

Abstract
Genetic variants in TMEM106B are a major risk factor for several neurodegenerative diseases including frontotemporal degeneration, limbic‐predominant age‐related TDP‐43 encephalopathy, Parkinson's disease, late‐onset‐Alzheimer's disease and constitute a genetic determinant of differential aging. TMEM106B encodes an integral lysosomal membrane protein but its precise physiological function in the central nervous system remains enigmatic. Presently, we aimed to increase understanding of TMEM106B contribution to general brain function and aging. We analyzed an aged cohort of Tmem106b knockout‐, heterozygote and wild‐type mice in a behavioral test battery including assessments of motor function as well as, social, emotional and cognitive function. Aged Tmem106b knockout (KO) mice displayed diverse behavioral deficits including motor impairment, gait defects and reduced startle reactivity. In contrast, no prominent deficits were observed in social, emotional or cognitive behaviors. Histologically, we observed late‐onset loss of Purkinje cells followed by reactive gliosis in the cerebellum, which likely contributed to progressive decline in motor function and gait defects in particular. Reactive gliosis was not restricted to the cerebellum but observed in different areas of the brain including the brain stem and parts of the cerebral cortex. Surviving Purkinje cells showed vacuolated lysosomes in the axon initial segment, implicating TMEM106B‐dependent lysosomal trafficking defects as the underlying cause of axonal and more general neuronal dysfunction contributing to behavioral impairments. Our experiments help to elucidate how TMEM106B affects spatial neuronal homeostasis and exemplifies a critical role of TMEM106B in neuronal cells for survival.
Keywords: behavior, cerebellar degeneration, FTD, lysosomes, purkinje cells, TMEM106B
TMEM106B is a disease‐modifying gene in several neurodegenerative diseases including frontotemporal dementia. We analyzed aged Tmem106b knockout mice by a battery of behavioral tests and histology and found cerebellar dysfunction accompanying motor dysfunction at later ages.

Abbreviations
- AIS
axon initial segment
- BAEPs
Brainstem auditory‐evoked potentials
- FMN
facial motor nucleus
- FTD
frontotemporal disease
- HET
heterozygote
- KO
knockout
- PPI
prepulse inhibition
- WT
wildtype
Introduction
Lysosomes play a pivotal role in neuronal homeostasis under physiological as well as pathophysiological conditions. The fusion of autophagosomes with hydrolytic enzyme‐containing lysosomes is an essential event for completing the removal of misfolded and aggregated proteins engulfed by autophagosomes, for example, in neuronal stem cells, and also terminally differentiated neurons (5, 15). The importance of proper lysosomal function in neurons is highlighted by pathophysiological conditions observed in several neurodegenerative disorders, including Alzheimer´s disease, Parkinson's disease, amyotrophic lateral sclerosis and frontotemporal disease (FTD) [reviewed in (36, 39, 44)]. In familial cases of these disorders, genetic variants or mutations in genes coding for lysosomal proteins were shown to cause disease, drastically increase the risk to develop the disease or modulate disease pathogenesis, respectively. This is particularly the case for FTD, the second most common type of presenile dementia affecting up to 15 per 100,000 in people 45–64 years of age (29). A prominent neuropathological feature of FTD is the aggregation of ubiquitinated TAR DNA‐binding protein 43 kDa (TDP‐43) in neuronal cytosolic inclusion, which is found in approximately 50% of cases with clinically diagnosed FTD (2, 19, 25). Moreover, endo‐/lysosomal defects are a primary cause of neuronal dysfunction in FTD (9, 10).
Heterozygous missense mutations in GRN (coding for Progranulin) lead to Progranulin haploinsufficiency and constitute a major genetic risk factor for FTD. Progranulin is most highly expressed in microglia, and also localizes to neuronal lysosomes, while its precise function is still unclear (27). Importantly, additional modifying genes appear to affect FTD progression. TMEM106B was identified as one of those major risk genes (41). Variations in TMEM106B were shown to affect the course of disease in both GRN‐ and C9orf72 carriers (7, 8, 28, 40, 42). Likewise, variants in TMEM106B have been associated with other neurodegenerative diseases such as hippocampal sclerosis, Limbic‐predominant age‐related TDP‐43 encephalopathy, Parkinson's disease, late‐onset Alzheimer's disease and chronic traumatic encephalopathy (3, 12, 13, 22, 23, 24, 31, 38, 48). Furthermore, TMEM106B was shown to play a role in the dissociation of cognition and neuropathology in elderly people (45) and TMEM106B was identified as a key genetic determinant of differential aging in the cerebral cortex (30, 47). Finally, a dominant mutation in TMEM106B leads to a familial hypomyelinating leukodystrophy (35, 46). This variety of apparent genetic links implicates TMEM106B as a major risk factor for neurodegenerative diseases. How TMEM106B is contributing to such a broad spectrum of neurological diseases, remains, however, enigmatic.
TMEM106B is a lysosomal transmembrane protein with a large, highly glycosylated luminal domain, a single transmembrane domain and a cytosolic amino‐terminus (14). In cultured cortical and hippocampal neurons, Tmem106b knockdown leads to altered microtubule‐dependent lysosomal positioning in dendrites (34, 37). However, analysis of Tmem106b knockout (KO) mice revealed that positioning and trafficking of lysosomes are drastically altered in myelinated axons of selected motoneuron nuclei. Pronounced pathology was particularly observed in the facial motor nucleus (FMN). Motoneurons of Tmem106b KO mice show drastically swollen vacuoles at the distal end of the axon initial segment (AIS), which proved positive for the lysosomal marker protein LAMP1 (18). Interestingly, this phenotype was already observed in young mice starting at the age of 4 weeks. Vacuolization is accompanied by dysfunction of facial motoneuron‐dependent innervated muscles such as the Musculus orbicularis orbis. Additionally, impaired autophagic cargo degradation was observed, which likely results from impaired axonal transport of LAMP1‐positive organelles (18).
In the present study, we aimed to increase understanding of TMEM106B involvement in general brain function and aging. Specifically, we analyzed the functional consequences of TMEM106B deficiency using a cohort of Tmem106b wildtype (WT), heterozygote (HET) and homozygous (KO) mice. Starting at 14 months of age these mice were subjected to an extended behavioral test battery. We aimed to provide a broad characterization but based on previous observations and theoretical relevance, we increasingly focused on tests detailing motor function and behaviors reminiscent of human FTD. Tmem106b KO mice displayed diverse behavioral deficits including motor impairment, gait alterations and reduced startle reactivity, but no prominent changes in social, emotional or cognitive behavior. Gait changes and more general sensorimotor dysfunction were histologically corroborated by late‐onset loss of Purkinje cells and subsequent reactive cerebellar gliosis. Surviving Purkinje cells, just like motoneurons at earlier stages, often showed vacuolated lysosomes in the AIS, implicating generalized lysosomal trafficking defects as the underlying cause for the (progression of) behavioral deficits and more general neuronal dysfunction.
Material and Methods
Mice
Tmem106b KO mice generated by CRISPR/Cas9 in a C57/Bl6N background were described previously (18). Mice were housed at standard laboratory conditions (12 hours’ light/dark cycle, constant room temperature and humidity). Behavioral testing was initiated at 14 months of age in Tmem106b WT (n = 14), Tmem106b HET (n = 11) and Tmem106b KO (n = 14) mice—all females. We chose female mice for these tests because behavioral readouts are generally more affected by territorial fighting and differences in the social hierarchy in socially housed males (1). In contrast, it has been well established that female mice are much more affiliative than males. Behavioral testing took place during the light phase of the cycle. Food and water were available ad libitum. Experimental protocols were approved by the ethical research committee of the KU Leuven according to EC guidelines/approved by local German authorities (Ministerium für Energiewende, Landwirtschaft, Umwelt und ländliche Räume, Kiel, V 242‐13648/2018).
Antibodies and chemicals
The following antibodies were used for immunofluorescence on brain sections and immunoblots: Glial Fibrillary Acidic Protein (GFAP) (1:500; mouse monoclonal; clone G‐A‐5, G3893, Sigma‐Aldrich); CD68 (1:500; rat monoclonal; clone FA‐11 (MCA1957, AbD Serotec), Calbindin D‐28 K (1:1000, mouse monoclonal; CB‐955, ascites fluid; Sigma‐Aldrich), Myelin Basic Protein (MBP) (1:1000; rabbit polyclonal; GTX22404, GeneTex), LAMP1 (1:500; rat monoclonal; clone 1D4B, Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA, USA) and p62/SQSTM1 (rabbit polyclonal, BML‐PW9860; Enzo). Fluorophore‐conjugated secondary antibodies against the corresponding primary antibody species (AlexaFluor 488, AlexaFluor 594 and AlexaFluor 647) were purchased from Invitrogen/Molecular Probes and were diluted 1:500. Analytical grade chemicals were purchased, if not stated otherwise, from Sigma‐Aldrich (MO, USA).
Preparation of brain lysates and immunoblotting
Brain lysates were prepared by homogenization of fresh or frozen brain material with 10–20 strokes at 1000 rounds per minute using a Glass homogenizer (B. Braun type 853202) in 15 volumes of lysis buffer (PBS supplemented with cOmplete™ Protease Inhibitor (Sigma‐Aldrich) and 1% (w/v) Triton X‐100). After homogenization the lysates were centrifuged for 20 minutes at 1000 × g at 4°C followed by ultrasonification twice for 20 seconds at 4°C using a Branson Sonifier 450 (level seven in a cup horn, Emerson Industrial Automation) and lysed on ice for approximately 60 minutes. The cell suspension was centrifuged at 16 000 × g for 15 minutes at 4°C and the protein concentration of the supernatant was determined using the Pierce BCA (bicinchoninic Acid) Assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Protein lysates were prepared for SDS‐PAGE in sample buffer (125 mM Tris/HCl pH 6.8, 10% (v/v) glycerol, 1% (w/v) SDS, 1% (v/v) β‐mercaptoethanol and traces of bromophenol blue) and were denatured for 10 minutes at 95°C. Western blot was carried out according to standard procedures. After washing, the membranes in TBS‐T buffer, horseradish peroxidase activity was detected by using an ImageQuant LAS 4000 (GE Healthcare). The intensity of the signal was quantified using Image J software. Before incubation with different antibodies, the membranes were stripped using 0.2 M NaOH. Incubations of 5 minutes at room temperature and gentle shaking were performed in distilled water, followed by 0.2 M NaOH, rinsing with distilled water, 0.2 M NaOH followed by distilled water, and finally, TBS‐T. Next, the membranes were incubated in 5% (w/v) milk powder in 1x TBS‐T buffer for 1 hour at room temperature followed by incubation with the first antibody.
Immunofluorescence of brain sections
Deeply anaesthetized mice were transcardially perfused with 0.1 M phosphate buffer (PB) (pH 7.4) followed by 4% paraformaldehyde (PFA). The brains were removed and postfixed by immersion for another 4 hours in 4% PFA. PFA was removed and replaced by 30% sucrose (w/v) in 0.1 M PB. After incubation overnight, brains were cut sagittally and 35 µm thick. Free‐floating sections were prepared with a Leica 9000s sliding microtome (Leica, Wetzlar, Germany). The sections were blocked in blocking solutions (0.5% Triton‐X 100, 4% normal goat serum in 0.1 M PB pH 7.4), incubated in blocking solution containing the primary antibody/antibodies at 4°C overnight. After washing three times with wash solution (0.1 M PB pH 7.4 containing 0.25% Triton‐X 100), sections were incubated for 2 hours in secondary antibody in solution, washed again three times in wash solution containing 4′,6‐diamidin‐2‐phenylindol (DAPI) and finally brought on glass slides and embedded in Mowiol/DABCO. Images were analyzed with a Leica DMi8 fluorescence microscope equipped with an automated stage and the LAS X software or an Olympus FV1000D Laser Scanning Confocal Microscope (model: FV10‐292‐115) with a 60x lens (UPLSAPO). Image acquisition with the Olympus confocal microscope was performed with the FV10‐ASW 4.2 Viewer software (Olympus, Germany).
For quantification of the CD68‐positive area, images were processed to 8‐bit images in ImageJ‐software, a region of interest (ROI) was specified and a threshold for the signal intensity was defined. The number of pixels was calculated/per ROI. For representation, the average of the CD68‐positive area of the WT animals was set as 1 and the CD68‐positive area for each animal is represented.
Behavioral tests
Cage activity
Home cage activity was recorded in transparent cages with an area of 20 × 30 cm, placed between three infrared beams. Activity patterns were evaluated by recording total number of beam crossings during 30 minutes intervals for 23 hours using customized software.
Grip strength
Forelimb grip strength was evaluated by allowing mice to spontaneously grab a T‐shaped bar. Connection of the bar to a digital dynamometer (Ugo Basile, Gemonio, Italy) allowed quantification of strength which was averaged over 10 trials.
Rotarod
The rotarod test was included as a measure of motor coordination and equilibrium. Animals first received 2 minutes of training at a fixed speed of 5 rpm on the apparatus (Ugo Basile, Gemonio, Italy). Following training four test trials (10) were conducted with an accelerating rotation from 5 to 40 rpm during 5 minutes. Drop latency was registered up to the 5 minutes cutoff. For the comparison between young animals and old animals (14 months), data for the young cohort (3 months) was used from a previous study (18).
Gait analysis
Footprint gait analysis was performed via ventral plane video tracking during treadmill walking for automatic quantification of gait patterns (DigiGait, Mouse Specifics Inc, MA, USA). Mice were habituated to walking on the treadmill at 10 cm/s. Belt speed was subsequently increased to 12 cm/s from the moment which video data was acquired for 45 seconds. The amount of times mice could not keep pace with the treadmill speed was registered (further referred to as the amount of errors). A fragment of the video where mice were consistently walking above the camera was selected for analysis. Different parameters were extracted from the video data using DigiGait analysis software, including fore versus hind gait symmetry (the ratio of forelimb stepping frequency to hind limb stepping frequency). Spatial parameters comprised stance widths (distance between contralateral paws), stride lengths (distance between subsequent placements of the same paw). Temporal aspects of gait were also evaluated by analyzing stance (time paw is in contact with the belt), brake (part of stance from initial contact to maximum contact), propulsion (part of stance from maximum contact to swing), swing (time paw is not in contact with the belt) and stride (sum of stance and swing duration) times. Stance and swing times were analyzed relative to stride time. Brake and propulsion times were analyzed relative to stance time.
Open field
Open field locomotion was recorded to evaluate exploratory behavior. Following 30 minutes dark adaptation, mice were placed into a transparent 50 × 50 cm arena. Subsequent to 1 minutes habituation, animal movement was recorded for 10 minutes using ANY‐mazeTM Video Tracking System software (Stoelting Co., IL, USA). Total path length, percentage of path length in the center (=30 cm circle) and time in the periphery (<5 cm from the walls) were extracted. For each genotype, an illustrative ANY‐maze heatmap was generated using the animals’ center points, reflecting average time spent in each area of the maze.
Elevated plus maze
Exploration of an elevated plus maze was assessed to characterized anxiety‐related behavior. The arena consisted of a plus‐shaped maze, elevated 50 cm above surface, with two arms (5 cm wide) closed by side walls and two open arms. Mice could freely explore for 10 minutes following 1 minute habituation. Four IR beams recording open and closed arm entries, and one recording the percentage of time per minute spent in the open arms were connected to a computerized activity logger.
Sociability and preference for social novelty
Sociability and preference for social novelty (SPSN) was evaluated in a three‐chamber Plexiglas setup. The two side chambers (10 × 10 cm) were separated from the center compartment (40 × 10 cm) by Plexiglas walls containing holes (diameter 0.8 cm) to allow social interaction and odor transfer between experimental and stimulus mice. First, an acclimation trial was performed during which mice were allowed to explore the central chamber for 5 minutes. During the 10 minutes sociability trial, one side chamber contained a stranger mouse. The side of the social chamber was randomized. Preference for social novelty was subsequently evaluated by placing a second stranger mouse into the previously empty side chamber for 10 minutes. Animal movement was recorded using ANY‐mazeTM Video Tracking System software (Stoelting Co., IL, USA). The time the animal’s head was closer than 1 cm to the chamber walls was measured to allow quantification of preferential exploration in sociability and social novelty phases.
Passive avoidance
Passive avoidance learning was examined in a step‐through box with a small illuminated compartment and a larger dark compartment containing a grid‐floor. After 30 minutes dark adaptation, the mouse was placed in the light compartment for a training trial. After 5 seconds, the sliding door to the dark compartment was opened and step‐through latency was manually recorded up to a 300‐sec cutoff. When all four paws were placed on the grid, a mild electric shock (0.3 mA, 1 seconds) was delivered with a constant current shocker (MED Associates Inc., St. Albans, Vermont, USA). Retention memory was tested 24 hours later using the same procedure without shock delivery.
Tail suspension
Depression‐like behavior was assessed during the tail suspension test. Mice were suspended by their tails with tape in a suspension box for 5 minutes. Escape‐oriented movement was recorded using ANY‐mazeTM Video Tracking System software (Stoelting Co., IL, USA). Distance moved, immobility time and latency to the first immobile episode were extracted. Three mice (two WT and one Tmem106b HET) were excluded because of repeated tail climbing behavior.
Repetitive behavior
For assessment of spontaneous repetitive behaviors, mice were placed in a transparent cage with an area of 20 × 30 cm for 20 minutes. Following 10 minutes of habituation, bouts of self‐grooming and digging behavior were recorded and summed. Grooming behavior was defined as stroking, scratching or licking any body region.
Startle reactivity and prepulse inhibition (PPI)
Startle reactivity and PPI were measured using a Med Associates (St. Albans, VT, USA) acoustic startle box (MED‐ASR‐PRO1) and Med Associates Startle Reflex software. Mice were placed in a cubical restrainer (ENV‐406SM‐8), which was mounted on a motion‐sensitive platform inside a sound‐attenuating box. Acoustic stimuli and background noise were delivered through speakers located inside the sound‐attenuating box. The first peak pressure value occurring within 200 ms after onset of startle pulse (SP) was taken as peak startle value. Mice habituated for 5 minutes inside the cubicle prior to administration of the first stimulus. Following acclimation, mice were presented with five white noise startle pulses (115 dB SPL; 38 ms duration plus 1 ms rise/fall time; SP alone). Thereafter, 10 stimulus trial blocks were administered. Each block contained seven stimulus trials, with each stimulus trial belonging to one of three trial types: SP alone, prepulse (PP) followed by SP or PP alone. Startle pulses were white noise bursts (SP: 115 dB; 38 ms plus 1 ms rise/fall time and PP: 70, 75 or 80 dB; 18 ms plus 1 ms rise/fall time). Time interval between onset of PP and onset of SP was 120 ms; intertrial intervals were 10–15 seconds. Peak values after SP administration were averaged for each trial type. Percentage PPI values (%PPI) were calculated according to the standard formula: %PPI = [1 − (startle peak at SP after PP)/(startle peak at SP alone)] × 100.
Brainstem auditory‐evoked potentials (BAEPs)
Brainstem auditory‐evoked potentials (BAEPs) were recorded on a Myos 4 plus digital EMG/EP recorder (Schwarzer, Munich, Germany). Mice were anesthetized with Nembutal (6 mg/ml i.p. at 1% body weight). A needle electrode was placed subcutaneously behind each ear and referenced to a common electrode near the base of the tail; a fourth electrode, also placed near the base of the tail, was used as a ground. Robust five‐peak tracings were obtained by averaging 2000 responses evoked by 85–86 dB clicks [measured with a Brüel & Kjær (Norcross, GA) sound intensity meter] emitted by a speaker placed 1 cm in front of the animal's head. For each of the tracings, latencies of the first five peaks (numbered I through V) were measured.
Statistics
Behavioral data are presented as mean + standard error of the mean (SEM). Shapiro–Wilk and Brown–Forsythe tests were used to determine normality and variance homogeneity. Group comparisons were performed through one‐way ANOVA, Kruskal–Wallis ANOVA on ranks, or two‐ or three‐way repeated‐measures ANOVA. Post hoc comparisons were performed upon omnibus test significance. Multiple comparisons and p‐value adjustments were executed according to the Dunn–Bonferroni method for Kruskal–Wallis ANOVA and the Holm–Sidak method for parametric ANOVA. One‐sample t‐tests or one‐sample signed rank tests were performed to compare group performances to coincidence levels. The number of animals per group is WT (n = 14), HET (n = 11) and KO (n = 14), unless otherwise specified. The significance threshold was set at α = 0.05. The level of statistical significance is indicated with asterisks as follows: not significant (ns) = P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Results
TMEM106B deficiency causes reactive gliosis, cerebellar atrophy and Purkinje cell death
Young Tmem106b KO mice (<3 months) develop early‐onset axonal vacuolization of motoneurons and progressive motor defects (18). To investigate the effect of aging on the general neurological phenotype, we analyzed the brains of WT, Tmem106b HET and Tmem106b KO animals by immunohistochemistry at the age of 18 months. Immunohistochemistry for the microglia markers CD68 and Iba1 (Figure 1A) revealed widespread but not generalized microgliosis in different brain regions. Microglia of Tmem106b KO mice extensively expressed CD68, a marker of activated microglia (11). Furthermore, they showed the typical morphology of activated and amoeboid microglia, while resting microglia in WT mice expressed little CD68 (Figure 1A). Areas with the highest CD68‐immunoreactivity included the hindbrain (pontine grey, pons and medulla) and spinal cord, distinct layers of the dorsal cerebral cortex (visual area) and the cerebellum (Figure 1B). Quantification of the CD68‐signal indicated a statistically significant increase of the CD68 area in these regions (Figure 1B). Activated microglia were particularly found in the white matter and in the molecular layer of the cerebellum. Staining for the astrocytic marker GFAP (Figure 1C) revealed hypertrophic astroglia at sites that were also affected by microgliosis, including the visual area of the cerebral cortex and the cerebellum.
Figure 1.
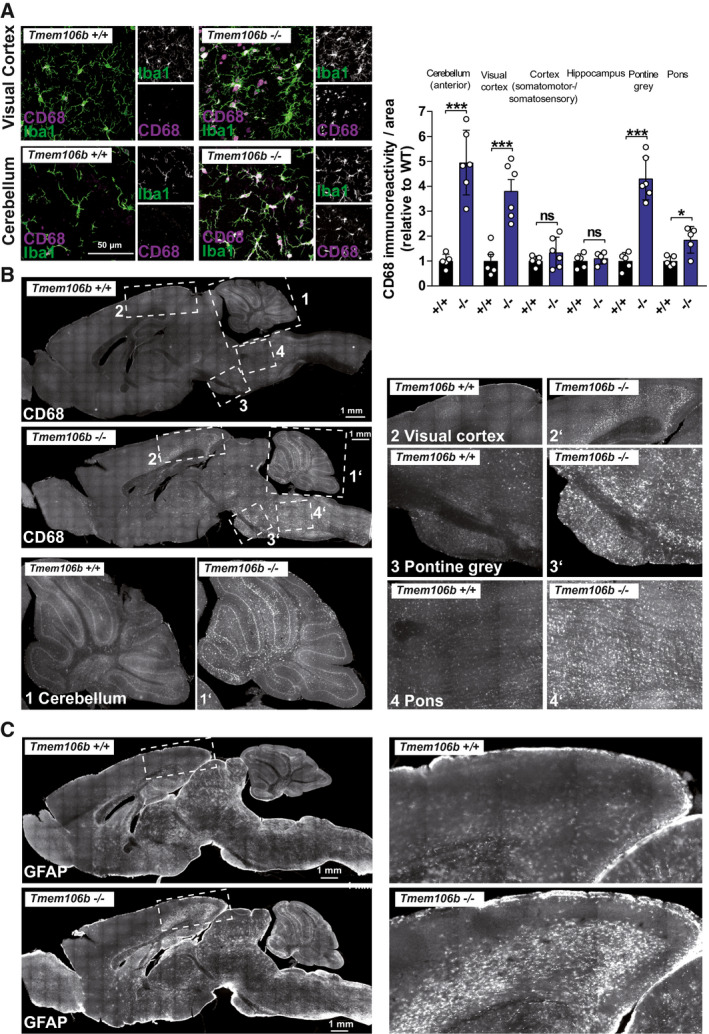
Aged Tmem106b KO mice show widespread reactive microgliosis and astrogliosis. (A) Immunohistochemistry staining of 18‐month‐old WT am Tmem106b KO mice with antibodies against the microglia marker CD68 (magenta) and Iba1 (green). (B) Immunohistochemistry staining of 18‐month‐old WT and Tmem106b KO mice with an antibody against the microglia marker CD68. Numbered boxed areas are depicted enlarged. Image‐based quantification (CD68‐positive area of selected region of interest, relative to the average CD68‐area of the WT) of selected brain areas is given (mean ± SEM, n = 5–6 ***P < 0.001). (C) Immunohistochemistry staining of 18‐month‐old WT am Tmem106b KO mice with an antibody against the astrocyte‐marker GFAP.
Among the brain regions with the most pronounced gliosis was the cerebellum. Examination of the brains revealed a reduced size of the cerebellum at the age of 18 months, indicating mild atrophy. Accordingly, the weight of the cerebellum was statistically significantly reduced in Tmem106b KO mice in comparison to WT animals (Figure 2A). No differences were observed for Tmem106b HET animals. Immunohistochemistry staining of brain sections for the Purkinje cell marker Calbindin‐D‐28K revealed a loss of Purkinje cells that was pronounced in the anterior lobules of the cerebellum of Tmem106b KO animals (Figure 2B). Co‐immunofluorescence of Calbindin‐D‐28K with CD68 (Figure 2C) revealed activated microglia in the molecular layer of the affected lobules (most pronounced observed in layer V/VI) and the white matter tracts. Microgliosis was accompanied by severe hypertrophy of Bergmann glia at sites of Purkinje cell death and astrogliosis of the white matter. Astrogliosis was confirmed by immunoblot and we observed a ~3.5 fold increase of GFAP levels in cerebellar lysates from Tmem106b KO mice (18‐month‐old) in comparison to WT animals (Figure 2D). In summary, TMEM106B deficiency causes widespread reactive gliosis and cell death in the cerebellum at late stages. We did not observe differences between male and female animals in any of the parameters analyzed by histology, indicating that both genders are equally affected.
Figure 2.
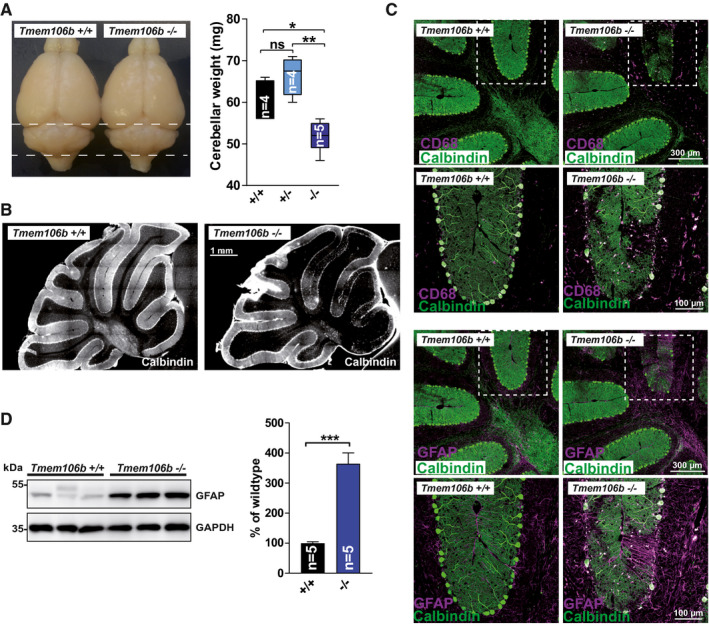
Cerebellar degeneration in aged Tmem106b KO mice. (A) Photos of representative brains of 18‐month‐old WT and Tmem106b KO mice. Cerebellar wet‐weight of WT, Tmem106b HET and Tmem106b KO mice is depicted (age: 18 months). (mean ± SEM, n = 4–5; *P < 0.05, **P < 0.01). (B) Representative immunofluorescence staining of cerebellar sections for the Purkinje cell marker calbindin‐D28‐K of WT and Tmem106b KO mice (age: 18 months). (C) Triple Co‐immunofluorescence staining of cerebellar sections from WT and Tmem106b KO mice for calbindin‐D28‐K (green) and the microglia/macrophage marker CD68 (magenta) (upper panel) and calbindin‐D28‐K (green) and the astrocyte‐marker GFAP (magenta), (lower panel). (D) Immunoblot of cerebellar extracts from WT and Tmem106b KO mice with an antibody against GFAP. GAPDH is depicted as a loading control. Quantification of the GFAP, normalized to GAPDH is shown. The mean of the values for the WT animals was set as 100%. (mean ± SEM, n = 4–5 ***P < 0.001).
The most notable early pathologic feature of Tmem106b KO mice is the prominent appearance of giant vacuoles in the proximal axon of facial motoneurons which are positive for the lysosomal marker LAMP1 (18). Therefore, we analyzed if vacuolization of Purkinje cells might be causative for their subsequent degeneration. For this purpose, we used brains of 9‐month‐old Tmem106b KO animals, an age preceding prominent loss of Purkinje cells. Co‐immunofluorescence staining of these brains with the AIS marker Ankyrin G revealed the conspicuous appearance of single enlarged lysosomes (typically 2–2.5 µm diameter) within the proximal axon/AIS in numerous Purkinje cells (Figure 3A). In contrast, no lysosomes were observed in the AIS of WT mice. These findings resemble the observations in motoneurons of young animals. However, it should be noted that these large lysosomes were considerably smaller compared to vacuoles found in the facial motor nucleus (with a size ranging up to >20–35 µm). In 18‐month‐old Tmem106b KO animals, we regularly observed vacuoles in the granular layer in very close proximity to Purkinje cells, with striking resemblance to those found early (2–4 month of age) in facial motoneurons with a diameter up to 15 µm (Figure 3B).
Figure 3.
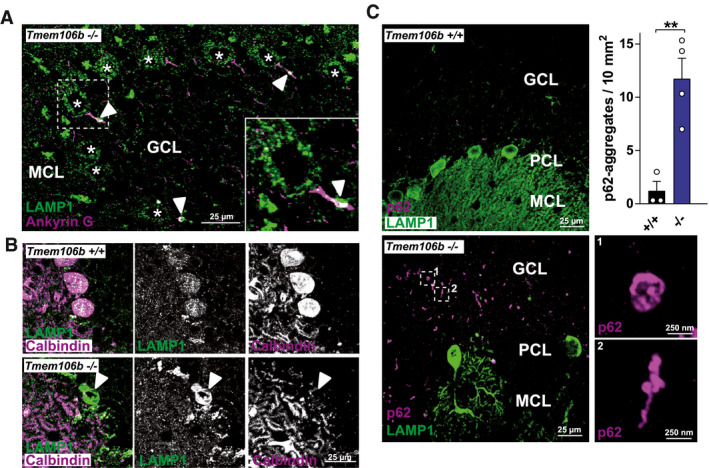
Vacuolization of TMEM106B‐deficient Purkinje cells in the cerebellum and p62‐pathology in the cerebellum of Tmem106b KO mice. (A) Co‐immunofluorescence staining of brain/cerebellar sections from a 9‐month‐old Tmem106b KO mouse for LAMP1 (green) and Ankyrin G (magenta). LAMP1‐positive vacuoles within the AIS are marked with arrowheads. Purkinje cells are marked with asterisks. (B) Co‐immunofluorescence staining of brain/cerebellar sections from 20‐month‐old WT and Tmem106b KO mouse for LAMP1 (green) and calbindin‐D28‐K (magenta). A LAMP1‐positive vacuole is marked with an arrowhead. (C) Co‐immunofluorescence staining of brain/cerebellar sections from 20‐month‐old WT and Tmem106b KO mice for calbindin‐D28‐K (green) and p62 (magenta). (A, C): MCL = molecular cell layer; GCL = granular cell layer, PCL = Purkinje cell layer. The number of p62‐positive aggregates/10 mm2 of the granular layer/white matter is depicted (mean ± SEM, n = 3–4 **P < 0.01).
In addition to LAMP1‐positive vacuoles, we observed pronounced p62 aggregates in the cerebellum of Tmem106b KO mice at an age >18 months (Figure 3C). p62‐pathology was indistinguishable between male and female mice. Small p62‐positive aggregates/inclusions were found throughout the granular cell layer and the white matter tracts of the Tmem106b KO mice. These aggregates were sized from 250 nm to 1 µm and appeared in various shapes, ranging from spherical to small filamentous or skein‐like shapes. Rare p62‐positive aggregates in the molecular layer were restricted to those layers that showed extensive Purkinje cell death (not shown). The appearance of p62‐positive aggregates in the granular cell layer suggests, involvement of granular cerebellar cells in addition to Purkinje cell death.
TMEM106B deficiency causes impaired treadmill performance and gait defects
The degenerative changes observed by histology prompted us to characterize the behavior of aged Tmem106b KO mice in detail. Considering the specific late‐stage vulnerability of the cerebellum to TMEM106B deficiency, our primary focus was on the motor function of these animals. We started our survey by determining general locomotor behavior. Home cage activity showed no significant difference between the genotypes in total or time‐dependent cage activity (P = 0.854 & P = 0.609) (Figure S1A). Like in 3‐month‐old animals (18), no significant difference was detectable for grip strength (P = 0.196) (Figure S1B). Drop latencies on the accelerating rotarod tended to be lower in Tmem106b KO mice, but there was no significant difference between the genotypes on average (P = 0.122) or dependent on the trial (P = 0.543) (Figure S1C). However, in comparison to earlier assessment [3 months, (18)], rotarod performance appeared to remain stable in aged Tmem106b HET and WT mice (14 months), while there was evidence for deterioration in Tmem106b KO animals. Indeed, Tmem106b KO mice showed significantly reduced motor performance during retesting at older in comparison to younger age (t(13) = 3.490, P = 0.004), while WT and Tmem106b HET mice showed similar performance (P = 0.671 & P = 0.371). (Figure 4A). These results suggest worsening of early‐onset motor impairment, putatively related to progressive cerebellar pathology in Tmem106b KO mice. As disturbance of cerebellar integrity typically results in gait alterations, mice were subjected to gait analysis during treadmill locomotion. There was a significant genotype effect on treadmill performance (H(2) = 13.012, P = 0.001). Tmem106b KO mice made significantly more errors than WT and Tmem106b HET mice (P = 0.019 & P = 0.002) (Figure 4B), confirming reduced motor performance. Mice were ventrally video‐tracked during treadmill walking to allow parameter extraction for gait analysis. Animal size measures were not significantly different between the genotypes (animal length: P = 0.243 & animal width: P = 0.106) (Figure S2A,B). Eight mice were excluded for gait analysis as video footage did not allow reliable parameter extraction. However, the remaining animals (N = 9–12) still allowed statistical evaluation. Fore versus hind gait symmetry (the ratio of forelimb stepping frequency to hind limb stepping frequency) tended to vary between the genotypes, but this effect did not reach statistical significance (P = 0.061) (Figure 4C). However, the symmetry of Tmem106b KO mice significantly differed from unity (t(10) = 3.081, P = 0.011), while this was not the case for Tmem106b HET mice (P = 0.742) and WT mice (P = 0.764) (Figure 4C). This indicates an asymmetric gait in which hind limbs of Tmem106b KO mice stepped more frequently in comparison to their forelimbs. There were no significant differences in fore and hind stance widths (gen P = 0.152 & genxpaws P = 0.716) (Figure 4D) or step angles (Figure S2C). There was, however, a significant difference between genotypes in stride length of the forepaws (F(2,29) = 4.301, P < 0.023). Tmem106b KO mice showed increased stride lengths in comparison to WT and Tmem106b HET mice (p = .049 & p = .036), indicating larger steps taken by these mice. A similar trend was observed for the hind paw stride length (P = 0.110) (Figure 4E). Analogous results were obtained for the stride times (P < 0.022 & P < 0.116) (Figure 4F). Considering this genotype difference in stride times, temporal gait parameters were analyzed using relative instead of raw measures. There were no differences between the genotypes in the relative contribution of swing and stance times to the total stride time of the fore and hind paws (gen P = 0.225 & genxpaws P = 0.550) (Figure 4G). There was a significant difference between the genotypes concerning the specific contributions of the brake and propulsion components to the stance time of the forepaws (genxpaws interaction F(2,63) = 5.550, P < 0.009) (Figure 4H). Tmem106b KO mice showed a relative increase in brake time (and consequently reduced propulsion time) in comparison to WT mice (P < 0.014). In summary, gait changes were evident in various parameters. Progressive motor impairment and gait deficits in Tmem106b KO mice are consistent with immunohistological observations of progressive cerebellar pathology and conjoining early‐onset motor neuron disease features.
Figure 4.
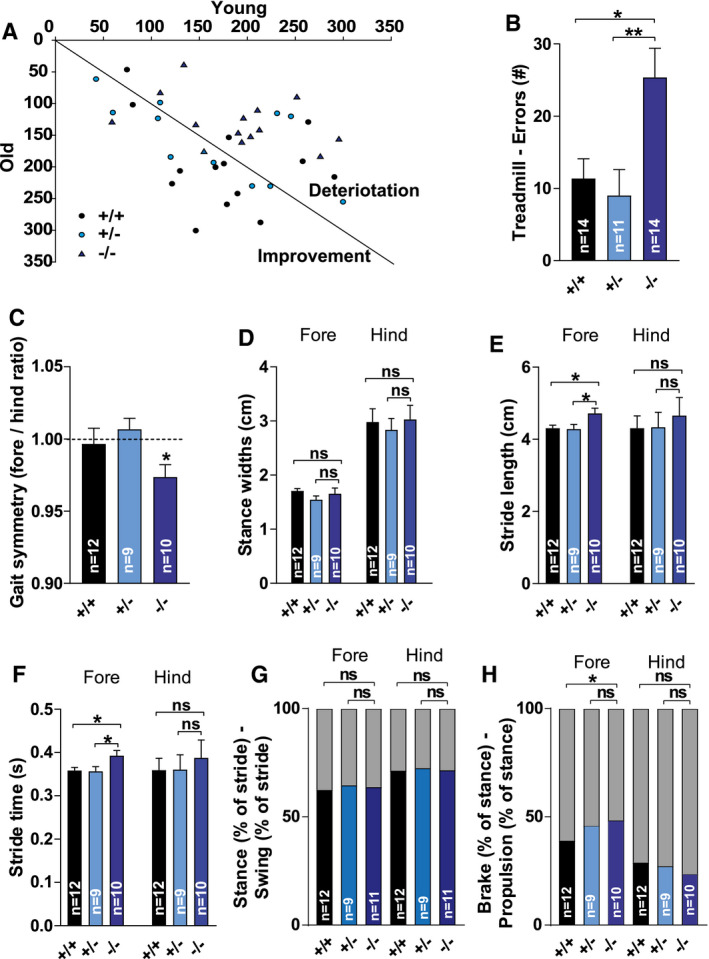
Progressive motor deficits, impaired treadmill performance and gait changes in aged Tmem106b KO mice. (A) Scatter plot representing average drop latencies on the rotarod during retesting of young animals [3 months, (18)] versus old animals (14 months). (B) Treadmill performance depicted as the number of errors. (C) Fore versus hind gait symmetry (the ratio of forelimb stepping frequency to hind limb stepping frequency) extracted from the treadmill walking video sequences. (D) Fore and hind stance widths. (E) Fore and hind stride length (F) Stride length and Fore and hind stride times. (G) Contributions of stance (lower bars) and swing (upper bars) components to the stride time of the fore and hind paws (H) Contributions of the brake (lowers bars) and propulsion (upper bars) components to the stance time of the fore and hind paws. (C–H): Eight mice were excluded for gait analysis as video footage did not allow reliable parameter extraction (final n; WT: n = 12; HET: n = 9; KO: n = 11). (B–H): ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01.
TMEM106B deficiency leads to reduced acoustic startle reactivity without affecting brainstem auditory‐evoked potentials (BAEP)
We previously described thalamic vacuolization in Tmem106b KO mice (18). Given the importance of the thalamus as the obligatory station through which nearly all sensory information must pass before reaching the cerebral cortex (21), we aimed to assess sensorimotor gating by measuring prepulse inhibition of the acoustic startle response (PPI) (Figure 5A,B). The acoustic startle reflex was evaluated using startle peak values from “startle alone” trials. Peak values during the null period and the startle period were compared. There were significant differences between the genotypes in startle peak values depending on the period (genotype × phase interaction (F(2,36) = 16.823, P < 0.001). WT and Tmem106b HET mice showed clearly increased peak values during the startle period in comparison to the null period (both P < 0.001), while Tmem106b KO mice did not (P = 0.547), suggesting severely reduced acoustic startle reactivity (Figure 5A). Indeed, startle peak values were significantly higher in WT and Tmem106b HET mice in comparison to Tmem106b KO mice (both P < 0.001), while there was no difference in null peak values. Furthermore, Tmem106b HET mice showed decreased startle peak values in comparison to WT mice (P = 0.002). PPI calculations showed significant genotype differences for all levels (H(2) = 13.855 & 16.195 & 19.222, all P < 0.001) (Figure 5B). No differences were observed between WT and Tmem106b HET mice. Tmem106b KO mice showed reduced PPI percentages in comparison to WT (P = 0.002, P < 0.001, P < 0.001) and Tmem106b HET mice (P = 0.009, P = 0.005, P = 0.006) for each prepulse level. However, the weaker baseline startle reflex in Tmem106b KO mice might have confounded these measurements. If the average null peak value is considered as the floor startle reactivity level, maximum attainable PPI for WT, Tmem106b HET and Tmem106b KO mice was 80.3%, 68.0% and 21.5%, respectively. This would mean WT, Tmem106b HET and Tmem106b KO mice reached a largely similar 70.0%, 77.6% and 71.8% of their maximum attainable PPI. Therefore, we cannot conclude sensorimotor gating is impaired in Tmem106b KO mice. A possible explanation for the strongly reduced acoustic startle reactivity could be impaired auditory function. We have previously shown that vacuolization in Tmem106b KO mice primarily affects motoneuron nuclei of the brainstem and the thalamus (18), but phenotypes might not be restricted to motoneuron nuclei and affect the brainstem in general. We recorded brainstem auditory‐evoked potentials in our mice (Figure 5C), which represent neuronal activity along the pontomedullary pathway following auditory stimulation. One Tmem106b HET animal was excluded from final analysis as no reliable signal could be recorded. Other animals showed a detectable five‐peak pattern. Peak latencies showed no significant differences between the genotypes (genotype P = 0.583, gen × peak P = 0.776) (Figure 5C). In summary, these data suggest that the reduced acoustic startle reflex in Tmem106b KO mice is not related to the impaired auditory processing, but could reflect reduced motor output on the level of spinal motoneurons.
Figure 5.
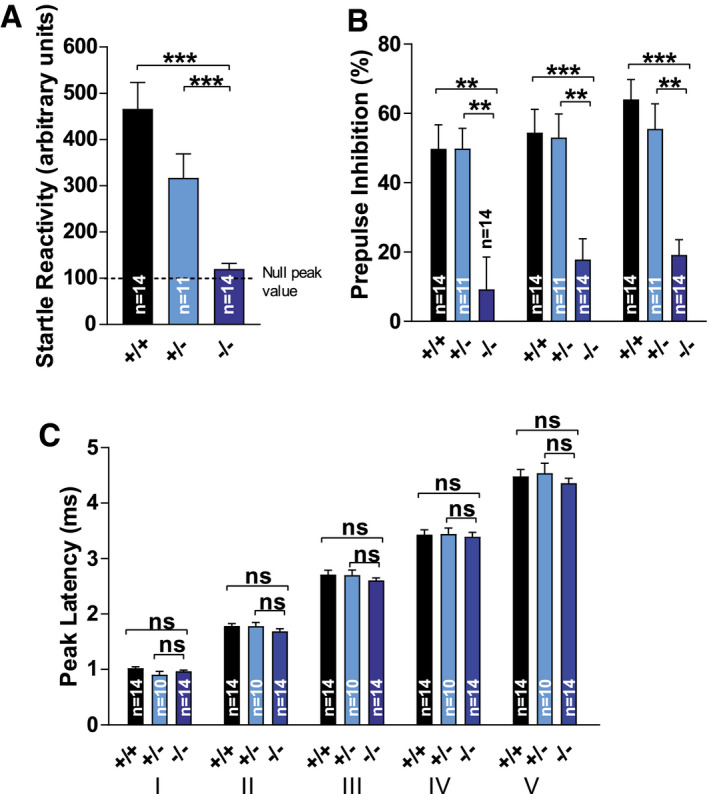
Reduced acoustic startle reactivity but normal brainstem auditory evoked potentials in Tmem106b KO mice. (A) Acoustic startle reflex was evaluated using startle peak values from “startle alone” trials. Peak values during the null period and the startle period were compared. (B) Prepulse inhibition calculations from acoustic startle reflexes. (C) Brainstem auditory evoked potentials (BAEP). One Tmem106b HET animal was excluded from final analysis as no reliable signal could be recorded. Other animals showed a detectable five‐peak pattern (I–V). (A)–(C): ns = P > 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Deficiency of TMEM106B leads to exploration deficits
Open field locomotion was analyzed to characterize exploratory behavior (Figure 6A–E). There was no significant difference between the genotypes for total distance traveled (P = 0.071), although Tmem106b KO mice tended to have decreased path length (Figure 6B). However, there was a significant difference between genotypes for the time spent in the periphery of the arena (F(2,36) = 8.083, P = 0.001). Tmem106b KO mice showed signs of thigmotaxis in comparison to WT and Tmem106b HET mice (P = 0.010 & P = 0.002) (Figure 6A, C). This was further confirmed by genotype differences in the average distance from the center (F(2,36) = 7.343, P = 0.002) (Figure 6D) and the percentage of path length in the center (F(2,36) = 8.362, P = 0.001) (Figure 6E). Tmem106b KO mice showed reduced exploration of the center in comparison to WT & Tmem106b HET mice (P = 0.025 & P = 0.002 for average distance from the center; P = 0.012 & P = 0.001 for percentage of path length in the center). These data show clear changes in exploratory tendencies and may suggest affective‐like changes Tmem106b KO mice. However, it is unclear to which extent motor deficits could have contributed to these differences.
Figure 6.
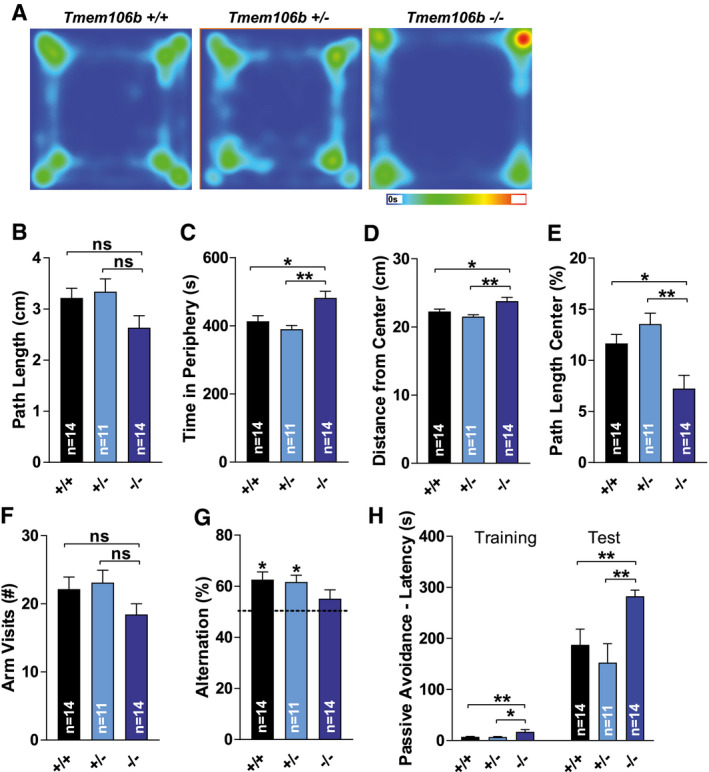
Aged Tmem106b KO mice show exploratory deficits. (A) Heatmap representation of the cumulative time spent in each area of the open field for each genotype. The scale in seconds is depicted below the heatmap. (B) Total distance traveled in the open field. (C) Time spent in the periphery of the arena of the open field. (D) Average distance from the center of the open field. (E). Percentage of path length in the center of the open field. (F) The number of arm visits in the Y‐maze. (G) The percentage of alternations in the Y‐maze test. (H) The step‐through latency during the training‐ and the test‐phase in the passive avoidance test are depicted in seconds. (A–H): Age 14 months. (B)–(C): ns = P > 0.05; *P ≤ 0.05; **P ≤ 0.01.
Similar to the open field, a trend for reduced exploratory activity in Tmem106b KO mice was noted in the Y‐maze test. There was, however, no significant difference between genotypes for the number of arm visits (P = 0.142). Spatial working memory was assessed through assessment of arm visit alternation (Figure 6F–G). One‐sample t‐tests indicated that WT and Tmem106b HET mice alternated significantly more than expected by coincidence (t(13) = 4.208, P = 0.001 & t(10) = 4.348, P = 0.001), while this was not the case for Tmem106b KO mice (P = 0.160). However, a direct comparison of spontaneous alternation percentages showed no significant difference between genotypes (P = 0.182) (Figure 6F–G). Memory assessment was subsequently extended by evaluation of fear memory in the passive avoidance task (Figure 6H). There were significant differences between the genotypes in step‐through latencies during the training and test phase of the passive avoidance task (H(2) = 12.156, P = 0.002 & H(2) = 12.702, P = 0.002). Tmem106b KO mice showed significantly increased step‐through latencies during training (Tmem106b KO vs. WT: P = 0.006, Tmem106b KO vs. Tmem106b HET: P = 0.014) and testing (Tmem106b KO vs. Tmem106b HET: P < 0.004). Differences in baseline step‐through behavior confounded assessment of performance, but Tmem106b KO mice clearly did not show reduced, but rather increased fear memory. By and large, lack of robust cognitive deficits in Tmem106b KO mice was consistent with the absence of obvious pathology in telencephalic regions typically involved in these behaviors, including the hippocampus.
Tmem106b KO mice do not exhibit obvious signs of abnormal repetitive, social, disinhibition‐ or depression‐related behavior
Finally, we tested the aged cohort of mice for equivalents of prominent human FTD features, including abnormal social behavior, repetitive behaviors, disinhibition and depression‐like behavior(4) (Figure 7A–H; Figure S3A–E). There were no significant differences between the genotypes for the distance traveled during the acclimation phase (P = 0.944) (Figure 6A), the sociability phase (P = 0.136) (Figure 6B) or the preference for social novelty phase (P = 0.908) (Figure 6C) of the SPSN test. Furthermore, during the sociability and social novelty phases, there were no differences between genotypes in time spent in proximity of the wire cages (P = 0.439 & P = 0.786), confirming baseline exploratory behavior did not appear to differ in this test. Likewise, social interaction behavior appeared intact in Tmem106b KO mice. A relative measure of sociability again showed no significant differences between the genotypes, on average (P = 0.296) or time‐dependent (genotype × time interaction, P = 0.279) (Figure 6D). Similarly, there were no significant differences in the relative preference for social novelty, on average (P = 0.229) or time‐dependent (genotype × time interaction, P = 0.748) (Figure 6E). During observations for assessment repetitive behavior, there were no significant differences between the genotypes in the time spent self‐grooming (P = 0.299) or the amount of digging behavior (P = 0.731) (Figure S3A,B). Other possible repetitive behaviors such as bar‐mouthing, jumping and somersaulting were rarely observed. There was no difference between the genotypes for the total amount of beam crossings in the elevated plus maze (P = 0.350) (Figure 6F). Furthermore, they showed similar relative activity in the open arms (P = 0.503) (Figure S3C) and spent a similar relative amount of time in the open arms (P = 0.666) (Figure 6G), results suggesting similar anxiety‐related behavior and no signs of a lack of inhibition in Tmem106b KO mice. Finally, we tested depression‐related behavior with the tail suspension test. There were no significant differences between the genotypes for the amount of distance moved (P = 0.973) (Figure S3D), the immobility time (P = 0.918) (Figure 6H) nor the latency to the first immobile episode (P = 0.808) (Figure S3E). In summary, TMEM106B deficiency does not seem to cause FTD‐like behavioral characteristics such as abnormal repetitive or social behaviors, disinhibition‐ or depression‐related behavior in mice.
Figure 7.
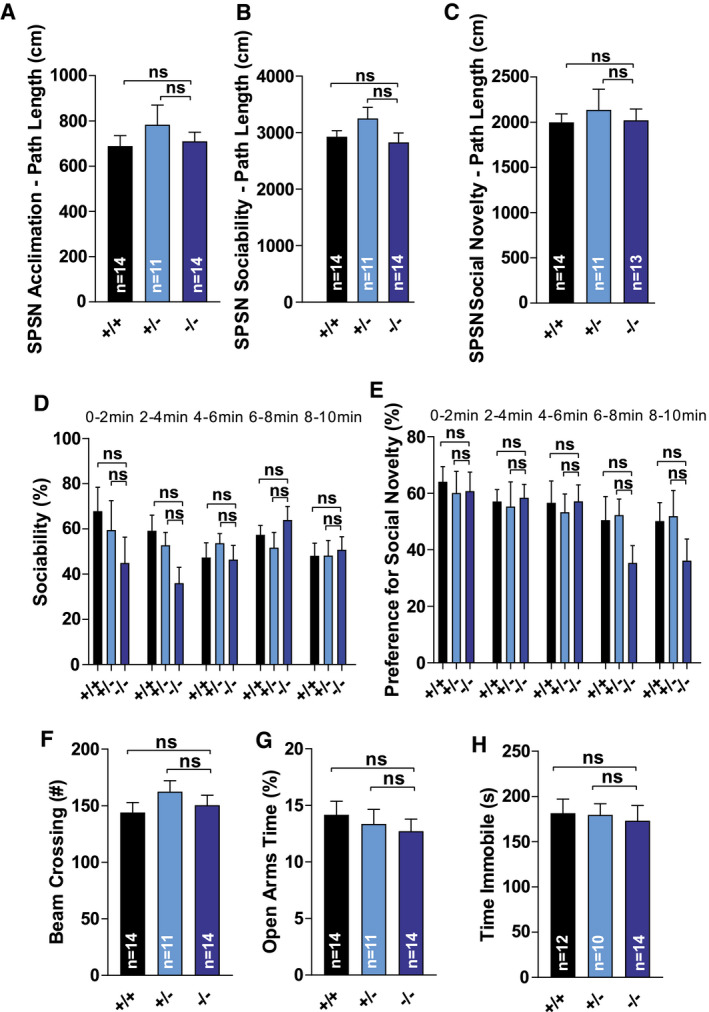
Tmem106b KO mice do not show altered social behavior, anxiety‐related or depression‐related deficits. (A) Distance traveled during the acclimation phase of the SPSN‐test. (B) Distance traveled during the sociability phase of the SPSN‐test. (C) Distance traveled during the social novelty phase of the SPSN‐test. (D) Relative measure of sociability. Data are depicted for each 2‐minute interval. (E) Relative preference for social novelty. Data are depicted for each 2‐minute interval. (F) Total number of beam crossings in the elevated plus maze. (G) Relative activity in the open arms in the elevated plus maze (% of total time). (H) Immobility time in the tail suspension test. Three mice (2 WT – 1 Tmem106b HET) were excluded because of repeated tail climbing behavior. (A)–(H): ns = P > 0.05.
Discussion
Variants in TMEM106B have initially been identified as an important genetic risk factor for FTD (41). Subsequently, genetic linkages were identified with a much broader spectrum of other neurodegenerative disorders including limbic‐predominant age‐related TDP‐43 encephalopathy, Parkinson's disease and late‐onset Alzheimer's disease (22, 23, 38). Moreover, genetic links were also confirmed with dissociation of cognitive performance in elderlies and differential aging in humans (16, 30, 45, 47) as well as with increased neuronal proportion in Alzheimer’s disease (16). Recently, a mutation in TMEM106B was identified as the underlying cause for a hypomyelinating leukodystrophy (35, 46, 51). Elucidation of how a single gene can affect disease severity and pathology of such a broad spectrum of diseases and cognitive function remains a major and important challenge. Better understanding of the pathogenic cascades and the specific role of TMEM106B could contribute to the development of therapeutic options by targeting TMEM106B‐related pathways.
In a first effort toward understanding the physiological function of TMEM106B, we generated Tmem106b KO mice and found drastic proximal axonal swelling in different motoneuron nuclei and the thalamus, caused by giant LAMP1‐positive vacuoles. Notably, this phenotype was already observed by 2‐month of age and was accompanied by mild motor dysfunction but without neuronal death (18). Presently, we extend our analysis of Tmem106b KO mice with an aged cohort and a detailed behavioral survey to understand age‐related phenotypes. Given the close genetic linkage of variants in TMEM106B with aging and cognition in humans (30), our findings of behavioral deficits in aged Tmem106b KO animals support the finding of a major role of TMEM106B in normal neuronal homeostasis, substantiate the findings of a genetic link of TMEM106B and aging observed in the human studies and justify the use of the Tmem106b KO mouse as a good model for studying disease‐ and age‐related pathology.
A dominant mutation in TMEM106B in humans causes a hypomyelinating leukodystrophy and recently, subtle changes in myelin‐related proteins and myelination were described in Tmem106b KO mice under basal conditions (50) and in a demyelination/re‐myelination model in Tmem106b KO mice (6). While we cannot rule out the observed behavioral and pathological deficits are caused by subtle changes in myelination rather than neuronal changes, our previous data of impaired lysosomal axonal transport in isolated cultured neurons suggest a cell‐intrinsic pathology (18). In vivo experiments, such as the generation of conditional knockout mice, might be a valuable tool to further determine the extent to which these phenotypes are due to neuronal changes or altered myelination.
Our current set of experiments reveal a TMEM106B‐dependent progressive development of neuronal dysfunction that is not (almost) exclusively restricted to motoneurons as observed in young Tmem106b KO animals (18). This observation is highlighted by the late onset of Purkinje cell death that is accompanied by mild gait abnormalities and progression of motor impairment. The robust but still relatively mild motor phenotype is in good agreement with the fact that even the oldest mice tested showed only partial loss of Purkinje cells. The finding of rare vacuoles at the distal end of the AIS of Purkinje cell axons suggests that these alterations are the underlying cause for their subsequent degeneration. The high susceptibility of Purkinje cells to any pathological challenge is well documented in numerous lysosomal storage disorders in which they often represent the neuronal subtype that primarily undergoes cell death (32, 33). It should be noted that the pattern of Purkinje cell death in the Tmem106b KO mice was not random, but followed a pattern observed previously in other genetic disorders like Niemann‐Pick type C and A/B (32, 33). While the Purkinje cell layer of the posterior lobules was still fully intact at 2 years of age, Purkinje cells started to disappear from layer V/IV and layer III at 9–12‐month of age and were strikingly reduced at 2 years of age. Similar to Tmem106b KO mice, Niemann‐Pick mouse models develop motor coordination deficits and gait abnormalities (17, 20, 26) Given the similar pattern of cell death in Tmem106b KO mice and other mouse models with cerebellar Purkinje cell degeneration, it seems likely that differential cell vulnerability is not specifically explained by changes in TMEM106B expression, but other (genetic) factors that are protective for surviving Purkinje cells. It should, however, be noted that other regions of the central nervous system relevant for motor behavior (e.g., spinal cord) are affected Tmem106b KO mice, which likely contribute to the observed gait defects as well. In summary, progressive motor impairment and gait deficits in aged Tmem106b KO mice are consistent with progressive cerebellar pathology exacerbating the consequences of early‐onset motor neuron pathology.
We observed widespread but not fully generalized microgliosis and astrogliosis in aged Tmem106b KO mice. Even though we cannot rule out a cell‐intrinsic relevant function of TMEM106B in microglia, the course of events and the spatial distribution of activated microglia (e.g., the appearance of activated microglia at sites of Purkinje cell death) point to reactive activation rather than microglia‐intrinsic activation. These findings suggest that unlike many other disease‐modifying genes (e.g., TREM2 or APOE in Alzheimer’s disease), TMEM106B does not convey its important disease‐modulating function in microglia, but cell‐autonomous in neurons.
Genetic variants in TMEM106B convey the highest disease‐modifying effect on diseases that mostly affect the frontal brain and the cerebral cortex, including FTD, Alzheimer's disease, chronic traumatic encephalopathy and limbic‐predominant age‐related TDP‐43 encephalopathy (16, 41, 49). However, phenotypes we observed in the Tmem106b KO mice (LAMP1‐positive vacuoles, micro‐ and astrogliosis, cell death) were most pronounced in the cerebellum, brainstem, midbrain and the hindbrain. These findings are supported by the behavioral experiments that did not appear to reveal major deficits in social, emotional or cognitive function, but clear deficits in reflexive behavior and coordinated movement. Open field assessment did reveal changes in exploratory behavior, more specifically indicating thigmotaxis in Tmem106b KO animals. Reduced center exploration could reflect an inert or anxious phenotype, for which, however, no further evidence was found in tail suspension or elevated plus maze behavior. Step‐through latencies in the passive avoidance task indicated a baseline delay in Tmem106b KO mice to enter a dark compartment. It is unclear to which extent the established motor defects contributed to changes in step‐through and exploratory movement. In combination with observation of apparently unaltered behaviors in isolation and during interaction with conspecifics, our results indicate that Tmem106b KO animals appear to show no typical FTD‐like signs such as apathy/inertia, lack of inhibition, repetitive behavior or social disinterest (43). The cause of this apparent contradiction remains an important question. Differences between humans and mice, for example, in TMEM106B expression, provide a possible explanation. Alternatively, changes in TMEM106B expression could convey a higher impact on neurons that are already affected by the primary disease‐cause as a first hit, like protein aggregates typically appearing the indicated disease entities.
Reduced acoustic startle reactivity was one of the most striking differences observed between WT and Tmem106b KO animals. We found no indication of impaired auditory processing in recordings of brainstem auditory‐evoked potentials, suggesting this difference could reflect reduced motor output on the level of spinal motor neurons. Deficient reflexes likely arise from partially dysfunctional motoneuron units of the corresponding facial nerves and motoneurons, underscoring axonal deficits (18). Notably, Tmem106b HET mice showed signs of reduced startle reactivity as well. We did not observe any LAMP1‐positive vacuoles in Tmem106b HET mice, but reduced TMEM106B might have functional relevance before the formation of the giant vacuoles observed in the Tmem106b KO animals. These results underscore that TMEM106B‐levels need to be tightly balanced for proper function of the lysosomal system in neurons.
A major open question regarding the role of TMEM106B in aging and neurodegenerative diseases is how a single protein can affect such a broad spectrum of diseases. The slowly progressive phenotype of the Tmem106b KO mice finally yielding in mild (lysosomal) dysfunction and eventually cell death in a subset of neurons suggests a mechanism in which a primary (genetic) predisposition determines disease onset and variants in TMEM106B leading to altered levels or function act like a second hit, by which the course of the disease is modulated. It will be interesting to determine the effect of TMEM106B deficiency or haploinsufficiency in mouse models for common neurodegenerative diseases like Parkinson's disease, Alzheimer’s disease or other age‐related neurodegenerative diseases.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
S.S. and M.D. conducted the experiments, S.S., R.H. and M.D. designed the experiments, and S.S. and M.D. wrote the paper. All authors read and approved the final manuscript.
Ethics Approval and Consent to Participate
Experimental protocols were approved by the ethical research committee of the KU Leuven according to EC guidelines/approved by local German authorities (Ministerium für Energiewende, Landwirtschaft, Umwelt und ländliche Räume, Kiel, V 242‐13648/2018).
Supporting information
Fig S1‐S3
Figure S1. Aged Tmem106b KO mice show no differences in cage activity, grip strength, or rotarod performance. (A) Total number of beam crossings during 30 minutes intervals for 23 h in home cages placed between three infrared beams. Dark (d1‐24) and light‐phases (l1‐22) are indicated. n = 14; Tmem106b HET: n = 11; Tmem106b KO: n = 14). (B) Forelimb grip strength evaluated by allowing mice to spontaneously grab a T‐shaped bar. (C) Drop latencies on the accelerating rotarod. n = 14; Tmem106b HET: n = 11; Tmem106b KO: n = 14). (B)–(C): ns = P > 0.05
Figure S2. Aged Tmem106b KO mice show no differences in size measures and step angles during gait analysis. (A) Animal lengths and animal widths (B) were not significantly different between the genotypes. (C) Fore‐ and hind step angles are depicted. (A)–(B): ns = P > 0.05
Figure S3. Aged Tmem106b KO mice show no obvious abnormal repetitive, anxiety‐, or depression‐related behavior. (A) Time spent self‐grooming during a 10 minutes observation time. (B) Amount of digging behavior during a 10 minutes observation time. (C) Activity in the open arms in the elevated plus maze test. The number of beam crossings is depicted. (D) Amount of distance moved during the tail suspension test. (E) The latency to the first immobile episode during tail suspension test. (A)–(E): ns = P > 0.05.
Acknowledgments
Maike Langer and Leen Van Aerschot are acknowledged for excellent technical assistance. Paul Saftig is acknowledged for discussion and ongoing support. Funding was provided by the intramural funding program of the Medical Faculty of the Christian‐Albrechts‐University Kiel.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Arakawa H (2018) Ethological approach to social isolation effects in behavioral studies of laboratory rodents. Behav Brain Res 341:98–108. [DOI] [PubMed] [Google Scholar]
- 2. Chen‐Plotkin AS, Lee VM, Trojanowski JQ (2010) TAR DNA‐binding protein 43 in neurodegenerative disease. Nat Rev Neurol 6:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cherry JD, Mez J, Crary JF, Tripodis Y, Alvarez VE, Mahar I et al (2018) Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol Com 6:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christidi F, Migliaccio R, Santamaria‐Garcia H, Santangelo G, Trojsi F (2018) Social cognition dysfunctions in neurodegenerative diseases: neuroanatomical correlates and clinical implications. Behav Neurol 2018:1849794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damme M, Suntio T, Saftig P, Eskelinen EL (2015) Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta Neuropathol 129:337–362. [DOI] [PubMed] [Google Scholar]
- 6. Feng T, Sheng RR, Sole‐Domenech S, Ullah M, Zhou X, Mendoza CS et al (2020) A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 143:2255–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus‐Hernandez M et al (2011) TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC et al (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 127:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghazi‐Noori S, Froud KE, Mizielinska S, Powell C, Smidak M, Fernandez de Marco M et al (2012) Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 135(Pt 3):819–832. [DOI] [PubMed] [Google Scholar]
- 10. Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G et al (2014) Common pathobiochemical hallmarks of progranulin‐associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 127:845–860. [DOI] [PubMed] [Google Scholar]
- 11. Graeber MB, Streit WJ, Kiefer R, Schoen SW, Kreutzberg GW (1990) New expression of myelomonocytic antigens by microglia and perivascular cells following lethal motor neuron injury. J Neuroimmunol 27:121–132. [DOI] [PubMed] [Google Scholar]
- 12. Hokkanen SRK, Kero M, Kaivola K, Hunter S, Keage HAD, Kiviharju A et al (2020) Putative risk alleles for LATE‐NC with hippocampal sclerosis in population‐representative autopsy cohorts. Brain Pathol 30:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Katsumata Y, Nelson PT, Ellingson SR, Fardo DW (2017) Gene‐based association study of genes linked to hippocampal sclerosis of aging neuropathology: GRN, TMEM106B, ABCC9, and KCNMB2. Neurobiol Aging 53:193.e17–193.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D et al (2012) Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 287:19355–19365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA et al (2018) Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359:1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Z, Farias FHG, Dube U, Del‐Aguila JL, Mihindukulasuriya KA, Fernandez MV et al (2020) The TMEM106B FTLD‐protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol 139:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loftus SK, Erickson RP, Walkley SU, Bryant MA, Incao A, Heidenreich RA, Pavan WJ (2002) Rescue of neurodegeneration in Niemann‐Pick C mice by a prion‐promoter‐driven Npc1 cDNA transgene. Hum Mol Genet 11:3107–3114. [DOI] [PubMed] [Google Scholar]
- 18. Luningschror P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D et al (2020) The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep 30:3506–19.e6. [DOI] [PubMed] [Google Scholar]
- 19. Mann DMA, Snowden JS (2017) Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype. Brain Pathol 27:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maue RA, Burgess RW, Wang B, Wooley CM, Seburn KL, Vanier MT et al (2012) A novel mouse model of Niemann‐Pick type C disease carrying a D1005G‐Npc1 mutation comparable to commonly observed human mutations. Hum Mol Genet 21:730–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCormick DA, Bal T (1994) Sensory gating mechanisms of the thalamus. Curr Opin Neurobiol 4:550–556. [DOI] [PubMed] [Google Scholar]
- 22. Milind N, Preuss C, Haber A, Ananda G, Mukherjee S, John C et al (2020) Transcriptomic stratification of late‐onset Alzheimer's cases reveals novel genetic modifiers of disease pathology. PLoS Genet 16:e1008775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K et al (2019) Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain 142:1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nelson PT, Estus S, Abner EL, Parikh I, Malik M, Neltner JH et al (2014) ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol 127:825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 26. Otterbach B, Stoffel W (1995) Acid sphingomyelinase‐deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann‐Pick disease). Cell 81:1053–1061. [DOI] [PubMed] [Google Scholar]
- 27. Paushter DH, Du H, Feng T, Hu F (2018) The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol 136:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Premi E, Grassi M, van Swieten J, Galimberti D, Graff C, Masellis M et al (2017) Cognitive reserve and TMEM106B genotype modulate brain damage in presymptomatic frontotemporal dementia: a GENFI study. Brain 140:1784–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58:1615–1621. [DOI] [PubMed] [Google Scholar]
- 30. Rhinn H, Abeliovich A (2017) Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst 4:404–415.e5. [DOI] [PubMed] [Google Scholar]
- 31. Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA et al (2012) TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79:717–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R (2003) Patterned Purkinje cell degeneration in mouse models of Niemann‐Pick type C disease. J Comp Neurol 456:279–291. [DOI] [PubMed] [Google Scholar]
- 33. Sarna J, Miranda SR, Schuchman EH, Hawkes R (2001) Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur J Neurosci 13:1873–1880. [DOI] [PubMed] [Google Scholar]
- 34. Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K et al (2014) The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J 33:450–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simons C, Dyment D, Bent SJ, Crawford J, D'Hooghe M, Kohlschutter A et al (2017) A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 140:3105–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sirkis DW, Geier EG, Bonham LW, Karch CM, Yokoyama JS (2019) Recent advances in the genetics of frontotemporal dementia. Curr Genet Med Rep 7:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM (2014) Lysosome size, motility and stress response regulated by fronto‐temporal dementia modifier TMEM106B. Mol Cell Neurosci 61:226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J et al (2019) TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol 85:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Acker ZP, Bretou M, Annaert W (2019) Endo‐lysosomal dysregulations and late‐onset Alzheimer's disease: impact of genetic risk factors. Mol Neurodegener 14:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC et al (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 127:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Deerlin VM, Sleiman PM, Martinez‐Lage M, Chen‐Plotkin A, Wang LS, Graff‐Radford NR et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP‐43 inclusions. Nat Genet 42:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R et al (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134(Pt 3):808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vernay A, Sellal F, Rene F (2016) Evaluating Behavior in Mouse Models of the Behavioral Variant of Frontotemporal Dementia: Which Test for Which Symptom? Neurodegener Dis 16:127–139. [DOI] [PubMed] [Google Scholar]
- 44. Wallings RL, Humble SW, Ward ME, Wade‐Martins R (2019) Lysosomal dysfunction at the centre of Parkinson's disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci 42:899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. White CC, Yang HS, Yu L, Chibnik LB, Dawe RJ, Yang J et al (2017) Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med 14:e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yan H, Kubisiak T, Ji H, Xiao J, Wang J, Burmeister M (2018) The recurrent mutation in TMEM106B also causes hypomyelinating leukodystrophy in China and is a CpG hotspot. Brain 141:e36. [DOI] [PubMed] [Google Scholar]
- 47. Yang HS, White CC, Klein HU, Yu L, Gaiteri C, Ma Y et al (2020) Genetics of gene expression in the aging human brain reveal TDP‐43 proteinopathy pathophysiology. Neuron 107:496‐508.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA (2015) The TMEM106B locus and TDP‐43 pathology in older persons without FTLD. Neurology 84:927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang L, Chen Y, Liu M, Wang Y, Peng G (2019) TDP‐43 and limbic‐predominant age‐related TDP‐43 encephalopathy. Front Aging Neurosci 11:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou X, Nicholson AM, Ren Y, Brooks M, Jiang P, Zuberi A et al (2020) Loss of TMEM106B leads to myelination deficits: implications for frontotemporal dementia treatment strategies. Brain 143:1905–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou X, Rademakers R (2017) TMEM106B and myelination: rare leukodystrophy families reveal unexpected connections. Brain 140:3069–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S3
Figure S1. Aged Tmem106b KO mice show no differences in cage activity, grip strength, or rotarod performance. (A) Total number of beam crossings during 30 minutes intervals for 23 h in home cages placed between three infrared beams. Dark (d1‐24) and light‐phases (l1‐22) are indicated. n = 14; Tmem106b HET: n = 11; Tmem106b KO: n = 14). (B) Forelimb grip strength evaluated by allowing mice to spontaneously grab a T‐shaped bar. (C) Drop latencies on the accelerating rotarod. n = 14; Tmem106b HET: n = 11; Tmem106b KO: n = 14). (B)–(C): ns = P > 0.05
Figure S2. Aged Tmem106b KO mice show no differences in size measures and step angles during gait analysis. (A) Animal lengths and animal widths (B) were not significantly different between the genotypes. (C) Fore‐ and hind step angles are depicted. (A)–(B): ns = P > 0.05
Figure S3. Aged Tmem106b KO mice show no obvious abnormal repetitive, anxiety‐, or depression‐related behavior. (A) Time spent self‐grooming during a 10 minutes observation time. (B) Amount of digging behavior during a 10 minutes observation time. (C) Activity in the open arms in the elevated plus maze test. The number of beam crossings is depicted. (D) Amount of distance moved during the tail suspension test. (E) The latency to the first immobile episode during tail suspension test. (A)–(E): ns = P > 0.05.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.