

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder affecting both the upper and lower motor neurons. Although ALS typically leads to death within 3 to 5 years after initial symptom onset, approximately 10% of patients with ALS live more than 10 years after symptom onset. We set out to determine similarities and differences in clinical presentation and neuropathology in persons with ALS with long vs. those with standard duration. Participants were United States military Veterans with a pathologically confirmed diagnosis of ALS (n = 179), dichotomized into standard duration (<10 years) and long‐duration (≥10 years). The ALS Functional Rating Scale‐Revised (ALSFRS‐R) was administered at study entry and semi‐annually thereafter until death. Microglial density was determined in a subset of participants. long‐duration ALS occurred in 76 participants (42%) with a mean disease duration of 16.3 years (min/max = 10.1/42.2). Participants with long‐duration ALS were younger at disease onset (P = 0.002), had a slower initial ALS symptom progression on the ALSFRS‐R (P < 0.001) and took longer to diagnose (P < 0.002) than standard duration ALS. Pathologically, long‐duration ALS was associated with less frequent TDP‐43 pathology (P < 0.001). Upper motor neuron degeneration was similar; however, long‐duration ALS participants had less severe lower motor neuron degeneration at death (P < 0.001). In addition, the density of microglia was decreased in the corticospinal tract (P = 0.017) and spinal cord anterior horn (P = 0.009) in long‐duration ALS. Notably, many neuropathological markers of ALS were similar between the standard and long‐duration groups and there was no difference in the frequency of known ALS genetic mutations. These findings suggest that the lower motor neuron system is relatively spared in long‐duration ALS and that pathological progression is likely slowed by as yet unknown genetic and environmental modifiers.
Keywords: amyotrophic lateral sclerosis, long‐duration, microglia, military Veterans, motor neuron disease, neuroinflammation, neuropathology, survival
Introduction
Amyotrophic lateral sclerosis (ALS) is usually an adult‐onset, progressive neurodegenerative disorder that affects both the upper and lower motor neurons. Neurodegeneration of motor neurons leads to progressive loss of gross motor, fine motor, bulbar and respiratory function that eventually leads to death within 3 to 5 years after initial symptom onset (1, 16). Epidemiological studies have estimated ALS incidence in the United States (US) to be between 1 and 2 cases per 100 000 people (16, 25). Approximately 10% of ALS cases are classified as familial ALS, in which the disorder tends to be inherited in an autosomal dominant fashion (16, 40), and numerous contributory genes have been identified (1). Other risk factors for the development of ALS include age (6), male gender (16) and military service (7, 38, 43, 50, 88). Despite differences in etiology, sporadic and familial ALS possess similar patterns of neuropathological degeneration (2).
Across cohort studies and clinical experience in ALS, one of the most striking observations is the extreme variability in disease duration. Previous studies have noted that 50% of patients die within the first 3 years after disease onset, and only a small fraction of cases (~10%) survive 10 years or longer (24, 63, 74). Furthermore, large database and population‐based studies have reported the median death in the US and Europe as approximately 3 years after disease onset (7, 41, 44, 73). Previous studies have shown most cases of ALS have survival rates of less than 10 years and suggest that patients with ALS that survive 10 years or more represent a group with altered biology (23, 87). Numerous clinical, genetic and neurobiological factors have been examined as moderators of disease duration variability including healthcare accessibility, comorbidities, ventilation, genetic mutations, neuroinflammation and other neurodegenerative markers in central nervous system tissues (7, 13, 22, 31, 66, 71, 74, 75, 78, 83, 84, 89). Specifically, prolonged survival has been linked with a younger age at diagnosis, extremity onset of initial symptoms and a longer diagnostic latency after symptom onset (74). Additionally, percutaneous endoscopic gastrostomy and multidisciplinary clinical care have also been linked to longer survival with ALS (29, 61, 74).
Some neuropathological features have been associated with disease progression in ALS. For instance, the presence and extent of TDP‐43 pathology have been evaluated in relation to survivability (13, 27, 47, 68, 89). Although the decreased severity of TDP‐‐43 pathology has been reported for some cases of long‐lasting ALS (68), other studies have found non‐significant or even opposite associations (13, 14, 27, 35). In addition, increased neuroinflammation with microglial activation within the motor system is a consistent feature in ALS (3, 33, 34, 39, 53, 56, 65, 77) and mouse models demonstrate such inflammation occurs early before symptom onset, suggesting a role in disease pathogenesis (48, 58, 60, 76). Microglial pathology was also found to be more extensive in participants with ALS with less than two‐year survival compared to those with longer survival durations (15). However, pathological associations with rarer, long‐lived (≥10 years) disease are unknown.
In this study, we examined clinical and neuropathological differences in a cohort of US military Veterans with a pathologically confirmed diagnosis of ALS from the Department of Veterans Affairs Biorepository Brain Bank (VABBB). The VABBB is a central nervous system tissue biobank founded in 2006 in response to previous findings indicating an increased prevalence of ALS among US military Veterans (28, 38, 43, 88). The VABBB ALS cohort is a unique population whose median survival is nearly double the 4.7 years from symptom onset observed in the VA National Registry (12, 72). Thus, this cohort provides a unique opportunity to examine clinical and neuropathological factors related to unusually long disease duration. We hypothesized long‐duration cases would have an initial slow progression and an altered and less severe motor neuron neuropathology at death. We further tested the hypothesis that marked long‐duration ALS was associated with decreased microglial density and activation.
Methods
Participants
Participants were selected from the Department of Veterans' Affairs (VA) Biorepository Brain Bank (VABBB). The VABBB is longitudinal observational study and central nervous system tissue biorepository that collects clinical, genetic and neuropathological data from United States military Veterans (12). Qualifying cases for this study had a neuropathological diagnosis of ALS determined by a trained neuropathologist. This included a total of 179 participants with ALS, which were then classified as either standard or long‐duration based on the total number of years between initial symptom onset and death. The standard duration was defined as less than 10 years from symptom onset to death (n = 103), and the long‐duration was defined as greater than or equal to 10 years from symptom onset to death (n = 76). In order to account for the prolongation of disease due to assisted ventilation, participants with and without the use of ventilation were examined separately. The methods for data acquisition, neuropathological examination, clinical assessment/progression and genetic analysis have been described previously (12, 86).
Data acquisition
Clinical and demographic data were collected via VA electronic medical records, participant self‐report or report by a participant healthcare proxy. In addition to the VABBB database, clinical and demographic data were obtained through the VA National ALS Registry, Cooperative Studies Program (CSP) 500A, for any VABBB tissue donors who were previously enrolled in the ALS Registry (n = 132). The VA National ALS Registry provided longitudinal data including disease progression, trauma history and motor functionality (49).
Clinical analyses
Demographic information included sex, ethnicity and race. Additionally, military information including service branch, years of service and deployment history were included for analysis. Information pertaining to the history of ALS included age of symptom onset, site of symptom onset, age at disease diagnosis, familial history of ALS and family history of neurological disease was obtained via the VA National ALS registry or VABBB databases. The site of symptom onset was classified as upper or lower extremity, bulbar or respiratory. For ventilation status, cases were considered positive for ventilation if they received 24‐hour invasive or non‐invasive respiratory maintenance for a period greater than or equal to three months prior to death. ALS symptom/disease progression was obtained through semiannual administrations of the ALS Functional Rating Scale‐Revised (ALSFRS‐R) administered by trained research staff at the VABBB and CSP500A (20). The ALSFRS‐R is a well‐validated measure used widely throughout different research institutions and clinical practices to quantify the degree of motor dysfunction due to ALS (55). Administration of the ALSFRS‐R by trained non‐clinicians has been shown to have good inter‐rater, intra‐rater and telephone‐administered reliability (51). The ALSFRS‐R provides a score across four domains of physical function including gross motor function, fine motor function, bulbar function and respiratory function. The maximum score of the ALSFRS‐R is 48, while the maximum score in each domain is 12 (19, 20, 86). For analysis, we used ALSFRS‐R data as an indicator of initial disease progression. Functional disease progression of ALS as measured by the ALSFRS‐R has been denoted by past research as a ΔFS score defined as [Total score of first ALSFRS‐R − 48]/months from onset to first ALSFRS‐R assessment.(57). Additionally, we compared ALSFRS‐R domain scores between the two groups at the first visit and last visit before death in order to test for differences in function between the short and long‐duration groups.
Neuropathological analysis
Brain and spinal cord tissues were processed and assessed as described previously (12). Briefly, a diagnosis of ALS required degeneration of both upper and lower motor neurons, degeneration of the lateral corticospinal tracts and anterior horn cell loss from all levels of the spinal cord (17, 37). TDP‐43 stage was determined using the criteria of Brettschneider et al (14) as well as Nishihira Type 1 or Type 2 (69). Motor cortex, anterior horn of the spinal cord, medulla, inferior frontal gyrus and hippocampus were accessed for the presence of phosphorylated TDP‐43, including neuronal and glial cytoplasmic inclusions and threads. Neuropathological changes in Alzheimer's disease were determined by the NIA‐Alzheimer Association's guidelines (64). Thal phase for amyloid‐β presence (80), Braak staging of neurofibrillary tangles (10, 11), CERAD neuritic plaque density (62) and Lewy body disease type were determined as per NIA guidelines (64). Frontotemporal lobar degeneration diagnosis was determined by atrophy of the frontal and temporal lobes and the presence of phosphorylated tau or phosphorylated TDP‐43 inclusions (8, 18, 59). The overall level of degeneration was evaluated on Luxol fast blue‐hematoxylin and eosin‐stained sections and defined by the degree of myelin loss and gliosis in the corticospinal tract and the ventral spinal roots and the degree of neuronal loss and gliosis in the motor cortex, spinal anterior horns and hypoglossal nuclei. Ratings of degeneration were made by the neuropathologists using a 0–3 scale, with 0 = none and 3 = severe as previously described (4).
Genetic analyses
A total of 158 of the analyzed 179 (88%) participants were genotyped for known ALS mutations. Genotyping was conducted at the National Institute on Aging in Bethesda, Maryland. Samples were genotyped using the OmniExpress chip and NeuroChip. OmniExpress tests for 730 525 single nucleotide polymorphisms. NeuroChip provides genotyping on 179 467 genetic mutations associated with different neurological diseases (9). Targeted next‐generation sequencing (NGS) and repeat‐primed polymerase chain reaction were used for C9orf72 hexanucleotide repeat expansion analysis. We obtained data for the following genes associated with ALS: optineurin (OPTN), senataxin (SETX), dynactin subunit 1 (DCTN1), NIMA‐related kinase 1 (NEK1), superoxide dismutase 1 (SOD1), TAR DNA binding protein (TARDBP), profilin 1 (PFN1) and chromosome 9 open reading frame 72 (C9orf72). Additional data for kinesin family member 5A (KIF5A) mutations were obtained for 87 participants.
Immunohistochemistry and digital slide scanning
The inflammatory markers ionized calcium‐binding adapter molecule 1 (Iba1) and cluster of differentiation 68 (CD68) were used to determine potential differences in neuroinflammatory profile between standard duration and long‐duration ALS cases. Tissue was fixed in periodate‐lysine‐paraformaldehyde and tissue blocks were paraffin‐embedded and cut at 10 μm (diagnostic histochemistry) or 20 μm (for microglia cell density quantification). Antigen retrieval was performed by boiling sections in citrate buffer (pH 6.0) for 10 minutes. Sections were incubated at 4℃ overnight with antibodies to anti‐Iba1 (Wako, 1:500), anti‐PHF‐tau (AT8) (Pierce Endogen, 1:2000) and anti‐CD68 (Vector, 1:500). For TDP‐43 immunohistochemistry, anti‐TDP‐43, phospho Ser409/410 mAb (clone 11‐9) (Cosmo Bio USA, 1:2000), was used following treatment with formic acid (88%) for 2 minutes. Sections were treated with biotinylated secondary antibodies then labeled with a 3‐amino‐9‐ethylcarbazol HRP substrate kit (Vector Laboratories). Sections were counter‐stained with Gill's Hematoxlin (Vector Laboratories H‐3401) and coverslipped with Permount mounting medium.
For microglial cell quantification, cervical spinal cord and medulla from a randomly selected subset of participants from long (n = 20) and standard duration (n = 15) cases were immunostained using markers for total and activated microglia and cellular densities were quantitated. Additional sub‐analyses did not demonstrate any significant differences in measurements of clinical or neuropathological features (eg, duration, TDP‐43 pathology, upper and lower motor neuron loss) of the long and standard duration subgroups when compared to their complete respective cohort. Tissue blocks from the cervical spinal cord and medulla were embedded in paraffin, cut at 20 μm, immunostained for Iba1 and CD68, and scanned at 20x magnification with a Leica Aperio Scanscope (Leica Biosystems, Richmond, IL) as previously described (21). ImageScope (Leica Biosystems) was used to separately highlight the anterior horn, pyramids at the level of the inferior olives and hypoglossal nuclei. Leica's image analysis and automated counting software (Aperio positive pixel count, Version 9) was calibrated for staining intensity to detect Iba1 or CD68‐positive cells within the region of interest. The cellular number was normalized to the area measured and presented as positive cells per mm2. To address errors in slide analysis due to artifacts, data points outside two‐ and one‐half standard deviations from group means were omitted from the analysis. One tissue block was available for each region analyzed. At least one section was analyzed per block based on availability. On average, 1.76 sections were analyzed per case. In addition, the total number of motor neurons in the anterior horn of the cervical spinal cord was counted. Two trained researchers performed independent manual motor neuron counts for standard duration cases (n = 14) and long‐duration cases (n = 20). The researchers were blind to both the neuropathologist ranking and the duration group of each tissue sample. The researcher counts were averaged and divided by the total area of the anterior horn.
Gene expression
Glial fibrillary acidic protein (GFAP) gene expression was used to further examine differences in neuroinflammatory profile between long and standard ALS durations. RNA was isolated from the cervical spinal‐cord using a modified TRIzol protocol with a QIAGEN RNeasy mini column clean‐up. One microgram of RNA was used to create cDNA using high‐capacity cDNA reverse transcription kit (Applied Biosystem, Waltham, MA) and PTC‐200 DNAEngine cycler (BioRad, Hercules, CA). cDNA was used to select endogenous controls using TaqMan universal buffer mix and an endogenous control TaqMan low‐density array. Ct‐values were obtained using Applied Biosystem SDS software and reference‐gene stability and optimal number of reference genes were determined with geNorm (Biogazelle qBase + software). Probes for GFAP (BioRad, qHsaCID0022307) were used, and qPCR was performed using SsoAdvanced universal SYBR green supermix (BioRad) and QuantStudio 7 Flex Real‐Time PCR system (Applied Biosystem). Thresholds were normalized across plates, triplicate Ct‐values were assessed, outliers removed, samples were normalized on endogenous controls PGK1 & PPIA, and ΔCt‐values were calculated with PrimePCR analysis software (BioRad).
Statistical analysis
All statistical analyses were completed using IBM SPSS Statistics for Windows, version 26 (IBM Corp., Armonk, NY). A Shapiro–Wilk test was performed for all scalar data to determine normality. An independent sample T‐test was performed for normally distributed scalar data. Equality of variances was determined using Levene's Test. Non‐parametric independent samples Mann‐Whitney U tests were performed between groups for all categorically ranked data and scalar data measurements with non‐normal distributions. Pearson chi‐squared tests of variance with Bonferroni corrections were conducted for multiple group comparisons on categorical data counts. Fisher exact tests of variance were applied when the expected sample size was less than five. An alpha level of 0.05 was used for all statistical tests.
Results
Cohort overview
A total of 179 participants with ALS that came to autopsy at the Department of Veterans Affairs Biorepository Brain Bank were included in the study. The group was largely male (98%) and Caucasian (97%). Participants were dichotomized into a standard duration group with survival time less than 10 years (n = 103) and a long‐duration group with survival time greater than or equal to 10 years (n = 76). The proportion of participants with long‐duration ALS in this cohort of Veterans was therefore 42%, which is substantially higher than the approximately 10% reported in other ALS cohorts (24, 63, 74). Of the 76 participants with long‐duration ALS, 44 had a duration between 10 and 15 years, 15 had a duration between 15 and 20 years, 12 had a duration between 20 and 30 years, and 5 had a duration greater than 30 years. The five longest duration cases in our cohort reached survival times of 31, 34, 34, 36 and 42 years respectively. Each of the 30 year or greater duration cases were limb onset; none had a familial history of ALS, and no known mutations were found during genetic analysis.
Comparison of duration groups
There were clear clinical distinctions between standard and long‐duration groups (Table 1). The long‐duration group on average had a younger age of onset (P = 0.002), slower initial disease progression (P < 0.001) and older age at death (P < 0.001) compared with the standard duration group. There was a substantially longer period between symptom onset and diagnosis in long‐duration compared to standard duration ALS (2.7 vs. 1.2 years, P = 0.002). Additionally, there was a trend toward an altered site of onset between groups such that there was increased frequency of bulbar onset in standard duration ALS (P = 0.071). Finally, there was a trend (P = 0.054) for an increased frequency of traumatic brain injury in long‐duration participants.
Table 1.
Demographic and clinical features of participants with standard and long‐duration ALS.
| Standard (n = 103) | Long (n = 76) | P‐alue | |
|---|---|---|---|
| Male/Female, ± (%) | 98/3 (98%) | 72/4 (95%) | 0.465‡ |
| Race | 0.494‡ | ||
| White, n (%) | 98 (98%) | 73 (96%) | |
| Black or African American, n (%) | 2 (2%) | 2 (3%) | |
| American Indian, n (%) | 0 (0%) | 1 (1%) | |
| Hispanic or Latino, ± (%) | 1/98 (1%) | 1/75 (1%) | 1.000‡ |
| Disease Duration, mean ± SEM | 5.29 ± 0.27 | 16.32 ± 0.82 | <0.001 † |
| Age at Onset, mean ± SEM | 61.65 ± 1.02 | 55.87 ± 1.51 | 0.002 † |
| Age at diagnosis, mean ± SEM | 63.33 ± 1.03 | 58.54 ± 1.56 | 0.073† |
| Symptom Onset to Diagnosis, mean ± SEM | 1.24 ± 0.12 | 2.67 ± 0.31 | 0.002 † |
| Age at Death, mean ± SEM | 67.28 ± 0.94 | 72.19 ± 1.42 | <0.001 † |
| Site of Onset | 0.071‡ | ||
| Lower extremity, n (%) | 42 (42%) | 42 (58%) | |
| Upper extremity, n (%) | 32 (32%) | 23 (32%) | |
| Bulbar, n (%) | 24* (24%) | 7* (10%) | |
| Respiratory, n (%) | 1 (1%) | 1 (1%) | |
| A Care, ± (%) | 90/13 (87%) | 68/8 (89%) | 0.815 |
| Continuous ventilation, ± (%) | 29/64 (31%) | 19/53 (40%) | 0.605‡ |
| Continuous ventilation, yrs, mean ± SEM | 0.72 ± 0.15 | 1.77 ± 0.46 | 0.954† |
| ΔFS, mean ± SEM | −0.64 ± 0.06 | −0.29 ± 0.03 | <0.001 † |
| Familial Neurologic Disease, ± (%) | 16/32 (33%) | 15/23 (40%) | 0.619‡ |
| Familial ALS, ± (%) | 4/81 (5%) | 8/60 (12%) | 0.135‡ |
| Genetic Mutations ± | 10/90 (10%)§ | 6/66 (8%)¶ | 0.710‡ |
| C9orf72 ± (%) | 3/97 (3%) | 1/71 (1%) | 0.641‡ |
| Historyof TBI, ± (%) | 12/91 (13%) | 17/59 (29%) | 0.054‡ |
P < 0.05 following Bonferroni correction.
Mann‐Whitney U Test.
χ 2 test of variance.
Known mutations included C9orf72, TARDBP, SETX, KIF5A, OPTN, SOD1, FUS.
Known mutations included C9orf72, TARDBP, SETX, PFN1, NEK1, DCTN1.
Bold values indicate significance of P < 0.05
In cases with available genetic testing, the frequency of common ALS‐associated mutations was not significantly different between groups. Of the 158 genotyped participants, 16 were positive for genetic mutations (10%). Among the standard duration cohort, there were 10 donors with known mutations out of the 90 genotyped cases (11%), involving C9orf72 mutations (n = 3), TARDBP (n = 2), and FUS, KIF5A, OPTN, SETX, NEK1 and SOD1 (n = 1 each). One donor screened positive for two mutations (OPTN and SETX). As for the long‐duration cohort, six participants were positive for genetic mutations (9%) involving C9orf72, DCTN1, NEK1, PFN1, SETX and TARDBP (n = 1 each). Of interest, only one case in the VABBB cohort was found to have a KIF5A mutation, belonging in the standard duration group with a disease duration of 1.4 years.
Furthermore, no significant differences between the duration groups were found for race, ethnicity, sex or familial history of neurological disease and ALS. In addition, the length of time with continuous invasive or non‐invasive ventilation did not significantly differ between groups (Table 1). When considering military demographics, there were no significant differences between duration groups for the number of years, branch, or era of military service or for participant deployment overseas (Table 2). Tissue quality measures were similar between standard and long‐duration groups, including Post‐mortem interval, as measured by time of death to the removal of the brain and spinal cord (3.9 vs. 3.4 h, P = 0.382), tissue pH (6.3 vs. 6.3, P = 0.967) and RNA integrity number (3.7 vs. 5.9, P = 0.459).
Table 2.
History of military service in standard and long‐duration ALS groups.
| Standard | Long | P‐value | |
|---|---|---|---|
| Years in Military, mean ± SEM | 6.49 ± 0.75 | 5.96 ± 0.77 | 0.971† |
| Military Branch, n (%) | 0.808‡ | ||
| Army | 47 (48%) | 37 (48%) | |
| Navy | 21 (21%) | 19 (25%) | |
| Air Force | 23 (23%) | 14 (19%) | |
| Marines | 5 (5%) | 3 (4%) | |
| Coast Guard | 1 (1%) | 0 (0%) | |
| Merchant Marines | 0 (0%) | 1 (1%) | |
| Multiple Branches | 2 (2%) | 2 (3%) | |
| Military Era, n (%) | 0.676‡ | ||
| WWII | 5 (5%) | 7 (9%) | |
| Korean | 5 (5%) | 6 (8%) | |
| Post‐Korean | 1 (1%) | 0 (0%) | |
| Vietnam | 63 (64%) | 42 (55%) | |
| Post‐Vietnam | 7 (7%) | 7 (9%) | |
| Gulf War | 8 (8%) | 4 (5%) | |
| Multiple Eras | 10 (10%) | 10 (13%) | |
| Deployed Overseas, ± (%) | 67/13 (84%) | 61/14 (81%) | 0.692‡ |
Mann‐Whitney U test.
χ 2 test of variance.
The total ALSFRS‐R score at initial assessment was not significantly different between groups. Additionally, we found no significant differences in the initial assessment of ALSFRS‐R domain scores during the initial assessment. Both participants with standard and long‐duration ALS were severely functionally impaired near death with similar ALSFRS‐R domain scores when last assessed before death (Table 3).
Table 3.
Comparison of ALSFRS and subdomain scores between standard and long‐duration ALS groups.
| Initial | Standard (n = 70) | Long (n = 73) | P‐value† | |
|---|---|---|---|---|
| ALSFRS‐R (mean ± SEM) | 28.63 ± 1.43 | 26.53 ± 1.39 | 0.229 | |
| Gross Motor (mean ± SEM) | 5.69 ± 0.46 | 4.63 ± 0.39 | 0.099 | |
| Fine Motor (mean ± SEM) | 6.33 ± 0.45 | 5.23 ± 0.44 | 0.098 | |
| Bulbar, (mean ± SEM) | 8.84 ± 0.41 | 7.93 ± 0.46 | 0.137 | |
| Respiratory (mean ± SEM) | 8.54 ± 0.42 | 8.64 ± 0.45 | 0.594 |
| Death | Standard (n = 66) | Long (n = 74) | P‐value† | |
|---|---|---|---|---|
| ALSFRS‐R, (mean ± SEM) | 12.39 ± 0.86 | 13.04 ± 1.04 | 0.732 | |
| Gross Motor, (mean ± SEM) | 1.06 ± 0.22 | 1.16 ± 0.22 | 0.736 | |
| Fine Motor, (mean ± SEM) | 1.36 ± 0.27 | 1.74 ± 0.27 | 0.242 | |
| Bulbar, (mean ± SEM) | 5.29 ± 0.44 | 5.07 ± 0.43 | 0.732 | |
| Respiratory, (mean ± SEM) | 5.21 ± 0.41 | 5.80 ± 0.48 | 0.367 |
Mann‐Whitney U test.
For comparisons of neuropathological features, we conducted separate analyses based on 24‐hour ventilation status prior to death to adjust for potential discrepancies caused by artificial respiratory support. Comparisons of the neuropathological findings between groups without mechanical ventilation revealed several similarities and differences. Participants with long‐duration ALS had the classical pathological findings of motor neuron loss in both the motor cortex and whole spinal cord and had similar severe involvement of the upper motor neuron system, including degeneration of the motor cortex and lateral corticospinal tracts. However, global semi‐quantitative severity scores of spinal motor neuron degeneration were lower in long‐duration compared to standard duration ALS (Table 4, P < 0.001), suggesting less severe involvement of the spinal cord despite the long disease duration. Additional analyses of degeneration of the anterior horns, lateral corticospinal tracts and ventral roots for cervical, thoracic and lumbar spinal cord showed that all regions were similarly affected. Degeneration of lateral corticospinal tracts between standard and long‐duration groups trended toward significance (P = 0.060) for all regions of the spinal cord, while ventral root degeneration was significantly different between groups for all spinal cord levels (P's < 0.05). Degeneration of the anterior horns was significant in the cervical and lumbar spinal cord (P's < 0.05) and approached significance in the thoracic spinal cord (P = 0.060). A subset of standard and long‐duration ALS participants were selected for motor neuron quantification within the anterior horn of the cervical spinal cord and showed an increased density of motor neurons in the long‐duration group (13.7 motor neurons per mm2) compared to the standard duration group (11.1 motor neurons per mm2) although the difference was not significant (P = 0.151) likely due to decreased power in this subset.
Table 4.
Neuropathological features in standard and long‐duration ALS.
| Non‐ventilated§ | Ventilated§ | |||||
|---|---|---|---|---|---|---|
| Standard (n = 66) | Long (n = 54) | P‐value | Standard (n = 30) | Long (n = 20) | P‐value | |
| Loss of Anterior Horn Cells§, Mean ± SEM | 2.86 ± 0.47 | 2.31 ± 0.12 | <0.001 † | 2.89 ± 0.62 | 2.88 ± 0.12 | 0.612† |
| Degeneration of Ventral Roots¶, Mean ± SEM | 2.61 ± 0.13 | 2.04 ± 0.19 | 0.016 † | 2.69 ± 0.12 | 2.62 ± 0.24 | 0.736† |
| Motor Cortex Degeneration, Mean ± SEM | 2.30 ± 0.15 | 2.20 ± 0.14 | 0.566† | 2.21 ± 0.26 | 2.18 ± 0.33 | 0.976† |
| Lateral Cortical Spinal Tracts Degeneration¶, Mean ± SEM | 2.14 ± 0.14 | 1.71 ± 0.16 | 0.060† | 2.42 ± 0.22 | 2.15 ± 0.30 | 0.424† |
| Hypoglossal Nerve Degeneration, Mean ± SEM | 1.91 ± 0.15 | 1.74 ± 0.15 | 0.528† | 2.32 ± 0.17 | 2.00 ± 0.22 | 0.269† |
| TDP‐43 Positive, ± (%) | 58/5 (92%) | 25/28 (47%) | <0.001 ‡ | 24/5 (83%) | 12/7 (63%) | 0.117‡ |
| TDP‐43 Stage¶, Mean ± SEM | 1.59 ± 0.19 | 1.33 ± 0.33 | 0.373† | 1.50 ± 0.34 | 1.33 ± 0.33 | 0.819† |
| Nishihira TDP‐43 Type† | 0.656‡ | 0.584‡ | ||||
| Type 1 | 45 (94%) | 21 (91%) | 18 (90%) | 8 (72%) | ||
| Type 2 | 3 (6%) | 2 (9%) | 2 (10%) | 3 (28%) | ||
| Lewy Body Disease, ± (%) | 4/39 (9%) | 10/36 (22%) | 0.147‡ | 2/19 (10%) | 3/11 (21%) | 0.369‡ |
| Arteriolosclerosis, Mean ± SEM | 1.66 ± 0.12 | 1.79 ± 0.10 | 0.404† | 1.71 ± 0.15 | 2.14 ± 0.18 | 0.084† |
| Atherosclerosis, Mean ± SEM | 0.66 ± 0.14 | 0.82 ± 0.15 | 0.455† | 0.47 ± 0.19 | 0.33 ± 0.17 | 0.829† |
| Braak Neurofibrillary Stage†, Median (25th, 75th) | II (I, III) | II (II, III) | 0.083† | I, (0, II) | II (0, II) | 0.467† |
| CERAD§§, Median (25th, 75th) | 0 (0, I) | 1 (0, 1) | 0.368† | 0 (0, 0) | 0 (0, 1) | 0.085† |
| Pathological changes in AD¶¶, ± (%) | 23/22 (51%) | 23/19 (55%) | 0.831‡ | 6/14 (30%) | 4/9 (31%) | 1.000‡ |
Mann‐Whitney U test.
χ2 test of variance.
Cases where ventilation status prior to death could not be determined were excluded from analysis
Analysis conducted using global scores of cervical, thoracic and lumbar spinal cord regions
Analysis only includes cases positive for TDP‐43 inclusions and assessed in all regions
Braak neurofibrillary tangle staging was rated as 0‐VI based on established guidelines.
CERAD plague density was rated as none (0), sparse (1), moderate (2) or frequent (3).
Neuropathological changes in Alzheimer disease were determined by the NIA‐Alzheimer Association's guidelines. Cases were coded as positive if the degree of Alzheimer pathology was low, intermediate, or high.
Bold values indicate significance of P < 0.05
In addition, the frequency of TDP‐43 pathology was reduced in long‐duration ALS (P < 0.001). For those with TDP‐43 pathology, the severity of TDP‐43 inclusions as assessed by either Brettschneider staging (14) or Nishihira types (69) were not significantly different between duration groups. No significant differences were noted for brain weight or comorbid neurodegenerative disease, including neuropathological changes in Alzheimer's disease, Lewy body disease, or neurovascular disease. Additionally, participants with long‐duration ALS who had received mechanical ventilation had more TDP‐43 pathology along with more severe lower motor neuron degeneration analogous to that of standard duration ALS, suggesting that ventilation may have prolonged what would have otherwise been standard duration ALS (Table 4). Pastula et al (72) suggest defining survival time from diagnosis to date of death or first significant ventilation (ie, more than 15 h of ventilatory support per day for at least two weeks). A secondary analysis of the above neuropathological and clinical variables using the Pastula et al criteria for survival time showed similar findings, including significant differences in anterior horn cell degeneration, presence of TDP‐43, ventral root degeneration and degeneration of the lateral corticospinal tracts.
Altered microglia in long‐duration ALS
Increased and activated microglia are a hallmark of ALS pathology and are likely contributors to motor neuron degeneration. To test the hypothesis that long‐duration ALS is associated with decreased microglial density, we quantitated the density of immunoreactive cells for total Iba1 and activated CD68 in a subset of participants with standard and long‐duration ALS. Iba1 protein is expressed in monocytic cell lineages including all types of microglia (45), while CD68 is a lysosomal protein expressed in activated microglia and macrophages (42). GFAP is present within astrocytes and elevated in reactive cells (32). There were no significant differences in clinical or neuropathological data between the overall cohort groups and the subset examined for additional cell density analysis. Consistent with our hypothesis, we found significantly lower Iba1 positive cellular density in the anterior horns (P = 0.008) and medullar pyramids (P = 0.012) in long‐duration ALS cases (Table 5 and Figure 1). In order to adjust for the effects of age, an age‐corrected linear regression found a similar negative association between Iba1 positive cellular density and ALS duration in the pooled group of participants with both standard and long‐duration ALS (P = 0.003).
Table 5.
Comparison of total (Iba1) and activated (CD68) microglia cell densities between standard and long‐duration ALS.
| Region | Stain | Standard | Long | P‐value† | |
|---|---|---|---|---|---|
| Anterior Horn | |||||
| Iba1 | |||||
| Positive cells per mm2 (mean ± SEM) | 270 ± 22 | 162 ± 23 | 0.008 | ||
| CD68 | |||||
| Positive cells per mm2 (mean ± SEM) | 265 ± 45 | 178 ± 32 | 0.111 | ||
| Medullary Pyramids | |||||
| Iba1 | |||||
| Positive cells per mm2 (mean ± SEM) | 292 ± 16 | 226 ± 17 | 0.012 | ||
| CD68 | |||||
| Positive cells per mm2 (mean ± SEM) | 179 ± 38 | 217 ± 35 | 0.471 | ||
| Hypoglossal Nucleus | |||||
| Iba1 | |||||
| Positive cells per mm2 (mean ± SEM) | 255 ± 29 | 231 ± 20 | 0.492 | ||
| CD68 | |||||
| Positive cells per mm2 (mean ± SEM) | 61 ± 15 | 52 ± 11 | 0.619 |
Independent Samples T‐test.
Bold values indicate significance of P < 0.05
Figure 1.
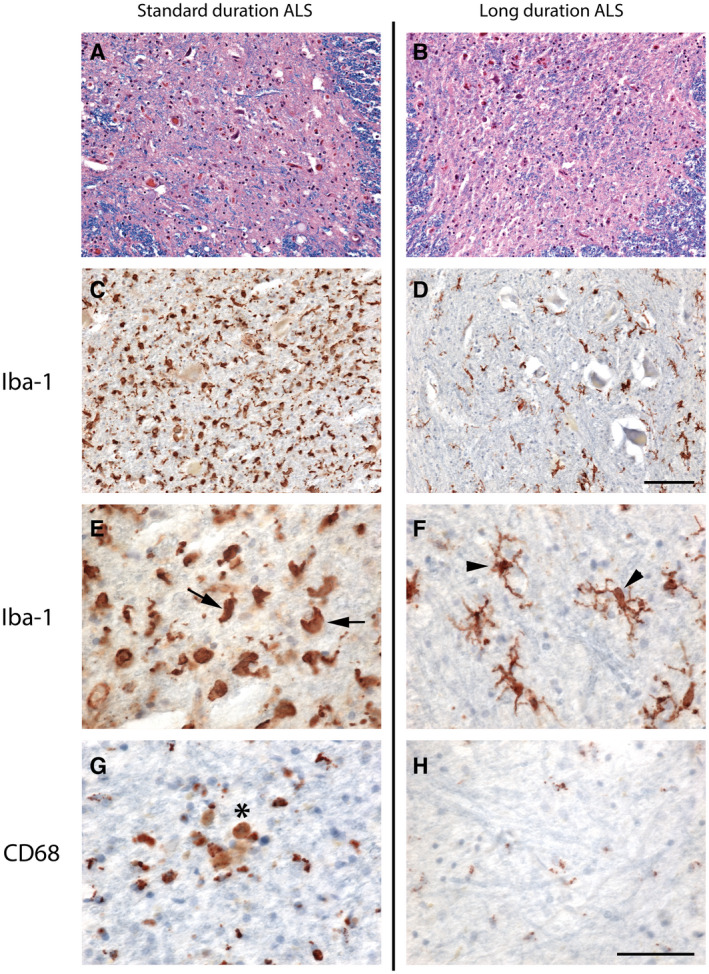
Decreased microglial cell density and resting‐type morphology within the spinal cord in long‐duration ALS. (A) and (B) Luxol fast blue/hematoxylin and eosin staining demonstrates severe degeneration and loss of motor neurons in both standard (A) and long (B) duration ALS. (C–F) Immunostaining for Iba‐1 shows increased numbers of Iba‐1‐positive microglia in standard (C) compared to long (D) duration ALS. The morphology of the microglia is altered toward a reactive phenotype with a larger, ameboid shape in standard duration ALS (E, arrows) compared to the resting phenotype of a small cell body with ramified processes in long‐duration ALS (F, arrowheads). (G and H) Foci of CD68‐positive macrophages are more frequent in standard (G, asterisk) than in long‐duration ALS (H). Scale bar in A–D, 100 μm; E‐H, 50 μm.
We also noted differences in microglial morphology between groups such that standard duration ALS tended to contain microglia with a reactive phenotype with a larger, ameboid shape, while microglia in long‐duration ALS possessed the resting phenotype of a small cell body with ramified processes (Figure 1A–F). Although total numbers of CD68‐positive cells were not significantly different between groups (Table 5), we observed frequent clusters of CD68‐positive macrophages within the anterior horn in standard, but not long, duration ALS cases (Figure 1G–H). There was no significant difference in spinal cord GFAP gene expression between duration groups (standard: ΔCt = 1.25 ± 0.20, long: ΔCt = 1.78 ± 0.28, P = 0.198). These results demonstrate that while total microglia density is lower in long‐duration ALS, GFAP levels are relatively unchanged, suggesting similar levels of astrocytosis between duration groups.
Discussion
In the largest group of neuropathologically confirmed long‐duration ALS (≥10 years) reported to date, we found numerous clinical and neuropathological distinctions with standard duration ALS. In long‐duration ALS the disease course was slow from the beginning and showed steady progression. Although both long and standard duration ALS had classic upper and lower motor neuron degeneration, participants with long‐duration ALS had less severe lower motor neuron degeneration as well as decreased frequency of TDP‐43 positive inclusions in the central nervous system. Further, the total density of microglia was decreased within the spinal cord of long‐duration ALS. Altogether these findings suggest that ALS disease duration is associated with an altered pattern of neurodegeneration and neuroinflammation.
The VABBB cohort has an unusually high number of participants with long‐duration ALS, with 76 of the 179 (42%) reviewed cases having disease durations lasting longer than ten years. The markedly increased frequency of long‐duration ALS in this cohort may be due to a variety of factors. The majority of participants were enrolled from the VA National Registry and therefore represent prevalent rather than incident cases which likely presented a selection bias for longer duration cases. Previous work suggests an altered incidence of ALS among United States military Veterans compared to that of the civilian population (7, 38, 43, 50, 88). Additionally, implementation of detailed health screening at the Military Entrance Processing Station (MEPS) and the physical demands of basic training could also render those with some ALS variants ineligible for service. We found no difference among the length of service, military branch, war of service, and deployment status between standard and long‐duration ALS. However, other factors of military service may be associated with slow progression of ALS.
A potential factor contributing to the longevity of this United States Military Veteran population is the availability of ALS multidisciplinary clinics. Enrollment of patients with ALS in multidisciplinary clinics or tertiary centers has been demonstrated to improve survivability when compared to ALS patients receiving care at general neurology clinics (22, 81). In our long‐duration group, 68 Veterans were enrolled in the VA (89%), while 90 Veterans (87%) in the standard duration group received VA care. Since 2008, the VA has treated ALS as a 100% service‐connected illness (28). The benefits for those with service‐connected illnesses bears healthcare policy considerations related to the accessibility and availability of resources. For example, any Veteran with ALS is eligible to receive VA healthcare, home assistance, home modification and medications at no out of pocket cost. This reduced financial and resource burden available to Veterans may also be contributing to the increased survival. However, in the present study no differences in utilization of VA care between duration groups were observed.
The frequency of TDP‐43 pathology was altered such that there was less frequent TDP‐43 pathology in the long‐duration group. Many of these participants also lacked known ALS‐associated genetic mutations and it is unclear whether the lack of TDP‐43 pathology is due to immunohistochemical detection limits, unknown genetic variants, or other mechanisms. In those with TDP‐43 pathology, the TDP‐43 stage as proposed by Brettschneider (14) and type proposed by Nishihira (69) were not significantly reduced in the long‐duration group. This is inconsistent with some previous reports demonstrating decreased TDP‐43 pathology in long‐lasting ALS (68); however, other reports have shown either no or an opposite association with TDP‐43 stage and severity (13, 14, 35). The discrepancy between this study and others may be due to the smaller number of participants and the lack of adjustment for mechanical ventilation in previous studies.
In fact, the frequency and length of usage of ventilation may influence comparisons between duration groups. It has been suggested that ALS duration should be measured as onset of symptoms until date of 24‐hour mechanical ventilation due to extension of disease duration by artificial respiratory support with no change in disease severity (72). In this study, the frequency of ventilation was not significantly different between standard and long‐duration ALS groups. Even when analyzing only cases without ventilation, we noted a preservation of the relatively less severe spinal motor neuron degeneration observed in the long‐duration cases. Moreover, ventilated long‐duration participants still had less frequent TDP‐43 pathology than standard duration ALS although the difference lost significance and was not as great as within non‐ventilated subjects. This is likely due to the inclusion of what would otherwise have been standard duration ALS had they not been ventilated.
Relatively few studies have examined the differences in degeneration across varying ALS durations. One study discussed the degeneration of upper motor neurons manifesting in a milder manner for cases of longer duration (46). Specifically, mild degeneration of Betz cells within the motor cortex, mild myelin pallor in the corticospinal tracts, but severe degeneration within the anterior horns was noted in three cases of long‐duration. Other studies have identified a slight increase in long‐term survival rate in cases in which involvement is predominantly in the upper or lower motor neurons (36, 54, 79). We found significantly decreased levels of lower motor neuron degeneration, but similar levels of upper motor neuron degeneration in long compared to standard duration ALS. These findings are consistent with the long survival times described in primary lateral sclerosis (36) and might suggest a relative resistance of lower motor neurons to degeneration in long‐duration ALS.
Microgliosis within the regions of neurodegeneration is a consistent feature of ALS (3, 33, 34, 39, 53, 56, 65, 77). Various microglial phenotypes have been described. Iba1 is widely used as an immunoreactive marker for all microglia, although it may also have additional reactivity to some types of activated microglia (85). In comparison, CD68 labels activated microglia and macrophages with increased lysosomes (30). Increased CD68 reactivity may also be indicative of less efficient phagocytosis and a detrimental inflammatory state (91). Murine disease models demonstrate increased activated microglia prior to motor neuron degeneration and motor symptoms (48, 58, 60), suggesting thar microglia are involved in disease pathogenesis. Furthermore, imaging studies suggest that activation and recruitment of microglia begins early in ALS and persists throughout the disease course (5, 26, 82). Increased total and activated microglia have been shown in the motor cortex and spinal cord of rapidly progressive ALS (<2 years) (15, 90), but microgliosis in long‐duration cases has not been quantitated. We found that the total density of microglia cells was decreased within the anterior horn in long‐duration ALS. Density measurements of CD68‐positive cells were also decreased in the anterior horn in long‐duration ALS, but the difference was not significant perhaps due to decreased power in the subset of immunostained cases. Furthermore, the microglial morphology was suggestive of a less activated, resting phenotype in long‐duration ALS. This decreased microglial density and altered morphology correlated with the decreased level of neurodegeneration in the spinal cord. Overall, these data suggest an altered neuroinflammatory phenotype in long‐duration ALS that may play a role in the slow disease progression.
Moreover, the hypoglossal nuclei were relatively preserved in both standard and long‐duration ALS participants. There was also a decreased density of CD68‐positive cells in this region when compared to the pyramids and spinal anterior horns. We suspect that this decrease in activated microglia/macrophages is related to the relative preservation of bulbar functionality in both long and standard duration cases. At death, both long and standard duration ALS cases had higher scores of bulbar function on the ALSFRS when compared to fine motor and gross motor functions (Table 3). This functional preservation may be explained in part by the reduced neuroinflammatory state observed in both duration groups.
As demonstrated in our analysis of ALSFRS‐R assessments, participants with long and standard duration ALS had similar functional abilities in the terminal stage of their diseases. This suggests that patients with ALS have a similar end stage phenotype regardless of overall disease progression and neurodegenerative profile when using the ALSFRS‐R as a guided scale of disability (52, 55). We did not note any differences in initial overall ALSFRS‐R assessment score between duration groups. This was likely due to standard cases having an initial assessment closer to first symptom onset of ALS. The differences in time from symptom onset to initial ALSFRS‐R assessments among cases with an available ALSFRS‐R score limit the significance of this finding. Further analysis is needed to determine the relationship between ALS progression and the predictive capabilities of the ALSFRS‐R at initial assessment.
It is likely that multiple factors converge to mediate disease progression in ALS. For instance, KIF5A is a recently discovered, but rare genetic mutation associated with long‐duration ALS (67). In the present study, one case had a KIF5A mutation, but this was a standard duration case with a disease duration of 1.4 years. Additional unknown genetic as well as environmental moderating factors such as social support and the resilience of neuronal reserve may be involved in decelerating the pathological progression of ALS resulting in extended survival (22, 31, 71, 84). Future studies focused on genetic variation, gene expression and clinical care and support will be necessary to uncover mechanisms of disease progression in ALS.
Limitations
The VABBB cohort may be subject to referral bias as participants were informed of the study in VA neurology clinics, online and through word of mouth beginning in 2011 (12, 86). Enrollment into the VABBB is voluntary and therefore may not reflect the overall population of Veterans with ALS. We also recognize that support (eg, the initiation and duration of ventilation support) available to Veterans with ALS varies by region, healthcare accessibility and personal choice, thus affecting potential treatment outcomes. As noted previously, the majority of participants were enrolled from the VA National Registry and therefore represent prevalent rather than incident cases thus selecting for longer duration cases. Regardless, this presents an advantage to examine correlates of very long‐duration ALS in a relatively large cohort. Although previous studies have shown that semi‐quantitative assessments of white matter degeneration are related to clinical outcomes (4), future studies utilizing densitometry may also be helpful in evaluating white matter degeneration. The VA ALS Biorepository does not accept donations from civilians currently, limiting the generalizability of the VABBB population to the general ALS population. For example, the physical demands of the military may screen out more rapidly progressive variants that exist in the civilian population. Future collaborative studies would be able to address this issue. Although no statistically significant differences in overall military profile were found, it is possible we did not capture other factors characteristic of military service that have been shown to increase ALS incidence. These factors include but are not limited to chemical exposures, head trauma and environmental exposures (7, 12). Previous studies have also reported a positive correlation between body mass index and ALS survivability (70), and these data were not available in the current study. Last, we acknowledge that the distinction between long and short‐term survival in ALS is not biologically based and instead reflects current disease survival guidelines which may change over time.
Conclusions
In the largest reported collection of ALS cases with disease durations lasting longer than 10 years, we demonstrated pathological differences related to disease duration. Veterans with long‐duration ALS had an earlier age of symptom onset, slower initial disease progression and took longer to diagnose compared to Veterans with standard duration ALS. long‐duration ALS was associated with decreased lower motor neuron degeneration, less frequent TDP‐43 pathology and decreased microglial density within the motor system. Overall, an altered neuroinflammatory phenotype in long‐duration ALS may play a role in the slow disease progression. Elucidation of genetic and environmental modifiers of ALS disease duration may facilitate the discovery of targets to extend survival.
Authors contributions
Study design and conception: NWK, CBB, TDS. Acquisition and analysis of data: KRS, ZWF, NAR, DC, LG, JGA, SEW, IR, JDC, VEA, BRH, ACM, NWK, CBB, TDS. Drafting of the manuscript: KRS, ZWF, CBB, TDS. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
IRB approval for the brain donation program was obtained through the VA Boston Healthcare System.
Acknowledgements
This work was supported by the Department of Veterans Affairs, Veterans Health Administration, Biomedical Laboratory Research and Development Merit Awards, Veterans Affairs Biorepository (BX002466); Gulf War Veterans' Illnesses Biorepository (BX003063); Clinical Sciences Research and Development Merit Award (I01‐CX001038); National Institute of Aging (RF1AG054156, R56AG057768); National Institute of Aging Boston University AD Center (P30AG13846; supplement 0572063345‐5). We are grateful to Dr. Bryan Traynor (NIH—National Institute on Aging) for providing genetic testing on VABBB tissue samples, and to the Cooperative Studies Program (CSP) and CSP #500A, National Registry of Veterans with Amyotrophic Lateral Sclerosis for providing data on those participants who were previously enrolled in CSP #500A that later enrolled in the Veterans Affairs Biorepository Brain Bank. We gratefully acknowledge Kerry Cormier, Rebecca Mathias and Caroline Kubilus for the histological and immunohistochemical work as well as all the veterans and their families whose participation and contributions made this work possible. All content, statements, opinions or views are solely of the author(s) and do not reflect official views of the Department of Veterans Affairs or the National Institutes of Health.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9:617–628. [DOI] [PubMed] [Google Scholar]
- 2. Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH (2012) The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 124:339–352. [DOI] [PubMed] [Google Scholar]
- 3. Alexianu ME, Kozovska M, Appel SH (2001) Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 57:1282–1289. [DOI] [PubMed] [Google Scholar]
- 4. Alosco ML, Stein TD, Tripodis Y, Chua AS, Kowall NW, Huber BR et al (2019) Association of white matter rarefaction, arteriolosclerosis, and tau with dementia in chronic traumatic encephalopathy. JAMA Neurol 76:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alshikho MJ, Zurcher NR, Loggia ML, Cernasov P, Reynolds B, Pijanowski O et al (2018) Integrated magnetic resonance imaging and [(11) C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol 83:1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ (2016) Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun 7:12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beard JD, Engel LS, Richardson DB, Gammon MD, Baird C, Umbach DM et al (2017) Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis survival. PLoS One 12:e0185751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bigio EH (2008) Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blauwendraat C, Faghri F, Pihlstrom L, Geiger JT, Elbaz A, Lesage S et al (2017) NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol Aging 57:e9–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 12. Brady CB, Trevor KT, Stein TD, Deykin EY, Perkins SD, Averill JG et al (2013) The Department of Veterans Affairs Biorepository Brain Bank: a national resource for amyotrophic lateral sclerosis research. Amyotroph Lateral Scler Frontotemporal Degener 14:591–597. [DOI] [PubMed] [Google Scholar]
- 13. Brettschneider J, Arai K, Del Tredici K, Toledo JB, Robinson JL, Lee EB et al (2014) TDP‐43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol 128:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al (2013) Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM, Trojanowski JQ (2012) Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 7:e39216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377:162–172. [DOI] [PubMed] [Google Scholar]
- 17. Brownell B, Oppenheimer DR, Hughes JT (1970) The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry 33:338–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cedarbaum JM, Stambler N (1997) Performance of the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) in multicenter clinical trials. J Neurol Sci 152(Suppl 1):S1–S9. [DOI] [PubMed] [Google Scholar]
- 20. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A (1999) The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 169:13–21. [DOI] [PubMed] [Google Scholar]
- 21. Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH et al (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chio A, Bottacchi E, Buffa C, Mutani R, Mora G, Parals (2006) Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. J Neurol Neurosurg Psychiatry 77:948–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chio A, Calvo A, Moglia C, Mazzini L, Mora G, Group Ps (2011) Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 82:740–746. [DOI] [PubMed] [Google Scholar]
- 24. Chio A, Logroscino G, Hardiman O, Swingler R,Mitchell D, Beghi E et al (2009) Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler 10:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41:118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J et al (2012) Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One 7:e52941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cykowski MD, Powell SZ, Peterson LE, Appel JW, Rivera AL, Takei H et al (2017) Clinical significance of TDP‐43 neuropathology in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 76:402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Department of Veterans A (2009) Presumption of service connection for amyotrophic lateral sclerosis. Final rule. Fed Regist 74:57072–57074. [PubMed] [Google Scholar]
- 29. Diagnosis ETFo , Management of Amyotrophic Lateral S , Andersen PM, Abrahams S, Borasio GD, de Carvalho M et al (2012) EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol 19:360–375. [DOI] [PubMed] [Google Scholar]
- 30. Doorn KJ, Moors T, Drukarch B, van de Berg W, Lucassen PJ, van Dam AM (2014) Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun 2:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eisen A, Schulzer M, MacNeil M, Pant B, Mak E (1993) Duration of amyotrophic lateral sclerosis is age dependent. Muscle Nerve 16:27–32. [DOI] [PubMed] [Google Scholar]
- 32. Eng LF, Ghirnikar RS, Lee YL (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem Res 25:1439–1451. [DOI] [PubMed] [Google Scholar]
- 33. Engelhardt JI, Appel SH (1990) IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol 47:1210–1216. [DOI] [PubMed] [Google Scholar]
- 34. Engelhardt JI, Tajti J, Appel SH (1993) Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 50:30–36. [DOI] [PubMed] [Google Scholar]
- 35. Fatima M, Tan R, Halliday GM, Kril JJ (2015) Spread of pathology in amyotrophic lateral sclerosis: assessment of phosphorylated TDP‐43 along axonal pathways. Acta Neuropathol Commun 3:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, Rowland LP (2006) The natural history of primary lateral sclerosis. Neurology 66:647–653. [DOI] [PubMed] [Google Scholar]
- 37. Greenfield JG, Love S, Louis DN, Ellison D (2008) Greenfield's neuropathology, 8th edn. London: Hodder Arnold. [Google Scholar]
- 38. Haley RW (2003) Excess incidence of ALS in young Gulf War veterans. Neurology 61:750–756. [DOI] [PubMed] [Google Scholar]
- 39. Hall ED, Oostveen JA, Gurney ME (1998) Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23:249–256. [DOI] [PubMed] [Google Scholar]
- 40. Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W et al (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17085. [DOI] [PubMed] [Google Scholar]
- 41. Haverkamp LJ, Appel V, Appel SH (1995) Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 118 (Pt 3):707–719. [DOI] [PubMed] [Google Scholar]
- 42. Holness CL, Simmons DL (1993) Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81:1607–1613. [PubMed] [Google Scholar]
- 43. Horner RD, Kamins KG, Feussner JR, Grambow SC, Hoff-Lindquist J, Harati Y et al (2003) Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology 61:742–749. [DOI] [PubMed] [Google Scholar]
- 44. Huisman MH, de Jong SW, van Doormaal PT, Weinreich SS, Schelhaas HJ, van der Kooi AJ et al (2011) Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J Neurol Neurosurg Psychiatry 82:1165–1170. [DOI] [PubMed] [Google Scholar]
- 45. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57:1–9. [DOI] [PubMed] [Google Scholar]
- 46. Iwanaga K, Hayashi S, Oyake M, Horikawa Y, Hayashi T, Wakabayashi M et al (1997) Neuropathology of sporadic amyotrophic lateral sclerosis of long duration. J Neurol Sci 146:139–143. [DOI] [PubMed] [Google Scholar]
- 47. Janssens J, Wils H, Kleinberger G, Joris G, Cuijt I, Ceuterick-de Groote C et al (2013) Overexpression of ALS-associated p. M337V human TDP‐43 in mice worsens disease features compared to wild-type human TDP‐43 mice. Mol Neurobiol 48:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jara JH, Genc B, Stanford MJ, Pytel P, Roos RP, Weintraub S et al (2017) Evidence for an early innate immune response in the motor cortex of ALS. J Neuroinflammation 14:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kasarkis EJ, Dominic K, Oddone EZ (2004) The National Registry of Veterans with Amyotrophic Lateral Sclerosis: Department of Veterans Affairs Cooperative Studies Program (CSP) #500a. Amyotroph Lateral Scler Other Motor Neuron Disord 5(Suppl 1):129–132. [DOI] [PubMed] [Google Scholar]
- 50. Kasarskis EJ, Lindquist JH, Coffman CJ, Grambow SC, Feussner JR, Allen KD et al (2009) Clinical aspects of ALS in Gulf War veterans. Amyotroph Lateral Scler 10:35–41. [DOI] [PubMed] [Google Scholar]
- 51. Kaufmann P, Levy G, Montes J, Buchsbaum R, Barsdorf AI, Battista V et al (2007) Excellent inter‐rater, intra-rater, and telephone-administered reliability of the ALSFRS‐R in a multicenter clinical trial. Amyotroph Lateral Scler 8:42–46. [DOI] [PubMed] [Google Scholar]
- 52. Kaufmann P, Levy G, Thompson JL, Delbene ML, Battista V, Gordon PH et al (2005) The ALSFRSr predicts survival time in an ALS clinic population. Neurology 64:38–43. [DOI] [PubMed] [Google Scholar]
- 53. Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 140:691–707. [PMC free article] [PubMed] [Google Scholar]
- 54. Kim WK, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, Mitsumoto H (2009) Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology 73:1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kollewe K, Mauss U, Krampfl K, Petri S, Dengler R, Mohammadi B (2008) ALSFRS‐R score and its ratio: a useful predictor for ALS-progression. J Neurol Sci 275:69–73. [DOI] [PubMed] [Google Scholar]
- 56. Kushner PD, Stephenson DT, Wright S (1991) Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J Neuropathol Exp Neurol 50:263–277. [DOI] [PubMed] [Google Scholar]
- 57. Labra J, Menon P, Byth K, Morrison S, Vucic S (2016) Rate of disease progression: a prognostic biomarker in ALS. J Neurol Neurosurg Psychiatry 87:628–632. [DOI] [PubMed] [Google Scholar]
- 58. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH (2012) Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol 237:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCauley ME, Baloh RH (2019) Inflammation in ALS/FTD pathogenesis. Acta Neuropathol 137:715–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W et al (2009) Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 73:1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 63. Mitchell JD, Borasio GD (2007) Amyotrophic lateral sclerosis. Lancet 369:2031–2041. [DOI] [PubMed] [Google Scholar]
- 64. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nagy D, Kato T, Kushner PD (1994) Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J Neurosci Res 38:336–347. [DOI] [PubMed] [Google Scholar]
- 66. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 67. Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R et al (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nishihira Y, Tan CF, Hoshi Y, Iwanaga K, Yamada M, Kawachi I et al (2009) Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP‐43 pathology. Acta Neuropathol 117:45–53. [DOI] [PubMed] [Google Scholar]
- 69. Nishihira Y, Tan CF, Onodera O, Toyoshima Y, Yamada M, Morita T et al (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP‐43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182. [DOI] [PubMed] [Google Scholar]
- 70. Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM (2011) Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 44:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Paganoni S, Karam C, Joyce N, Bedlack R, Carter GT (2015) Comprehensive rehabilitative care across the spectrum of amyotrophic lateral sclerosis. NeuroRehabilitation 37:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pastula DM, Coffman CJ, Allen KD, Oddone EZ, Kasarskis EJ, Lindquist JH et al (2009) Factors associated with survival in the National Registry of Veterans with ALS. Amyotroph Lateral Scler 10:332–338. [DOI] [PubMed] [Google Scholar]
- 73. Pugliatti M, Parish LD, Cossu P, Leoni S, Ticca A, Saddi MV et al (2013) Amyotrophic lateral sclerosis in Sardinia, insular Italy, 1995–2009. J Neurol 260:572–579. [DOI] [PubMed] [Google Scholar]
- 74. Pupillo E, Messina P, Logroscino G, Beghi E, Group S (2014) long‐term survival in amyotrophic lateral sclerosis: a population-based study. Ann Neurol 75:287–297. [DOI] [PubMed] [Google Scholar]
- 75. Saberi S, Stauffer JE, Schulte DJ, Ravits J (2015) Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin 33:855–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sanagi T, Yuasa S, Nakamura Y, Suzuki E, Aoki M, Warita H et al (2010) Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J Neurosci Res 88:2736–2746. [DOI] [PubMed] [Google Scholar]
- 77. Schiffer D, Cordera S, Cavalla P, Migheli A (1996) Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139(Suppl):27–33. [DOI] [PubMed] [Google Scholar]
- 78. Scotter EL, Chen HJ, Shaw CE (2015) TDP‐43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12:352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670. [DOI] [PubMed] [Google Scholar]
- 80. Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 81. Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O (2003) Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996–2000. J Neurol Neurosurg Psychiatry 74:1258–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ et al (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 15:601–609. [DOI] [PubMed] [Google Scholar]
- 83. Van Damme P, Robberecht W, Van Den Bosch L (2017) Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech 10:537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van den Berg JP, Kalmijn S, Lindeman E, Veldink JH, de Visser M, Van der Graaff MM et al (2005) Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology 65:1264–1267. [DOI] [PubMed] [Google Scholar]
- 85. Walker DG, Lue LF (2015) Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Walt GS, Burris HM, Brady CB, Spencer KR, Alvarez VE, Huber BR et al (2018) Chronic traumatic encephalopathy within an amyotrophic lateral sclerosis brain bank cohort. J Neuropathol Exp Neurol 77:1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wei QQ, Chen Y, Chen X, Cao B, Ou R, Zhang L et al (2018) Clinical and prognostic features of ALS/MND in different phenotypes-data from a hospital-based registry. Brain Res Bull 142:403–408. [DOI] [PubMed] [Google Scholar]
- 88. Weisskopf MG, O'Reilly EJ, McCullough ML, Calle EE, Thun MJ, Cudkowicz M, Ascherio A (2005) Prospective study of military service and mortality from ALS. Neurology 64:32–37. [DOI] [PubMed] [Google Scholar]
- 89. Yousef A, Robinson JL, Irwin DJ, Byrne MD, Kwong LK, Lee EB et al (2017) Neuron loss and degeneration in the progression of TDP‐43 in frontotemporal lobar degeneration. Acta Neuropathol Commun 5:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang R, Gascon R, Miller RG, Gelinas DF, Mass J, Hadlock K et al (2005) Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol 159:215–224. [DOI] [PubMed] [Google Scholar]
- 91. Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S et al (2013) Inflammatory components in human Alzheimer's disease and after active amyloid-beta42 immunization. Brain 136(Pt 9):2677–2696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.