

Abstract
Pediatric ependymomas frequently develop in the cerebellum and are currently treated using non‐specific therapies, in part, because few somatically mutated driver genes are present, and the underlying pathobiology is poorly described. Circular RNAs (circRNAs) constitute as a large class of primarily non‐coding RNAs with important roles in tumorigenesis, but they have not been described in pediatric ependymomas. To advance our molecular understanding of ependymomas, we performed Next Generation Sequencing of rRNA‐depleted total RNA of 10 primary ependymoma and three control samples. CircRNA expression patterns were correlated to disease stage, outcome, age, and gender. We found a profound global downregulation of circRNAs in ependymoma relative to control samples. Many differentially expressed circRNAs were discovered and circSMARCA5 and circ‐FBXW7, which are described as tumor suppressors in glioma and glioblastomas in adults, were among the most downregulated. Moreover, patients with a dismal outcome clustered separately from patients with a good prognosis in unsupervised hierarchical cluster analyses. Next, NanoString nCounter experiments were performed, using a custom‐designed panel targeting 66 selected circRNAs, on a larger cohort that also included medulloblastomas and pilocytic astrocytomas. These experiments indicated that circRNA expression profiles are different among distinct pediatric brain tumor subtypes. In particular, circRNAs derived from RMST, LRBA, WDR78, DRC1 and BBS9 genes were specifically upregulated in ependymomas. In conclusion, circRNAs have different expression profiles in ependymomas relative to controls and between survivors and patients with a dismal outcome, suggesting that circRNAs could be exerted as diagnostic and prognostic biomarkers in the future if further validated in larger cohorts.
Keywords: alternative splicing, circular RNA, medulloblastoma, NanoString nCounter, non‐coding RNA, pediatric ependymoma, pilocytic astrocytoma, RNA‐sequencing
Ependymomas are rare cancers of the central nervous system (CNS), however, one of the commonest primary CNS tumors among children. Ependymomas mostly occur intracranially (supratentorial brain and posterior fossa) in children between 0 and 4 years of age, but they are also observed in older children and adults. Clinical management is challenging, and pediatric patients with intracranial ependymomas have high morbidity and mortality rates. Among nine distinct molecular subgroups of ependymomas based on DNA methylation analyses, pediatric Posterior Fossa PF‐EPN‐A and Supratentorial RELA fusion subgroups are characterized by a dismal outcome. Moreover, PF‐EPN‐A subgroup are diagnosed at a markedly younger age (median age is 3 years) compared to the PF‐EPN‐B subgroup. In the majority of cases, the underlying oncogenic drivers are unknown and the underlying pathobiology of ependymoma is poorly described (1, 2).
Recently, circular RNAs (circRNAs) have emerged as a large class of endogenous RNAs with mainly non‐coding functions, which play key roles in development and disease (3, 4). They exhibit tissue‐specific expression patterns and constitute a significant amount of cellular RNA, particularly in the brain (5). Importantly, most of these molecules are extremely stable and generally situated in the cytoplasm where they may bind other cellular molecules, such as microRNAs or proteins, and regulate their functions (3, 4). In ependymomas nothing is currently known about the expression and potential deregulation of circRNAs. Moreover, circRNAs are emerging as important oncogenic drivers and tumor suppressors in glioma and glioblastoma of adults.
Here, we first employed Next Generation Sequencing of rRNA‐depleted total RNA (RNA‐seq) for an unbiased identification and profiling of circRNAs in a cohort of 10 pediatric patients diagnosed with ependymoma and three controls (resected from temporal lobe) (Supporting Information and Table 1). We detected 11,217 unique circRNAs supported by at least two backsplicing junction‐spanning reads in a single sample; 6975 in the ependymomas and 8479 in the controls (Figures 1A,B and S1A) (Table S1). Next, we defined a high abundance set of circRNAs (Supporting Information) giving rise to a total of 1167 circRNAs of which 263 were detected in the ependymomas and 1126 in the controls (Figure S1B and Table S2). The low number of circRNAs detected in ependymomas was not due to a difference in sample quality/sequencing depth, and most of these circRNAs (231 of 263 and 1033 of 1126) were also detected by an alternative detection algorithm (Table S2). Interestingly, circSH3KBP1 was only detected in the controls and circLRRC55‐as was only detected in the ependymomas (Table S2). Moreover, 62 (23.6%) and 309 (27.4%) of the high abundance circRNAs were on average expressed at higher levels than their cognate linear host genes in the ependymomas and in the controls, respectively (Figure S1C,D and Table S2), indicating that many of the circRNAs are not merely side‐products of aberrant splicing.
TABLE 1.
Clinical characteristics of the ependymoma patients used for RNA‐sequencing.
| Patient number | Anatomical site | WHO grade | Age at diagnosis | Clinical follow‐up time (years) | Survival status |
|---|---|---|---|---|---|
| 1 | Posterior fossa | II | 2 | 3 | Deceased |
| 2 | Posterior fossa | II | 5 | 20 | Alive |
| 3 | Posterior fossa | II | 5 | 20 | Alive |
| 4 | Posterior fossa | II | 2 | 9 | Unknown |
| 5 | Posterior fossa | II | 1 | 3 | Deceased |
| 6 | Posterior fossa | II | 13 | 17 | Alive |
| 7 | Posterior fossa | III | 8 | 22 | Alive |
| 8 | Posterior fossa | III | 2 | 3 | Deceased |
| 9 | Posterior fossa | III | 2 | 6 | Deceased |
| 10 | Posterior fossa | III | 1 | 17 | Deceased |
FIGURE 1.
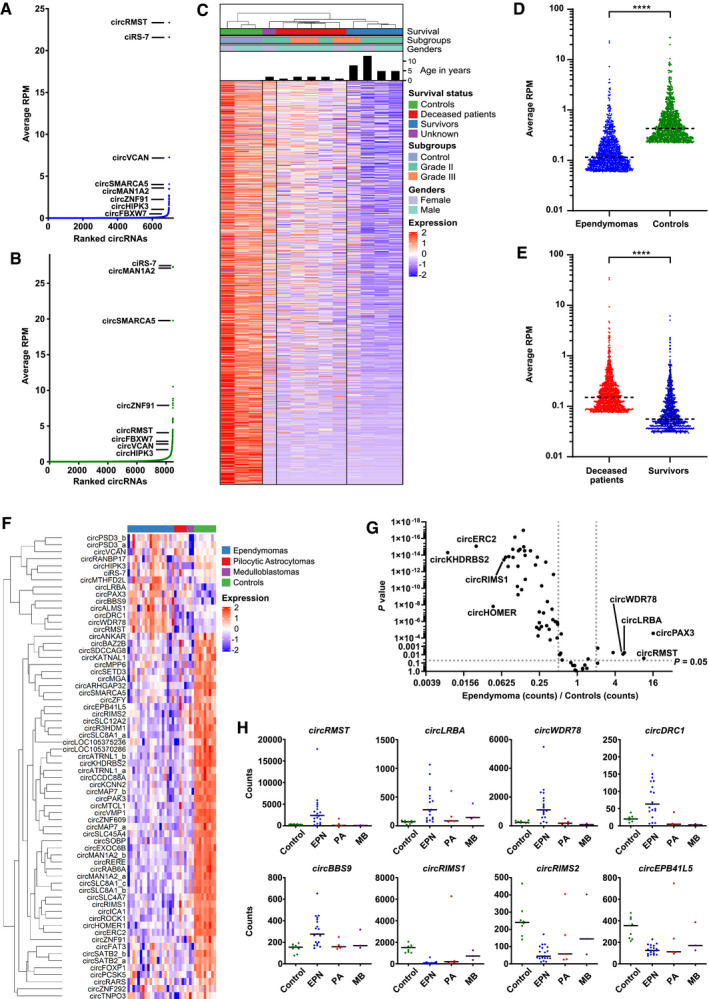
Identification and characterization of circular RNAs in ependymoma. (A,B) Scatter plots of unique circRNAs that are supported by at least two RNA‐sequencing reads (BSJ > 2) and ranked according to average expression level (average RPM) in ependymomas (A) and in healthy controls (B). (C) Heatmap and unsupervised hierarchical cluster analysis using RNA‐sequencing data for the high abundance circRNAs. Age, WHO Grade (Grade II vs. Grade III), survival status (survivors, deceased or unknown) and gender (male vs. female) of the ependymoma samples are indicated. Each row in the heatmap corresponds to a unique circRNA and each column corresponds to a patient or control sample. Each sample is annotated below the dendrogram in the top. (D) Scatter plot of the top 1000 circRNAs identified in ependymoma (n = 10) and control (n = 3) samples. Mann–Whitney U test was used for the statistical calculation. (E) Scatter plot of the top 1000 circRNAs identified in samples from deceased patients (n = 5) and survivors (n = 4). Mann–Whitney U test was used for the statistical calculation. (F) Heatmap representing the expression of 66 circRNAs quantified using NanoString nCounter technology in ependymomas (blue, n = 19), pilocytic astrocytomas (red, n = 5), medulloblastomas (purple, n = 3) and healthy control samples (green, n = 9). Each sample is annotated in the top. (G) Volcano plot comparing differences of 66 circRNAs between ependymomas and controls. One unpaired t test for each circRNA was used to generate the P values. (H) Column scatter plots of the expression levels of circRMST and circRNAs derived from LRBA, WDR78, DRC1, BBS9, RIMS1, RIMS2, and EPB41L5 genes. EPN, ependymomas (n = 19); MB, medulloblastomas (n = 3); PA, pilocytic astrocytomas (n = 5); Control, healthy control samples (n = 9). Median is shown. ****P < 0.0001. RPM, reads‐per‐million
We then performed unsupervised hierarchical cluster analysis using all the 1167 high abundance circRNAs detected in the ependymomas and in the controls (Figure S1B) and observed that the ependymomas clustered separately due to a marked downregulation of the majority of the circRNAs (Figures 1C,D and S1E and Table S3). This finding is in line with previous studies indicating that rapidly proliferating cells generally contain fewer circRNAs, possibly due to a dilution effect preventing the highly stable circRNAs from reaching steady‐state levels (6). Interestingly, the circRNA expression profiles from deceased patients were distinct from the profiles of patients who survived. This sub‐clustering according to outcome could not be explained by gender, tumor location (all the tumors were located in Posterior Fossa) or disease grade, but the deceased patients were diagnosed at a marked younger age (Figure 1C). As stated above, only the PF‐EPN‐A subgroup is characterized by a dismal outcome and a markedly younger median age among Posterior Fossa tumors. Therefore, it is highly likely that the subgroup with dismal outcome, which we identified by circRNA profiling, corresponds to PF‐EPN‐A (1, 2). However, we were not able to confirm this as several attempts to perform 850K DNA methylation analyses failed due to poor DNA quality.
Many of the circRNAs that displayed a marked downregulation in the ependymoma samples relative to the controls were statistically significant (Figure S1E and Table S3). Among the circRNAs that previously have been shown to be aberrantly expressed in adult brain tumors, circSMARCA5 and circ‐FBXW7 were found to be significantly downregulated in the ependymoma samples. This is consistent with the previous studies describing these circRNAs as tumor suppressors in gliomas (7). Likewise, circVCAN and circRMST were more abundant in ependymomas relative to the controls, similar to what has been observed in glioblastoma (8).
As expected from the heatmap (Figure 1C), we observed a significant overall upregulation of circRNAs in deceased patients relative to survivors (Figures 1E, S1F and Table S4). The many upregulated circRNAs in the deceased patients included cZNF292, consistent with a previous study describing an oncogenic role of cZNF292 in glioma (9) and circRMST.
Most differentially expressed circRNAs between ependymomas and controls were changed independent of their cognate linear host genes, whereas this was true for a smaller fraction of the circRNAs when comparing the deceased patients with the survivors (Figure S1G,H and Tables S3 and S4). Together, these analyses indicate that the majority of the identified differentially expressed circRNAs cannot be explained as passenger events due to gene expression changes in the cognate linear hosts.
To validate and further investigate circRNA expression profiles in ependymoma, we collected another independent cohort of 19 ependymomas as well as five pilocytic astrocytomas, three medulloblastomas and 9 control samples (Supporting Information). Because these samples were derived from Formalin‐Fixed Paraffin‐embedded (FFPE) tissues, we decided to quantitate 66 unique circRNAs (selected based on differential expression and abundance in the RNA‐seq data) (Table S5) using NanoString nCounter technology (Supporting Information), as this method has proved to work well for circRNA quantitation in highly degraded RNA samples (10). First, we observed a strong correlation between the NanoString nCounter data and the RNA‐seq data; the Pearson correlation coefficient (R 2) was 0.74 with p < 0.0001 when comparing log2 fold change values between ependymoma and control samples (Figure S2A). Then, as expected based on how the circRNAs were selected, the ependymoma samples were distinguished from the control samples in unsupervised hierarchical cluster analyses (Figure 1F) and the circRNAs were on average much more abundant in control samples compared to ependymoma samples (Figures 1F and S2B). In these analyses, we also observed that individual circRNAs derived from the same host genes tended to cluster together (e.g., circRNAs from PSD3, SATB2 and SLC8A1) (Figure 1F). Moreover, all ependymoma samples were grouped separately from the pilocytic astrocytoma and medulloblastoma samples with the exception of one pilocytic astrocytoma sample according to principal component analysis (PCA) (Figure S2C), indicating that circRNA expression profiles might be used to distinguish ependymoma from other pediatric brain tumors. Of note, the circRNAs were not selected for this purpose and the separation could be expected to be even more pronounced had this been the case. Therefore, these preliminary data warrant further investigation into the diagnostic biomarker potential of circRNAs in pediatric brain tumors.
Next, in a search for individual circRNAs that may distinguish ependymoma and control samples based on their expression levels, we generated a volcano plot and found that many of the downregulated circRNAs were highly statistically significant (Figure 1G). Four circRNAs were more than fourfold upregulated and were derived from the PAX3, RMST, LRBA, and WDR78 genes, whereas the top four most downregulated circRNAs were derived from the KHDRBS2, ERC2, HOMER1, and RIMS1 genes (Table S6). Among the upregulated circRNAs, the circRNAs derived from PAX3, RMST, LRBA, WDR78, DRC1, and BBS9 genes seem to be specifically upregulated in ependymomas (not in other tumor entities) relative to the control samples, and among the downregulated, the circRNAs derived from RIMS1, RIMS2, and EPB41L5 genes seem to be specifically downregulated in ependymomas (Figure 1H and Table S6).
In conclusion, we found a marked global downregulation of circRNAs in ependymoma relative to control samples, and in patients with a good prognosis relative to patients with a dismal prognosis. In addition, we found that expression levels of individual circRNAs could potentially be used to distinguish ependymoma from healthy brain tissue as well as other pediatric brain tumor entities. Thus, the data presented here, suggest that circRNAs could be utilized as diagnostic and prognostic biomarkers in the future if further validated. Finally, future research should aim at investigating the functional relevance of individual deregulated circRNAs in tumorigenesis and progression of pediatric ependymoma. We find upregulated circRNAs, such as circRMST, circLRBA circWDR78, circDRC1, and circBBS9, to be of particular interest to study further, as upregulation, despite the aforementioned dilution effect of circRNAs, may indicate an active selection for these events due to a potential oncogenic function. Nevertheless, many of the downregulated circRNAs are promising as diagnostic or prognostic biomarkers in pediatric brain tumors and should also be investigated and validated further in larger cohorts.
ETHICAL APPROVAL
This study has been conducted according to the Declaration of Helsinki and Danish legislation and approval from the national ethics committee has been granted and waived the requirement for obtaining informed consent or parental permission for participating in this retrospective study (approval number: 1707758).
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
LSK, HB, and JK conceived the study. HB and AMS collected the clinical samples and the clinical data. MMB, KKE, and LSK carried out the laboratory work. UA and LSK analyzed the data and prepared the figures and tables. LSK and JK provided reagents and materials. UA and LSK wrote the manuscript, which was revised for important intellectual content and approved by all authors.
Supporting information
FIGURE S1 Characterization of circular RNAs in ependymoma by RNA‐sequencing. (A,B) Venn diagrams of unique circRNAs supported by at least two backsplicing junction‐spanning reads (BSJ > 2) in ependymomas (blue circle) and healthy controls (green circle) (A), and high abundance circRNAs (average RPM >0.2) in ependymoma (black circle) and healthy controls (red circle) (B). (C,D) Correlation between average CTL ratios and average RPM values with corresponding linear regression statistics and R 2 values for the high abundance circRNAs in ependymomas (C) and in healthy controls (D). CircRNAs with a CTL ratio >1 are expressed at higher levels than their cognate linear host genes. Simple linear regression test was used to determine R and P values. (E,F) Volcano plots of 1167 high abundance circRNAs showing a high number of differentially expressed circRNAs between ependymoma (n = 10) and control (n = 3) samples (E), and between deceased patients (n = 5) and survivors (n = 4) (F). (G,H) Scatter plots of fold change in CTL ratios against fold change in RPM values for circRNAs with corresponding linear regression statistics and R‐squared values between ependymoma (n = 10) and control (n = 3) samples (G) and between deceased patients and survivors (H). CircRNAs between the dotted blue lines are considered changed independent of their respective host genes. BSJ, backsplicing junction; CTL, circular‐to‐linear ratio; RPM, reads‐per‐million
FIGURE S2 Validation of ependymoma‐related circRNA expression changes using NanoString nCounter. (A) Correlation between log2 fold change values between ependymoma samples and control samples in two independent cohorts using two different methods. (B) Column scatter plot of average circRNA expression in ependymomas (n = 19), pilocytic astrocytomas (n = 5), medulloblastomas (n = 3) and healthy control samples (n = 9). Mann–Whitney U test was used for statistical calculations. (C) PCA plot of ependymoma (n = 19, green dots), pilocytic astrocytomas (n = 5, purple dots), medulloblastomas (n = 3, blue dots) and healthy control (n = 9, red dots) samples. ****P < 0.0001. Fc, fold change; PCA, principal component analysis
TABLE S1 The list of the unique circRNAs samples, supported by at least two sequencing reads (BSJ > 2), are displayed from RNA‐seq analyses of ependymoma (n = 10) and control (n = 3). BSJ, backsplicing junction
TABLE S2 The list of 1167 high abundance circRNAs quantified by RNA‐sequencing of ependymoma (n = 10) and control (n = 3) samples. RPM, read‐per‐million
TABLE S3 The list of the differentially expressed circRNAs between ependymoma samples (n = 10) and healthy control samples (n = 3). One unpaired t test was used to calculate the P values. Correction for multiple testing was performed to calculate adjacent P values using the Holm‐Sidak method. CTL, circular‐to‐linear; RPM, read‐per‐million
TABLE S4 The list of the differentially expressed circRNAs between deceased patients (n = 5) and survivors (n = 4). One unpaired t test was used to calculate the P values. Correction for multiple testing was performed to calculate adjacent P values using the Holm‐Sidak method. CTL, circular‐to‐linear; RPM, read‐per‐million
TABLE S5 The list and the expression levels of the circRNAs selected for the NanoString nCounter analysis. The expression of the circRNAs was obtained from the NanoString nCounter data of ependymoma samples (n = 19), pilocytic astrocytoma (n = 5), medulloblastoma (n = 3) and healthy control (n = 3) samples
TABLE S6 Average expression levels of 66 circRNAs in ependymoma samples (n = 19), pilocytic astrocytoma (n = 5), medulloblastoma (n = 3) and healthy control (n = 3) samples analyzed by the NanoString nCounter. One unpaired t test was used to calculate the P values. Correction for multiple testing was performed to calculate adjacent P values using the Holm‐Sidak method. Control, samples from healthy individuals; CTL, circular‐to‐linear; Epn, ependymoma; Fc, fold change; MB, medulloblastoma; PA, pilocytic astrocytoma; RPM, read‐per‐million
Supplementary Material
ACKNOWLEDGMENTS
We thank Linea Cecilie Melchior for her technical help and Morten Trillingsgaard Venø for his support in bioinformatic analyses and Børnecancerfonden and Dagmar Marshalls Fond for funding this project.
DATA AVAILABILITY STATEMENT
Our raw data cannot be submitted to publicly available databases because the patients did not provide consent to share their raw data, which can potentially identify the individuals, but are available from the corresponding author on reasonable request.
REFERENCES
- 1. Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, et al. Epigenomic alterations define lethal CIMP‐positive ependymomas of infancy. Nature. 2014;506:445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across All CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27:728–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–91. [DOI] [PubMed] [Google Scholar]
- 4. Kristensen LS, Hansen TB, Veno MT, Kjems J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene. 2018;37:555–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rybak‐Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–85. [DOI] [PubMed] [Google Scholar]
- 6. Bachmayr‐Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner‐Hofmann T, et al. Correlation of circular RNA abundance with proliferation—exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun J, Li B, Shu C, Ma Q, Wang J. Functions and clinical significance of circular RNAs in glioma. Mol Cancer 2020;19:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song X, Zhang N, Han P, Moon BS, Lai RK, Wang K, et al. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res 2016;44:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang P, Qiu Z, Jiang Y, Dong L, Yang W, Gu C, et al. Silencing of cZNF292 circular RNA suppresses human glioma tube formation via the Wnt/beta‐catenin signaling pathway. Oncotarget. 2016;7:63449‐63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dahl M, Daugaard I, Andersen MS, Hansen TB, Gronbaek K, Kjems J, et al. Enzyme‐free digital counting of endogenous circular RNA molecules in B‐cell malignancies. Lab Invest. 2018;98:1657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Characterization of circular RNAs in ependymoma by RNA‐sequencing. (A,B) Venn diagrams of unique circRNAs supported by at least two backsplicing junction‐spanning reads (BSJ > 2) in ependymomas (blue circle) and healthy controls (green circle) (A), and high abundance circRNAs (average RPM >0.2) in ependymoma (black circle) and healthy controls (red circle) (B). (C,D) Correlation between average CTL ratios and average RPM values with corresponding linear regression statistics and R 2 values for the high abundance circRNAs in ependymomas (C) and in healthy controls (D). CircRNAs with a CTL ratio >1 are expressed at higher levels than their cognate linear host genes. Simple linear regression test was used to determine R and P values. (E,F) Volcano plots of 1167 high abundance circRNAs showing a high number of differentially expressed circRNAs between ependymoma (n = 10) and control (n = 3) samples (E), and between deceased patients (n = 5) and survivors (n = 4) (F). (G,H) Scatter plots of fold change in CTL ratios against fold change in RPM values for circRNAs with corresponding linear regression statistics and R‐squared values between ependymoma (n = 10) and control (n = 3) samples (G) and between deceased patients and survivors (H). CircRNAs between the dotted blue lines are considered changed independent of their respective host genes. BSJ, backsplicing junction; CTL, circular‐to‐linear ratio; RPM, reads‐per‐million
FIGURE S2 Validation of ependymoma‐related circRNA expression changes using NanoString nCounter. (A) Correlation between log2 fold change values between ependymoma samples and control samples in two independent cohorts using two different methods. (B) Column scatter plot of average circRNA expression in ependymomas (n = 19), pilocytic astrocytomas (n = 5), medulloblastomas (n = 3) and healthy control samples (n = 9). Mann–Whitney U test was used for statistical calculations. (C) PCA plot of ependymoma (n = 19, green dots), pilocytic astrocytomas (n = 5, purple dots), medulloblastomas (n = 3, blue dots) and healthy control (n = 9, red dots) samples. ****P < 0.0001. Fc, fold change; PCA, principal component analysis
TABLE S1 The list of the unique circRNAs samples, supported by at least two sequencing reads (BSJ > 2), are displayed from RNA‐seq analyses of ependymoma (n = 10) and control (n = 3). BSJ, backsplicing junction
TABLE S2 The list of 1167 high abundance circRNAs quantified by RNA‐sequencing of ependymoma (n = 10) and control (n = 3) samples. RPM, read‐per‐million
TABLE S3 The list of the differentially expressed circRNAs between ependymoma samples (n = 10) and healthy control samples (n = 3). One unpaired t test was used to calculate the P values. Correction for multiple testing was performed to calculate adjacent P values using the Holm‐Sidak method. CTL, circular‐to‐linear; RPM, read‐per‐million
TABLE S4 The list of the differentially expressed circRNAs between deceased patients (n = 5) and survivors (n = 4). One unpaired t test was used to calculate the P values. Correction for multiple testing was performed to calculate adjacent P values using the Holm‐Sidak method. CTL, circular‐to‐linear; RPM, read‐per‐million
TABLE S5 The list and the expression levels of the circRNAs selected for the NanoString nCounter analysis. The expression of the circRNAs was obtained from the NanoString nCounter data of ependymoma samples (n = 19), pilocytic astrocytoma (n = 5), medulloblastoma (n = 3) and healthy control (n = 3) samples
TABLE S6 Average expression levels of 66 circRNAs in ependymoma samples (n = 19), pilocytic astrocytoma (n = 5), medulloblastoma (n = 3) and healthy control (n = 3) samples analyzed by the NanoString nCounter. One unpaired t test was used to calculate the P values. Correction for multiple testing was performed to calculate adjacent P values using the Holm‐Sidak method. Control, samples from healthy individuals; CTL, circular‐to‐linear; Epn, ependymoma; Fc, fold change; MB, medulloblastoma; PA, pilocytic astrocytoma; RPM, read‐per‐million
Supplementary Material
Data Availability Statement
Our raw data cannot be submitted to publicly available databases because the patients did not provide consent to share their raw data, which can potentially identify the individuals, but are available from the corresponding author on reasonable request.