secreted antibodies could potentially further help microglia and macrophages during phagocytosis of damaged tissue components.
Infection and Brain Inflammation Poststroke
As mentioned above, stroke is associated with an increased risk of infection. Studies in stroke patients have shown that notably the occurrence of pneumonia can increase autoimmune responses in the blood, and thus, also influence stroke outcome (8). Besides this effect on the peripheral, adaptive immune response, infections can also have an important impact on the innate immune response after stroke. It is well established now that peripheral infection can lead to activation and alteration of microglia phenotypes. Many different studies in animals showed that peripheral administration of LPS or killed/live bacteria can induce microglia activation as measured by increased expression of Iba‐1, MHCII, and pro‐inflammatory cytokines TNFα, IL‐1β, and IL‐6. Furthermore, similar experiments also showed an increased permeability of the blood–brain barrier after peripheral challenge with LPS or bacteria. Studies in sepsis patients revealed that systemic infection leads to a pro‐inflammatory activation of microglia shown by increased expression of CD68, iNOS, HLA‐DR, and CD86 on histological sections (120). Similar mechanisms could play a role during stroke and stroke‐associated infections. The peripheral immune challenge could further activate microglia, thereby increasing the inflammatory reaction, and fostering autoreactive immune responses in the brain after stroke.
In addition, it was shown that exposure to bacterial lipopolysaccharide (LPS) before the induction of ischemia can train microglia to produce higher amounts of IL‐1β. Microglia activation and neuronal damage was also increased in this model 7 days after stroke (115). Similar training effects are also observed in peripheral myeloid cells (47, 76, 77). Conversely, repeated LPS injections before the induction of stroke resulted in tolerized microglia with reduced expression of IL‐1β and reduced neuronal damage 7 days after ischemia (115). Therefore, a short‐term peripheral inflammatory stimulus such as stroke‐associated pneumonia could boost the immune reaction in the brain, whereas chronic stimulation likely leads to tolerance induction.
Conclusion
Neuroinflammation is a crucial part of stroke pathology in the acute and the chronic phase of the disease. Activation of resident immune cells as well as infiltration of peripheral leukocytes affects infarct development, resolution of the acute inflammation, tissue reorganization as well as motor and cognitive functional outcome (Figure 1). In this process, myeloid cells play a crucial role not only as initiators and perpetuators of inflammation, but also in the resolution phase. Recent advances in single‐cell technologies have revealed the whole spectrum of resting and activated myeloid cells after stroke with different beneficial and detrimental functions in mice and humans (48, 56, 59, 68, 95, 102, 106). Especially the recently described disease‐associated microglia (DAM) seem to represent a phenotype that is commonly observed in different neurodegenerative/neuroinflammatory disorders such as Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and their animal models as well as during aging. It is proposed that these cells arise through a sequential two‐step differentiation, resulting in microglia cells that upregulate genes involved in phagocytosis, lysosomal degradation, and lipid metabolism. Their proposed function is to remove dying cells and cell debris in order to contain and resolve damage (24, 59). Whether DAMs are also generated after stroke, how they influence the acute phase of stroke as well as the chronic adaptive immune response is unknown to date. Furthermore, diversity of infiltrating myeloid cells is likely just as big as that of resident microglia and many of their functions probably remain understudied in the field of stroke immunology. To date, it is clear that following stroke, a complex immune reaction occurs in the ischemic brain. This reaction is influenced by the cross talk between innate and adaptive, peripheral and CNS‐resident immune cells. Furthermore, systemic events such as infections can modulate the inflammatory response following stroke. As outlined above, the function of different immune cells can change during the course of the poststroke immune reaction. In order to identify suitable therapeutic strategies that block deleterious effects and enhance protective and pro‐regenerative functions of immune cells, a detailed, molecular characterization of the different cell populations as well as their spatiotemporal interaction after stroke remains an essential topic to be studied in the future.
Figure 1.
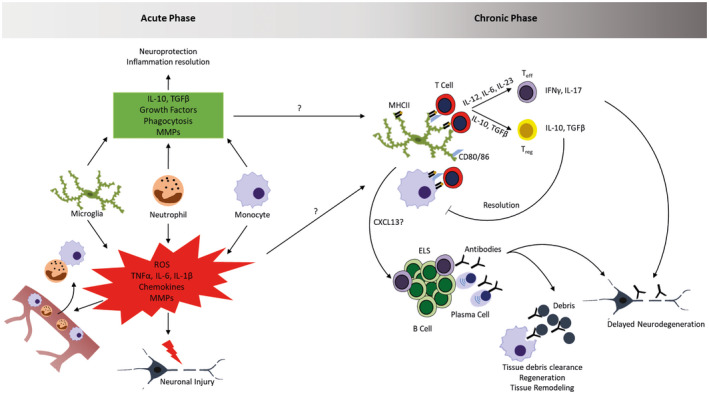
Inflammation in acute and chronic stroke lesions. Upon cerebral ischemia, tissue damage triggers the activation of microglia and the early infiltration of peripheral myeloid cells (neutrophils, monocytes). These cells contribute to tissue damage by the production of inflammatory mediators, reactive oxygen species (ROS) and MMPs, but also dampen this reaction and influence regeneration by releasing anti‐inflammatory mediators, growth factors, remove tissue debris and contribute to tissue remodeling. The mediators and molecular mechanisms influencing the transition from acute to chronic phase are currently unknown and a matter of active research. In this chronic phase where lymphocytes start infiltrating, myeloid cells can influence their function through antigen‐presentation and cytokine production. This leads to the generation of pro‐inflammatory T effector cells and anti‐inflammatory regulatory T cells. Through the release of chemokines, myeloid cells can potentially also influence the generation of B cell‐rich ectopic lymphoid structures (ELS) in which antibodies are produced. The generation of different T cell subsets and antibodies can promote delayed neurodegeneration, but potentially also contributes to tissue debris clearance and regenerative processes.
Acknowledgment
This work was supported by the German Research Foundation (SFB‐TRR167), the Einstein Foundation (Grant Number A‐2017‐406), and Leducq Foundation (Grant Number 19CVD01).
References
- 1. Accarias S, Genthon C, Rengel D, Boullier S, Foucras G, Tabouret G (2016) Single‐cell analysis reveals new subset markers of murine peritoneal macrophages and highlights macrophage dynamics upon Staphylococcus aureus peritonitis. Innate Immun. 22:382‐392. [DOI] [PubMed] [Google Scholar]
- 2. Aloisi F, Pujol‐Borrell R (2006) Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol 6:205–217. [DOI] [PubMed] [Google Scholar]
- 3. Appel SH (2009) CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J Clin Invest 119:13–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arumugam TV, Granger DN, Mattson MP (2005) Stroke and T‐cells. Neuromolecular Med 7:229–242. [DOI] [PubMed] [Google Scholar]
- 5. Bagaeva LV, Rao P, Powers JM, Segal BM (2016) CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol 176:7676‐7685. [DOI] [PubMed] [Google Scholar]
- 6. Barone F, Bombardieri M, Rosado MM, Morgan PR, Challacombe SJ, De Vita S et al (2008) CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjögren’s syndrome and MALT lymphoma: association with reactive and malignant areas of lymphoid organization. J Immunol 180:5130–5140. [DOI] [PubMed] [Google Scholar]
- 7. Becker KJ, Kalil AJ, Tanzi P, Zierath DK, Savos AV, Gee JM et al (2011) Autoimmune responses to the brain after stroke are associated with worse outcome. Stroke 42:2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Becker KJ, Zierath D, Kunze A, Fecteau L, Lee B, Skerrett S (2016) The contribution of antibiotics, pneumonia and the immune response to stroke outcome. J Neuroimmunol 295–296:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benakis C, Garcia‐Bonilla L, Iadecola C, Anrather J (2014) The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci 8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berchtold D, Weitbrecht L, Meisel C, Meisel A (2019) Friend or foe? ‐ B cells in stroke. Neuroforum 25:3. [Google Scholar]
- 11. Brommer B, Engel O, Kopp MA, Watzlawick R, Müller S, Prüss H et al (2016) Spinal cord injury‐induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain 139:692–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924. [DOI] [PubMed] [Google Scholar]
- 13. Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P (2004) Monocyte‐like and mature macrophages produce CXCL13 (B cell‐attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood 104:3021–3027. [DOI] [PubMed] [Google Scholar]
- 14. Chabot S, Charlet D, Wilson TL, Yong V (2001) Cytokine production consequent to T cell‐microglia interaction: the PMA/IFNγ‐treated U937 cells display similarities to human microglia. J Neurosci Methods 105:111–120. [DOI] [PubMed] [Google Scholar]
- 15. Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R (2012) The immunology of acute stroke. Nat Rev Neurol 8:401–410. [DOI] [PubMed] [Google Scholar]
- 16. Chen Y, Bodhankar S, Murphy SJ, Vandenbark AA, Alkayed NJ, Offner H (2012) Intrastriatal B‐cell administration limits infarct size after stroke in B‐cell deficient mice. Metab Brain Dis 27:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi I‐A, Yun JH, Kim J‐H, Kim HY, Choi D‐H, Lee J (2019) Sequential transcriptome changes in the penumbra after ischemic stroke. Int J Mol Sci 20:6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chuaqui R, Tapia J (1993) Histologic assessment of the age of recent brain infarcts in man. J Neuropathol Exp Neurol 52:481–489. [DOI] [PubMed] [Google Scholar]
- 19. Cohen KW, Dugast AS, Alter G, McElrath MJ, Stamatatos L (2019) HIV single‐stranded RNA induces CXCL13 secretion in human monocytes via TLR7 activation and plasmacytoid dendritic cell ‐ derived type I IFN. J Immunol 194:2769–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corsiero E, Bombardieri M, Manzo A, Bugatti S, Uguccioni M, Pitzalis C (2012) Role of lymphoid chemokines in the development of functional ectopic lymphoid structures in rheumatic autoimmune diseases. Immunol Lett 145:62–67. [DOI] [PubMed] [Google Scholar]
- 21. Corsiero E, Nerviani A, Bombardieri M, Pitzalis C (2016) Ectopic lymphoid structures: powerhouse of autoimmunity. Front Immunol 7:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA et al (2013) N2 neutrophils, novel players in brain inflammation after stroke: modulation by the pparγ agonist rosiglitazone. Stroke 44:3498–3508. [DOI] [PubMed] [Google Scholar]
- 23. Dambinova SA, Khounteev GA, Izykenova GA, Zavolokov IG, Ilyukhina AY, Skoromets AA (2003) Blood test detecting autoantibodies to N‐methyl‐D-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin Chem 49:1752–1762. [DOI] [PubMed] [Google Scholar]
- 24. Deczkowska A, Keren‐Shaul H, Weiner A, Colonna M, Schwartz M, Amit I (2018) Disease‐associated microglia: a universal immune sensor of neurodegeneration. Cell 173:1073–1081. [DOI] [PubMed] [Google Scholar]
- 25. Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA et al (2007) Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 27:1941–1953. [DOI] [PubMed] [Google Scholar]
- 26. Derkow K, Krüger C, Dembny P, Lehnardt S. Microglia induce neurotoxic IL‐17+ γδ T cells dependent on TLR2, TLR4, and TLR9 activation. PLoS One 2015;10:e0135898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV (2007) Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38:1345–1353. [DOI] [PubMed] [Google Scholar]
- 28. Doyle KP, Buckwalter MS (2017) Does B lymphocyte‐mediated autoimmunity contribute to post‐stroke dementia? Brain Behav Immun 64:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Doyle KP, Quach LN, Solé M, Axtell RC, Nguyen T‐VV, Soler‐Llavina GJ et al (2015) B‐lymphocyte‐mediated delayed cognitive impairment following stroke. J Neurosci 35:2133–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C et al (2018) Thrombus neutrophil extracellular traps content impair tPA‐induced thrombolysis in acute ischemic stroke. Stroke 49:754–757. [DOI] [PubMed] [Google Scholar]
- 31. Dziewulska D, Mossakowski MJ (2003) Cellular expression of tumor necrosis factor α and its receptors in human ischemic stroke. Clin Neuropathol 22:35–40. [PubMed] [Google Scholar]
- 32. Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V et al (2013) Microglial activation milieu controls regulatory t cell responses. J Immunol 191:5594–5602. [DOI] [PubMed] [Google Scholar]
- 33. Engel O, Akyüz L, da Costa Goncalves AC, Winek K, Dames C, Thielke M et al (2015) Cholinergic pathway suppresses pulmonary innate immunity facilitating pneumonia after stroke. Stroke 46:3232–3240. [DOI] [PubMed] [Google Scholar]
- 34. Enzmann G, Mysiorek C, Gorina R, Cheng YJ, Ghavampour S, Hannocks MJ et al (2013) The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol 125:395–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Esen N, Rainey‐Barger EK, Huber AK, Blakely PK, Irani DN (2014) Type‐I interferons suppress microglial production of the lymphoid chemokine, CXCL13. Glia 62:1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA et al (2014) Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet 383:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Felger JC, Abe T, Kaunzner UW, Gottfried‐Blackmore A, Gal‐Toth J, McEwen BS et al (2010) Brain dendritic cells in ischemic stroke: Time course, activation state, and origin. Brain Behav Immun 24:724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL (2005) Neuroprotection by IL‐10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci 233:125–132. [DOI] [PubMed] [Google Scholar]
- 39. Garcia‐Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J et al (2016) Spatio‐temporal profile, phenotypic diversity, and fate of recruited monocytes into the post‐ischemic brain. J Neuroinflammation 13:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garcia‐Bonilla L, Moore JM, Racchumi G, Zhou P, Butler JM, Iadecola C et al (2014) Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J Immunol 193:2531–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. García‐Culebras A, Durán‐Laforet V, Peña‐Martínez C, Moraga A, Ballesteros I, Cuartero MI et al (2019) Role of TLR4 (Toll‐like receptor 4) in N1/N2 neutrophil programming after stroke. Stroke 50:2922–2932. [DOI] [PubMed] [Google Scholar]
- 42. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe C‐U, Siler DA et al (2009) Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 43. Gliem M, Mausberg AK, Lee J‐I, Simiantonakis I, van Rooijen N, Hartung H‐P et al (2012) Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 71:743–752. [DOI] [PubMed] [Google Scholar]
- 44. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T et al (2005) Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 11:328–334. [DOI] [PubMed] [Google Scholar]
- 45. Gülke E, Gelderblom M, Magnus T (2018) Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord 11:1756286418774254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo Z, Yu S, Xiao L, Chen X, Ye R, Zheng P et al (2016) Dynamic change of neutrophil to lymphocyte ratio and hemorrhagic transformation after thrombolysis in stroke. J Neuroinflammation 13:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haley MJ, Brough D, Quintin J, Allan SM (2019) Microglial priming as trained immunity in the brain. Neuroscience 405:47–54. [DOI] [PubMed] [Google Scholar]
- 48. Hammond TR, Dufort C, Dissing‐Olesen L, Giera S, Young A, Wysoker A et al (2019) Single‐cell rna sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell‐state changes. Immunity 50:253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hannawi Y, Hannawi B, Rao CPV, Suarez JI, Bershad EM (2013) Stroke‐associated pneumonia: major advances and obstacles. Cerebrovasc Dis 35:430–443. [DOI] [PubMed] [Google Scholar]
- 50. Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C (2002) Monocyte chemoattractant protein‐1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab 22:308–317. [DOI] [PubMed] [Google Scholar]
- 51. Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN (2004) Platelet–leukocyte–endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab 24:907–915. [DOI] [PubMed] [Google Scholar]
- 53. Jin W‐N, Gonzales R, Feng Y, Wood K, Chai Z, Dong J‐F et al (2018) Brain ischemia induces diversified neuroantigen‐specific T‐cell responses that exacerbate brain injury. Stroke 49:1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin WN, Gonzales R, Feng Y, Wood K, Chai Z, Dong JF et al (2018) Brain ischemia induces diversified neuroantigen‐specific T‐Cell responses that exacerbate brain injury. Stroke 49:1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jin W‐N, Shi SX‐Y, Li Z, Li M, Wood K, Gonzales RJ et al (2017) Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab 37:2224–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jordão MJC, Sankowski R, Brendecke SM, Sagar, Tai Y‐H et al (2019) Single‐cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 363:eaat7554. [DOI] [PubMed] [Google Scholar]
- 57. Kalev‐Zylinska ML, Symes W, Little KCE, Sun P, Wen D, Qiao L et al (2013) Stroke patients develop antibodies that react with components of N‐methyl‐D-aspartate receptor subunit 1 in proportion to lesion size. Stroke 44:2212–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL et al (2015) Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci 35:3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169:1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 60. Klehmet J, Hoffmann S, Walter G, Meisel C, Meisel A (2016) Stroke induces specific alteration of T memory compartment controlling auto‐reactive CNS antigen‐specific T cell responses. J Neurol Sci 368:77–83. [DOI] [PubMed] [Google Scholar]
- 61. Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T et al (2010) Early detrimental T‐cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 115:3835–3842. [DOI] [PubMed] [Google Scholar]
- 62. Laridan E, Denorme F, Desender L, François O, Andersson T, Deckmyn H et al (2017) Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol 82:223–232. [DOI] [PubMed] [Google Scholar]
- 63. Liesz A, Kleinschnitz C (2016) Regulatory T cells in post‐stroke immune homeostasis. Transl Stroke Res 7:313–321. [DOI] [PubMed] [Google Scholar]
- 64. Liesz A, Suri‐Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S et al (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15:192–199. [DOI] [PubMed] [Google Scholar]
- 65. Liu YL, Lu JK, Yin HP, Xia PS, Qiu DH, Liang MQ et al (2020) High Neutrophil‐to-lymphocyte ratio predicts hemorrhagic transformation in acute ischemic stroke patients treated with intravenous thrombolysis. Int J Hypertens. 2020:5980261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ma W, Cojocaru R, Gotoh N, Gieser L, Villasmil R, Cogliati T et al (2013) Gene expression changes in aging retinal microglia: relationship to microglial support functions and regulation of activation. Neurobiol Aging 34:2310–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Martinez‐Sanchez ME, Huerta L, Alvarez‐Buylla ER, Luján CV (2018) Role of cytokine combinations on CD4+ T cell differentiation, partial polarization, and plasticity: continuous network modeling approach. Front Physiol 9:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar et al (2019) Spatial and temporal heterogeneity of mouse and human microglia at single‐cell resolution. Nature 566:388–392. [DOI] [PubMed] [Google Scholar]
- 69. Meisel C, Meisel A (2011) Suppressing immunosuppression after stroke. N Engl J Med 365:2134–2136. [DOI] [PubMed] [Google Scholar]
- 70. Mena H, Cadavid D, Rushing EJ (2004) Human cerebral infarct: a proposed histopathologic classification based on 137 cases. Acta Neuropathol 108:524–530. [DOI] [PubMed] [Google Scholar]
- 71. Meng H, Zhao H, Cao X, Hao J, Zhang H, Liu Y et al (2019) Double‐negative T cells remarkably promote neuroinflammation after ischemic stroke. Proc Natl Acad Sci 116:5558–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Michaud J‐P, Pimentel‐Coelho PM, Tremblay Y, Rivest S (2014) The impact of Ly6C low monocytes after cerebral hypoxia‐ischemia in adult mice. J Cereb Blood Flow Metab 34:e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Miró‐Mur F, Urra X, Gallizioli M, Chamorro A, Planas AM (2016) Antigen presentation after stroke. Neurotherapeutics 13:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Monson NL, Ortega SB, Ireland SJ, Meeuwissen AJM, Chen D, Plautz EJ et al (2014) Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflammation 11:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Morrison HW, Filosa JA (2013) A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG et al (2016) Trained immunity: a program of innate immune memory in health and disease. Science 352:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Netea MG, Latz E, Mills KHG, O’Neill LAJ. (2015) Innate immune memory: A paradigm shift in understanding host defense. Nature Immunol 16:675–679. [DOI] [PubMed] [Google Scholar]
- 78. Neumann J, Sauerzweig S, Rönicke R, Gunzer F, Dinkel K, Ullrich O et al (2008) Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci 28:5965–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ortega SB, Noorbhai I, Poinsatte K, Kong X, Anderson A, Monson NL et al (2015) Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med 19:381–392. [PMC free article] [PubMed] [Google Scholar]
- 80. Ortega SB, Torres VO, Latchney SE, Whoolery CW, Noorbhai IZ, Poinsatte K et al (2020) B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc Natl Acad Sci 117:4983–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Otxoa‐de-Amezaga A, Miró‐Mur F, Pedragosa J, Gallizioli M, Justicia C, Gaja‐Capdevila N et al (2019) Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 137:321–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Patel AR, Ritzel R, McCullough LD, Liu F (2013) Microglia and ischemic stroke: a double‐edged sword. Int J Physiol Pathophysiol Pharmacol 5:73–90. [PMC free article] [PubMed] [Google Scholar]
- 83. Pedragosa J, Miró‐Mur F, Otxoa‐de-Amezaga A, Justicia C, Ruíz‐Jaén F, Ponsaerts P et al (2020) CCR2 deficiency in monocytes impairs angiogenesis and functional recovery after ischemic stroke in mice. J Cereb Blood Flow Metab. 10.1177/0271678X20909055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pedragosa J, Salas‐Perdomo A, Gallizioli M, Cugota R, Miró‐Mur F, Briansó F et al (2018) CNS‐border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol Commun 6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Peña‐Martínez C, Durán‐Laforet V, García‐Culebras A, Ostos F, Hernández‐Jiménez M, Bravo‐Ferrer I et al (2019) Pharmacological modulation of neutrophil extracellular traps reverses thrombotic stroke tPA (tissue‐type plasminogen activator) resistance. Stroke 50:3228–3237. [DOI] [PubMed] [Google Scholar]
- 86. Pendlebury ST, Rothwell PM (2009) Prevalence, incidence, and factors associated with pre‐stroke and post‐stroke dementia: a systematic review and meta‐analysis. Lancet Neurol 8:1006–1018. [DOI] [PubMed] [Google Scholar]
- 87. Pitzalis C, Jones GW, Bombardieri M, Jones SA (2014) Ectopic lymphoid‐like structures in infection, cancer and autoimmunity. Nat Rev Immunol 14:447–462. [DOI] [PubMed] [Google Scholar]
- 88. Planas AM, Gómez‐Choco M, Urra X, Gorina R, Caballero M, Chamorro Á (2012) Brain‐derived antigens in lymphoid tissue of patients with acute stroke. J Immunol 188:2156–2163. [DOI] [PubMed] [Google Scholar]
- 89. Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T et al (2003) Stroke‐induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1‐like immunostimulation. J Exp Med 198:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Prüss H, Iggena D, Baldinger T, Prinz V, Meisel A, Endres M et al (2012) Evidence of intrathecal immunoglobulin synthesis in stroke. Arch Neurol 69:752–759. [DOI] [PubMed] [Google Scholar]
- 91. Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD et al (2011) Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 31:8556–8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ren X, Akiyoshi K, Grafe MR, Vandenbark AA, Hurn PD, Herson PS et al (2012) Myelin specific cells infiltrate MCAO lesions and exacerbate stroke severity. Metab Brain Dis 27:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ritzel RM, Al Mamun A, Crapser J, Verma R, Patel AR, Knight BE et al (2019) CD200‐CD200R1 inhibitory signaling prevents spontaneous bacterial infection and promotes resolution of neuroinflammation and recovery after stroke. J Neuroinflammation 16:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Romer C, Engel O, Winek K, Hochmeister S, Zhang T, Royl G et al (2015) Blocking stroke‐induced immunodeficiency increases CNS antigen‐specific autoreactivity but does not worsen functional outcome after experimental stroke. J Neurosci 35:7777–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram E et al (2019) Mapping microglia states in the human brain through the integration of high‐dimensional techniques. Nat Neurosci 22:2098–2110. [DOI] [PubMed] [Google Scholar]
- 96. Schmidt A, Strecker JK, Hucke S, Bruckmann NM, Herold M, MacK M et al (2017) Targeting different monocyte/macrophage subsets has no impact on outcome in experimental stroke. Stroke 48:1061‐1069. [DOI] [PubMed] [Google Scholar]
- 97. Schuhmann MK, Langhauser F, Kraft P, Kleinschnitz C (2017) B cells do not have a major pathophysiologic role in acute ischemic stroke in mice. J Neuroinflammation 14:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Selvaraj UM, Stowe AM (2017) Long‐term T cell responses in the brain after an ischemic stroke. Discov Med 24:323–333. [PMC free article] [PubMed] [Google Scholar]
- 99. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I et al (2009) Pivotal role of cerebral interleukin‐17–producing γδT cells in the delayed phase of ischemic brain injury. Nat Med 15:946–950. [DOI] [PubMed] [Google Scholar]
- 100. Shimakura A, Kamanaka Y, Ikeda Y, Kondo K, Suzuki Y, Umemura K (2000) Neutrophil elastase inhibition reduces cerebral ischemic damage in the middle cerebral artery occlusion. Brain Res 858:55–60. [DOI] [PubMed] [Google Scholar]
- 101. Song Q, Li Y, Wang Y, Wei C, Liu J, Liu M (2019) Increased neutrophil‐to-lymphocyte ratios are associated with greater risk of hemorrhagic transformation in patients with acute ischemic stroke. Curr Neurovasc Res 15:326–335. [DOI] [PubMed] [Google Scholar]
- 102. Sousa C, Golebiewska A, Poovathingal SK, Kaoma T, Pires‐Afonso Y, Martina S et al (2018) Single‐cell transcriptomics reveals distinct inflammation‐induced microglia signatures. EMBO Rep 19:e46171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Stowe AM, Adair‐Kirk TL, Gonzales ER, Perez RS, Shah AR, Park TS et al (2009) Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis 35:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Stubbe T, Ebner F, Richter D, Engel O, Randolf Engel O, Klehmet J et al (2013) Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab 33:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Szalay G, Martinecz B, Lénárt N, Környei Z, Orsolits B, Judák L et al (2016) Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7:11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tay TL, Sagar, Dautzenberg J, Grün D, Prinz M (2018) Unique microglia recovery population revealed by single‐cell RNAseq following neurodegeneration. Acta Neuropathol Commun 6:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tsementzis SA, Chao SW, Hitchcock ER, Gill JS, Beevers DG (1986) Oligoclonal immunoglobulin G in acute subarachnoid hemorrhage and stroke. Neurology 36:395–397. [DOI] [PubMed] [Google Scholar]
- 108. Tsuyama J, Nakamura A, Ooboshi H, Yoshimura A, Shichita T (2018) Pivotal role of innate myeloid cells in cerebral post‐ischemic sterile inflammation. Semin Immunopathol 40:523–538. [DOI] [PubMed] [Google Scholar]
- 109. Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA (2010) Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A 107:11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wan YY. Multi‐tasking of helper T cells. Immunology 130:166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang S, Zhang H, Xu Y (2016) Crosstalk between microglia and T cells contributes to brain damage and recovery after ischemic stroke. Neurol Res 38:495–503. [DOI] [PubMed] [Google Scholar]
- 112. Watanabe M, Masaki K, Yamasaki R, Kawanokuchi J, Takeuchi H, Matsushita T et al (2016) Th1 cells downregulate connexin 43 gap junctions in astrocytes via microglial activation. Sci Rep 6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J et al (2016) Monocyte‐derived macrophages contribute to spontaneous long‐term functional recovery after stroke in mice. J Neurosci 36:4182–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Weissman JD, Khunteev GA, Heath R, Dambinova SA (2011) NR2 antibodies: risk assessment of transient ischemic attack (TIA)/stroke in patients with history of isolated and multiple cerebrovascular events. J Neurol Sci 300:97–102. [DOI] [PubMed] [Google Scholar]
- 115. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G et al (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Westendorp WF, Nederkoorn PJ, Vermeij J‐D, Dijkgraaf MG, van de Beek D (2011) Post‐stroke infection: a systematic review and meta‐analysis. BMC Neurol 11:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. WHO . Disease Burden and Mortality Estimates. WHO; 2018. [cited 2019 Mar 21]; Available from: https://www.who.int/healthinfo/global_burden_disease/estimates/en/. [Google Scholar]
- 118. Xie L, Li W, Hersh J, Liu R, Yang S‐H (2019) Experimental ischemic stroke induces long‐term T cell activation in the brain. J Cereb Blood Flow Metab 39:2268–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhao SC, Ma LS, Chu ZH, Xu H, Wu WQ, Liu F (2017) Regulation of microglial activation in stroke. Acta Pharmacol Sin 38:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zrzavy T, Höftberger R, Berger T, Rauschka H, Butovsky O, Weiner H et al (2019) Pro‐inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol Appl Neurobiol 45:278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zrzavy T, Machado‐Santos J, Christine S, Baumgartner C, Weiner HL, Butovsky O et al (2017) Dominant role of microglial and macrophage innate immune responses in human ischemic infarcts. Brain Pathol. 28:791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. van Zwam M, Huizinga R, Melief M‐J, Wierenga‐Wolf AF, van Meurs M, Voerman JS et al (2009) Brain antigens in functionally distinct antigen‐presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med 87:273–286. [DOI] [PubMed] [Google Scholar]