

Abstract
The aggressive nature of malignant gliomas and their genetic and clinical heterogeneity present a major challenge in their diagnosis and treatment. Development of targeted therapy brought attention on detecting novel gene fusions, since they represent promising therapeutic targets (eg, TRK inhibitors in NTRK fusion‐positive tumors). Using targeted next‐generation sequencing, we prospectively analyzed 205 primary brain tumors and detected a novel PTPRZ1‐ETV1 fusion transcript in 11 of 191 (5.8%) gliomas, including nine glioblastomas, one anaplastic oligodendroglioma and one pilocytic astrocytoma. PTPRZ1‐ETV1 fusion was confirmed by RT‐PCR followed by Sanger sequencing, and in‐silico analysis predicted a potential driver role. The newly detected fusion consists of the PTPRZ1 promoter in frame with the highly conserved DNA‐binding domain of ETV1 transcription factor. The ETV1 and PTPRZ1 genes are known oncogenes, involved in processes of tumor development. ETV1 is a member of the ETS family of transcription factors, already known oncogenic drivers in Ewing sarcoma, prostate cancer and gastrointestinal stromal tumors, but not in gliomas. Its overexpression contributes to tumor growth and more aggressive tumor behavior. PTPRZ1 is already considered to be a tumor growth promoting oncogene in gliomas. In 8%–16% of gliomas, PTPRZ1 is fused to the MET oncogene, resulting in a PTPRZ1‐MET fusion, which is associated with poorer prognosis but is also a positive predictive biomarker for treatment with kinase inhibitors. In view of the oncogenic role that the two fusion partners, PTPRZ1 and ETV1, exhibit in other malignancies, PTPRZ1‐ETV1 fusion might present a novel potential therapeutic target in gliomas. Although histopathological examination of PTPRZ1‐ETV1 fusion‐positive gliomas did not reveal any specific or unique pathological features, and the follow‐up period was too short to assess prognostic value of the fusion, careful monitoring of patients and their response to therapy might provide additional insights into the prognostic and predictive value of this novel fusion.
Keywords: ETV1, gliomas, novel fusion, PTPRZ1, RNA‐sequencing, oncogenes
Introduction
The formation of fusions between oncogenes is one of the hallmarks of cancer development. With the development and advances in high‐throughput sequencing techniques, more and more gene fusions and other genomic rearrangements are being detected, helping us to increase our knowledge of tumor biology and understand the mechanisms and functions of present aberrations. Malignant gliomas, especially glioblastomas, are aggressive primary brain tumors and one of the most clinically and genetically heterogeneous group of neoplasms, presenting major challenge in their treatment. In the majority of cases, gliomas recur due to diffuse growth and incomplete tumor resection, and patient survival is poor 14, 31.
Gene fusions are frequent genomic abnormalities in gliomas, especially in glioblastomas; previous studies have shown that fusions in gliomas most frequently occur at two major specific genomic hotspots; chromosomes 7p and 12q, and chromosomes 1, 4, 6 and 19 41, 51. Although most fusions appear to be passenger genomic changes, they are also promising treatment targets.
Chromosome arm 7p harbors one of the most characterized and common fusions in high‐grade gliomas (detected in a third of glioblastoma cases)—the EGFRvIII mutation (deletion of exons 2‐7 and fusion of exon 1 with exon 8), which results in truncated, constitutively active EGF receptors and dramatically enhances the malignancy of the glioblastoma cells 30, 48. Fusions of the EGFR gene with various fusion partners, such as SEPT14 (4% of glioblastomas) or PSHS (2.2% of glioblastomas), are the most frequent recurrent fusions in glioblastomas 2, 16.
Other potentially targetable fusions in malignant gliomas are the FGFR‐TACC fusions (FGFR1 or FGFR3, and TACC1 or TACC3, respectively) of two oncogenes at a close chromosome location. FGFR3‐TACC3 fusion protein promotes tumorigenesis and enhances tumor progression through different signaling pathways (MAPK, ERK, PI3K and STAT3), and is more frequent in glioblastomas (in up to 8%) 29, 44.
Fusions of NTRK genes, encoding the tropomyosin receptor kinase (TRK) receptor family, have been reported to occur in 1.2%–1.7% of adult malignant glioma, but are more frequent (around 40%) in high‐grade gliomas in the cerebral hemispheres of infants less than 3 years of age 50, 51. NTRKs play an important role in nervous system development, regulating cell proliferation, differentiation, apoptosis and survival of neurons in both central and peripheral nervous systems. NTRK has several different partner genes, including NFASC and BCAN (both expressed in neuronal tissue), EML4, TPM3 and also CHTOP and ARHGEF2 26, 53. Rearrangements of NTRKs play an important oncogenic role in many types of tumors, including gliomas, non‐small cell lung cancer, colon cancer and papillary thyroid cancer 47, and several drugs targeting cancers harboring the NTRK fusion are under clinical trials 51.
Several other gene fusions have been detected, including FIG‐ROS1, as one of the first fusions detected in glioblastoma cell lines, and also fusions involving non‐coding genes such as RP11‐745C15.2‐LANCL2 10, 41. Last in the line of identified fusions in gliomas is the PTPRZ1‐MET fusion, resulting in the constitutive activation of MET. The fusion induces expression and phosphorylation of the MET oncoprotein and is associated with a poor prognosis. The PTPRZ1‐MET fusion is found in around 10% of pediatric and up to 16% of adult high‐grade gliomas (anaplastic astrocytoma and secondary glioblastoma) 6, 11, 20, 52. In glioblastomas, activation of MET signaling has been proposed as one of the mechanisms of resistance to EGFR‐inhibitors 4.
There are several promising ongoing preliminary clinical trials of molecularly targeted treatment for patients with glioma, but there is still a substantial gap and the need for better understanding the mechanisms of gliomas' development. Moreover, there is an active search to identify additional druggable fusions across different glioma subtypes for more personalized treatment 51. Herein, we report the presence of a novel fusion detected in gliomas—the PTPRZ1‐ETV1 fusion.
Material and Methods
Patients and samples
We prospectively analyzed tumor tissue samples from 205 patients with diagnosed primary brain tumor who had undergone tumor resection from March 2017 until March 2019. All tumors were classified according to the WHO 2016 Classification of Tumours of the Central Nervous System 31. The cohort of analyzed tumors consisted of 191 gliomas (8 pilocytic astrocytomas, 11 diffuse astrocytomas, 18 anaplastic astrocytomas, 8 anaplastic oligodendrogliomas, 1 anaplastic oligoastrocytoma, 143 glioblastomas, 2 diffuse midline gliomas—H3 K27 mutant) and 14 other primary brain tumors (five medulloblastomas, two gangliogliomas, one ganglioneuroblastoma, one atypical teratoid/rhabdoid tumor, one choroid plexus papilloma, one neuroblastoma, one craniopharyngioma, one pineal parenchymal tumor of intermediate differentiation and one embryonal tumor, NOS).
DNA and RNA isolation
Total DNA and RNA were isolated from formalin‐fixed, paraffin embedded (FFPE) tumor tissue samples. Most samples were enriched in the region of tumor tissue using a 0.6‐mm needle (punch) (Manual Tissue Arrayer MTA, Beecher, Estigen). Isolation of DNA and RNA was carried out on the Promega automated system using a Maxwell RSC FFPE Plus DNA Purification kit and Maxwell RSC RNA FFPE kit (Promega), or the manual magnetic beads isolation approach with a MagMAX DNA/RNA Ultra FFPE kit (Thermo Fisher Scientific). The quality and concentration of isolated DNAs and RNAs were assessed both spectrophotometrically on NanoDrop‐1000 and fluorometrically on Qubit 3.0 (both from Thermo Fisher Scientific).
NGS analysis
We performed targeted, amplicon‐based DNA and RNA‐sequencing analysis using the Oncomine Focus Assay test (Thermo Fisher Scientific), which focuses on detection of hotspots and other single nucleotide variants, small indels, copy number variations, and gene fusions across 52 genes relevant to solid tumors and with clinical significance. All steps of library preparation were performed according to the manufacturer's instructions. Briefly, we used 30 ng of total RNA for the reverse transcription (RT) reaction (SuperScript VILO cDNA Synthesis kit, Thermo Fisher Scientific) and further cDNA amplification. Thermal cycling was performed according to the manufacturer's recommendations and consisted of 30 cycles of amplification for the cDNA reactions (RNA libraries). For DNA library preparation, we used 30 ng of DNA and 20 cycles for target amplification. The prepared DNA and RNA libraries were quantified using an Ion Library Quantitation kit (Thermo Fisher Scientific). After quantification, libraries were mixed proportionally in 33‐pM concentrations. Preparation of template ion sphere particles, enrichment PCR (emulsion PCR) and chip loading was carried out on the automated Ion Chef system, sequencing analysis and further raw data processing of DNA/RNA libraries were performed on the Ion S5 system (Thermo Fisher Scientific). Output files were uploaded onto Ion Reporter software (Thermo Fisher Scientific) for variant annotation and bioinformatical analysis.
Validation of fusion by RT‐PCR and Sanger sequencing
We used 100 ng of total RNA for a 10‐µL RT reaction (SuperScript VILO cDNA Synthesis kit, Thermo Fisher Scientific) following the manufacturer's protocol. We used an RNA‐seq‐negative FFPE tumor sample as PTPRZ1‐ETV1 fusion negative control and a non‐template control for RT‐PCR and Sanger sequencing validation control.
For PCR amplification, we added cDNA to 10% of the reaction's volume and one of the two primer pairs (location of primer annealing in parenthesis):
pair 1 (88 bp): PTPRZ1‐ETV1 forward 5′‐GTGTTTGCCGCCTGGTG‐3′ (annealing on the junction) and ETV1 reverse 5′‐AAAGGGCTGTTCTTGACTG‐3′ (exon 5);
pair 2 (121 bp): PTPRZ1 forward primer 5′‐CATTCAGCTCCTCTGTGTTT‐3′ (exon 1) and ETV1 reverse primer 5′‐AAAGGGCTGTTCTTGACTG‐3′ (exon 5). We also performed a control PCR for verifying the success of the RT reaction, by amplifying the GAPDH gene.
PCR products were then purified of all excess dNTPs and primers using the ExoSAP‐IT enzymatic approach (Applied Biosystems) according to the manufacturer′s protocol. We used 2 µL of purified PCR product, 1 µL of designated primer (5‐μM concentration) and a BigDye Terminator Cycle Sequencing kit (v1.1, Thermo Fisher Scientific) for the sequencing reaction. The ethanol precipitation approach was used for purification of the sequencing reactions. Sanger sequencing analysis was carried out on the SeqStudio system (Thermo Fisher Scientific), and sequences were analyzed using SeqScanner software (Thermo Fisher Scientific) and the Ensembl genome browser. The obtained sequences were uploaded onto the BLASTN tool for homology search.
Prediction of the oncogenic potential of gene fusions
In order to predict the oncogenic potential of the novel gene fusion, we performed in‐silico analysis using a Bayesian classifier that provides accurate classification of driver and passenger fusion genes, as described by Shugay and colleagues and Panigrahi and colleagues 36, 43. Briefly, the Oncofuse bioinformatical pipeline predicts the oncogenic potential of fusion genes by calculating the Bayesian probability that a fusion sequence behaves as an oncogenic driver (P‐values <0.5 are classified as “driver”), based on features present in known oncogenic fusions.
Results
A novel PTPRZ1‐ETV1 fusion detected by targeted RNA‐sequencing
Targeted RNA‐sequencing analysis of 205 primary brain tumors revealed the presence of a novel PTPRZ1‐ETV1 fusion in 11 of 191 gliomas (5.8%) (Figure 1), of which nine were glioblastomas, one anaplastic oligodendroglioma and one pilocytic astrocytoma (Table 1). Since the fusion was discovered only in gliomas, cases of other primary brain tumor types (fourteen in total) were not considered in further analyses.
Figure 1.
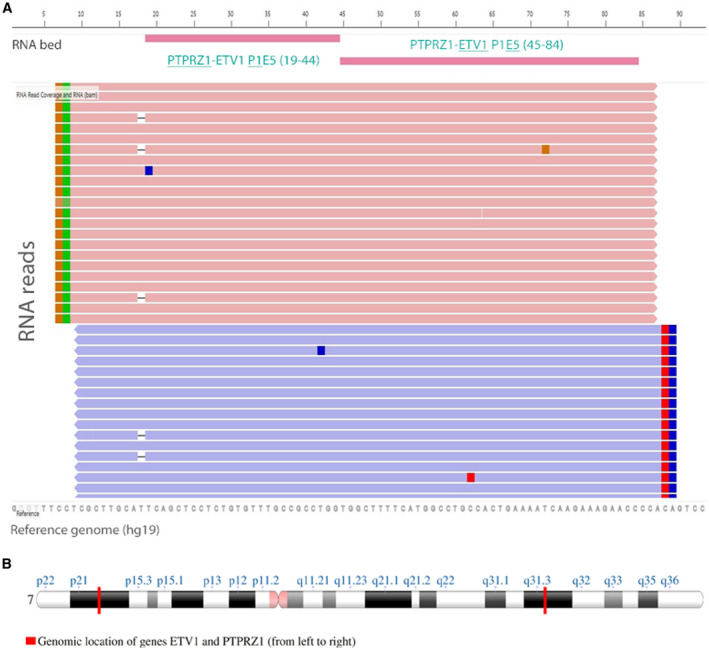
Novel fusion gene PTPRZ1‐ETV1 detected by amplicon‐based RNA‐sequencing analysis. A. IGV‐Light visualization of sequence read pileup for the fusion detected. B. Genomic location of genes ETV1 and PTPRZ1 on chromosome 7.
Table 1.
Patient demographics and histopathological characteristics of gliomas harboring the PTPRZ1‐ETV1 fusion.
| Case number | Diagnosis | Tumor location | Gender | Age at diagnosis (years) | Primary or recurrent tumor | Multifocal tumor | Other neoplasms |
|---|---|---|---|---|---|---|---|
| 1 | Pilocytic astrocytoma | Cerebellum | Male | 7 | Primary | Yes | Neurofibromatosis type 1 |
| 2 | Glioblastoma | Left frontal lobe | Male | 61 | Primary | Yes | None |
| 3 | Glioblastoma | Left temporal lobe | Male | 68 | Primary | No | None |
| 4 | Anaplastic oligodendroglioma | Left frontotemporal lobe | Male | 43 | Primary | No | Testicular teratocarcinoma in 2000 |
| 5 | Glioblastoma | Left cerebellum | Male | 41 | Recurrent (radiotherapy and Temodal) | No | None |
| 6 | Glioblastoma | Right temporal lobe | Female | 69 | Primary | No | None |
| 7 | Glioblastoma | Right parietal lobe | Male | 50 | Primary | No | None |
| 8 | Glioblastoma | Left frontal lobe | Female | 84 | Primary | No | None |
| 9 | Glioblastoma | Left frontal lobe | Male | 69 | Primary | Yes | Basal cell carcinoma on the cheek and cutaneous squamous cell carcinomas on the neck and forehead in 2017, treated by complete surgical excisions |
| 10 | Glioblastoma | Frontal lobe | Male | 48 | Primary | Yes | None |
| 11 | Glioblastoma | Bilateral in parietal lobes | Female | 58 | Primary | Yes | None |
RNA‐sequencing software detected two distinct in‐frame fusion transcripts, with the lowest read count of 258 reads (the default threshold of reads for intergenic fusion is ≥20) (Table 2). The two transcripts involved two breaking points within the ETV1 coding sequence (exon 4 or 5, respectively). In contrast, the breakpoints in the PTPRZ1 coding sequence were located at the same junction (at the end of exon 1). All other variants detected on DNA and RNA levels are listed in Table 2.
Table 2.
Molecular data of gliomas harboring the PTPRZ1‐ETV1 fusion, detected by immunohistochemistry and amplicon‐based NGS analysis. NA = not available; WT = wild type; MT = mutated; IHC = immunohistochemistry.
| Case number | PTPRZ1‐ETV1 fusion detected by NGS RNA‐seq | Read count (threshold >20) | RT‐PCR validation of PTPRZ1‐ETV1 fusion | IDH mutation (NGS) | 1p/19 co‐deletion | MGMT promoter methylation | Other variants detected with Oncomine Focus assay | IHC TP53 | IHC ATRX |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Exon 1–exon 5 in‐frame | 1795 | Exon 1–exon 5 | WT | NA | Not methylated | None | NA | WT |
| 2 | Exon 1–exon 5 in‐frame | 389 | Exon 1–exon 5 | WT | NA | Methylated |
|
WT | WT |
| 3 | Exon 1–exon 5 in‐frame | 275 | Exon 1–exon 5 | WT | NA | Not methylated | None | Questionable | WT |
| 4 | Exon 1–exon 5 in‐frame | 258 | Exon 1–exon 5 | R132H | Very likely | Methylated | None | WT | WT |
| 5 | Exon 1–exon 5 in‐frame | 403 | Exon 1–exon 5 | R132H | NA | Methylated |
|
MT | MT |
| Exon 1–exon 4 | |||||||||
| 6 | Exon 1–exon 5 in‐frame | 266 | Exon 1–exon 5 | WT | NA | Methylated |
|
WT | WT |
| 7 | Exon 1–exon 5 in‐frame | 457 | Exon 1–exon 5 | WT | NA | Not methylated |
|
WT | WT |
| 8 | Exon 1–exon 4 in‐frame | 480 | Exon 1–exon 5 | WT | NA | Methylated |
|
MT | WT |
| Exon 1–exon 4 | |||||||||
| 9 | Exon 1–exon 4 in‐frame | 462 | Exon 1–exon 5 | WT | NA | Methylated |
|
WT | WT |
| 10 | Exon 1–exon 4 in‐frame | 389 | No PCR amplification | WT | NA | Methylated |
|
WT | WT |
| 11 | Exon 1–exon 4 in‐frame | 304 | No PCR amplification | WT | NA | Not methylated |
|
WT | WT |
Successful validation of the PTPRZ1‐ETV1 fusion in gliomas
To confirm the presence of the detected fusion, we designed two primer pairs. The forward primer of one pair was designed as a fusion‐specific primer, flanking exon 1 of PTPRZ1 and exon 5 of ETV1 (Figure 2A). The other pair was designed in such way that one of the primers flanked exon 1 of the PTPRZ1 gene and the other exon 5 of the ETV1 gene (Figure 2A), which also amplifies the exon1‐exon4 transcript of the PTPRZ1‐ETV1 fusion.
Figure 2.
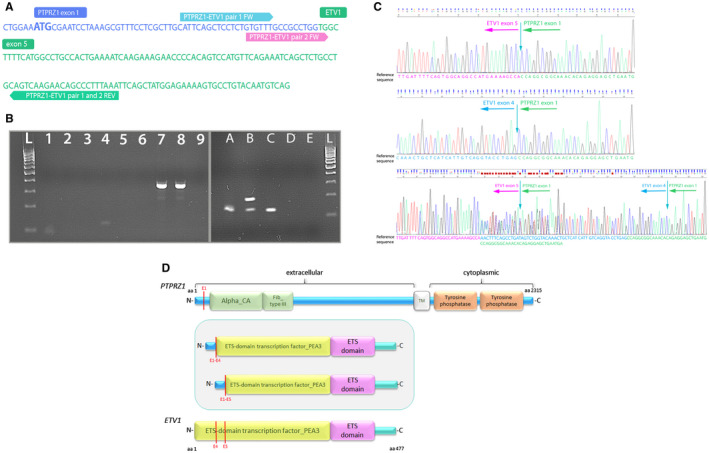
RT‐PCR validation of novel PTPRZ1‐ETV1 fusion. A. Nucleotide sequence of fused PTPRZ1 exon 1 and ETV1 exon 5 with primers designed for RT‐PCR and direct sequencing. B. RT‐PCR for validation of PTPRZ1‐ETV1 fusion present (lane L—DNA 100bp ladder; lanes 1‐3—PCR reactions with primer pair 1 (fusion‐positive sample (88 bp), fusion‐negative sample and no template control (NTC)); lanes 4–6—PCR reactions with primer pair 2 (fusion‐positive sample (121 bp), fusion‐negative sample and no template control (NTC); lanes A–D—PCR validation with primer pair 2, lane E—NTC). Sample B shows two amplified fragments and Sanger sequencing showed it harbors both fusion transcripts, P1E4 (175 bp long fragment) and P1E5 (121 bp long fragment). Sample D showed no amplified product. C. Chromatogram of the RT‐PCR amplified fusion PTPRZ1(exon1)‐ETV1(exon5), PTPRZ1(exon1)‐ETV1(exon4) and both fusion transcripts present (for Sanger sequencing, we used the reverse primer of pair 2). D. Schematic depiction of predicted fusion protein (in the frame) involving PTPRZ1 and ETV1 genes. Red lines represent exons of the PTPRZ1 and ETV1 gene, and also the point of break and fusion. aa = amino acid; Alpha_CA = alpha carbonic anhydrase; Fib_Type III = fibronectin type‐III; TM = transmembrane domain.
At the transcription level, we successfully validated both fusion variants using the RT‐PCR approach followed by Sanger sequencing. The presence of PCR products was visualized on 2% agarose gel electrophoresis, which showed an individual band of amplified products for the primer pair used, approximately 90 bp (primer pair 1) and 120 bp long (primer pair 2) (Figure 2B, left image). Surprisingly, using primer pair 2, we also observed two fragments (sample B, right image), which corresponded to the two fusion transcripts present, PTPRZ1(exon 1)‐ETV1(exon4) and PTPRZ1(exon1)‐ETV1(exon5). Sequence analysis and further homology search (BLASTN) in the genome database confirmed that the PCR product consisted of the PTPRZ1 gene fused to the ETV1 gene (Figure 2D). The presence of fusion was confirmed in 9 of 11 tumor samples; we were not able to obtain a confirmatory PCR product in two samples, which may be due to a lower frequency of fusion present or a poorer quality of the RNA sample.
Demographic, histopathological and molecular features of gliomas harboring the PTPRZ1‐ETV1 fusion
The average age of patients with a glioma harboring the PTPRZ1‐ETV1 fusion at the time of diagnosis was 55 years (median 58.7 years; range 7–84 years), and female to male ratio was 1:2.7 (Table 1). Histopathological assessment of gliomas with the PTPRZ1‐ETV1 fusion did not show any specific or unique pathological features, other than those characteristic of a particular glial tumor. With the exception of Case 1, all were high‐grade gliomas. Case 1 was a 7‐year‐old patient, diagnosed with pilocytic astrocytoma, which showed no variants in the BRAF gene (eg, V600E mutation or KIAA1549‐BRAF fusion) or in other genes of the MAPK signaling pathway, often detected in pilocytic astrocytoma. However, the patient had been previously diagnosed with neurofibromatosis type 1 (NF1), which is associated with an increased risk of the occurrence of gliomas, pilocytic astrocytomas being the most frequent type, which occurs in about 15% of these patients. Pilocytic astrocytoma in patients with neurofibromatosis tend not to harbor BRAF mutations 18, 32.
There was no association between the presence of PTPRZ1‐ETV1 fusion and glioma biomarkers, such as IDH1/2 mutation status or methylation of MGMT promoter (Table 2). The IDH1/2 mutation was present in two of 11 tumors, one was an anaplastic oligodendroglioma and one glioblastoma, both carrying an R132H mutation in the IDH1 gene. The MGMT promoter was methylated in seven of 11 tumors.
Sanger sequencing analysis showed the presence of both PTPRZ1‐ETV1 fusion transcripts—PTPRZ1(exon1)‐ETV1(exon5) and PTPRZ1(exon1)‐ETV1(exon4)—in two tumors, although RNA‐sequencing detected only one of the two transcripts (Table 2, cases 5 and 8). Patocs and colleagues observed multiple splice variants of EWSR1‐ETS fusion transcripts co‐existing within the same tumor. The most probable explanation for the occurrence of multiple variants of one fusion event is alternative mRNA splicing. Nevertheless, there is also a chance of occurrence of two different chromosomal rearrangements in the same patient 39. Interestingly, both tumors also harbored the PTPRZ1‐MET fusion and mutated TP53. EGFRvIII and EGFR amplification (detected by NGS analysis) were the most common additional genetic alterations, present in five glioblastomas. In one glioblastoma, RNA‐sequencing detected the exon1‐exon4 fusion transcript, but we detected only the exon1‐exon5 transcript with RT‐PCR validation (Table 2, case 9). Such discrepancies could be caused by a mispriming event or amplification bias.
In‐silico prediction of PTPRZ1‐ETV1 oncogenic potential
In‐silico prediction analysis, using the Oncofuse classifier and the Bayesian probability of oncogenic potential for specific fusion transcripts, revealed a high probability (P‐value = 0.0008) that the novel PTPRZ1‐ETV1 fusion acts as a driver in the oncogenic process with a completely retained 3′‐Ets domain (Winged helix‐turn‐helix DNA‐binding domain). The complete results of Oncofuse classification are listed in Supplemental Table S1.
Discussion
Over the past decade, molecular profiling of numerous cancer types has been incorporated into cancer diagnostics, including gliomas, allowing for a more personalized, targeted therapy approach 9. To the best of our knowledge, this is the first report of a PTPRZ1‐ETV1 fusion in general, not just in glial tumors. Furthermore, the newly described fusion is predicted to have an oncogenic role, retaining a highly conserved ETS domain, as established by in‐silico prediction analysis. Both PTPRZ1 and ETV1 genes are known fusion partners in other cancer types, for example, TMPRSS2‐ETV1 in prostate cancer and EWSR1‐ETV1 in Ewing sarcoma and, as already mentioned, PTPRZ1‐MET in gliomas 6.
The PTPRZ1 gene is located on chromosome 7q31.32 and encodes member Z of the receptor protein tyrosine phosphatase family. It is preferentially expressed in the central nervous system and is one of the most abundant tyrosine phosphatases expressed in oligodendrocyte precursor cells 28. As such, it may be involved in the regulation of specific developmental processes in the central nervous system. Kubojama and colleagues indicated that PTPRZ1, as part of the phosphatase family, acts as a negative regulator of oligodendrocyte differentiation and myelination of embryonic mice white matter 27, 28. Protein tyrosine phosphatases are the enzymatic counterparts of protein tyrosine kinases and are important regulators of developmental and disease‐related signaling pathways. Multiple members are directly linked to malformation syndromes and tumorigenesis 35, 46. In glioblastoma, PTPRZ1 receptors were found preferentially expressed in glioma stem cells, predicting a poor prognosis 33, 42. They bound pleiotropin (PTN), an inhibitory ligand with a protective role, which has been suggested to promote myelination by inhibiting PTPRZ1 28. PTN is a neurotrophic factor, secreted by infiltrating tumor‐associated macrophages, which have multiple functions in either inhibiting or promoting tumor progression, and are correlated with increased intra‐tumor heterogeneity 42. In addition, Bao and Wang showed that PTPRZ1‐MET signaling contributes to malignant progression in gliomas by recruiting tumor‐associated macrophages 7. A study by Shi and colleagues showed that disrupting PTPRZ1 expression abrogated glioma stem cell maintenance and their tumorigenic potential. Blocking PTN‐PTPRZ1 signaling in animal models by shRNA or anti‐PTPRZ1 antibody potently suppressed glioblastoma tumor growth and prolonged animal survival 42. The selective inhibition of PTPRZ1 is a promising approach for glioma therapy 17, 38.
The ETV1 gene is also located on chromosome 7 (7p21.2) and encodes a member of the ETS (E twenty‐six) family of transcription factors, which regulate many target genes that modulate biological processes, such as cell growth, angiogenesis, migration, proliferation and differentiation 25. All ETS proteins contain an ETS DNA‐binding domain in the C‐terminal region that binds to DNA sequences. At the N‐terminal region, the ETV1 protein contains a conserved short acidic transactivation domain, which is a feature of the small PEA3 subfamily of ETS proteins. The PEA3 subfamily consists of three ETS‐translocation variants (ETV): ETV1 (Ets‐related protein or ER81), ETV4 (polyoma enhancer activator 3 or PEA3) and ETV5 (Ets‐related molecule or ERM) 25, 34. PEA3 transcription factors have a role in morphogenesis and neuronal differentiation 1, 13, 24, 37. The involvement of ETV1 in cancer was initially observed in Ewing sarcoma, in which a chromosomal translocation involving the Ewing sarcoma gene (EWSR1) and ETV1 gene results in the EWS‐ETV1 fusion protein, which exerts oncogenic properties 23, 34. ETV1 is involved in chromosomal translocations underlying prostate cancer development, having numerous fusion gene partners, including TMPRSS2 (which is more often fused to the ERG gene of the ETS family in prostate cancer), SLC45A3, HERV‐K_22q11.23, C15orf21 and HNRPA2B1 45. ETV1 drives the androgen receptor transcriptional response associated with aggressive prostate cancer 5. Amplification of ETV1 is found in approximately 40% of melanomas, and ETV1 has been shown to promote melanoma cell growth 21. ETV1 overexpression has been detected in early stages of breast cancer development, and was found to be significantly associated with amplification of HER2/neu 15, 49. Amplification of ETV1 has also been detected in gastrointestinal stromal tumors 12, 22. Heeg and colleagues found that ETV1 is involved in epithelial‐mesenchymal transition during pancreatic development in mice and that tumor cells with elevated ETV1 expression have increased invasive capacity, contributing to metastatic progression of pancreatic cancer 19.
As observed in cases of childhood high‐grade gliomas with PTPRZ1‐MET fusion, expression of full‐length MET is driven from the highly active PTPRZ1 promoter, leading to MET overexpression 8. In view of the same PTPRZ1 exon‐1 breakpoint in both PTPRZ1‐MET and PTPRZ1‐ETV1 fusions, the activated PTPRZ1 promoter may also affect the expression of ETV1. As elevated expression of ETV1 has been shown to contribute to increased invasiveness of tumor cells in other cancer types, gliomas harboring the PTPRZ1‐ETV1 fusion could potentially exhibit a more aggressive phenotype. In the PTPRZ1‐ETV1 fusion, the ETS DNA‐binding domain of ETV1 gene is retained, which would suggest that the activation of ETV1 affects the transcription of its target genes. Regarding the role of ETV1 as a transcription factor, even a small change in its expression can have a significant biological impact. However, the biological function of the PTPRZ1‐ETV1 fusion and its contribution to tumor development remains undetermined. Since gliomas with PTPRZ1‐ETV1 fusion in our series represent recent cases with a very short follow‐up, the prognostic significance of the fusion remains unknown.
Although ETV1 as a transcription factor lacks enzymatic activity, its increasingly recognized function in oncogenic signaling pathways of various tumor types makes it a possible target for future therapeutic approaches 40. Akgül and colleagues showed that glial tumors are polyclonal tumors, creating a dynamic environment consisting of diverse tumor elements and treatment responses. Designing targeted therapies based on a range of tumor‐specific molecular profiles (stratified and precise therapy) may be a more effective strategy for diagnosing and assigning a prognostic subgroup, and minimizing treatment resistance, recurrence and metastasis 3.
Conclusions
Fusion PTPRZ1‐ETV1 presents in about 6% of gliomas and represents a novel potential therapeutic target due to the fact that both genes exhibit oncogenic effects in the development and progression of various tumor types. The presence of the PTPRZ1‐ETV1 fusion does not seem to be associated with some unique morphologic features of gliomas. A longer follow‐up and careful monitoring of patients' response to therapy might provide additional insights into the prognostic and predictive roles of this novel fusion.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Table S1. Oncofuse in‐silico prediction analysis of newly described PTPRZ1ETV1 fusion.
Acknowledgment
The authors acknowledge financial support from the Slovenian Research Agency (research core funding no. P3‐0054).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Abe H, Okazawa M, Nakanishi S (2011) The Etv1/Er81 transcription factor orchestrates activity‐dependent gene regulation in the terminal maturation program of cerebellar granule cells. Proc Natl Acad Sci 108:12497–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Addeo R, Zappavigna S, Parlato C, Caraglia M (2014) Erlotinib: early clinical development in brain cancer. Expert Opin Investig Drugs 23:1027–1037. [DOI] [PubMed] [Google Scholar]
- 3. Akgul S, Patch AM, D'Souza RCJ, Mukhopadhyay P, Nones K, Kempe S et al (2019) Intratumoural heterogeneity underlies distinct therapy responses and treatment resistance in glioblastoma. Cancers 11:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. An Z, Aksoy O, Zheng T, Fan Q‐W, Weiss WA (2018) Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 37:1561–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y et al (2013) ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev 27:683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K, Ye WL et al (2014) RNA‐seq of 272 gliomas revealed a novel, recurrent PTPRZ1‐MET fusion transcript in secondary glioblastomas. Genome Res 24:1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bao Z, Wang Z (2018) PTPRZ1‐MET signaling promotes glioma progression through stimulation the transformation from M1 to M2 macrophage. Neuro‐Oncology 20:vi199. [Google Scholar]
- 8. Bender S, Gronych J, Warnatz HJ, Hutter B, Gröbner S, Ryzhova M et al (2016) Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 22:1314–1320. [DOI] [PubMed] [Google Scholar]
- 9. Bonner ER, Bornhorst M, Packer RJ, Nazarian J (2018) Liquid biopsy for pediatric central nervous system tumors. NPJ Precis Oncol 2:29–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Charest A, Lane K, McMahon K, Park J, Preisinger E, Conroy H et al (2003) Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosom Cancer 37:58–71. [DOI] [PubMed] [Google Scholar]
- 11. Chen HM, Yu K, Tang XY, Bao ZS, Jiang T, Fan XL et al (2015) Enhanced expression and phosphorylation of the MET oncoprotein by glioma‐specific PTPRZ1‐MET fusions. FEBS Lett 589:1437–1443. [DOI] [PubMed] [Google Scholar]
- 12. Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T et al (2010) ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467:849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chotteau‐Lelievre A, Montesano R, Soriano J, Soulie P, Desbiens X, de Launoit Y (2003) PEA3 transcription factors are expressed in tissues undergoing branching morphogenesis and promote formation of duct‐like structures by mammary epithelial cells in vitro. Dev Biol 259:241–257. [DOI] [PubMed] [Google Scholar]
- 14. Cohen AL, Colman H (2015) Glioma biology and molecular markers. Cancer Treat Res 163:15–30. [DOI] [PubMed] [Google Scholar]
- 15. Eid W, Abdel‐Rehim W. Genome‐wide analysis of ETV1 targets: Insights into the role of ETV1 in tumor progression. J Cell Biochem 2019;120:8983–8991. [DOI] [PubMed] [Google Scholar]
- 16. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F et al (2013) The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 45:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujikawa A, Nagahira A, Sugawara H, Ishii K, Imajo S, Matsumoto M et al (2016) Small‐molecule inhibition of PTPRZ reduces tumor growth in a rat model of glioblastoma. Sci Rep 6:20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gutmann DH, McLellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS et al (2013) Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1‐associated pilocytic astrocytoma. Genome Res 23:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heeg S, Das KK, Reichert M, Bakir B, Takano S, Caspers J et al (2016) ETS‐transcription factor ETV1 regulates stromal expansion and metastasis in pancreatic cancer. Gastroenterology 151:540–553.e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J et al (2018) Mutational landscape of secondary glioblastoma guides MET‐targeted trial in brain tumor. Cell 175:1665–1678.e1618. [DOI] [PubMed] [Google Scholar]
- 21. Jané‐Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, Lin WM et al (2010) An oncogenic role for ETV1 in melanoma. Can Res 70:2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jang BG, Lee HE, Kim WH (2015) ETV1 mRNA is specifically expressed in gastrointestinal stromal tumors. Virchows Arch 467:393–403. [DOI] [PubMed] [Google Scholar]
- 23. Janknecht R (2005) EWS‐ETS oncoproteins: the linchpins of Ewing tumors. Gene 363:1–14. [DOI] [PubMed] [Google Scholar]
- 24. Kandemir B, Caglayan B, Hausott B, Erdogan B, Dag U, Demir O et al (2014) Pea3 transcription factor promotes neurite outgrowth. Front Mol Neurosci 7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kar A, Gutierrez‐Hartmann A (2013) Molecular mechanisms of ETS transcription factor‐mediated tumorigenesis. Crit Rev Biochem Mol Biol 48:522–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J, Lee Y, Cho HJ, Lee YE, An J, Cho GH et al (2014) NTRK1 fusion in glioblastoma multiforme. PLoS ONE 9:e91940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuboyama K, Fujikawa A, Masumura M, Suzuki R, Matsumoto M, Noda M (2012) Protein tyrosine phosphatase receptor type z negatively regulates oligodendrocyte differentiation and myelination. PLoS ONE 7:e48797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuboyama K, Fujikawa A, Suzuki R, Noda M (2015) Inactivation of protein tyrosine phosphatase receptor type Z by pleiotrophin promotes remyelination through activation of differentiation of oligodendrocyte precursor cells. J Neurosci 35:12162–12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lasorella A, Sanson M, Iavarone A (2017) FGFR‐TACC gene fusions in human glioma. Neuro Oncol 19:475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Louis DN (2006) Molecular pathology of malignant gliomas. Annu Rev Pathol 1:97–117. [DOI] [PubMed] [Google Scholar]
- 31. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. [DOI] [PubMed] [Google Scholar]
- 32. Marko NF, Weil RJ (2012) The molecular biology of WHO grade I astrocytomas. Neuro‐Oncology 14:1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Navis AC, van den Eijnden M, Schepens JTG, Hooft van Huijsduijnen R, Wesseling P, Hendriks WJAJ (2010) Protein tyrosine phosphatases in glioma biology. Acta Neuropathol 119:157–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oh S, Shin S, Janknecht R (2012) ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta 1826:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ostman A, Hellberg C, Bohmer FD (2006) Protein‐tyrosine phosphatases and cancer. Nat Rev Cancer 6:307–320. [DOI] [PubMed] [Google Scholar]
- 36. Panigrahi P, Jere A, Anamika K (2018) FusionHub: A unified web platform for annotation and visualization of gene fusion events in human cancer. PLoS ONE 13:e0196588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paratore C, Brugnoli G, Lee HY, Suter U, Sommer L (2002) The role of the Ets domain transcription factor Erm in modulating differentiation of neural crest stem cells. Dev Biol 250:168–180. [DOI] [PubMed] [Google Scholar]
- 38. Pastor M, Fernandez‐Calle R, Di Geronimo B, Vicente‐Rodriguez M, Zapico JM, Gramage E et al (2018) Development of inhibitors of receptor protein tyrosine phosphatase beta/zeta (PTPRZ1) as candidates for CNS disorders. Eur J Med Chem 144:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patocs B, Nemeth K, Garami M, Arato G, Kovalszky I, Szendroi M et al (2013) Multiple splice variants of EWSR1‐ETS fusion transcripts co‐existing in the Ewing sarcoma family of tumors. Cell Oncol 36:191–200. [DOI] [PubMed] [Google Scholar]
- 40. Pop MS, Stransky N, Garvie CW, Theurillat J‐P, Hartman EC, Lewis TA et al (2014) A small molecule that binds and inhibits the ETV1 transcription factor oncoprotein. Mol Cancer Ther 13:1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shah N, Lankerovich M, Lee H, Yoon JG, Schroeder B, Foltz G (2013) Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genom 14:818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS et al (2017) Tumour‐associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun 8:15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shugay M, Ortiz de Mendibil I, Vizmanos JL, Novo FJ (2013) Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 29:2539–2546. [DOI] [PubMed] [Google Scholar]
- 44. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A et al (2012) Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS et al (2007) Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448:595–599. [DOI] [PubMed] [Google Scholar]
- 46. Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7:833–846. [DOI] [PubMed] [Google Scholar]
- 47. Vaishnavi A, Le AT, Doebele RC (2015) TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 5:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ (2010) Exciting new advances in neuro‐oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60:166–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Y, Wang L, Chen Y, Li L, Yang X, Li B et al (2011) ER81 Expression in Breast Cancers and Hyperplasia. Patholog Res Int 2011:980513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu T, Wang H, Huang X, Li W, Huang Q, Yan Y et al (2018) Gene Fusion in malignant glioma: an emerging target for next‐generation personalized treatment. Transl Oncol 11:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zeng AL, Yan W, Liu YW, Wang Z, Hu Q, Nie E et al (2017) Tumour exosomes from cells harbouring PTPRZ1‐MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 36:5369–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD et al (2014) Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med 20:1479–1484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oncofuse in‐silico prediction analysis of newly described PTPRZ1ETV1 fusion.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.