

Abstract
Background
Cisplatin/gemcitabine is standard first‐line treatment for patients with advanced biliary tract cancer (ABC). NUC‐1031 (phosphoramidate transformation of gemcitabine) is designed to enhance efficacy by maximizing intratumoral active metabolites.
Methods
Patients with untreated ABC, Eastern Cooperative Oncology Group performance status 0–1 received NUC‐1031 (625 or 725 mg/m2) and cisplatin (25 mg/m2) on days 1 and 8, every 21 days. Primary objectives were safety and maximum tolerated dose; secondary objectives were objective response rate (ORR), pharmacokinetics, progression‐free survival (PFS), and overall survival (OS).
Results
Twenty‐one patients (median age 61 years, n = 13 male; 17 cholangiocarcinoma, 2 ampullary, and 2 gallbladder cancer) received NUC‐1031 625 mg/m2 (n = 8 and expansion n = 7; median six cycles) or 725 mg/m2 (n = 6; median 7.5 cycles). Treatment was well tolerated; most common treatment‐emergent grade 3–4 adverse events occurring in more than one patient with 625 mg/m2 NUC‐1031 were increased gamma‐glutamyl transferase (GGT), 40%; alanine aminotransferase, 20%; bilirubin, 13%; neutropenia, 27%; decreased white cell count, 20%; thrombocytopenia, 13%; nausea, 13%; diarrhea, 13%; fatigue, 13%; and thrombus, 20% and with 725 mg/m2, increased GGT, 67%, and fatigue, 33%. NUC‐1031 725 mg/m2 was selected as the recommended dose with cisplatin in ABC. ORR was 33% (one complete response, six partial responses), DCR was 76%, median PFS was 7.2 months (95% confidence interval [CI], 4.3–10.1), and median OS was 9.6 months (95% CI, 6.7–13.1). The median estimates of area under the plasma concentration–time curve from time 0 to last measurable time and maximum concentration were highest for NUC‐1031 (218–324 μg•h/mL and 309–889 μg/mL, respectively) and lowest for di‐fluoro‐deoxycytidine (0.47–1.56 μg•h/mL and 0.284–0.522 μg/mL, respectively).
Conclusion
This is the first study reporting on the combination of NUC‐1031 with cisplatin in ABC and demonstrated a favorable safety profile; 725 mg/m2 NUC‐1031 in combination with cisplatin is undergoing phase III trial evaluation in ABC. (ClinicalTrials.gov ID: NCT02351765; EudraCT ID: 2015‐000100‐26).
Implications for Practice
The prognosis for patients with advanced biliary tract cancer (ABC) is approximately 1 year, and new treatment options are required. The cisplatin/gemcitabine combination is standard first‐line treatment for patients with ABC. NUC‐1031 is a phosphoramidate transformation of gemcitabine and is designed to enhance efficacy by maximizing intratumoral active metabolites. This phase Ib study (ABC‐08) demonstrated a favorable safety profile of NUC‐1031 in combination with cisplatin for the first‐line treatment of patients with ABC, and 725 mg/m2 NUC‐1031 was recommended in combination with cisplatin for phase III trial evaluation; the NuTide:121 global randomized study is currently enrolling.
Keywords: Biliary tract cancer, Advanced disease, NUC‐1031, Cisplatin, Phase Ib
Short abstract
Standard of care for patients with advanced biliary tract cancer is combination therapy with cisplatin and gemcitabine. New therapeutic options are needed. This article assesses the safety of NUC‐1031 in combination with cisplatin and evaluates its antitumor activity in patients with advanced biliary tract cancer.
Introduction
Standard‐of‐care first‐line systemic treatment for patients with advanced biliary tract cancer (ABC) is the cisplatin/gemcitabine combination [1]. The median overall survival (OS) for patients receiving this combination is approximately 1 year [1]; new therapeutic options are required.
The activity of gemcitabine is limited by inherent and acquired cancer cell resistance mechanisms associated with transport, activation, and breakdown [2]. Through the application of ProTide technology, a new agent, NUC‐1031, has been designed to overcome the key resistance mechanisms associated with gemcitabine. NUC‐1031 is a phosphoramidate transformation of gemcitabine, and, like gemcitabine, the cytotoxic effect on cancer cells is largely attributed to the generation of the triphosphate form of the nucleotide analog (di‐fluoro‐deoxycytidine triphosphate [dFdCTP]) [3].
NUC‐1031 is designed specifically to generate and maintain higher concentrations of dFdCTP inside the tumor cell compared with gemcitabine. The phosphoramidate moiety enables NUC‐1031 to enter the cancer cell, independent of the presence of nucleoside transporters. Once NUC‐1031 has entered the cell, the protective group is cleaved off and releases an activated, monophosphorylated form of gemcitabine (dFdCMP). The delivery of dFdCMP obviates the need for the activating enzyme, deoxycytidine kinase, which drives the rate limiting phosphorylation of gemcitabine. dFdCMP is rapidly converted to di‐fluoro‐deoxycytidine diphosphate and then the key anticancer metabolite, dFdCTP. Moreover, NUC‐1031 is not subject to breakdown by cytidine deaminase (CDA) [3]. As a result of overcoming all three key resistance mechanisms, NUC‐1031 achieves much higher levels of the active anticancer metabolite, dFdCTP, than gemcitabine [3]. This mechanism of action has been illustrated in previous publications on NUC‐1031 [4, 5].
The results of a phase I dose‐escalation study of NUC‐1031 monotherapy in 68 patients with advanced solid tumors who had progressed on standard therapy concluded that it was well tolerated, with the most common grade 3–4 adverse events (AEs) being neutropenia, lymphopenia, and fatigue [4]. It also demonstrated clinically significant antitumor activity in patients with prior gemcitabine exposure, including patients refractory to prior gemcitabine treatment (patients who developed progression while on gemcitabine) [4]; in 49 patients with an evaluable response (patients who had completed at least two cycles of NUC‐1031 and had at least one follow‐up radiographic assessment to measure changes in tumor size), there was a 78% disease control rate reported, with 33 patients having stable disease (SD) and 5 achieving a partial response (PR) (patients with primary cancers of the cervix, lung, fallopian tube, pancreas, and unknown primary achieved PRs) [4].
In this study [4], NUC‐1031 was detected in plasma up to 24 hours from the end of infusion (estimated half‐life [t1/2] was 8.3 hours). For the analytes analyzed, the median plasma Area under the curve (AUC0–t) and maximum concentration (Cmax) values on day 1 were highest for NUC‐1031 (269 μM/h and 710 μM, respectively). They were intermediate for 2′,2′‐difluorodeoxyuridine (dFdU) (76.0 μM/h and 5.11 μM, respectively), and lowest for di‐fluoro‐deoxycytidine (dFdC) (2.92 μM/h and 1.82 μM, respectively). The intracellular concentrations of the active anticancer moiety dFdCTP remained high throughout the 24‐hour pharmacokinetic (PK) sampling period. Urine samples were analyzed from 46 patients, and 21.7% and 27.3% of the NUC‐1031 was excreted via the urine as dFdU over the 24 hours after the dose on days 1 and 15, respectively. In total, less than 1% of the dose was excreted as either NUC‐1031 or dFdC [4].
The recommended phase II dose (RP2D) of NUC‐1031 as a single agent was reported as 825 mg/m2 on days 1, 8, and 15 of a 28‐day cycle [4]. This study included seven patients with ABC, all of whom had received prior cisplatin/gemcitabine treatment; of these, six patients had response‐evaluable disease; the best response to therapy in five of these patients was SD (three had target lesion size reduction; percentage not stated in publication), and one had progressive disease (PD) [4].
The aim of this phase Ib, multicenter, open‐label study was to assess the safety of NUC‐1031 in combination with cisplatin in patients with ABC (ABC‐08) and to define the RP2D, in addition to evaluating its antitumor activity, including PK analyses.
Subjects, Materials, and Methods
Study Design
This was a single‐arm, open‐label, multicenter, phase Ib dose‐escalation study conducted under the auspices of the National Cancer Research Institute Upper Gastrointestinal Clinical studies group (hepatobiliary subgroup) to assess the safety and to determine the RP2D of NUC‐1031 in combination with cisplatin in patients with locally advanced/metastatic biliary tract cancer (all centers were high volume, receiving tertiary referrals, with multidisciplinary pathological input).
All patients provided written informed consent approved by North West Liverpool Central Research Ethics Committee prior to any study‐related procedures. The study was performed in accordance with Good Clinical Practice guidelines and the principles of the 1964 Declaration of Helsinki and subsequent revisions (North West Liverpool Central Research Ethics Committees reference 15/NW/0160).
The number of patients per cohort during the dose‐escalation phase was determined according to a 3 + 3 classic design. The starting dose level, 625 mg/m2, of NUC‐1031 was administered via a central venous catheter (CVC) after 25 mg/m2 cisplatin on days 1 and 8 of a 21‐day cycle. The starting dose was selected as approximately 75% of the recommended monotherapy dose of NUC‐1031 (825 mg/m2) [4], with a plan to explore up to four dose levels (625 mg/m2, 725 mg/m2, 825 mg/m2, 925 mg/m2) in combination with a fixed dose of cisplatin.
The primary objectives were determination of safety and maximum tolerated dose (MTD) of NUC‐1031 in combination with cisplatin in patients with ABC. Secondary objectives included assessment of progression‐free survival (PFS), OS, objective response rate (ORR) and PK profile of NUC‐1031.
Dose‐limiting toxicities (DLTs) were determined by clinical and laboratory toxicity assessments performed (day 1 and day 8) during the first 21‐day treatment cycle, and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.
The following were considered DLTs if at least possibly related to either NUC‐1031 or cisplatin: grade 4 neutropenia >7 days, febrile neutropenia defined as a disorder characterized by an absolute neutrophil count <1,000/mm3 and a single temperature of >38.3°C (101°F) or a sustained temperature of ≥38°C (100.4°F) for more than 1 hour, grade 4 thrombocytopenia, grade ≥ 3 nausea and vomiting despite optimal supportive medication, grade ≥ 3 laboratory abnormality or toxicity, delay of >21 days to start cycle 2 treatment due to treatment‐related toxicity, and any isolated or recurrent (e.g., cardiac, renal, neurologic) toxicity that was judged by the investigator and Trial Management Group (TMG) to be a DLT. After a protocol amendment, grade ≥ 3 alanine aminotransferase (ALT) or aspartate aminotransferase (AST) were only considered a DLT if they did not resolve to grade ≤ 2 within 7 days (as not considered clinically significant), and grade > 3 laboratory abnormalities were only considered a DLT if deemed to be clinically significant by the investigator and TMG.
Patient Eligibility
Patients aged ≥18 years with a life expectancy >3 months with histologically/cytologically verified, nonresectable or recurrent/metastatic cholangiocarcinoma (intrahepatic, hilar, distal bile duct), gallbladder cancer, or ampulla of Vater carcinoma (biliary subtype) (radiological diagnosis was allowed for recurrent disease if previously histologically/cytologically verified), who had received no prior systemic therapy for ABC, were eligible for inclusion in this study. Prior adjuvant treatment was allowed if completed more than 6 months prior to enrolment. Other inclusion criteria included an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–1, adequate renal, hematological, and liver function, and adequate biliary drainage, with no evidence of ongoing infection. Supplemental online material 1 contains full eligibility criteria.
Dose‐Escalation Design
A period of at least 48 hours was mandated before recruiting the second and subsequent patients to a single dose cohort. Patients who did not receive both day 1 and day 8 of treatment during cycle 1 for reasons other than toxicity were not considered evaluable for DLT assessments and were replaced. Dose escalation followed a 3 + 3 design until the MTD was determined, defined as the highest dose level for which at least one of six (or < 33%) patients experience a DLT.
A computed tomography scan of the thorax, abdomen, and pelvis was performed within 28 days prior to registration and every 12 weeks (±7 days) on study (assessed using RECIST version 1.1) [6]. Response was determined by experienced radiologist review at individual recruiting sites (investigator review). If clinically indicated, magnetic resonance imaging (MRI) was permitted. It was recommended that the imaging method used for assessment at baseline was used at all subsequent time points, if clinically appropriate. Treatment continued until intolerable toxicity or progressive disease or until withdrawal of consent for other reasons. Patients could continue to receive NUC‐1031 alone, at the investigator's discretion, if cisplatin had to be discontinued for reasons of intolerable toxicity.
Dose Modifications and Delays
Treatment interruptions up to 21 days were allowed for patients to meet retreatment criteria before commencing their next cycle. Dose modifications adhered to those from the ABC‐02 phase III randomized trial of cisplatin/gemcitabine versus gemcitabine for the first‐line treatment of patients with ABC [1]. Supplemental online material 2 has details.
Pharmacokinetic Analysis
Samples for PK analysis were taken on day 1 only, at baseline (prior to chemotherapy administration), and 30 minutes, 60 minutes, and 240 minutes after CVC flush at the completion of NUC‐1031 administration. Blood (6 mL) was collected using heparinized blood collection tubes spiked with tetrahydrouridine (25 μg/mL) in order to inhibit CDA activity. Plasma samples were assayed for NUC‐1031, dFdC, and dFdU using a previously described liquid chromatography–tandem mass spectrometry method [4]. Pharmacokinetic parameters were estimated for ABC‐08 using PK samples and a PK model developed with clinical study data from the phase I dose‐escalation monotherapy study to assess the safety, efficacy, and PK of NUC‐1031 in patients with advanced solid tumors [4].
Statistical Analysis
Adverse events were summarized by maximum toxicity grade and causality assessment for each initial dose level of NUC‐1031 and cisplatin.
Efficacy was determined in all patients with measurable disease who received at least one cycle of NUC‐1031 with cisplatin and who had at least one follow‐up radiographic assessment. Objective response was defined as a participant who achieved a best response of PR or complete response (CR) on study treatment. Objective response rates were calculated as the percent of evaluable participants who achieved CR or PR, and confidence intervals (CIs) were constructed. Progression‐free survival was defined as the time from registration until the date of radiological or clinical disease progression or death (from any cause in the absence of progression), regardless of whether the subject withdrew from therapy or received another anticancer therapy prior to progression. The OS was calculated from the date of registration to date of death. The date of registration as time of origin for PFS and OS calculation was protocol‐defined. Overall survival and PFS were analyzed using Kaplan‐Meier curves. No formal statistical analyses were planned or performed on safety, PK, or efficacy data.
Results
Patient Characteristics
A total of 21 patients with ABC were enrolled at five centers in the U.K. and received cycle 1 day 1 of treatment between February 2, 2016, and March 14, 2018. There was a 12‐month period of patient follow‐up from the date of the last patient registered onto the study. The median age was 61 years (range, 47–78) and 13 (62%) were male. Seven and 14 patients had an ECOG PS of 0 and 1, respectively. The primary tumor site was hilar cholangiocarcinoma in seven patients, intrahepatic and distal bile duct cholangiocarcinoma in five patients each, and gallbladder cancer and ampulla of Vater carcinoma in two patients each. Seventeen and four patients had metastatic and locally advanced disease, respectively (Table 1). Eight patients had recurrent disease, with two patients having received previous adjuvant chemotherapy (capecitabine or cisplatin/gemcitabine), completed >6 months from enrolment. No prior radiotherapy was recorded.
Table 1.
Baseline demographics and clinical characteristics of patients enrolled in ABC‐08
| Characteristic | NUC‐1031 a 625 mg/m2 (n = 15) | NUC‐1031 a 725 mg/m2 (n = 6) |
|---|---|---|
| Age, median (range), years | 61 (55–78) | 59.5 (47–71) |
| Gender | ||
| Male | 10 | 3 |
| Female | 5 | 3 |
| ECOG PS | ||
| 0 | 4 | 3 |
| 1 | 11 | 3 |
| Primary tumor site | ||
| Intrahepatic | 5 | 0 |
| Hilar | 3 | 4 |
| Distal bile duct | 3 | 2 |
| Gallbladder cancer | 2 | 0 |
| Ampulla of Vater carcinoma | 2 | 0 |
| Disease status | ||
| Locally advanced | 3 | 1 |
| Metastatic | 12 | 5 |
| Recurrent disease | ||
| No | 11 | 2 |
| Yes | 4 | 4 |
In combination with cisplatin 25 mg/m2 on days 1 and 8 (21‐day schedule).
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group performance status.
Dose Determination
Eight patients were treated at the starting dose of 625 g/m2 NUC‐1031 in combination with cisplatin 25 mg/m2 on days 1 and 8 of a 21‐day schedule. Two patients were not evaluable for DLTs because of omission of day 8 of treatment and so were replaced (one patient with hilar cholangiocarcinoma had disease‐related cholangitis requiring endoscopic retrograde cholangiopancreatography and was unable to restart treatment within 21 days; the other had grade 2 thrombocytopenia on cycle 1 day 8; a subsequent protocol amendment was enacted to align with ABC‐02 [1] permitting treatment on day 8 with NUC‐1031 at a 25% dose reduction, with no dose reduction required for cisplatin in the presence of grade 2 thrombocytopenia and/or grade 3 neutropenia).
One of the first three evaluable patients in the 625 mg/m2 cohort had a drug‐related rise in AST (grade 3) during cycle 1, which returned to grade ≤ 2 within 7 days. At that time, the decision of the TMG was to expand the 625 mg/m2 cohort to six patients, as a grade ≤ 3 AST/ALT was initially classified as a DLT. An additional patient in the expanded cohort had a rise in ALT (grade 3) during cycle 1, which also returned to grade ≤ 2 within 7 days.
The TMG noted that both the AST and ALT rise returned to grade ≤ 2 within 7 days and recommended a protocol amendment to clarify that patients who had a grade ≤ 3 ALT or AST (drug‐related) that returned to grade ≤ 2 within 7 days would not be classified as experiencing a DLT, as it would not be considered clinically significant [7]. The TMG decision was made to escalate to 725 mg/m2 NUC‐1031 in combination with cisplatin 25 mg/m2, days 1 and 8 of a 21‐day schedule.
One patient in the 725 mg/m2 cohort developed a drug‐related grade 3 gamma‐glutamyl transferase (GGT) rise during cycle 1. This was considered not clinically significant by the TMG. This dose cohort was expanded by an additional three patients as per the existing protocol version; therefore, six patients in total were included in the 725 mg/m2 NUC‐1031 cohort.
A protocol amendment was subsequently instituted to clarify the wording in the relevant criteria within the DLT definition to “Greater than or equal to grade 3 NUC‐1031–related or chemotherapy combination AE or laboratory abnormality that is deemed clinically significant by the investigator.”
After protocol clarifications, it was concluded by the TMG that no DLTs were experienced by the first 14 patients enrolled (eight and six patients in the 625 mg/m2 and 725 mg/m2 NUC‐1031 dose cohorts, respectively). On review of the available safety and PK data, it was determined that there was no discernible difference in terms of the safety or PK between the two dose cohorts of NUC‐1031 (625 mg/m2 and 725 mg/m2) in combination with cisplatin. Acknowledging that the RP2D for NUC‐1031 monotherapy was 825 mg/m2 [4], the decision was made not to escalate the dose of NUC‐1031 in combination with cisplatin further and to expand the 625 mg/m2 NUC‐1031 dose cohort by six additional patients.
No MTD was therefore defined. Seven patients were enrolled in the expanded 625 mg/m2 cohort (one patient was replaced as they only received cycle 1 day 1 of treatment because of disease‐related clinical deterioration). There were no DLTs in this expanded dose cohort.
On review of the complete safety, efficacy, and PK data, it was determined that there was no difference in safety or PK between the two cohorts. Initially, the 625 mg/m2 NUC‐1031 dose was being considered as the dose for phase III evaluation with cisplatin. However, as the data matured, there was an indication that patients in the 725 mg/m2 cohort maintained dose intensity and remained on treatment for longer than patients in the 625 mg/m2 cohort. At this time point, based on the available data from the monotherapy study [4] and the current study, the TMG decided that the NUC‐1031 725 mg/m2 dose should be selected as the dose (without further cohort expansion) to be given in combination with cisplatin 25 mg/m2, on days 1 and 8 of a 21‐day schedule in patients with ABC for phase III evaluation in the first‐line setting, additionally allowing greater scope for dose reduction, if required.
Safety and Tolerability
The most common treatment‐emergent grade 2–4 adverse events occurring in at least one patient enrolled in ABC‐08 are depicted in Table 2. The most common treatment‐emergent grade 3–4 AEs occurring in more than one patient in the 625 mg/m2 NUC‐1031 cohort were increased GGT (n = 6; 40%), neutropenia (n = 4; 27%), increased ALT (n = 3; 20%), decreased white blood cells (n = 3; 20%), nausea (n = 2; 13%), diarrhea (n = 2; 13%), fatigue (n = 2; 13%), increased bilirubin (n = 2; 13%), thrombocytopenia (n = 2; 13%), and thrombus (n = 3; 20%) (Table 2).
Table 2.
The most common treatment‐emergent grade 2–4 adverse events occurring in at least one patient enrolled in ABC‐08
| Adverse event: Grade a | NUC‐1031 b 625 mg/m2 (n = 15)n (%) | NUC‐1031 b 725 mg/m2 (n = 6)n (%) |
|---|---|---|
| GGT increase | ||
| 2 | 3 (20) | 1 (17) |
| 3 | 4 (27) | 4 (67) |
| 4 | 2 (13) | 0 (0) |
| Neutropenia | ||
| 2 | 1 (7) | 0 (0) |
| 3 | 3 (20) | 1 (17) |
| 4 | 1 (7) | 0 (0) |
| ALT increase | ||
| 2 | 1 (7) | 1 (17) |
| 3 | 3 (20) | 0 (0) |
| 4 | 0 (0) | 0 (0) |
| WCC decrease | ||
| 2 | 3 (20) | 1 (17) |
| 3 | 3 (20) | 0 (0) |
| 4 | 0 (0) | 0 (0) |
| Nausea | ||
| 2 | 3 (20) | 4 (67) |
| 3 | 2 (13) | 0 (0) |
| 4 | 0 (0) | 0 (0) |
| Vomiting | ||
| 2 | 2 (13) | 1 (17) |
| 3 | 1 (7) | 1 (17) |
| 4 | 0 (0) | 0 (0) |
| Diarrhea | ||
| 2 | 0 (0) | 0 (0) |
| 3 | 2 (13) | 1 (17) |
| 4 | 0 (0) | 0 (0) |
| Fatigue | ||
| 2 | 5 (33) | 3 (50) |
| 3 | 2 (13) | 2 (13) |
| 4 | 0 (0) | 0 (0) |
| Hyperbilirubinemia | ||
| 2 | 0 (0) | 0 (0) |
| 3 | 2 (13) | 0 (0) |
| 4 | 0 (0) | 0 (0) |
| Thrombocytopenia | ||
| 2 | 4 (27) | 3 (50) |
| 3 | 2 (13) | 0 (0) |
| 4 | 0 (0) | 0 (0) |
| Thrombus | ||
| 2 | 0 (0) | 0 (0) |
| 3 | 3 (20) | 0 (0) |
| 4 | 0 (0) | 0 (0) |
| Hb decrease: c 2 | 4 (27) | 1 (17) |
| Mg decrease: c 2 | 0 (0) | 1 (17) |
| Alk phos increase: c 2 | 3 (20) | 1 (17) |
| AST increase: c 2 | 1 (7) | 1 (17) |
| Abdominal pain: c 2 | 1 (7) | 0 (0) |
| Anorexia: c 2 | 2 (13) | 0 (0) |
| Dyspnea: c 2 | 0 (0) | 1 (17) |
| Infection: c 2 | 0 (0) | 1 (17) |
| Infusion reaction: c 2 | 1 (7) | 0 (0) |
| Neutropenia: c 2 | 1 (7) | 0 (0) |
| Urinary tract infection: c 2 | 1 (7) | 0 (0) |
According to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.
In combination with cisplatin 25 mg/m2 on day 1 and 8 (21‐day schedule). In total, there were 15 and 6 patients recruited to the NUC‐1031 625 mg/m2 and 725 mg/m2 cohorts, respectively.
No grade 3–4 adverse events.
Abbreviations: Alk phos, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma‐glutamyl transferase; Hb, hemoglobin; Mg, magnesium; WCC, white cell count.
The most common treatment‐emergent grade 3–4 AEs occurring in more than one patient in the 725 mg/m2 NUC‐1031 cohort were increased GGT (n = 4; 67%) and fatigue (n = 2; 33%) (Table 2).
There were two patients (13%) and one patient (7%) who experienced treatment‐emergent grade 4 increased GGT and grade 4 neutropenia in the 625 mg/m2 NUC‐1031 cohort, respectively. There were no treatment‐emergent grade 4 AEs recorded in the 725 mg/m2 NUC‐1031 cohort. The most relevant treatment‐emergent grade 1 adverse events occurring in at least four patients enrolled in ABC‐08 are described in supplemental online Table 1. The impression that there may be fewer overall toxicities in the 725 mg/m2 NUC‐1031 cohort may be a reflection of the smaller patient numbers.
Patients discontinued treatment for the following reasons: adverse events (n = 9), intercurrent illness (n = 1), investigator decision (n = 2), patient decision (n = 1), progressive disease (n = 6), and death (n = 2). The detailed reasons for treatment discontinuation are described in Table 3.
Table 3.
Reasons for treatment discontinuation in ABC‐08 (n = 21)
| Reason for treatment discontinuation | Patients (n) |
|---|---|
| Adverse events | |
| Cholangitis a | 1 |
| Biliary obstruction b | 2 |
| Small bowel perforation secondary to adhesions c | 1 |
| Perforated diverticulitis | 1 |
| Grade 3 GGT d | 1 |
| Brain metastases (clinical deterioration) e | 1 |
| Grade 2 vomiting and hematemesis secondary to cisplatin f | 1 |
| Radiological pneumonitis (grade 1) g | 1 |
| Intercurrent illness (preexisting cirrhosis) h | 1 |
| Investigator decision: (a) surgically resectable disease after ABC‐08 combination treatment and (b) inability to restart treatment within 21 days because of biliary obstruction | 2 |
| Patient decision (holiday) | 1 |
| Progressive disease | 6 |
| Death (underlying disease) | 2 |
Cholangitis related to biliary stent; unrelated to disease progression or treatment; patient not neutropenic.
Biliary obstruction; unrelated to disease progression or treatment; patients not neutropenic.
In a patient with recurrent distal bile duct cholangiocarcinoma.
Not clinically significant, but patient decided to commence cisplatin/gemcitabine locally.
Patient complained of right arm paresthesia after one cycle of treatment on ABC‐08, which on questioning was present prior to therapy commencement (brain metastases were identified on magnetic resonance imaging). The decision was made to proceed with treatment, given that patient was relatively asymptomatic, and the patient went on to achieve a partial response on body imaging.
Deemed related to cisplatin by treating investigator on assessment and review of relevant Summary of Product Characteristics.
Asymptomatic on a background of long‐standing fibrosis noted in lower lobes of lungs (radiological findings attributed to NUC‐1031/cisplatin).
Patient discontinued treatment because of decline in liver function (ascites accumulation) deemed unrelated to treatment; a partial response was noted on imaging.
Abbreviation: GGT, gamma‐glutamyl transferase.
Efficacy
The median number of received cycles was 6 (range, 1–12) for the cohort that received 625 mg/m2 NUC‐1031 (n = 15) and 7.5 (range, 1–14) for the 725 mg/m2 NUC‐1031 dose (n = 6) in combination with cisplatin (days 1 and 8 of a 21‐day schedule).
For the 625 mg/m2 NUC‐1031 cohort, the median cumulative relative dose was 78.8% (range, 45.3%–102.6%) for NUC‐1031 and 84.1% (range, 43.1%–104.6%) for cisplatin. For the 725 mg/m2 NUC‐1031 cohort, the median cumulative relative dose was 73.7% (range, 55.3%–98.1%) for NUC‐1031 and 68% (range, 56.5%–103.5%) for cisplatin. Details on dosing of NUC‐1031 and cisplatin received by individual patients enrolled in ABC‐08 are provided in supplemental online Table 2, including individual cumulative percentages.
There were four patients alive at the end of study follow‐up, with hilar cholangiocarcinoma (n = 2), intrahepatic cholangiocarcinoma (n = 1), and distal bile duct (n = 1) as the primary site of their tumors, after a median follow‐up time of 20.6 months (range, 16.2–36.0).
In the intention‐to‐treat (ITT) population (n = 21), the overall ORR was 33% (7/21) (one CR in the NUC‐1031 625 mg/m2 cohort and four and two PRs in the NUC‐1031 625 mg/m2 and 725 mg/m2 cohorts, respectively: two patients who had PRs did not have subsequent scans, as treatment was discontinued because of biliary obstruction and deteriorating liver function due to preexisting underlying cirrhosis; confirmation of PR was not mandated per protocol): 5 of 15 (33%) and 2 of 6 (33%) in the NUC‐1031 625 mg/m2 and 725 mg/m2 cohorts, respectively. Eight patients had stable disease as best response; one patient did not have measurable disease at baseline (nonevaluable), three had PD, and two did not have a second computed tomography scan to assess response (clinical deterioration after biliary obstruction and secondary to comorbidities respectively). For the efficacy evaluable cohort, two patients did not receive at least one cycle of NUC‐1031 with cisplatin because of cholangitis and grade 3 GGT (considered not clinically significant, but before protocol amendment); they switched to standard‐of‐care cisplatin/gemcitabine. Tumor control (PR, CR, or SD) was achieved in 16 of 21 patients (76%) who received NUC‐1031 and cisplatin. Of note, the two patients included with ampulla of Vater carcinoma had a CR and SD as best response and a median OS of 10.7 and 17.2 months, respectively. The two patients who received prior adjuvant treatment (capecitabine or cisplatin/gemcitabine) had SD as best response and a median OS of 17.2 and 21.1 months, respectively.
In the efficacy evaluable population (n = 16), the overall ORR was 44% (7/16) (Fig. 1).
Figure 1.
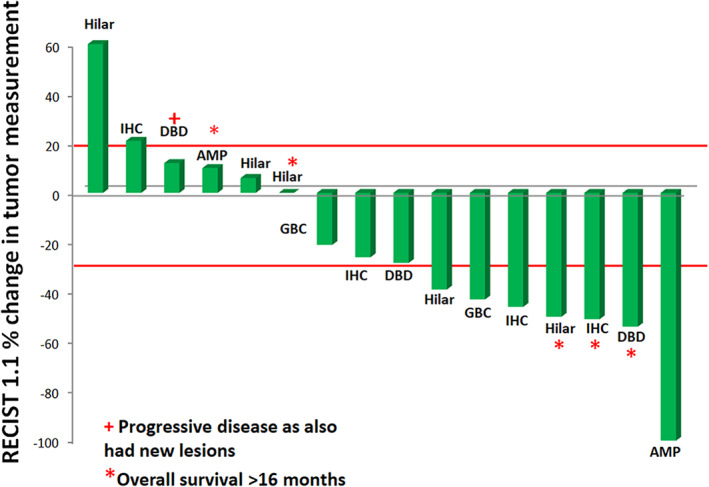
Waterfall plot of best response to therapy in ABC‐08. Sixteen patients with measurable disease were assessed for efficacy (received at least one cycle of NUC‐1031 with cisplatin and had at least one follow‐up radiographic assessment). In the efficacy evaluable population, the overall objective response rate was 44%.Abbreviations: AMP, ampulla of Vater carcinoma; DBD, distal bile duct cholangiocarcinoma; GBC, gallbladder cancer; IHC, intrahepatic cholangiocarcinoma.
The median time between registration and cycle 1 day 1 of treatment on ABC‐08 was 2 days. The median PFS (radiological) was 5.7 months (95% CI, 3.3–10) in the NUC‐1031 625 mg/m2 cohort and 8.6 months (95% CI, 2.6–not estimable) in the 725 mg/m2 NUC‐1031 cohort. The overall median PFS was 7.2 months (95% CI, 4.3–10.1).
The median OS was 9.6 months (95% CI, 4.7–10.7) in the NUC‐1031 625 mg/m2 cohort and 8.6 months (95% CI, 6.7–not estimable) in the NUC‐1031 725 mg/m2 cohort. The overall median OS was 9.6 months (95% CI, 6.7–13.1). Summary details of overall survival based on ORR, primary tumor site, and NUC‐1031 dose (mg/m2) (efficacy evaluable population) are provided in Table 4.
Table 4.
Summary details of overall survival based on objective response rate, primary tumor site, and NUC‐1031 dose (mg/m2) (efficacy evaluable population)
| Patient | Change in tumor volume (%) | Primary tumor site | NUC‐1031 a dose (mg/m2) | Overall survival (months) | Treatment after ABC‐08 | Status at end of trial follow‐up |
|---|---|---|---|---|---|---|
| 6 | −100 | AMP | 625 | 10.7 | No | Dead |
| 5 | −54 | DBD | 625 | 25.8 | Cisplatin/gemcitabine | Dead |
| 2 | −51 | IHC | 625 | 36.0 | SIRT | Alive |
| 12 | −50 | Hilar | 725 | 20.0 | No | Alive |
| 17 | −46 | IHC | 625 | 7.2 | No | Dead |
| 8 | −43 | GBC | 625 | 8.7 | No | Dead |
| 10 | −39 | Hilar | 725 | 6.7 | No | Dead |
| 15 | −28.3 | DBD | 625 | 6.4 | No | Dead |
| 18 | −26 | IHC | 625 | 10.0 | No | Dead |
| 21 | −21 | GBC | 625 | 9.6 |
Irinotecan/capecitabine/ trastuzumab |
Dead |
| 13 | 0 | Hilar | 725 | 16.3 | No | Alive |
| 16 | +6 | Hilar | 625 | 13.1 | No | Dead |
| 7 | +10 | AMP | 625 | 17.2 | Surgical resection + adjuvant gemcitabine | Dead |
| 9 | +12 | DBD | 725 | 8.0 | No | Dead |
| 4 | +21 | IHC | 625 | 10.1 | Oxaliplatin/5‐fluorouracil/folinic acid | Dead |
| 20 | +60 | Hilar | 625 | 4.7 | No | Dead |
Change in tumor volume (%) based on RECIST version 1.1 (efficacy evaluable population: 16 patients with measurable disease who received at least one cycle of NUC‐1031 with cisplatin and had at least one follow‐up radiographic assessment). An additional patient did not have measurable disease (best response based on nontarget lesion was noncomplete response/nonprogressive disease); two patients did not receive any treatment on ABC‐08 beyond cycle 1, day 1 because of cholangitis and raised gamma‐glutamyl transferase (surviving 5.0 and 21.2 months, respectively), and two patients did not have follow‐up imaging beyond baseline, having died at 1.5 and 3.6 months, respectively.
In combination with cisplatin 25 mg/m2 on days 1 and 8 (21‐day schedule).
Abbreviations: AMP, ampulla of Vater carcinoma; DBD, distal bile duct cholangiocarcinoma; IHC, intrahepatic cholangiocarcinoma; GBC, gallbladder cancer; SIRT, selective internal radiation therapy.
Treatment Beyond ABC‐08
One patient (with recurrent ampulla of Vater carcinoma) was deemed to have surgically resectable liver disease (after computed tomography scan, MRI, and 18F‐fluorodeoxyglucose positron emission tomography imaging) after receiving four cycles of NUC‐1031 625 mg/m2 in combination with cisplatin in the advanced setting; this patient had surgery and then went on to receive six cycles of adjuvant gemcitabine. This patient developed liver and lung recurrence approximately 11 months after resection and was not fit for further systemic treatment.
Further treatment lines in the advanced setting were given to five patients; two patients received cisplatin/gemcitabine, one received oxaliplatin/5‐fluorouracil/folinic acid, one received irinotecan/capecitabine/trastuzumab (this patient had human epidermal growth factor receptor 2 [HER2]–positive advanced gallbladder cancer), and one received selective internal radiation therapy, which was delivered immediately after discontinuation of ABC‐08 (because of grade I asymptomatic pneumonitis). Treatments given after ABC‐08 followed previously published literature [8, 9, 10, 11].
Only three patients had molecular testing in ABC‐08 in addition to the patient described above who had HER2 positivity; one had no targetable mutation (distal bile duct cholangiocarcinoma; survival 8.6 months); one had a breast cancer gene 1 genomic alteration and Erb‐B2 receptor tyrosine kinase 2 and type 2 topoisomerase alpha amplifications (hilar cholangiocarcinoma; survival 16.3 months); and one had mutations in epidermal growth factor receptor, serine/threonine kinase 11, Kirsten ras oncogene homolog, cyclin‐dependent kinase inhibitor 2A, and tumor suppressor TP53 (distal bile duct cholangiocarcinoma; survival 21.2 months).
Pharmacokinetics
Pharmacokinetic analysis was available for 17 patients: 12 from the 625 mg/m2 NUC‐1031 cohort and 5 from the 725 mg/m2 NUC‐1031 cohort (two patients did not have PK samples taken, and samples were misplaced for two patients).
Plasma PK of NUC‐1031, dFdC, and dFdU
The highest plasma exposures were seen for NUC‐1031; median NUC‐1031 area under the plasma concentration–time curve from time 0 to last measurable time (AUC0–24) and Cmax estimates ranged from 218 to 324 μg•h/mL and 309 to 889 μg/mL, respectively. The lowest overall plasma exposures were observed for dFdC; median plasma dFdC AUC0–24 and Cmax estimates ranged from 0.47 to 1.56 μg•h/mL and 0.284 to 0.522 μg/mL, respectively. The median plasma dFdU AUC0–24 and Cmax estimates ranged from 44.9 to 71.0 μg•h/mL and 2.65 to 3.56 μg/mL, respectively. The median terminal half‐life for NUC‐1031 (estimated from the t1/2,λ) and dFdU (estimated from the t1/2,β) was 2.0–3.3 hours and 5.0–13.6 hours, respectively.
Comparison of Plasma AUC0 –24 and Cmax Between NUC‐1031 Doses
AUC0–24 values for NUC‐1031 were similar with increasing dose, with median values of 261 and 282 μg•h/mL for the 625 mg/m2 and 725 mg/m2 doses, respectively. Cmax values for NUC‐1031 were also similar with increasing dose, with median values of 552 and 685 μg/mL for the 625 mg/m2 and 725 mg/m2 doses, respectively.
AUC0–24 values for dFdC, with increasing dose, were (median values) 0.863 and 1.17 μg•h/mL for the 625 mg/m2 and 725 mg/m2 doses, respectively. Cmax values for dFdC were similar, with median values of 0.432 and 0.423 μg/mL for the 625 mg/m2 and 725 mg/m2 doses, respectively.
AUC0–24 values for dFdU, with increasing dose, were (median values) 48.0 and 71.0 μg•h/mL for the 625 mg/m2 and 725 mg/m2 doses, respectively. Cmax values for dFdU were similar, with median values of 2.82 and 3.56 μg/mL for the 625 mg/m2 and 725 mg/m2 doses, respectively (Table 5). There was no formal correlation between PKs and toxicity. On review of individual day 1 PK parameters for two patients who received NUC‐1031 625 mg/m2 or 725 mg/m2 in combination with cisplatin (without any treatment omissions), the Cmax values and AUC0–24 were 793 μg/mL and 458 μg•h/mL, and 646 μg/mL and 282 μg•h/mL, respectively. In two patients who received NUC‐1031 625 mg/m2 or 725 mg/m2 in combination with cisplatin (with one omission within one cycle [day 1 or day 8]), the Cmax values and AUC0–24 were 700 μg/mL and 220 μg•h/mL, and 497 μg/mL and 473 μg•h/mL, respectively, demonstrating variability and no observable trend.
Table 5.
Summary statistics for individual, model‐derived plasma pharmacokinetic parameters by dose (mg/m2) for participants enrolled in ABC‐08
| Parameter | NUC‐1031 625 mg/m2 (n = 12), median (90% CI) | NUC‐1031 725 mg/m2 (n = 5), median (90% CI) |
|---|---|---|
| NUC‐1031 | ||
| Cmax (μg/mL) | 552 (244–1020) | 685 (527–897) |
| AUC0–24 (μg•h/mL) | 261 (176–588) | 282 (265–573) |
| AUC0–∞ (μg•h/mL) | 261 (176–588) | 282 (265–573) |
| t1/2,α (h) | 0.0250 (0.0126–0.0521) | 0.0254 (0.0218–0.0424) |
| t1/2,β (h) | 0.147 (0.103–0.682) | 0.155 (0.130–0.521) |
| t1/2,λ (h) | 2.38 (1.15–3.86) | 3.07 (2.74–5.66) |
| Vss (L) | 4.28 (2.69–7.81) | 4.19 (3.23–4.71) |
| CL (L/h) | 4.46 (2.08–6.62) | 4.37 (2.31–4.92) |
| dFdC | ||
| Cmax (μg/mL) | 0.432 (0.165–0.588) | 0.423 (0.363–0.499) |
| AUC0–24 (μg•h/mL) | 0.863 (0.358–2.07) | 1.17 (1.03–1.69) |
| AUC0–∞ (μg•h/mL) | 0.869 (0.358–2.14) | 1.18 (1.04–1.72) |
| t1/2,α (h) | 0.0771 (0.0567–0.117) | 0.0920 (0.0756–0.103) |
| t1/2,β (h) | 1.52 (0.634–2.90) | 1.98 (1.52–2.13) |
| t1/2,λ (h) | 1310 (1300–1350) | 1320 (1320–1330) |
| Vss (L) | 2820 (2590–3790) | 3000 (2760–3100) |
| CL (L/h) | 602 (208–1560) | 475 (339–577) |
| dFdU | ||
| Cmax (μg/mL) | 2.82 (2.21–3.58) | 3.56 (3.05–4.32) |
| AUC0–24 (μg•h/mL) | 48.0 (39.2–68.9) | 71 (52.1–78.2) |
| AUC 0–∞ (μg•h/mL) | 63.8 (43.9–110) | 98.6 (63.0–141) |
| t1/2,β (h) a | 7.57 (4.00–19.3) | 10.0 (5.39–14.7) |
| Vss (L) | 66.6 (43.4–161) | 67.2 (60.4–82.9) |
| CL (L/h) | 6.37 (4.13–9.22) | 5.41 (3.61–8.30) |
Note that the half‐life estimate for dFdU is listed under t1/2,β, as there is only one applicable half‐life for that analyte.
Abbreviations: AUC0–24, area under the plasma concentration–time curve from time 0 to last measurable time; AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; CI, confidence interval; CL, clearance; Cmax, maximum clearance; dFdC: di‐fluoro‐deoxycytidine, dFdU: 2′,2′‐difluorodeoxyuridine; t1/2: half‐life, Vss, volume of distribution at steady‐state.
Assessment of PK Interaction with Cisplatin
In the ABC‐08 expansion cohort, the median AUC0–24 and Cmax values for NUC‐1031 were found to be 218 μg•h/mL (range, 169–263 μg•h/mL) and 309 μg/mL (range, 225–400 μg/mL), respectively. In the ABC‐08 expansion cohort (NUC‐1031 625 mg/m2), the AUC0–24 and Cmax values for dFdU were found to be 49.1 μg•h/mL (range, 38.1–58.5 μg•h/mL) and 2.85 μg/mL (range, 2.38–3.19 μg/mL), respectively.
Discussion
This phase Ib study of NUC‐1031 combined with cisplatin for the first‐line treatment of patients with ABC demonstrated that the combination had a favorable safety profile and achieved good tumor control.
Treatment‐emergent grade 3–4 AEs were not unexpected and were similar to those previously reported for gemcitabine in this disease group, including fatigue, hematological toxicity, and altered liver function enzymes [1, 12].
The ORR (ITT) was 33%, and efficacy evaluable ORR was 44%, compared with 26.1% reported in the ABC‐02 study for the cisplatin/gemcitabine combination [1]. Tumor control and OS in ABC‐08 were 76% and 9.6 months (with an upper 95% CI of 13.1 months), respectively, similar to the cisplatin/gemcitabine combination in ABC‐02: 81.4% and 11.7 months [1]. Despite the potential biological heterogeneity of ABC primary tumor sites [13], responses seen in ABC‐08 were distributed across all five biliary tract cancer subtypes (intrahepatic, hilar, and distal bile duct cholangiocarcinoma, gallbladder cancer, and ampulla of Vater carcinoma).
The median number of cycles received in ABC‐08 was 6 and 7.5 in the NUC‐1031 625 mg/m2 and 725 mg/m2 dose groups, respectively, resembling the median duration of cisplatin/gemcitabine combination treatment in ABC‐02: seven cycles (21 weeks) [1]. Approximately 19% of patients went on to receive second‐line treatment in ABC‐08, which approximates with previously reported literature (25%) [14], and four patients were still alive at the end of follow‐up, each having survived >16 months (all with a diagnosis of cholangiocarcinoma), which reflects conditional probability of survival or landmark survival in patients with ABC: the longer a patient survives, the greater the chance that they will survive another year; this is potentially influenced by receipt of combination therapy and anatomic ABC primary site, with those with intrahepatic cholangiocarcinoma and nonspecified cholangiocarcinoma having superior landmark survival to those with a gallbladder cancer diagnosis in a recently reported study [15].
NUC‐1031 achieved higher concentrations than its metabolites dFdU and dFdC in this study. AUC0–24 values for NUC‐1031, dFdU, and dFdC were not similar with increasing dose (625 mg/m2 to 725 mg/m2). Cmax values for NUC‐1031, dFdU, and dFdC were similar with increasing dose. The Cmax of dFdU was around 50‐fold lower (2.82 and 3.56 μg/mL in 625 mg/m2 and 725 mg/m2, respectively) compared with reported levels for gemcitabine (121 μg/mL) [7], further supporting that NUC‐1031 is resistant to degradation by CDA. It has previously been reported that the estimated plasma t1/2 of NUC‐1031 was 8.3 hours, in comparison with the shorter reported plasma t1/2 of gemcitabine (up to 94 minutes), potentially allowing tumor cells to have a more prolonged exposure to dFdCTP (and so enhancing its activity) [4]. In this current study, the terminal half‐life for NUC‐1031 was 2.0–3.3 hours (still greater than gemcitabine), which may reflect that no PK samples were taken at 6 and 24 hours, as were analyzed in the monotherapy study [4], which would have allowed a more accurate comparison.
In general, the interpatient variability predicted from the PK model generated in the NUC‐1031 monotherapy study [4] was found to be in the region of 40% for NUC‐1031. Results for ABC‐08 showed that there was an approximate 26% decrease in AUC0–24 and a 36% increase in Cmax for NUC‐1031 after combination with cisplatin when compared with historical monotherapy data [4], and so the combination with cisplatin did not seem to alter the PK profile of NUC‐1031.
Conclusion
This is the first study reporting on the combination of NUC‐1031 with cisplatin in ABC. A dose of 725 mg/m2 of NUC‐1031 is recommended in combination with cisplatin 25 mg/m2 on a day 1 and 8 schedule every 3 weeks in patients with ABC. This regimen is currently being compared with cisplatin plus gemcitabine in the NuTide:121 study (NCT04163900), a global phase III randomized study in patients with ABC [16].
Author Contributions
Conception/design: Mairéad G. McNamara, Juan W. Valle
Provision of study material or patients: Mairéad G. McNamara, J. Bridgewater, D.H. Palmer, O. Faluyi, H. Wasan, T.R.J. Evans, Juan W. Valle
Collection and/or assembly of data: Mairéad G. McNamara, J. Bridgewater, D.H. Palmer, O. Faluyi, H. Wasan, A. Patel, W.D. Ryder, S. Barber, C. Gnanaranjan, E. Ghazaly, T.R.J. Evans, Juan W. Valle
Data analysis and interpretation: Mairéad G. McNamara, J. Bridgewater, D.H. Palmer, O. Faluyi, H. Wasan, A. Patel, W.D. Ryder, S. Barber, C. Gnanaranjan, E. Ghazaly, T.R.J. Evans, Juan W. Valle
Manuscript writing: Mairéad G. McNamara, J. Bridgewater, D.H. Palmer, O. Faluyi, H. Wasan, A. Patel, W.D. Ryder, S. Barber, C. Gnanaranjan, E. Ghazaly, T.R.J. Evans, Juan W. Valle
Final approval of manuscript: Mairéad G. McNamara, J. Bridgewater, D.H. Palmer, O. Faluyi, H. Wasan, A. Patel, W.D. Ryder, S. Barber, C. Gnanaranjan, E. Ghazaly, T.R.J. Evans, Juan W. Valle
Disclosures
Mairéad G. McNamara: Servier, Ipsen, NuCana plc (RF), Bayer, Ipsen, Novartis (other—travel), Pfizer, Ipsen, NuCana plc, Mylan (H), Celgene, Ipsen, Sirtex, Baxalta (C/A); John Bridgewater: Merck Serono, Roche, Sanofi, Bayer (H); Daniel H. Palmer: NuCana plc (H, RF); Harpreet Wasan: Eli Lilly & Co., Merck, Roche, Celgene (H), Sirtex, Pfizer (RF), Merck, Sirtex, Eli Lilly & Co., Celgene (other—travel); Chathunissa Gnanaranjan: NuCana plc (RF); Essam Ghazaly: NuCana plc (RF); T.R. Jeff Evans: Basilea, Bayer, AstraZeneca, Beigene, Celgene, Bristol‐Myers Squibb, Clovis, Eisai, Genentech, GlaxoSmithKline, Immunova, Nucana, Karus Therapeutics, Otsuka, Roche, MiNa Therapeutics, Pfizer, Sierra, Eli Lilly & Co., Novartis, Bicycle Therapeutics, Halozyme, Johnson & Johnson, Vertex, CytomX, Plexxikon, Boehringer, Athenex, Adaptimmune, Verastem, Immunocore, Iovance, Berg, BiolinerX, Merck Sharp & Dohme, Medivir (RF), Bristol‐Myers Squibb, Merck Sharp & Dohme, Celgene, Bayer (other—travel); Juan W. Valle: Ipsen, Novartis, AstraZeneca, Merck, Delcath Systems, Agios, Pfizer, PCI Biotech, Incyte, Keocyt, QED, Pieris Pharmaceuticals, Genoscience Pharma, Mundipharma EDO (C/A), Ipsen (H), Novartis, Ipsen, NuCana plc, Imaging Equipment Limited (other—speakers’ bureau). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplementary material 1 Full eligibility criteria for ABC‐08
Supplementary material 2: Criteria for treatment of patients enrolled onto ABC‐08
Supplementary Table 1 The most relevant treatment‐emergent grade 1 adverse events occurring in ≥4 patients enrolled in ABC‐08
Supplementary Table 2: Details on dosing of NUC‐1031 and cisplatin received by individual patients enrolled in ABC‐08
Acknowledgments
This study was sponsored by the Christie NHS Foundation Trust, Manchester, U.K. (sponsor reference: CFTSp096), managed by the Manchester Clinical Trials Unit and funded by an unrestricted educational grant from NuCana plc. The authors wish to thank the patients who took part in this study and their families. The study was carried out at the National Institute for Health Research (NIHR) Manchester Clinical Research facility. The views expressed are those of the authors and not necessarily those of NuCana plc, the National Health Service, the NIHR, or the Department of Health. Thanks also to Alison Backen (Project Manager, the Christie NHS Foundation Trust, Manchester, U.K.) for her contribution to initial protocol development. The authors would also like to thank the independent Trial Management Group members: Alan Anthoney, Sheela Rao, Paul Ross, and Tim Iveson. The data included in this study are available on reasonable request. E.G. is currently affiliated with the Pharmacokinetics Group, Medicines and Healthcare Products Regulatory Agency (MHRA), London, U.K.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Valle J, Wasan H, Palmer DH et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–1281. [DOI] [PubMed] [Google Scholar]
- 2. Nakano Y, Tanno S, Koizumi K et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer 2007;96:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Slusarczyk M, Lopez MH, Balzarini J et al. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC‐1031) in clinical development. J Med Chem 2014;57:1531–1542. [DOI] [PubMed] [Google Scholar]
- 4. Blagden SP, Rizzuto I, Suppiah P et al. Anti‐tumour activity of a first‐in‐class agent NUC‐1031 in patients with advanced cancer: Results of a phase I study. Br J Cancer 2018;119:815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kapacee ZA, Knox JJ, Palmer D et al. NUC‐1031, use of ProTide technology to circumvent gemcitabine resistance: Current status in clinical trials. Med Oncol 2020;37:61. [DOI] [PubMed] [Google Scholar]
- 6. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 7. Ciccolini J, Serdjebi C, Peters GJ et al. Pharmacokinetics and pharmacogenetics of gemcitabine as a mainstay in adult and pediatric oncology: An EORTC‐PAMM perspective. Cancer Chemother Pharmacol 2016;78:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doherty MK, McNamara MG, Aneja P et al. Long term responders to palliative chemotherapy for advanced biliary tract cancer. J Gastrointest Oncol 2017;8:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lamarca A, Galdy S, Barriuso J et al. The HER3 pathway as a potential target for inhibition in patients with biliary tract cancers. PLoS One 2018;13:e0206007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lamarca A, Palmer DH, Wasan HS et al. ABC‐06: A randomised phase III, multi‐centre, open‐label study of active symptom control (ASC) alone or ASC with oxaliplatin/5‐FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously‐treated with cisplatin/gemcitabine (CisGem) chemotherapy. J Clin Oncol 2019;37(suppl 15):4003a. [Google Scholar]
- 11. McNamara MG, Metran‐Nascente C, Knox JJ. State‐of‐the‐art in the management of locally advanced and metastatic gallbladder cancer. Curr Opin Oncol 2013;25:425–431. [DOI] [PubMed] [Google Scholar]
- 12. Okusaka T, Nakachi K, Fukutomi A et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br J Cancer 2010;103:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura H, Arai Y, Totoki Y et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–1010. [DOI] [PubMed] [Google Scholar]
- 14. Walter T, Horgan AM, McNamara M et al. Feasibility and benefits of second‐line chemotherapy in advanced biliary tract cancer: A large retrospective study. Eur J Cancer 2013;49:329–335. [DOI] [PubMed] [Google Scholar]
- 15. McNamara MG, Lopes A, Wasan H et al. Landmark survival analysis and impact of anatomic origin in prospective clinical trials of biliary tract cancer. J Hepatol 2020;73:1109–1117. [DOI] [PubMed] [Google Scholar]
- 16. McNamara MG, Goyal L, Doherty M et al. NUC‐1031/cisplatin versus gemcitabine/cisplatin in untreated locally advanced/metastatic biliary tract cancer (NuTide:121). Future Oncol 2020;16:1069–1081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplementary material 1 Full eligibility criteria for ABC‐08
Supplementary material 2: Criteria for treatment of patients enrolled onto ABC‐08
Supplementary Table 1 The most relevant treatment‐emergent grade 1 adverse events occurring in ≥4 patients enrolled in ABC‐08
Supplementary Table 2: Details on dosing of NUC‐1031 and cisplatin received by individual patients enrolled in ABC‐08