

Abstract
The phosphorylation states of RNA polymerase II coordinate the process of eukaryotic transcription by recruitment of transcription regulators. The individual residues of the repetitive heptad of the C-terminal domain (CTD) of the biggest subunit of RNA polymerase II are phosphorylated temporally at different stages of transcription. Intriguingly, despite similar flanking residues, phosphorylation of Ser2 and Ser5 in CTD heptads play dramatically different roles. The mechanism of how the kinases place phosphorylation on the correct serine is not well understood. In this paper, we use biochemical assays, mass spectrometry, molecular modeling, and structural analysis to understand the structural elements determining which serine of the CTD heptad is subject to phosphorylation. We identified three motifs in the activation/P+1 loops differentiating the intrinsic specificity of CTD in various CTD kinases. We characterized the enzyme specificity of the CTD kinases—CDK7 as Ser5-specific, Erk2 with dual specificity for Ser2 and Ser5, and Dyrk1a as a Ser2-specific kinase. We also show that the specificities of kinases are malleable and can be modified by incorporating mutations in their activation/P+1 loops that alter the interactions of the three motifs. Our results provide an important clue to the understanding of post-translational modification of RNA polymerase II temporally during active transcription.
Graphical Abstract
INTRODUCTION
The accurate placement of phosphorylation marks by kinases is essential for precision in cellular signal transduction. This is particularly true in eukaryotic transcription, in which the phosphorylation at the C-terminal domain (CTD) of the largest subunit of RNA polymerase II, Rpb1, correlates to different transcriptional stages.1–3 This domain is a unique region only found in RNA polymerase II but not I or III. The apparently simple heptad (Y1S2P3T4S5P6S7) is highly conserved throughout eukaryotes, with the number of repeats varying in organisms. Even though it does not carry out any enzymatic activity, the loss of CTD leads to cell death,4 indicating that the coordination and regulation of transcription mediated by CTD is crucial for the active transcription inside the cells.5
Precise positional phosphorylation on the heptad repeat is vital for the orchestration of active transcription.6,7 Phosphorylation at Ser2 or Ser5 of the heptad repeats is found in every single round of transcription mediated by RNA polymerase II.8 While RNA polymerase II free of phosphorylation binds to the promoter, Ser5 is phosphorylated during initiation of transcription and mRNA capping.9,10 During the transition from initiation to productive elongation, Ser5 sites gradually become dephosphorylated while Ser2 sites are phosphorylated when the promoter-proximal pausing is released.11–13 At the end of transcription, all remaining phosphate groups on the CTD are removed since nonphosphorylated CTD is required for RNA polymerase II to recycle and bind to promoters again.14 The timely phosphorylation and dephosphorylation of Ser2/Ser5 are essential for eukaryotic transcription as omission or defects in the proteins involved in the CTD modification directly lead to severe growth defects, even causing cell death.3
With the high demands for accurate phosphorylation on CTD during transcription, it is puzzling how CTD kinases distinguish the highly similar recognition motifs once they are recruited to RNA polymerase II. The enigma is particularly notable for Ser2 and Ser5, both of which are SP motifs recognized by cyclin-dependent kinases (Y1S2P3T4S5P6S7), yet their phosphorylation leads to recruitment of late and early transcription regulators, respectively.7 Obviously, an error in the placement of phosphate on Ser2 versus Ser5 will result in errors in the transcription process.15 Compounded with the high requirement for accuracy at Ser2 versus Ser5 phosphorylation, variation from the consensus sequence occurs during evolution at some positions in more complicated eukaryotes. While most of the 26 repeats in yeast match the consensus heptad, only 21 of the 52 heptad repeats match the canonical consensus sequence in humans, owing to a deviation most frequently occurring at the seventh position. This diverging stretch lies closer to the C-terminal of the CTD, called the distal region (Supporting Information Figure S1A).
To understand this high precision of phosphorylation at the molecular level, we investigated the substrate specificity of the CTD kinases. The kinase module of transcription factor II human (TFIIH), called Cyclin Dependent Kinase (CDK)-activating kinase (CAK), is recruited to RNA polymerase II at the beginning of transcription as part of the TFIIH complex.16 CAK is a heterotrimer complex formed by the kinase subunit CDK7 with its associated cyclin H and then stabilized by menage à trois homolog (MAT1).17 At the initiation stage of transcription as part of the general transcription TFIIH, CAK phosphorylates Ser5 and Ser7 of CTD to recruit the capping of mRNA and stimulate the polymerase to dissociate from the promoter after initiation of transcription.18 In addition to CDK7, mitogen-activated protein (MAP) kinase, Erk2, was identified to phosphorylate Ser5, priming developmental genes in mouse embryonic stem cells.19 Other kinases such as CDK9 and dual specificity tyrosine kinase 1A (Dyrk1a) are also reported to be responsible for the phosphorylation of Ser residues of CTD, even though discrepancy of their specificity exists.20,21
In this manuscript, we investigate the structural elements that determine the site of phosphorylation in CTD kinases. Previously, we have shown that CAK, the kinase module of TFIIH, has a strong preference toward Ser5 of CTD in the consensus sequence.15 Herein, we mapped the phosphorylation of CAK on human distal CTD (heptad number 26 to 52) and characterized the specificity of CAK on the distal region of human CTD using ultraviolet photodissociation (UVPD) mass spectrometry (MS) to provide single residue resolution of phosphorylation. Although the seventh residue in the distal CTD heptads tends to deviate from consensus, it does not seem to affect the placement of phosphate groups on Ser5 by CAK. Sequence and structural analyses of the kinase subunit CDK7 and Erk2 reveal three regions close to the activation/P+1 loops as major determinants affecting the substrate selection of Ser2/Ser5. Modeling and structural analysis predicted that novel CTD kinase Dyrk1a favors Ser2 over Ser5 as a substrate, which is validated by mass spectrometry experiments. Our work reveals key elements of CTD kinases that determine the substrate specificity for the CTD of RNA polymerase II.
RESULTS
Sites of Phosphorylation by CAK on Human CTD.
We previously have shown that CAK phosphorylates almost exclusively at Ser5 in consensus sequences of the CTD substrate.22 The high specificity is surprising since both Ser2 and Ser5 are within one SP motif.23,24 We speculate that the flanking residue of these two SP motifs might differentiate them upon CAK phosphorylation. The sequence of human CTD in the distal region (heptads 27–52) provides a compelling platform to evaluate the selectivity of CAK (Figure 1A). In humans, a variety of residues replaces the serine residue present at the seventh position of the heptad repeat (Figure 1A). Thus, we can test whether the CAK will exhibit altered specificity with different residues at the seventh position using human distal CTD as the substrate.
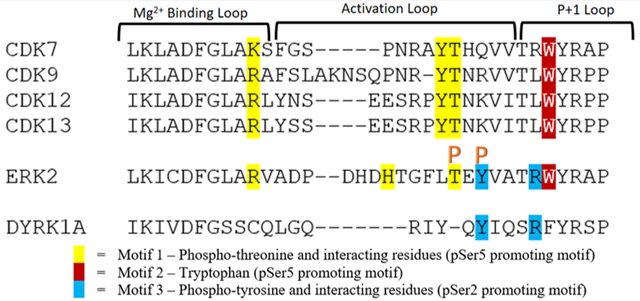
Figure 1.
Mapping the phosphorylation sites of human distal CTD treated with CDK7. (A) Human distal CTD sequence is arranged with each repeat vertically. Numbers on the vertical axis indicate residue numbers in every heptad of the CTD. Numbers on the horizontal axis indicate the repeat number of human distal CTD. Dark blue highlights major phosphorylation species. Light blue indicates minor phosphorylation species. Light gray shade shows regions not covered in mapping. Blue vertical solid line marks the boundaries of peptide fragments generated by trypsin digestion. (B) Liquid chromatography traces of each peptide showing monophosphorylated species. The numbers above each sequence and each chromatographic peak correspond to the specific repeats of the CTD sequence. Vertical dashes are used to demarcate each repeat of the CTD. The quantification for peptides is shown as a pie chart with a color-coded percentage for each peptide product. The bottom right panel shows two repeat numbers corresponding to the same sequence due to their identical fragmentation in the distal CTD map. Thus, the two diheptads would be indistinguishable from each other, and the resultant LC trace is a sum of both the diheptads.
To identify the site of phosphorylation by CDK7 in distal CTD, we used high-resolution mass spectrometry. Previously, the sites of CTD phosphorylation were identified using immunoblotting.6 However, antibodies cannot identify the exact positions of the repeat upon phosphorylation. Furthermore, since the antibodies are developed to recognize specific consensus sequences, the recognition of phosphorylation sites in the distal human region can vary in binding strength when the sequence deviates from consensus, making quantification unreliable. Finally, the amount of different phosphorylation species cannot be quantified using different antibodies. We developed a method based on liquid chromatographic separation of phosphorylated peptides followed by UVPD mass spectrometry to identify the specific position of each phosphorylation site.15,22 Traditional MS/MS methods used for peptide fragmentation in proteomic workflows, such as collision-induced dissociation (CID), typically promote cleavages of weak chemical bonds, ultimately causing preferential loss of labile modifications such as phosphorylation. UVPD uses high-energy photoabsorption for ion activation, resulting in the production of numerous diagnostic sequence ions and retention of labile modifications.25 The preservation of post-translational modifications allows them to be tracked as unambiguous mass shifts in the fragment ions, permitting confident localization during MS/MS analysis. We have successfully utilized this strategy to map the phosphorylation sites of multiple kinases toward S. cerevisiae, Drosophila, and human CTDs.15,22,26,27
We generated a construct of human distal CTD, including heptads 26–52, N-terminal-tagged with glutathione-S-transferase (GST) for solubility, and 6X His tag for purification purposes (Figure 1A). We treated the construct with CAK (the kinase module of TFIIH) in vitro, cleaved off the GST protein with 6X His tag, and digested the distal CTD into distinct peptides using trypsin. Unlike the consensus CTD heptad sequence, human distal CTD has multiple lysines and one arginine distributed along the sequence, creating six unique peptides after tryptic digestion (Figure 1A). UVPD mass spectrometry characterizes the phosphorylated peptides with nearly comprehensive coverage of repeats 26–49, with only heptads 39 and 40 missing from the final phosphoryl mapping (Figure 1A). The first glance of the phosphorylation identification reveals a high selectivity of Ser5 for phosphorylation for CAK (Figure 1A and Figures S2–S5). For all 22 sites of phosphorylation detected using LC–UVPD-MS, the only exception to Ser5 phosphorylation occurs for repeat 42, where a small amount of phosphoryl-Ser2 is detected instead (Figure 1A). This repeat is the only heptad where Ser5 is replaced by a Thr residue. This result indicates that the kinase module of TFIIH strongly prefers the phosphorylation of Ser5–Pro6 over Ser2–Pro3 as long as there is one Ser5 available, and Thr is a much less preferred substrate than Ser.
Direct quantification of the phosphorylation of each site over the whole series of 27 heptads is not possible. Yet, the trypsin digestion of the human distal CTD produces several peptides containing one to six heptads (Figure 1A). For each resulting peptide, the relative amount of each phosphorylated heptad can be quantified to provide the relative preference of Ser5 when different residues occupy the seventh position (summarized as percentages in the pie charts in Figure 1B). These percentages are highly consistent among biological duplicates. In human distal CTD, the residues locating at the seventh position of the heptad include Asn, Thr, Arg, Lys, Glu, and Val (Figure 1A). None of the amino acid replacements at the seventh position prevent Ser5 phosphorylation (Figure 1A). Yet, it seems that small polar amino acids in the seventh position are preferred for the phosphorylation of nearby Ser5. In all fragments, the priority for CDK7 phosphorylation is Ser5 with the flanking seventh residue as Ser or Thr (the only exception is repeat 37 with an E at the seventh position). When a bulky residue—such as Lys (in repeats 35, 38, 45, 47, and 49) or Val (in repeat 44)—occupies the seventh position, the preceding Ser5 is less favored to be phosphorylated (Figure 1A). The CAK phosphorylation on distal CTD generates a diverse landscape for the recruitment of transcription regulators with Ser5 phosphorylation in combination with various seventh residues.
We next asked whether the residue at the fourth position affects the phosphorylation of Ser5 in distal human CTD (Figure 1A). In all repeats of human distal CTD, the fourth position is always occupied by a small polar residue like Ser/Thr, with the only exception being at repeat 32, which contains a much bulkier Gln residue. The heptad-repeat-containing Gln at the fourth position still undergoes phosphorylation at Ser5, but we only observed the phosphorylated species when its flanking heptad repeats are phosphorylated first (Figure S6). Thus, neither the fourth nor the seventh residue on the heptad repeat seems to prevent CAK phosphorylation, at least for the physiologically relevant replacements. Since the first position of the arbitrary heptad repeat is consistently preserved as tyrosine in distal human CTD (and that of RNA polymerase II of most species), the effect of Tyr1 variation cannot be derived from this phosphorylation mapping.
Structural Analyses for the Specificity of CDK7 toward Ser5 of CTD.
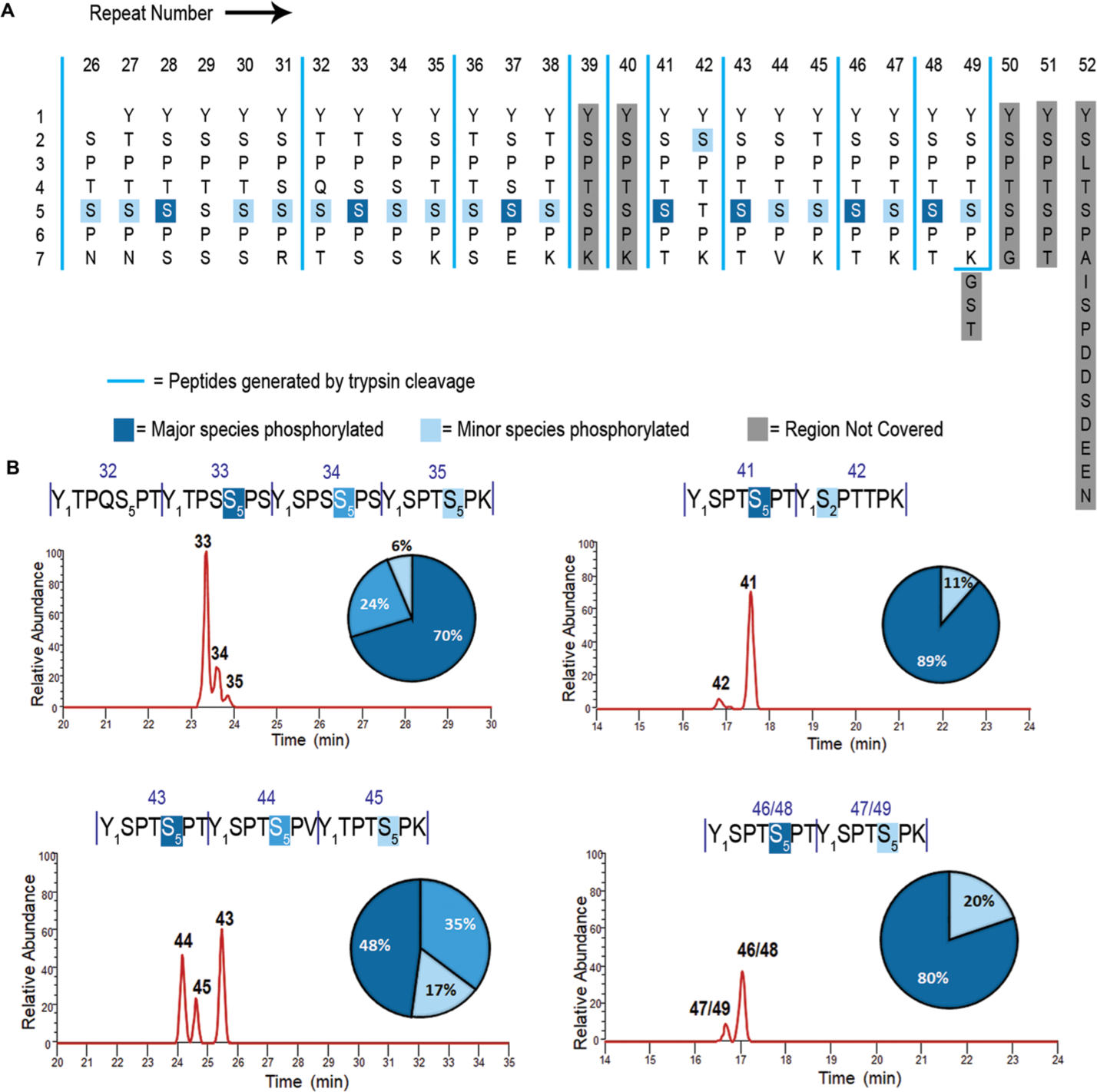
The kinase module of TFIIH (CAK) shows remarkable consistency in phosphorylating Ser5 in the human distal CTD indifferent to the nearby fourth or seventh residues. To understand this high selectivity, we analyzed the structure of the kinase subunit CDK7 and investigated the structural elements that determine this strong preference of Ser5 over Ser2. A tertiary complex of apo CAK, containing CDK7 with cyclin H and MAT1, has been determined (PDB code: 1UA2).28 However, the complex structure of CDK7 bound to peptide substrates is elusive due to multiple reasons. First, the interaction between kinases and their substrate ligands is weak and transient during the phosphoryl-transfer reaction, usually insufficient to be captured by X-ray crystallography that requires stable complex formation. Second, the purification of CAK through recombinant protein expression has been technically challenging. Thus, the CAK heterotrimer is only available through endogenous purification with limited quantity and purity. On the contrary, high sequence and structural conservation are maintained in cyclin-dependent kinases (CDKs). Thus, the conformational information on CDK7-substrate binding can be reliably derived from the interaction of other cyclin-dependent kinases. The superimposition of CDK7 with other cyclin-dependent kinases reveals that the overall fold is highly conserved within the kinase domains of CDK family, with the only structural variation occurring at the activation and P+1 loops at the transition of N- to C-terminal domains of the kinase where the putative substrate-binding site locates.29–31 The conformations of these consecutive loops are varied in inactive CDKs, but once activated, they adopt a similar configuration (Figure 2A).32 In the only available structure for CDK7 kinase, the activation loop adopts a conformation occupying substrate-binding groove and precluding substrate recognition (Figure 2B). Thus, it is reasonable to assume that the structure of CDK7, 1UA2, is that of an inactive form.28,32
Figure 2.
Structural and sequence analyses of CDKs. (A) Structural alignment of activated CDKs zoomed into the active sites: CDK2 (PDB code: 2CCI, light green), CDK9 (PDB code: 6GZH, orange), CDK12 (PDB code: 4CXA, teal), and CDK13 (PDB code: 5EFQ, pink). Structural elements such as the catalytic loop, activation loop, and the P+1 loops are highlighted. Catalytic residues lysine (K), asparagine (N), and phosphoryl-threonine (p-Thr) are shown as the sticks. (B) Structural alignment of CDK2 (PDB code: 2CCI, red) and CDK7 (PDB code: 1UA2, gray) showing differences in the conformation of activation and P+1 loops. The image on the right is a close-up view of the active site with a peptide ligand for CDK2 shown in sticks with carbon atoms colored yellow, nitrogen atoms colored blue, and oxygen atoms in red. A phospho-threonine residue is shown as sticks on both enzymes for comparing the structural differences. (C) Surface representation of the model for the substrate-binding groove active CDK7 with Ser5 phosphorylated. The color scheme for the substrate is identical to that of panel b. The inset is the sequence alignment of the activation/P+1 loops of CDK2 and CDK7: ‘|’ = fully conserved residue, ‘:’ = conservation of residues with strongly similar properties, ‘.’ = conservation of residues with weakly similar properties; no character indicates no conservation. Numbers indicate the positions of the initial and final amino acids.
A crystal structure of substrate ligand with CDK7 is elusive due to the weak and transient interaction between CDK7 and substrate. Furthermore, even if a crystal can be obtained, it is unlikely to capture the binding of CDK7 with nonfavorable substrate crystallographically. To directly compare the mode of recognition between favored versus unfavored substrates at the active state, we modeled the CTD peptides in the active site of CDK7 with either Ser5 or Ser2 as the residue subject to phosphorylation. To understand the binding of the CTD when CDK7 is active, we first aligned the CDK7 sequence with other cyclin-dependent kinases with active conformations (Figure 2A). We found that CDK2 has the most sequence similarity in the region where the substrate is bound (Figure 2C). A structure of CDK2 in its active configuration with substrate peptide bound has been published (PDB code: 2CCI).33 We used this consensus backbone configuration of this active site loop in CDK2 as a model of activated CDK7 (Figure 2C). The ligand-binding mode and orientation in CDK2 are identical to other peptide-bound CDKs (PDB code, 1QMZ34 and PDB code, 1GY335), indicating high conservation. Assuming CTD peptide bound to CDK7 using the same orientation as all other CDKs, we first manually modeled in the CTD sequence with either Ser5 or Ser2 as the residue subject to phosphorylation. The models were then energy minimized by Maestro36,37 assuming little change in the backbone position but with the side chains of amino acid flexible to adopt alternative conformations. Inspection of the models manually or with validation programs like MolProbity suggests that the model is reasonable with no steric clashes. In particular, when Ser5 is located at the active site as the phosphorylatable residue, the flanking proline residues locate in well-formed hydrophobic pockets (Figure 2C).
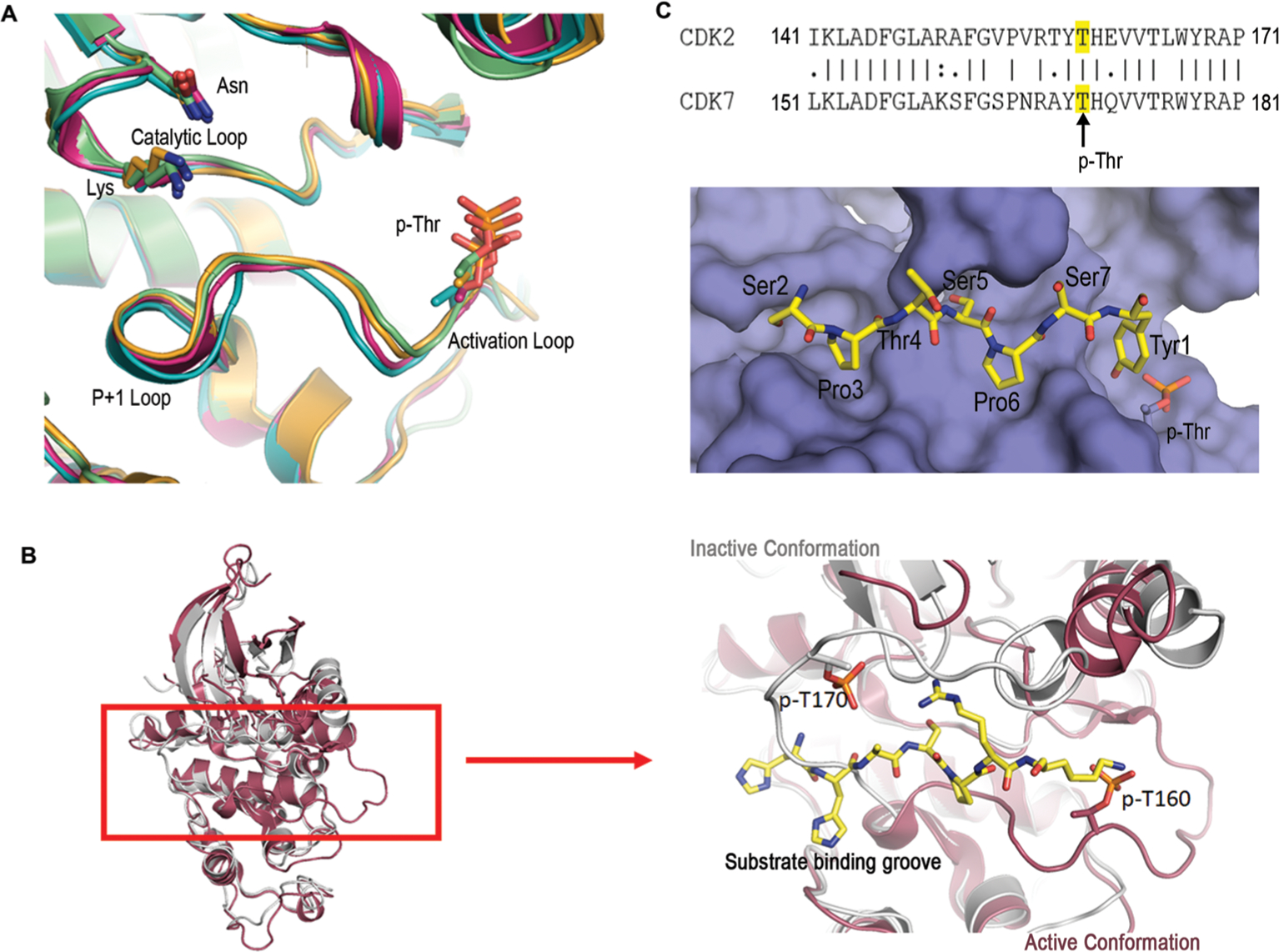
Careful inspection of the models reveals two significant potential interactions between CDK7 and the CTD when Ser5 is subject to phosphorylation (Figure 3A). First, Tyr1 in the next repeat is interacting with the side chain of the phosphorylated Thr in the activation loop (Figure 3A). This interaction is further reinforced with Lys150 forming a salt bridge to the phosphoryl-Thr in the activation loop (Figure 3B). In turn, the position of Lys150 forms a cation-π interaction with Tyr159 (Figure 3B). The triad formation of a favorable hydrophilic interaction network strengthens the favorable bond formation between Tyr1 in CTD and CDK7 activation Thr. However, this interaction is missing if Ser2 is the residue subject to phosphorylation with a serine residue at this position too far away to form favorable interaction with p-Thr170 (5.9 Å) (Figure 3C). The second major difference between the Ser5- and Ser2-bound models concerns tryptophan (Trp167) conserved in CDKs hydrophobically stacking with proline residue of the CTD when bound to Ser5 at the active site (Figure 3A). However, if Ser2 is subject to phosphorylation, this hydrophobic tryptophan would interact with the hydrophilic Ser7 (Figure 3C). This configuration would also extend Pro6 into a hydrophilic pocket formed by Glu99 and Lys102, a highly unfavorable environment for hydrophobic prolines (Figure S7). Therefore, our structural models suggest the different structural interactions between the CTD peptides and CDK7, thus explaining the differentiation of Ser5 over Ser2.
Figure 3.
Structural modeling and analysis of CDK7 with the CTD substrates. (A) Model of the active site interactions of CDK7 (violet) with a substrate primed for Ser5 phosphorylation. The modeled CTD peptide with Ser5 as the phosphorylable residue is shown as sticks with carbon atoms colored yellow, nitrogen atoms as blue, and oxygen atoms as red. Catalytic residues K139 and N141 are shown as sticks. (B) Interaction network stabilizing p-Thr in the activation loop. (C) Model of the active site interactions of CDK7 with a substrate primed for Ser2 phosphorylation. The modeled CTD peptide with Ser2 as the phosphorylable residue is shown as sticks with the same color scheme except the carbon atoms are colored orange. (D) Surface representation of CDK7 with a substrate primed for Ser5 phosphorylation with different residues at the seventh position. The color scheme for different side chains of the residues at the seventh positions is lysine (blue), aspartate (pale pink), glutamate (red), asparagine (magenta), and threonine (light blue). (E–G) MALDI-TOF analysis of five-repeat (5X) CTD variants treated with CDK7. Labels indicate the mass differences corresponding to the number of phosphates (P). (E) 5X CTD with consensus repeats treated with CDK7. (F) 5X CTD with all tyrosines mutated to phenylalanines treated with CDK7. (G) 5X CTD with all tyrosines mutated to histidines treated with CDK7.
Our CDK7 structural models rationalize the high specificity of CAK toward Ser5 of RNA polymerase II, disregarding the flanking seventh residue identity. In our mass spec phosphoryl-mapping, we noticed that the identity of the seventh position has little effect on the phosphorylation of Ser5 (Figure 1). In the model of Ser5 bound by CDK7, the side chain of any residue located at the seventh position extends outward of the enzyme (Figure 3D). Varying the identity of the seventh residue close to the Ser5 will not prevent the phosphorylation by CDK7, although a bulkier residue seems to be less favorable due to steric clashes (Figure 3D). This structure model adequately explains the high specificity of CAK that we observed in mass spec experiments and the slight preference for a small polar residue at the seventh position (Figure 1). The interactions formed by Tyr1 with phosphoryl-Thr in the activation loop and hydrophobic stacking of the proline of CTD with Trp in the P+1 loop seem key to the substrate recognition.
Tyrosine Is Essential for Substrate Recognition by the CDK7.
In our structural analysis of CDK7 binding to CTD, the interaction of Tyr1 and activation/P+1 loops of CDK7 is essential for substrate recognition (Figure 3B). To validate our model and evaluate the role of Tyr1 in substrate recognition by CDK7, we generated a series of CTD variants with five repeat peptides with every tyrosine replaced (Figure 3E–G). We monitored the number of phosphates added using matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry (Figure 3E–G). While CDK7 adds four phosphates to the wildtype five repeats CTD (Figure 3E), the variant with Tyr mutated to Phe reduced the number of phosphates added to the peptide by two when the reaction was carried out under identical conditions and for the same reaction time (Figure 3F). The reduction of activity is understandable since the removal of the hydroxyl side chain of Tyr loses the potential for strong polar interaction with phosphoryl-Thr in CDK7. Histidine replacement of Tyr1 resulted in only one phosphate added to the peptide (Figure 3G). Histidine mutation increases the distance between the first residue of heptad with phosphoryl-Thr from CDK7 to 3.6 Å, too far for the favorable hydrogen bond to form. This mutation result is consistent with our previous results when mutation of Tyr to Ala lost any phosphorylation on the CTD.22 Thus, the interaction between Tyr1 and CDK7 plays a critical role in Ser5 recognition, as predicted by our structural analysis.
CTD Residue Preferences by Erk2.
We next asked whether these structural elements exist in other CTD kinases. If so, we can use them to predict the Ser2/Ser5 preference for CTD kinases whose specificity is unknown. Both these two motifs are conserved in other CDKs that phosphorylate CTD (CDK9, CDK12, and CDK13) (Figure 4A). Indeed, it has been reported from multiple studies that in vitro these CDKs phosphorylate Ser5 in CTD substrate with no prior phosphorylation.15,22,44
Figure 4.
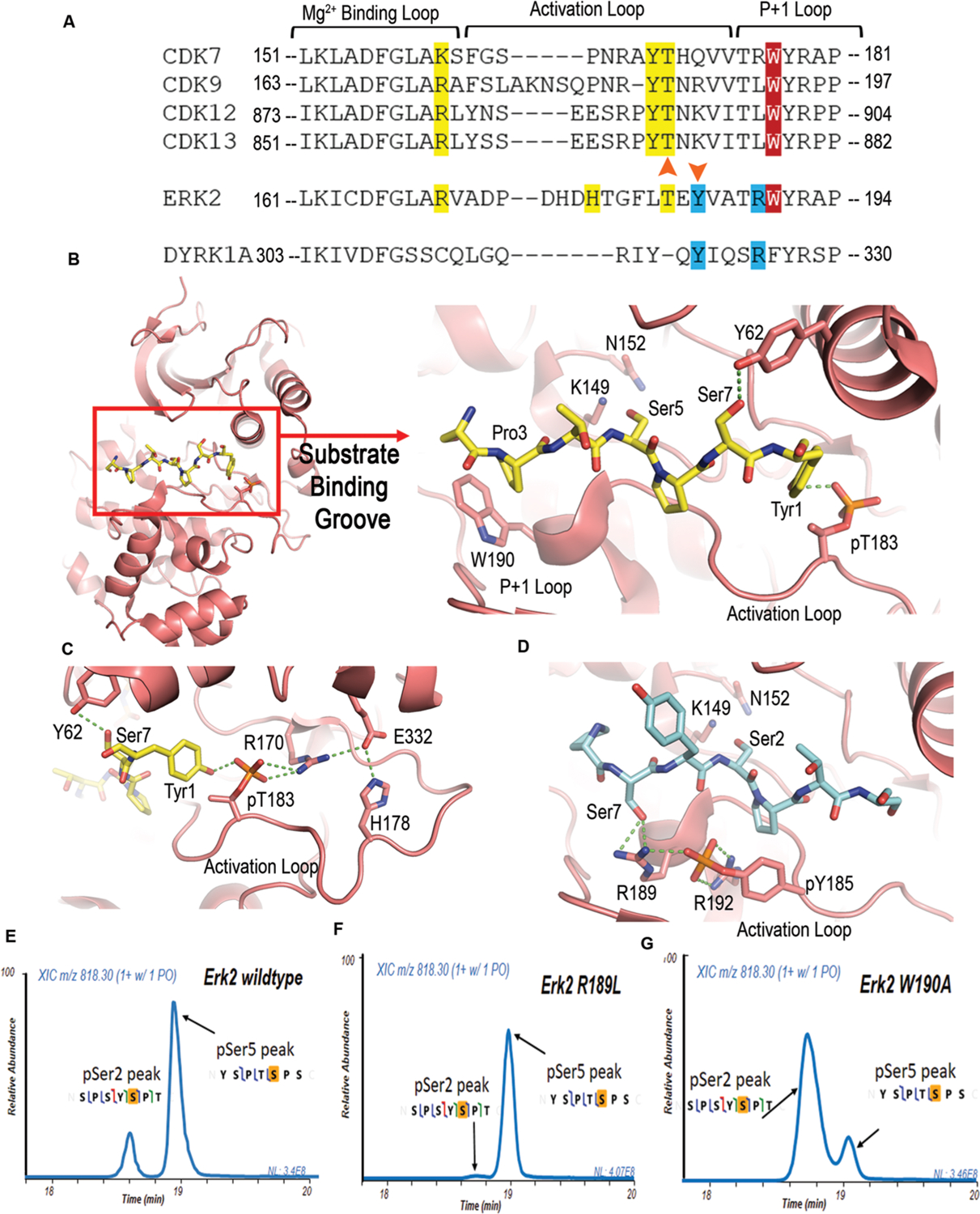
Sequence alignment and analysis of CDKs with Erk2 and Dyrk1a. (A) Sequence alignment of the active loop of CDKs with Erk2 and Dyrk1a, showing the three motifs in the activation/P+1 motifs for specificity. Motif one contains phosphoryl threonine and interacting residues and is colored in yellow. Motif two is highlighted in red and shows the conserved tryptophan. Motif 3 is colored in blue with phosphoryl tyrosine and interacting residues. Orange arrowheads highlight the phosphorylation sites. Amino acid numbers are labeled from the N-terminal to C-terminal. (B) Structure model of the active site interactions of Erk2, colored in salmon, bound to a CTD peptide with Ser5 as the phosphorylable residue. The peptide is shown as sticks with yellow color for carbons, blue color for nitrogen atoms, and red color for oxygen atoms. Catalytic residues K149 and N152 are shown as sticks. (C) Hydrogen bonding network of the interaction between CTD and Erk2 in the model of panel b. (D) Model of Erk2 recognizing Ser2 of CTD. The CTD peptide is shown as sticks with the same color scheme except with the carbon atoms colored light blue. (E) Liquid chromatography traces of 26 repeat CTD with consensus sequence treated with wild type and mutants—R189L and W190A Erk2 indicating phosphoryl-Ser2 and phosphoryl-Ser5 species. The MS/MS mapping is shown with the phosphorylated residue highlighted in orange.
These structural elements are also found in MAP kinase Erk2, which phosphorylates Ser2/5 of CTD during the development of embryos.19 Erk2 modulates transcription in a gene-specific fashion by priming Pol II at Erk2-targeted developmental genes. In the transcription of such genes, Erk2 replaces TFIIH in phosphorylate Ser5 and poises Pol II for their transcription.19 Previously, we have demonstrated by mass spectrometry and mutagenesis that Erk2 can phosphorylate both Ser2/Ser5 in vitro although it strongly prefers Ser5.22 The superimposition of Erk2 and CDK7 reveals that the two major regions for Ser5 recognition are both retained in Erk2 in sequence (Figure 4A). The superimposition of the active Erk2 structure with CDK shows an almost identical substrate-binding groove as activated CDKs (Figure 4B). The phosphoryl-Thr (p-Thr183) in the activation site loop can interact with Tyr1 when Ser5 is in the active site (Figure 4B,C). A salt bridge between p-Thr183 and Arg170 further stabilized the interaction (Figure 4C). The hydrophobic interaction between Trp190 in the P+1 loop of the kinase and Pro3 of CTD heptads is also preserved (Figure 4A,B). Both interactions are lost if Ser2 locates at the position of phosphorylation. Instead, a surprising interaction can be gained between the side chain of Ser7 with Arg189 in Erk2 (Figure 4D). A salt bridge with a phosphoryl-Tyr residue and Arg189 strengthens the interaction (Figure 4D). Unlike CDKs, Erk2 is a dual-activating kinase with two phosphorylation sites for the optimal activity (Figure 4A).19,38 Other than the Thr residue conserved with CDKs (p-Thr183 in Erk2), a phosphoryl-Tyr, a few residues downstream of p-Thr183 in the activation loop, stabilizes the active configuration of kinases (Figure 4A). The salt bridge of Arg189 and phosphoryl-Tyr185 indeed stabilizes the active site configuration of Erk2, promoting the recognition of Ser2 as the phosphorylation site (Figure 4D). On the basis of the model, we speculate that the interaction of Ser2 peptide with Arg189 partially compensates for the loss of favorable interaction found in the Ser5 peptide recognition, thus making Ser2 a possible substrate (although not a preferred one). This modeling analysis is consistent with our observation in mass spectrometry that Erk2 phosphorylates Ser5 but can use Ser2 as a substrate too. The existence of both activities suggests that by adjusting the interaction of peptide substrates with Erk2, the preference toward Ser5 over Ser2 can potentially be reversed.
We tested our models of kinase recognition by engineering Erk2 interaction with its substrates to alter its activity and/or specificity toward Ser2/5 of the CTD. The recognition mode of Erk2 toward Ser2 depends on the interaction of flanking Ser7 with Arg189 and its salt bridge with phosphoryl-Tyr185. We thus generated a leucine mutant replacing Arg189 to interrupt the recognition of Ser2 by Erk2 (Figure 4D). Leucine residue locates at this position in some of the CDKs, such as P-TEFb, CDK12, and CDK1 (Figure 4A). The Erk2 R189L variant is well folded and exhibits robust kinase activity. The products of the kinase reaction with GST-tagged yeast CTD were analyzed using mass spectrometry. Two products of monophosphorylated heptad were identified in both wild-type and R189L variant reactions (Figure 4E,F). The major peak is a Ser5 phosphorylated heptad, whereas the small peak is identified as a Ser2 phosphorylated heptad (Figure 4E,F). However, the phosphoryl-Ser2 product is substantially less in the R189L variant reaction with only 2% of the overall product (Figure 4F), whereas it accounts for ~20% of overall product in the wild-type reaction (Figure 4E). Thus, Erk2 R189L is highly specific toward Ser5 of the CTD. This result is consistent with our structural model in which the interaction between Erk2 Arg189 with the substrate is key to Ser2 recognition as the phosphorylation site.
Since wild-type Erk2 can recognize both Ser2 and Ser5 of the CTD as the substrate, we next asked whether we could rewire the interaction network and convert its preference from Ser5 to Ser2. Our design was to attenuate the favorable interaction in the Ser5-interacting configuration to promote the Ser2 phosphorylation. Since the activation loop is critical for the kinase activity of Erk2,38 we generated a mutation replacing Trp190 to Ala to weaken the hydrophobic interaction between Trp190 and Pro residues of the substrate (Figure 4B). The kinase reaction products of the Erk2 Trp190Ala variant were resolved by liquid chromatography, and then, the phosphorylation sites were identified using UVPD-mass spectrometry (Figure 4G). The same two peaks appear for the monophosphorylated products but showed a dramatically different profile from those observed upon reactions using wild-type Erk2 (Figure 4G). The major peak corresponds to a Ser2 phosphorylation heptad (~75% of the overall product), 3-fold higher in intensity than the heptad with Ser5 phosphorylation (Figure 4G). The Trp hydrophobic interaction with proline in the heptad affords a crucial interaction for the differentiation of Ser5/2. Importantly, the selectivity of Ser2/Ser5 in CTD kinases is malleable and has the potential to be altered and regulated inside the cells.
Dyrk1a is a Ser2-Specific Kinase.
The flexibility in Ser2/Ser5 preference in Erk2 motivated us to search for CTD kinases with different specificity. Dyrk1a is a newly identified CTD kinase whose function is associated with critical cellular functions.21,39,40 Dyrk1a belongs to a branch of the CMGC serine/threonine kinase family,41 famous for its implication in Down Syndrome as one of the proteins located on the “Down Syndrome Critical Region” of the chromosome 21 overexpressed in patients.42,43 Dyrk1a has an extensive portfolio of substrates with most located in the cytosol.44 Recently, its nuclear targets have started to be recognized, with RNA polymerase II being highly abundant.21 In vitro experiments showed that Dyrk1a could phosphorylate Ser2/Ser5 of RNA polymerase II CTD.21
We analyzed the sequence of Dyrk1a for its potential interaction with CTD (Figures 4A and 5A). Intriguingly, both motifs contributing to the Ser5 recognition in CDK7 are absent in Dyrk1a (Figures 4A and 5A). Instead of phosphoryl-Thr found in most CMGC kinases, only tyrosine is phosphorylated in Dyrk1a at the activation loop, corresponding to the second activation site in Erk2 that contributes to the stabilization of the active configuration for substrate recognition of Ser2. A complex structure of Dyrk1a bound to the substrate peptide (PDB code: 2WO6)45 reveals the orientation and configuration for substrate recognition. Assuming the CTD peptide would bind to the kinase in identical configuration, we manually replaced the peptide ligand with the CTD sequence. We then conducted Maestro energy minimization to optimize the configuration. The models show reasonable configuration with no steric clashes detected by MolProbity (Figure 5A,B). In the Dyrk1a structure, the Thr residue, whose phosphorylation is key to kinase activation in CDKs and other MAPK, is replaced by a tyrosine residue.45 This replacement prevents the possibility of the favorable hydrogen bonding between Tyr1 of CTD and phosphoryl-Thr on the kinase activation loop upon Ser5 phosphorylation (as found in CDKs and Erk2). The well-conserved Trp residue in CDKs and Erk2 is replaced by Phe, which significantly reduces the strength of hydrophobic interaction between CTD proline upon Ser5 recognition (Figures 4A and 5A). The loss of both factors in Dyrk1a eliminates any advantage for Ser5 as the site of phosphorylation in CTD binding (Figure 5A). On the contrary, Dyrk1a is activated by the phosphorylation of the Tyr residue, which forms a salt bridge with Arg325, an electrostatic interaction reminiscent of the interaction network of Erk2 when it recognizes Ser2 as a substrate (Figure 5B). The orientation of the Arg325 side chain is fastened by a triad of salt bridge formations between phosphoryl-tyrosine with Arg325 and Arg329. Thus, Arg325 interacts with Ser7 of CTD when Ser2 is the residue subject to phosphorylation, stabilizing the configuration for Ser2 rather than Ser5 recognition (Figure 5B). Compounded by these factors, we postulated that Dyrk1a strongly favors the recognition of Ser2 over Ser5 on the basis of the structural features of its active site.
Figure 5.
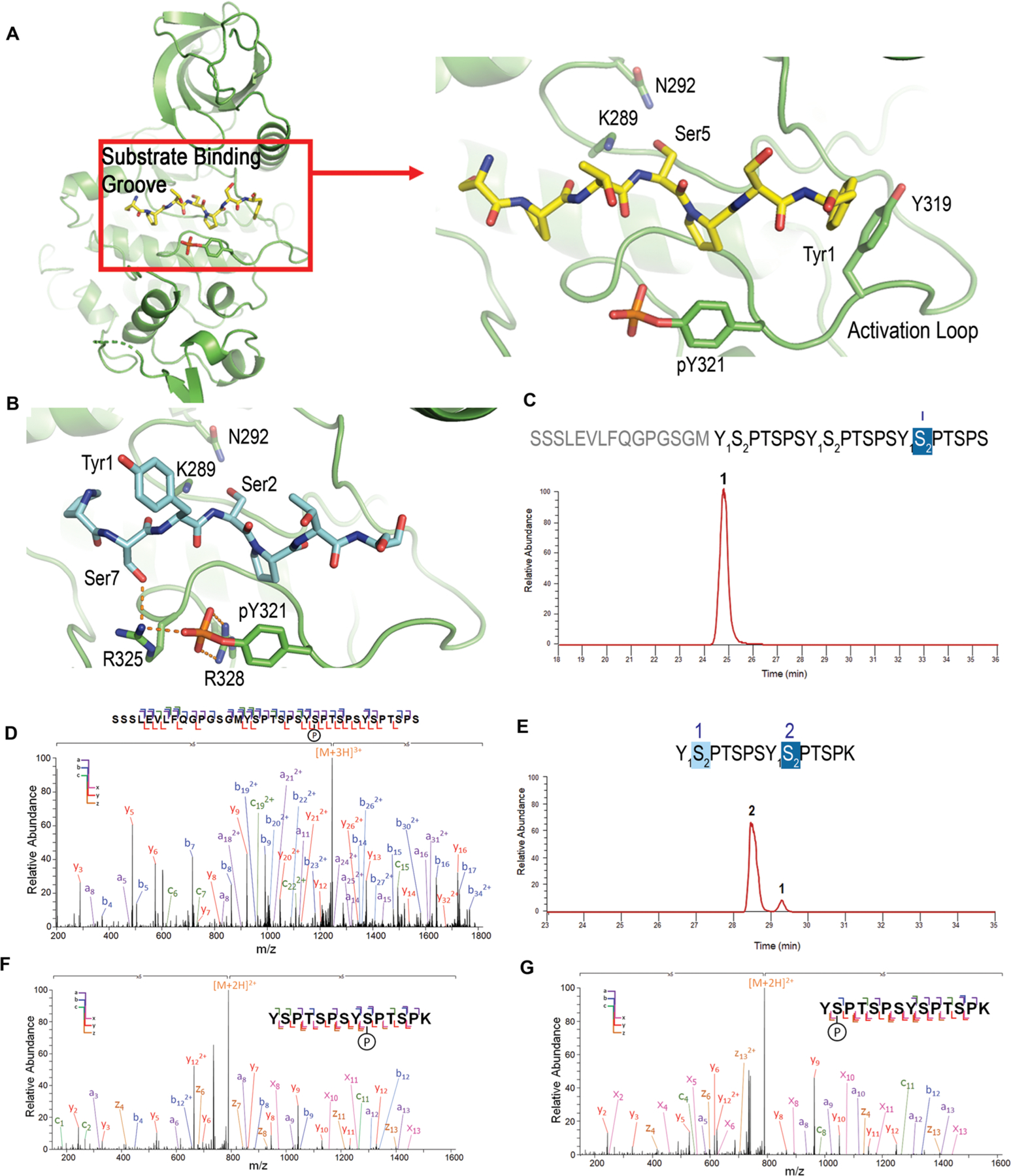
Modeling/structural analysis and mass spectrometric identification of Dyrk1a specificity. (A) Overall kinase structure of Dyrk1a, and zoomed in picture of the active site interactions of Dyrk1a (green) with the modeled CTD peptide. Ser5 is modeled as the phosphorylable residue. (B) Model of the active site interactions of Dyrk1a with Ser2 for phosphorylation (same color scheme except with carbon atoms in light blue). Catalytic residues K289 and N292 are shown as sticks. (C) Liquid chromatography trace of 3 repeat consensus CTD treated with Dyrk1a. The numbers above each sequence and each chromatographic peak correspond to the specific repeats of the CTD sequence. (D) MS/MS analysis of the peptide shown in panel c. (E) Liquid chromatography trace of 26 repeat S7K spaced CTD treated with Dyrk1a. The numbers above the sequence show the position of phosphates in the diheptad fragments generated from the 26 repeat S7K spaced CTD. The numbers above each chromatographic peak correspond to the specific repeats of the CTD sequence. (F and G) MS/MS analysis of the peptides shown in panel e.
To evaluate the hypothesis that Dyrk1a favors Ser2 phosphorylation on the basis of structural analysis, we characterized the Dyrk1a specificity on CTD using UVPD-mass spectrometry (Figure 5C–G). We treated a recombinant CTD peptide containing three heptad repeats of the consensus sequence with Dyrk1a (Figure 5C). The peptide was incubated with the enzyme with 2 mM ATP, and the products were then characterized by LC–UVPD-MS (Figure 5C). Under these reaction conditions, the majority of the product obtained was monophosphorylated at Ser2, in agreement with our prediction based on the structural analysis (Figure 5D). To test whether the specificity for Ser2 is consistent using the length of a physiologically relevant CTD, we made a long recombinant CTD with 26 repeats (same length as S. cerevisiae CTD) with every other Ser7 replaced by Lys, named S7K-spaced CTD (Figure 5E). The design of this engineered CTD has several advantages. First, the long CTD heptad mimics the eukaryotic CTD while at the same time eliminating the concern that short recombinant CTD fragments obviate kinase selectivity due to low activity. Second, Lys at Ser7 position is the most frequent replacement for Ser7 in human distal CTD (Figure S1). Finally, the introduction of Lys also allows the long CTD to be digested into diheptad fragments for further MS/MS analysis (Figure 5F,G). Two diheptide products were identified for monophosphorylated species (Figure 5E), both of which are diheptides with Ser2 phosphorylated (Figure 5F,G). A small amount of bis-phosphorylated heptad shows that both Ser2 is phosphorylated. This result reiterates that Dyrk1a is a specific Ser2 kinase for CTD, as predicted by our structural analysis and demonstrated by the UVPD-MS results of short CTD peptides.
Flanking Residues in the CTD Substrate Influence Dyrk1a Specificity.
Careful analysis of the modeled structure of Dyrk1a with Ser2 revealed that the hydrogen bonding between Ser7 of CTD and Arg325 is pivotal for substrate recognition. We thus speculated that the identity and post-translational modification of the seventh residue of the preceding heptad could affect the phosphorylation of Ser2. Indeed, in the experiment where Dyrk1a phosphorylates yeast CTD but with a Lys residue replacing Ser7 on every other heptad, we notice a preference of phosphorylation on Ser2 on the basis of the nearby seventh position residue (Figure 5E). We quantified the relative amount for each Ser2 population in monophosphorylated species. The one preceded with Ser7 shows high abundance, whereas the one following Lys7 is a minor product (Figure 5E). We tested how Lys at the seventh position affects Dyrk1a kinase activity, using a GST recombinant CTD containing three heptads with every seventh position replaced by a lysine residue (Figure 6A). The peaks corresponding to monophosphorylation revealed a mixture of peptides with Ser2 phosphorylation (Figure 6A and Figure S8). Thus, while a bulkier seventh residue will not change the specificity of Dyrk1a, the Ser2 following it is not favored for phosphorylation.
Figure 6.
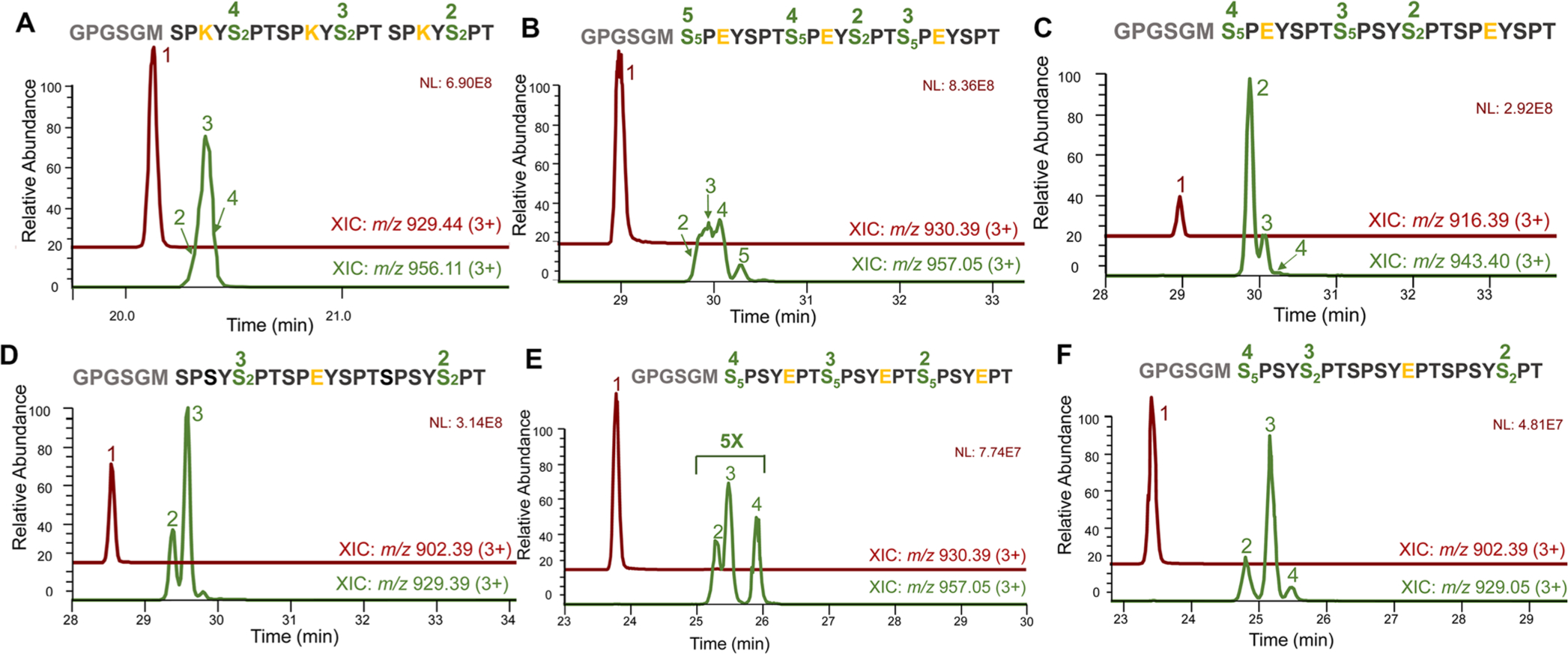
In-depth LC–MS/MS analysis of Dyrk1a’s specificity using 3X CTD as substrates. Green-colored LC traces indicate monophosphorylated products with peak numbers matching corresponding sites of phosphorylation. (A) 3X CTD S7K treated with Dyrk1a. (B) 3X CTD S7E treated with Dyrk1a. (C) 3X CTD S7E on the first and third repeats treated with Dyrk1a. (D) 3X CTD with S7E only on the middle repeat treated with Dyrk1a. (E) 3X CTD S2E treated with Dyrk1a. (F) 3X CTD with S2E only on the middle repeat treated with Dyrk1a. In all LC-traces, the unphosphorylated peptide is shown as peak 1 colored in maroon. The sequence for each CTD mutation variant is exhibited with green fonts highlighting sites of phosphorylation with numbers matching to corresponding peak intensities of monophosphorylated species. The residues varied from the consensus sequence are colored yellow.
Since Ser7 phosphorylation occurs extensively during active transcription,46 we questioned whether it would change the specificity of Dyrk1a. To answer this question, we treated a three heptad CTD peptide with all Ser7 replaced by glutamate to mimic its phosphorylation (Figure 6B). The resultant products were composed of a mixture of peptides with phosphorylation at both Ser2 and Ser5 (Figure 6B and Figure S9). To check if Dyrk1a has a preference for Ser2 or Ser5 when nearby Ser7 is phosphorylation, we tested the activity against two other three heptad CTD peptides, each with selected Ser7 replaced by Glu (Figure 6C,D). The observed products were heptads with phosphorylated Ser2 not Ser5. Dyrk1a preferably phosphorylates the Ser2 preceded by Ser7 rather than the Ser2 near the phosphoryl-mimick (Figure 6C,D and Figures S10 and S11). Our structural model rationalizes the preferences with the favorable interaction with Ser7 preceding Ser2 forming interaction with Arg325 (Figure 5B). We also wondered whether Dyrk1a could phosphorylate Ser5 if all the Ser2 were already phosphorylated. We used a three heptad CTD with every Ser2 mutated to phosphoryl-mimicking glutamate. Phosphorylation occurred, but the activity is low as indicated by the low abundance of products after phosphorylation with the majority of the peptide population observed as unreacted substrate peptides (Figure 6E and Figure S12). When we used a three heptad CTD with some heptad containing S2E while others remain unmutated, the Ser2-phosphorylated peptides constituted most of the products (Figure 6F and Figure S13). These results illustrate that Dyrk1a prefers the phosphorylation of Ser2 when flanking residues support the formation of favorable substrate recognition configuration.
DISCUSSION
Phosphorylation is a ubiquitous post-translational modification that can transmit signals reversibly. The accuracy of the signal transduction is critical, but the structural elements encoding the kinase specificity have been elusive. The issue of substrate specificity is particularly crucial for eukaryotic transcription since the phosphorylations on different Ser residues in the heptad repeats of CTD recruit different transcriptional regulatory factors to the active RNA polymerase II. Thus, the complex structures of kinases with substrate trapped in the active site are especially challenging to obtain due to the weak and transient interactions between CTD kinases and their substrates.
To understand how different CTD kinases differentiate Ser2 versus Ser5, we conducted sequence, structural, and mass spectrometric analysis. From our modeling, the active conformation of the activation/P+1 loops forms different interactions with the CTD substrate when Ser2 or Ser5 locate within the active site. In particular, our analyses reveal that three motifs are critical in the determination of the Ser5 or Ser2 subject to phosphorylation (Figure 5A). The first motif is the Thr phosphorylation, which is the critical residue for activation in most kinases. The orientation of this phosphoryl-Thr is anchored by a strong hydrophilic interaction network formed with Arg/Lys and Tyr/His residues (Figures 3B and 5C). The phosphoryl group of this Thr in kinases binds to the hydroxyl group of Tyr1 in the CTD heptad repeats when Ser5 is placed at the active site subject to phosphorylation. The second motif is the Trp in the P+1 loop, which forms a hydrophobic interaction with Pro residues when Ser5 is bound at the active site. The interaction between the substrate and both motifs will be lost if the Ser2 is at the active site. The conservation of both of these motifs explains the strong preference of CDKs and Erk2 for Ser5 as its substrate. Indeed, we have shown here that CAK (TFIIH kinase module) almost exclusively phosphorylates Ser5 of CTD. In other CDKs like P-TEFb (CDK9/cyclin T complex), CDK12 and CDK13 both show priority in phosphorylating Ser5 of CTD with no prephosphorylation.47 Erk2 also phosphorylates Ser5 first as the preferred substrate.22 The third motif that is a phosphoryl-Tyr residue found two residues downstream from the phosphoryl-Thr. The existence of this motif favors Ser2 phosphorylation, which places Ser7 close to an Arg residue that engages in salt bridging with the phosphoryl-Tyr. This phosphoryl-Tyr exists in Dyrk1a and dual-phosphorylation kinases such as Erk2. The combination of these three motifs in the activation/P+1 loops thus seems to play a key role in determining which Ser residue is phosphorylated in RNA polymerase II.
The high flexibility of the activation/P+1 loop implies that enhancing or disrupting the interaction of these three motifs with the substrate can alter the kinase specificity. Indeed, we showed that by eliminating the interaction of motif two, the hydrophobic effect between Trp in kinase and Pro residue in Pol II, the preference of Erk2 could be reversed from Ser5 to Ser2 (Figure 5E). The phosphorylation of flanking residues in CTD heptad can also interrupt the interaction between the motifs and the kinases, thus changing kinase specificity. The structural models of CTD kinases binding to substrate explain our previous observation that the phosphorylation states of CTD heptad appear to be combinatorial.15 We have shown previously that phosphorylation of Tyr1 promotes the Ser2 phosphorylation activity of P-TEFb.15 Our model provides a structural explanation for this experimental observation since the phosphorylation of Tyr1 interrupts its interaction with phosphoryl-Thr. Interestingly, the activation/P+1 loops of CDK9, CDK12, and CDK13 are longer than those of CDK7 (Figure 5A). All three kinases have been reported to phosphorylate Ser2 in cells, even though they all prefer Ser5 phosphorylation in vitro.15,22,47 It is thus intriguing to speculate that regulatory mechanisms altering the conformation of the activation/P+1 loop and interrupting the interaction at these three motifs can potentially change the specificity of these kinases and lead to different outcomes of transcription.
Using the three motifs, we predicted that the recently identified CTD kinase, Dyrk1a, is a Ser2-specific CTD kinase, which is demonstrated by mass spectrometry (Figure 5). The Dyrk1a gene is one of the overexpressed genes in Down syndrome due to its location on chromosome 21. Dyrk1a has a large clientele of protein substrates, so its activity is implicated in the pathological pathways in neurogenesis, immunity, even tumorigenesis.39,48 While most of the substrates located in the cytosol, its nuclear substrates have recently been identified, with RNA polymerase II and Histone H3 being the most highly represented. Indeed, knockdown of Dyrk1a shows the global reduction in Ser2 and Ser5 phosphorylation.21 However, considering the complicated network of proteins whose biological function is modified by Dyrk1a, it is hard to deconvolute direct and indirect effects. Using mass spectrometry and biochemistry, we establish that Dyrk1a is a Ser2-specific kinase. Using mutational analysis, we show that Dyrk1a does phosphorylate Ser5 but only when Ser2 is not available for phosphorylation or a negatively charged residue replaces the preceding Ser7. This discovery will help us to untangle the complicated signaling network involving Dyrk1a and illuminate its unique role during transcription, in particular, the cross-talk with histone phosphorylation.49
METHODS
Sequence Alignments.
The sequences of the CDKs, ERK, and Dyrk1a were obtained from NCBI. (CDK2 - P24941, CDK7 - P50613, CDK9 - P50750, CDK12 - Q9NYV4, CDK13 - Q14004, ERK2 - P28482, and Dyrk1a - Q13627). The sequences were aligned using the MUSCLE alignment program.50
Cloning and Purification of CTD Kinase Constructs.
All CTD constructs were ordered as primers, amplified, and cloned using ligation-independent cloning (SLIC).51 CTD (26x) constructs with Lys in every other repeat were ordered as synthetic genes (Genescript). Human distal CTD was cloned from a full-length human CTD construct from pYFP-RPB1-αAmr.52 The constructs were cloned into a PET28a vector (Novagene) containing a 6X histidine tag and glutathione-S-transferase (GST) tag with a 3C protease cleavage site added after the two tags. T4 DNA polymerase (NEB) was used to create 5′ overhangs for cloning into the vectors using SLIC.
Protein expression and purification were done as described previously.15,22,26,27 E. coli BL21 (DE3) cells were used as the protein expression system. The transformation was carried out by thawing the cells on ice for 10 min, adding the DNA and incubating on ice for 30 min, heat shocking at 42 °C for 1 min and cooling the cells on ice for 5 min. The cells were recovered in SOC medium for 1 h at 37 °C and were plated on Luria–Bertani agar plates with 50 μg/mL kanamycin for selection using the spread plate technique. Individual colonies were grown in Luria–Bertani medium at 37 °C containing 50 μg/mL kanamycin in 50 mL flasks. Inoculum (10 mL) was used for inoculating 1 L of terrific broth (Thermo Fisher), and the culture was grown to an OD of 0.4–0.6. IPTG was used to induce the expression at a final concentration of 0.5 mM. The cultures were pelleted by centrifugation after overnight growth (16 h at 16 °C), and the cells were lysed through sonication in a lysis buffer (50 mM Tris-Cl pH 8.0, 500 mM NaCl, 10% glycerol, 0.1% Triton-X 100, 15 mM imidazole, and 10 mM BME). Sonication of the samples was carried out on ice at 90 A for 3 min per cycle (1 s on and 5 s off) for five cycles with a 3 min break between each cycle. The lysate was cleared by centrifugation at 27000g for 45 min at 4 °C. The proteins were purified through affinity column chromatography using Ni2+/NTA beads (Qiagen). Briefly, the column was equilibrated with lysis buffer; then, the cleared lysate supernatant was run through the column. A wash (50 mM Tris-Cl pH 8.0, 500 mM NaCl, and 10 mM BME) was done before eluting with an elution buffer (50 mM Tris-Cl pH 8.0, 500 mM NaCl, 200 mM imidazole, and 10 mM BME). Proteins were dialyzed in a gel filtration buffer (50 mM NaCl, 20 mM Tris-Cl pH 8.0, and 10 mM BME) at 4 °C overnight with a suitable dialysis membrane. Proteins were concentrated using centrifugal filtration and cleaned up by size exclusion chromatography using a Superdex 200 column (GE Life Sciences). The integrity of the proteins was assessed by polyacrylamide gel electrophoresis (Coomassie Brilliant Blue Staining).
Human Dyrk1a was purified using the same protocol using a synthetic construct (Addgene). Dyrk1a was dialyzed and cleaned up by size exclusion chromatography in a different gel filtration buffer (50 mM Tris-Cl pH 8.0, 200 mM NaCl, and 10 mM BME). CDK7/CyclinH/MAT1 purified protein complex was purchased from PROQINASE. Homo sapiens Erk2 was expressed from pET-His6-ERK2-MEK1_R4F_coexpression vector as a gift from Melanie Cobb (Addgene plasmid #39212).53
Kinase Reaction Assays and Sample Preparation for UVPD MS/MS.
All kinase reaction assays were performed with the 2 mM ATP, 50 mM Tris pH 8, and 10 mM MgCl2. A mixture of 0.2 μM Cdk7/CycH/MAT1, 0.6 μM Dyrk1a, and 0.6 μM Erk2 was used to treat 1 mg mL−1 CTD substrate for 12 h (buffer containing 50 mM Tris-Cl pH 8.0 and 10 mM MgCl2). The reaction time of 12 h was optimized so that no further phosphorylation occurred on the substrate. The reactions were stopped through ion quenching using 10 mM ethylenediaminetetraacetic acid (EDTA). All CTD samples were digested using 3C-protease at a molar ratio of 100:1 protein/protease in a reaction volume of 100 μL. GST-26xCTD lysine constructs were prepared for bottom-up analysis by overnight digestion with trypsin at 37 °C using a 1:50 enzyme-to-substrate ratio. CTD digests were desalted on C18 spin columns and resuspended to 0.5 μg/μL with 2% acetonitrile in 0.1% formic acid in HPLC-grade water for LC–MS analysis. The CTD constructs with 26 repeats were additionally treated with trypsin to yield diheptad fragments. The human distal CTD was treated with trypsin to yield multiheptad fragments before UVPD MS/MS.
UVPD Tandem Mass Spectrometry and Data Analysis.
All peptides were separated using a Dionex Ultimate 3000 nano liquid chromatography system (Thermo Scientific) plumbed for direct injection into a 75 μm ID Picofrit analytical column (New Objective, Woburn, MA). One microliter of each sample was injected for separations using a 1.8 μm UChrom C18 analytical column (NanoLCMS Solutions, Oroville, CA) packed in-house to 20 cm. Mobile phases A and B were composed of HPLC-grade water and acetonitrile, respectively, each containing 0.1% formic acid. Separations were carried out using gradients that were optimized for various samples as follows: a linear gradient of 2% to 20% B in 40 min was used for 26-repeat S7K Sp and three-repeat CTD (S7K Sp). A stepwise gradient of 2% to 10% B for 5 min and then 10% to 25% B for another 40 min was used for the human distal CTD samples. The flow rate was maintained at 0.300 μL/min for all samples. Eluted peptides were analyzed in positive polarity mode using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using a NanoFlex electrospray source. The mass spectrometer was equipped with an excimer laser operated at 193 nm (Coherent, Santa Clara, CA) and modified to allow for UVPD in the dual linear ion trap as described previously.54 All spectra were acquired in the Orbitrap mass analyzer using resolution settings of 60 K and 30 K (at m/z 200) for MS1 and MS/MS events, respectively. Targeted peptides were activated using two laser pulses of 1.5 mJ for UVPD in the low-pressure ion trap.54
MS/MS spectra were deconvoluted in the XCalibur QualBrowser software using the Xtract algorithm with a signal-to-noise threshold of 3. Fragments were matched to the nine ion types observed from UVPD of peptides (a, a+1, b, c, x, x+1, y, y–1, z) using ProSight Lite. Phosphosites were localized by adding the mass of a phospho-group (+79.97 Da) at each of the possible Tyr or Ser residues to identify fragment ions that contained the moiety. The relative abundances of each phosphorylated species were calculated from the LC peak area of the eluting peptide. Specifically, the ion current of the phosphopeptide precursor ion was integrated across the elution profile and normalized to the total ion current for all phosphopeptide isomers. Due to the identical chemical composition of isobaric peptides, the ionization efficiencies were assumed to be the same for the compared species. The abundances of phosphorylated species should not be compared for peptides with different sequences.
Analysis of each CTD construct was performed identically to the previous analysis.15,22,26,27 All LC–UVPD-MS studies were conducted on at least two biological samples, and mass spectra were confirmed manually as well as using commercial search algorithms.
MALDI-TOF Analysis.
After quenching kinase reactions, protein samples were desalted using Pierce C18 zip tips (ThermoFisher) using recommended protocols. The samples were resuspended in 50% acetonitrile with 0.1% trifluoroacetic acid in water and analyzed using an AB Voyager-DE PRO MALDI-TOF at UT Austin’s proteomics facility. 2,5-Dihydroxybenzoic acid was used as the matrix at a 1:1 ratio (final concentration of 20 mg mL−1).
Structural Modeling and Docking.
The active configuration of CDK7 was generated by replacing residue 158 through residue 181 of CDK7 (PDB code: 1UA2) using residues 148–171 of the active kinase loop of CDK2 (PDB code: 2CCI). The model was then optimized with the substrate in the binding groove with Maestro (Schrödinger, LLC), which has a simple minimization routine on the basis of the OPLS_2005 force field.36,37 The model of Erk2 binding to CTD peptides was obtained by superimposing an active Erk2 structure (PDB code: 6OPG) to that of the CDK2 and then energy minimizing using Maestro. The model for Dyrk1a binding to CTD was obtained by virtual mutation of the substrate ligand of Dyrk1a complex structure (PDB code: 2WO6) to the CTD sequence and then energy minimizing using Maestro. Mutations were fit with the likeliest rotamer configuration. PyMOL (Schrödinger, LLC) was used as the graphic visual preparation software. All modifications on CTD substrate structures were carried out using WinCoot. The PDB codes of the kinases are as follows: Cdk2 = 2CCI, Cdk7 = 1UA2, Cdk9 = 6GZH, Cdk12 = 4CXA, Cdk13 = 5EFQ, Erk2 = 6OPG, and Dyrk1a = 2WO6.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by grants from the National Institutes of Health (R01GM104896 and R01GM125882 to Y.J.Z. and J.S.B.) and Welch Foundation (F-1778 to Y.J.Z. and F-1155 to J.S.B.). Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00474.
Figures of sequence logos of the conservation of yeast and human CTD, lists of fragment ions for peptides, LC–MS/MS trace, and modeling of CTD (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00474
The authors declare no competing financial interest.
Contributor Information
Mukesh Kumar Venkat Ramani, Department of Molecular Biosciences, The University of Texas at Austin, Austin, Texas 78712, United States.
Edwin E. Escobar, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States.
Jennifer S. Brodbelt, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States;.
Yan Jessie Zhang, Department of Molecular Biosciences and Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, Texas 78712, United States;.
REFERENCES
- (1).Buratowski S (2009) Progression through the RNA Polymerase II CTD Cycle. Mol. Cell 36 (4), 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Jeronimo C, Bataille AR, and Robert F (2013) The Writers, Readers, and Functions of the RNA Polymerase II C-Terminal Domain Code. Chem. Rev 113 (11), 8491–8522. [DOI] [PubMed] [Google Scholar]
- (3).Tombácz I, Schauer T, Juhász I, Komonyi O, and Boros I (2009) The RNA Pol II CTD Phosphatase Fcp1 Is Essential for Normal Development in Drosophila Melanogaster. Gene 446 (2), 58–67. [DOI] [PubMed] [Google Scholar]
- (4).Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, and Corden JL (1996) The C-Terminal Domain of the Largest Subunit of RNA Polymerase II Interacts with a Novel Set of Serine/Arginine-Rich Proteins. Proc. Natl. Acad. Sci. U. S. A 93 (14), 6975–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hsin J-P, and Manley JL (2012) The RNA Polymerase II CTD Coordinates Transcription and RNA Processing. Genes Dev. 26 (19), 2119–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Eick D, and Geyer M (2013) The RNA Polymerase II Carboxy-Terminal Domain (CTD) Code. Chem. Rev 113 (11), 8456–8490. [DOI] [PubMed] [Google Scholar]
- (7).Komarnitsky P, Cho E-J, and Buratowski S (2000) Different Phosphorylated Forms of RNA Polymerase II and Associated MRNA Processing Factors during Transcription. Genes Dev. 14 (19), 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fong N, Saldi T, Sheridan RM, Cortazar MA, and Bentley DL (2017) RNA Pol II Dynamics Modulate Co-Transcriptional Chromatin Modification, CTD Phosphorylation, and Transcriptional Direction. Mol. Cell 66 (4), 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Helenius K, Yang Y, Tselykh TV, Pessa HKJ, Frilander MJ, and Mäkelä TP (2011) Requirement of TFIIH Kinase Subunit Mat1 for RNA Pol II C-Terminal Domain Ser5 Phosphorylation, Transcription and MRNA Turnover. Nucleic Acids Res. 39 (12), 5025–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ho CK, and Shuman S (1999) Distinct Roles for CTD Ser2 and Ser-5 Phosphorylation in the Recruitment and Allosteric Activation of Mammalian MRNA Capping Enzyme. Mol. Cell 3 (3), 405–411. [DOI] [PubMed] [Google Scholar]
- (11).Marshall NF, Peng J, Xie Z, and Price DH (1996) Control of RNA Polymerase II Elongation Potential by a Novel Carboxyl-Terminal Domain Kinase. J. Biol. Chem 271 (43), 27176–27183. [DOI] [PubMed] [Google Scholar]
- (12).Li J, Liu Y, Rhee HS, Ghosh SKB, Bai L, Pugh BF, and Gilmour DS (2013) Kinetic Competition between Elongation Rate and Binding of NELF Controls Promoter-Proximal Pausing. Mol. Cell 50 (5), 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Adelman K, and Lis JT (2012) Promoter-Proximal Pausing of RNA Polymerase II: Emerging Roles in Metazoans. Nat. Rev. Genet 13 (10), 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hausmann S, Koiwa H, Krishnamurthy S, Hampsey M, and Shuman S (2005) Different Strategies for Carboxyl-Terminal Domain (CTD) Recognition by Serine 5-Specific CTD Phosphatases. J. Biol. Chem 280 (45), 37681–37688. [DOI] [PubMed] [Google Scholar]
- (15).Mayfield JE, Irani S, Escobar EE, Zhang Z, Burkholder NT, Robinson MR, Mehaffey MR, Sipe SN, Yang W, Prescott NA, Kathuria KR, Liu Z, Brodbelt JS, and Zhang Y (2019) Tyr1 Phosphorylation Promotes Phosphorylation of Ser2 on the C-Terminal Domain of Eukaryotic RNA Polymerase II by P-TEFb. eLife 8, No. e48725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fisher RP (2005) Secrets of a Double Agent: CDK7 in Cell-Cycle Control and Transcription. J. Cell Sci 118 (22), 5171–5180. [DOI] [PubMed] [Google Scholar]
- (17).Mäkelä TP, Tassan J-P, Nigg EA, Frutiger S, Hughes GJ, and Weinberg RA (1994) A Cyclin Associated with the CDK-Activating Kinase M015. Nature 371, 254–257. [DOI] [PubMed] [Google Scholar]
- (18).Wong KH, Jin Y, and Struhl K (2014) TFIIH Phosphorylation of the Pol II CTD Stimulates Mediator Dissociation from the Preinitiation Complex and Promoter Escape. Mol. Cell 54 (4), 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tee W-W, Shen SS, Oksuz O, Narendra V, and Reinberg D (2014) Erk1/2 Activity Promotes Chromatin Features and RNAPII Phosphorylation at Developmental Promoters in Mouse ESCs. Cell 156 (4), 678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yu D, Cattoglio C, Xue Y, and Zhou Q (2019) A Complex between DYRK1A and DCAF7 Phosphorylates the C-Terminal Domain of RNA Polymerase II to Promote Myogenesis. Nucleic Acids Res 47 (9), 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Di Vona C, Bezdan D, Islam ABMMK, Salichs E, López-Bigas N, Ossowski S, and de la Luna S (2015) Chromatin-Wide Profiling of DYRK1A Reveals a Role as a Gene-Specific RNA Polymerase II CTD Kinase. Mol. Cell 57 (3), 506–520. [DOI] [PubMed] [Google Scholar]
- (22).Mayfield JE, Robinson MR, Cotham VC, Irani S, Matthews WL, Ram A, Gilmour DS, Cannon JR, Zhang YJ, and Brodbelt JS (2017) Mapping the Phosphorylation Pattern of Drosophila Melanogaster RNA Polymerase II Carboxyl-Terminal Domain Using Ultraviolet Photodissociation Mass Spectrometry. ACS Chem. Biol 12 (1), 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, and Cantley LC (1994) Use of an Oriented Peptide Library to Determine the Optimal Substrates of Protein Kinases. Curr. Biol 4 (11), 973–982. [DOI] [PubMed] [Google Scholar]
- (24).Errico A, Deshmukh K, Tanaka Y, Pozniakovsky A, and Hunt T (2010) Identification of Substrates for Cyclin Dependent Kinases. Adv. Enzyme Regul 50 (1), 375–399. [DOI] [PubMed] [Google Scholar]
- (25).Brodbelt JS (2014) Photodissociation Mass Spectrometry: New Tools for Characterization of Biological Molecules. Chem. Soc. Rev 43 (8), 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Irani S, Sipe SN, Yang W, Burkholder NT, Lin B, Sim K, Matthews WL, Brodbelt JS, and Zhang Y (2019) Structural Determinants for Accurate Dephosphorylation of RNA Polymerase II by Its Cognate CTD Phosphatase during Eukaryotic Transcription. J. Biol. Chem 294 (21), 8592–8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Burkholder NT, Sipe SN, Escobar EE, Venkatramani M, Irani S, Yang W, Wu H, Matthews WM, Brodbelt JS, and Zhang Y (2019) Mapping RNAPII CTD Phosphorylation Reveals That the Identity and Modification of Seventh Heptad Residues Direct Tyr1 Phosphorylation. ACS Chem. Biol 14 (10), 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lolli G, Lowe ED, Brown NR, and Johnson LN (2004) The Crystal Structure of Human CDK7 and Its Protein Recognition Properties. Structure 12 (11), 2067–2079. [DOI] [PubMed] [Google Scholar]
- (29).Endicott JA, Noble MEM, and Johnson LN (2012) The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem 81 (1), 587–613. [DOI] [PubMed] [Google Scholar]
- (30).Bradley D, and Beltrao P (2019) Evolution of Protein Kinase Substrate Recognition at the Active Site. PLoS Biol. 17 (6), No. e3000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nolen B, Taylor S, and Ghosh G (2004) Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Mol. Cell 15 (5), 661–675. [DOI] [PubMed] [Google Scholar]
- (32).Wood DJ, and Endicott JA (2018) Structural Insights into the Functional Diversity of the CDK–Cyclin Family. Open Biol. 8 (9), 180112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cheng K-Y, Noble MEM, Skamnaki V, Brown NR, Lowe ED, Kontogiannis L, Shen K, Cole PA, Siligardi G, and Johnson LN (2006) The Role of the Phospho-CDK2/Cyclin A Recruitment Site in Substrate Recognition. J. Biol. Chem 281 (32), 23167–23179. [DOI] [PubMed] [Google Scholar]
- (34).Brown NR, Noble MEM, Endicott JA, and Johnson LN (1999) The Structural Basis for Specificity of Substrate and Recruitment Peptides for Cyclin-Dependent Kinases. Nat. Cell Biol 1 (7), 438–443. [DOI] [PubMed] [Google Scholar]
- (35).Cook A, Lowe ED, Chrysina ED, Skamnaki VT, Oikonomakos NG, and Johnson LN (2002) Structural Studies on Phospho-CDK2/Cyclin A Bound to Nitrate, a Transition State Analogue: Implications for the Protein Kinase Mechanism. Biochemistry 41 (23), 7301–7311. [DOI] [PubMed] [Google Scholar]
- (36).Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, and Sherman W (2010) Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput 6 (5), 1509–1519. [DOI] [PubMed] [Google Scholar]
- (37).Kaminski GA, Friesner RA, Tirado-Rives J, and Jorgensen WL (2001) Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides †. J. Phys. Chem. B 105 (28), 6474–6487. [Google Scholar]
- (38).Prowse CN, and Lew J (2001) Mechanism of Activation of ERK2 by Dual Phosphorylation. J. Biol. Chem 276 (1), 99–103. [DOI] [PubMed] [Google Scholar]
- (39).Menon VR, Ananthapadmanabhan V, Swanson S, Saini S, Sesay F, Yakovlev V, Florens L, DeCaprio JA, Washburn MP, Dozmorov M, and Litovchick L (2019) DYRK1A Regulates the Recruitment of 53BP1 to the Sites of DNA Damage in Part through Interaction with RNF169. Cell Cycle 18 (5), 531–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Roewenstrunk J, Di Vona C, Chen J, Borras E, Dong C, Arató K, Sabidó E, Huen MSY, and de la Luna S (2019) A Comprehensive Proteomics-Based Interaction Screen That Links DYRK1A to RNF169 and to the DNA Damage Response. Sci. Rep 9 (1), 6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Aranda S, Laguna A, and de la Luna S (2011) DYRK Family of Protein Kinases: Evolutionary Relationships, Biochemical Properties, and Functional Roles. FASEB J. 25 (2), 449–462. [DOI] [PubMed] [Google Scholar]
- (42).Dowjat WK, Adayev T, Kuchna I, Nowicki K, Palminiello S, Hwang YW, and Wegiel J (2007) Trisomy-Driven Overexpression of DYRK1A Kinase in the Brain of Subjects with Down Syndrome. Neurosci. Lett 413 (1), 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Delabar J-M, Theophile D, Rahmani Z, Chettouh Z, Blouin J-L, Prieur M, Noel B, and Sinet P-M (2017) Molecular Mapping of Twenty-Four Features of Down Syndrome on Chromosome 21. Eur. J. Hum. Genet 1 (2), 114–124. [DOI] [PubMed] [Google Scholar]
- (44).Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG, and Rao A (2006) A Genome-Wide Drosophila RNAi Screen Identifies DYRK-Family Kinases as Regulators of NFAT. Nature 441 (7093), 646–650. [DOI] [PubMed] [Google Scholar]
- (45).Soundararajan M, Roos AK, Savitsky P, Filippakopoulos P, Kettenbach AN, Olsen JV, Gerber SA, Eswaran J, Knapp S, and Elkins JM (2013) Structures of Down Syndrome Kinases, DYRKs, Reveal Mechanisms of Kinase Activation and Substrate Recognition. Structure 21 (6), 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Egloff S, O’Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, and Murphy S (2007) Serine-7 of the RNA Polymerase II CTD Is Specifically Required for SnRNA Gene Expression. Science 318 (5857), 1777–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bösken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, and Geyer M (2014) The Structure and Substrate Specificity of Human Cdk12/Cyclin K. Nat. Commun 5 (1), 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fotaki V, Dierssen M, Alcántara S, Martínez S, Martí E, Casas C, Visa J, Soriando E, Estivill X, and Arbonés ML (2002) Dyrk1A Haploinsufficiency Affects Viability and Causes Developmental Delay and Abnormal Brain Morphology in Mice. Mol. Cell. Biol 22 (18), 6636–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jang SM, Azebi S, Soubigou G, and Muchardt C (2014) DYRK1A Phoshorylates Histone H3 to Differentially Regulate the Binding of HP1 Isoforms and Antagonize HP1-Mediated Transcriptional Repression. EMBO Rep. 15 (6), 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Madeira F, Park Y. Mi, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, and Lopez R (2019) The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 47 (W1), W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Li MZ, and Elledge SJ (2007) Harnessing Homologous Recombination in Vitro to Generate Recombinant DNA via SLIC. Nat. Methods 4 (3), 251–256. [DOI] [PubMed] [Google Scholar]
- (52).Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, and Singer RH (2007) In Vivo Dynamics of RNA Polymerase II Transcription. Nat. Struct. Mol. Biol 14 (9), 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Khokhlatchev A, Xu S, English J, Wu P, Schaefer E, and Cobb MH (1997) Reconstitution of Mitogen-Activated Protein Kinase Phosphorylation Cascades in Bacteria: EFFICIENT SYNTHESIS OF ACTIVE PROTEIN KINASES. J. Biol. Chem 272 (17), 11057–11062. [DOI] [PubMed] [Google Scholar]
- (54).Klein DR, Holden DD, and Brodbelt JS (2016) Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 88 (1), 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.