

Abstract
We report on a woman with aggressive estrogen receptor‐positive, KRAS‐mutated ovarian cancer who achieved a remarkable response to combination therapy with the MEK inhibitor (trametinib) and the aromatase inhibitor (letrozole), even though the disease had failed to respond to a combination of a PI3K inhibitor and different MEK inhibitor, as well as to trametinib and the estrogen modulator, tamoxifen, and to letrozole by itself. The mechanism of action for exceptional response was elucidated by in vitro experiments that demonstrated that the fact that tamoxifen can have an agonistic effect in addition to antagonist activity, whereas letrozole results only in estrogen depletion was crucial to the response achieved when letrozole was combined with an MEK inhibitor. Our current observations indicate that subtle variations in mechanisms of action of outwardly similar regimens may have a major impact on outcome and that such translational knowledge is critical for optimizing a precision medicine strategy.
Key Points
This report describes the remarkable response of a patient with KRAS‐mutated, estrogen receptor‐positive low‐grade serous ovarian cancer treated with trametinib (MEK inhibitor) and letrozole (aromatase inhibitor), despite prior progression on similar agents including tamoxifen (estrogen modulator).
In vitro investigation revealed that tamoxifen can have agonistic in addition to antagonistic effects, which could be the reason for the patient not responding to the combination of trametinib and tamoxifen.
The current observations suggest that drugs with different mechanisms of action targeting the same receptor may have markedly different anticancer activity when used in combinations.
Keywords: KRAS, Estrogen, Personalized therapy, Combination
Short abstract
This article reports the case of a patient with aggressive estrogen receptor‐positive, KRAS‐mutated ovarian cancer who achieved a remarkable response to combination therapy with the MEK inhibitor trametinib and the aromatase inhibitor letrozole, despite earlier failures of treatment with other combination inhibitor treatment. This article focuses on the possibility that subtle variations in mechanisms of action of outwardly similar regimens may have major effects on patient outcomes.
Introduction
Ovarian cancer is the second most common gynecologic malignancy and the most frequent cause of gynecologic cancer death in the U.S. [1, 2]. Serous ovarian cancer is generally classified into low‐ or high‐grade serous carcinoma [3]. Classification of low‐ versus high‐grade serous ovarian cancer is clinically important because patients with low‐grade serous ovarian cancer (LGSOC) are known to have better progression‐free survival compared with patients with high‐grade disease [4]. However, LGSOC is often resistant to cytotoxic agents (response rate [RR] of ~4%) [5].
Understanding the tumor grade is also important because there are molecular differences between low‐ and high‐grade serous carcinoma. Notably, LGSOC is less commonly associated with TP53 alterations compared with high‐grade carcinoma. Meanwhile, KRAS is altered in 20% to 40% of LGSOC, whereas this mutation is rare among high‐grade histology [5]. Moreover, overexpression of estrogen receptor (ER) and progesterone receptor (PR) are frequent in LGSOC (ER/PR overexpression: 43%–58% vs. 17%–27% for low and high grade, respectively) [6]. Attempts have been made to use antihormone therapy for ER/PR‐positive cancers and MEK inhibitor for KRAS mutant cancers; however, low response rates were observed with these treatment approaches (RR of 9% and 15%, respectively, when given as a single agent) [7, 8]. In some cases, one patient may have multiple molecular alterations that individually suggest different drug options [9, 10]. Previous work reveals that patient outcomes are better when biomarkers are concurrently targeted with different agents [9, 11].
Herein, we describe a patient with heavily pretreated, widely metastatic LGSOC with overexpression of ER by immunohistochemistry (PR negative) and with a KRAS G12V mutation as the sole genomic alteration detected by tissue and circulating cell‐free DNA (cfDNA) next‐generation sequencing. This patient experienced a dramatic and durable response (19+ months) to the combination of the aromatase inhibitor letrozole with the MEK inhibitor trametinib after a series of related regimens had failed. We performed translational experiments to demonstrate that differences in the mechanisms of action of the antiestrogen tamoxifen versus letrozole might explain the remarkable response to letrozole together with trametinib after primary failure of tamoxifen combined with trametinib or letrozole alone.
Patient Story
Case Report
A 28‐year‐old woman initially presented with right‐sided pelvic pain and underwent right‐sided salpingo‐oophorectomy. Pathology detected a serous low‐malignant‐potential tumor. One year later, the patient experienced left‐sided pelvic pain, and imaging studies revealed an adnexal mass, which prompted a hysterectomy with left salpingo‐oophorectomy. Pathology confirmed serous low‐malignant‐potential tumor. Three years after initial diagnosis, imaging revealed a hepatic lesion. The lesion was resected, and pathology showed metastatic LGSOC. Subsequently, the patient received carboplatin and paclitaxel (six cycles), followed by carboplatin, gemcitabine, and bevacizumab (seven cycles). A computed tomography scan performed 9 months after completion of chemotherapy revealed new, widely metastatic disease, and the patient subsequently underwent surgery for tumor debulking. Approximately 1 year later, the patient underwent five cycles of doxorubicin for recurrence. At progression, she sought treatment on a clinical trial with PI3K and MEK inhibitors (PF‐04691502, 130 mg i.v. weekly, and PD‐0325901 [mirdametinib], initially with 4 mg p.o. b.i.d. 3 weeks on, 1 week off; dose reduction was required to 2 mg p.o. b.i.d. 3 weeks on, 1 week off because of intolerable grade 2 rash), which she continued for approximately 1 year until progression. She was then referred to the Precision Medicine Clinic.
Molecular profiling of tissue next‐generation sequencing as well as cfDNA revealed a single alteration—KRAS G12V. Immunohistochemistry was positive for ER (2+, 75% positive) but negative for PR. The patient was started on the MEK inhibitor trametinib for the KRAS G12V mutation and tamoxifen for ER positivity. Unfortunately, serial imaging and cancer antigen 125 (CA125) assessments showed progression at 5 months, and treatment was stopped (Fig. 1A). She was lost to follow‐up for approximately 9 months, at which time she presented again with bulky pelvic masses and a rapidly rising CA125 level (Fig. 1A). Based on ER positivity, she was started on letrozole as monotherapy; however, after 3 months of therapy, she was found to have rising CA125 levels (>15,000 U/mL) with radiographic progression (Fig. 1A and B, left panel).
Figure 1.
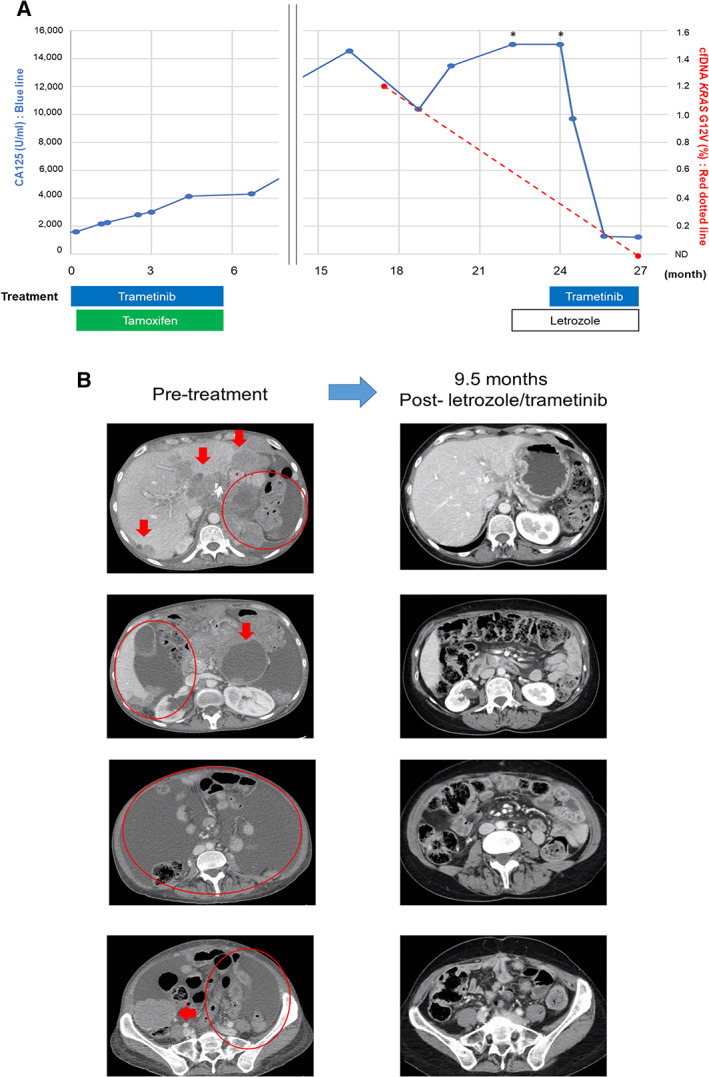
(A): Dynamic change in tumor marker (CA125) (blue line) and KRAS G12V cell‐free DNA (red dotted line) along with treatment course. (Note that the patient was lost to follow‐up for several months between treatments). (B): Computed tomography images before and after the combination of letrozole and trametinib. Reduction of solid tumor masses (arrows) and improvement in ascites (circles) were seen after the initiation of letrozole and trametinib. Therapy is ongoing at 19+ months. *, Over upper limit of detection (>15,000 U/mL). Abbreviations: CA125, cancer antigen 125; ND, not detected.
At that time, she was rechallenged with trametinib (for KRAS G12V) while continuing on letrozole. Surprisingly, after 2 months of therapy, the patient noted a reduction in abdominal pain and girth. Three months after the therapy began, CA125 level declined to 1,265 U/mL from >15,000 U/mL. KRAS G12V in cfDNA became undetectable. Restaging computed tomography scan performed 4 months after the initiation of letrozole and trametinib showed >50% tumor reduction (per RECIST version 1.1; Fig. 1A and B, right panel). She continues to do well with an ongoing progression‐free survival of 19+ months.
In Vitro Experiments: Combination of Estrogen Depletion with an MEK Inhibitor Sensitized ER‐Positive, RAS Pathway‐Activated Serous Ovarian Cancer Cell Lines Resistant to Tamoxifen and MEK Inhibitor Combination
Because both tamoxifen and letrozole target estrogen signaling, it was of interest to understand why our patient did not respond to tamoxifen combined with trametinib yet had an exceptional response to trametinib combined with letrozole. We considered that tamoxifen and letrozole have different mechanisms of action despite both targeting estrogen signaling. Tamoxifen is known to compete with 17β‐estradiol at the receptor site and to block the promotional role of 17β‐estradiol in breast cancer [12]. However, tamoxifen, can also act as a selective estrogen receptor modulator, and in some contexts, it can have partial agonist effects (i.e., mimicking the effects of estrogen) [13, 14, 15, 16]. In contrast, letrozole inhibits the enzyme aromatase and thereby reduces the production of estrogens; letrozole thereby acts purely as an estrogen antagonist [17].
We set out to experimentally determine whether we could reproduce the pattern of responsiveness seen in our patient. KRAS mutant/ER‐positive serous ovarian carcinoma models are not readily available. We therefore used two ER‐positive serous carcinoma cell lines (OVCAR 3 and SKOV 3) that are readily available. We hypothesized that the effects of the KRAS mutation reflected overactivation of the RAS signaling pathway, and for our experimental efforts we activated epidermal growth factor receptor (EGFR) to induce elevated levels of RAS signaling [18]. This would be consistent with our hypothesized mechanism (Fig. 2). We also used an ER‐negative cell line (OVCAR 5) as a control (supplemental online Methods).
Figure 2.
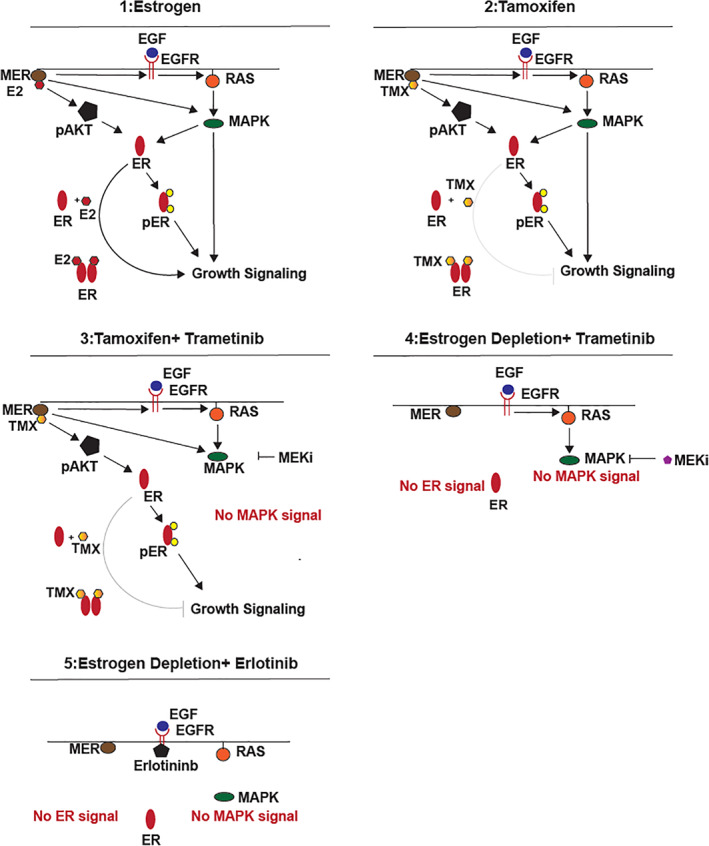
Noncanonical and canonical ER signaling axis. We hypothesized that the partial agonist activity of tamoxifen versus the estrogen depletion effect of letrozole might explain the observed and widely different clinical responses [13, 14, 15, 16]. We also hypothesized that noncanonical ER signaling might be involved, in which MER can drive noncanonical ER signaling [13, 15, 23]. We constructed a map of the signaling pathway that comprises the described interactions, and we considered each of the treatment scenarios and responses. We found that our hypothesized mechanisms could potentially explain the diverging responses to tamoxifen and letrozole when combined with trametinib in a patient with hyperactivated RAS pathway signaling. (Scenario 1): MER‐driven noncanonical ER signaling and RAS signaling (replicating baseline condition of current case report). MER/E2 activates AKT and ER, which leads to cell growth. Activation of EGFR subsequently activates RAS/MAPK pathway, which also leads to growth signaling (similar to KRAS mutation). (Scenario 2): TMX functions as a partial agonist for MER‐driven noncanonical ER signaling. Thus, no inhibitory effect is seen on ER signaling. (Scenario 3): Combination treatment with tamoxifen and trametinib is not sufficient to halt the cell growth despite the successful inhibition of RAS/MEK pathway with trametinib because ER signaling is not adequately reduced with tamoxifen. (Scenario 4): Combination of estrogen depletion (mimicking treatment with letrozole) and trametinib successfully inhibits both ER and RAS signaling, thus leading to the inhibition of cell growth. (Scenario 5): Combination of estrogen depletion (mimicking treatment with letrozole) and erlotinib (EGFR inhibitor to reduce RAS activation) successfully reduces both ER and RAS signaling, thus leading to the inhibition of cell growth. Abbreviations: E2, estradiol; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ER, estrogen receptor; MAPK, mitogen‐activated protein kinase; MEKi, MEK inhibitor; MER, membrane estrogen receptor; pAKT, phospho‐AKT; pER, phospho‐ER; TMX, tamoxifen.
We first tested if these cells were sensitive to estrogen depletion and found that all three cell lines grew just as well with estrogen depletion compared with when they were grown with estrogen (supplemental online Fig. 1A). Furthermore, we treated these cells with tamoxifen and found that they proliferated at approximately the same rate (supplemental online Fig. 1B). These observations suggest that targeting estrogen signaling alone with either estrogen depletion strategies (mimicking letrozole) or with a selective estrogen modulator (like tamoxifen) would be ineffective.
We then tested scenarios that replicated the patient's treatment schema. Cells were treated with trametinib, trametinib combined with tamoxifen, estrogen depletion, or estrogen depletion combined with trametinib. We found that ER‐positive cells maximally responded to estrogen depletion in combination with the MEK inhibitor trametinib. Interestingly, the ER‐negative cell line OVCAR 5 was sensitive to all treatments with MEK inhibition. No added benefit was observed when combining MEK inhibitor with tamoxifen or estradiol depletion (Fig. 3A). We hypothesized that the presence of ER may prove an alternate signaling pathway to survival when cells are treated with an MEK inhibitor.
Figure 3.
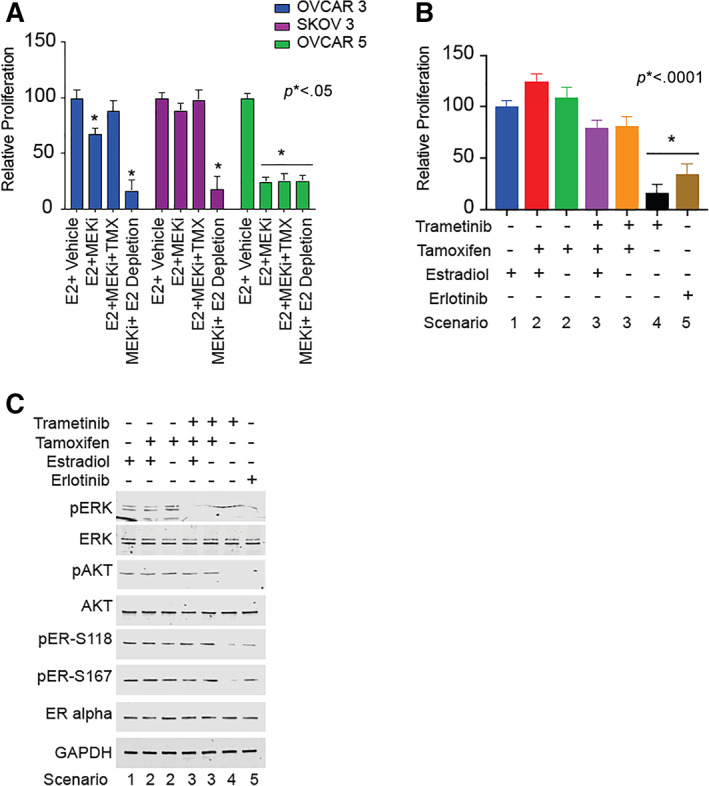
Mechanism for tamoxifen and trametinib resistance in OVCAR 3 cells. (A): OVCAR 3 (estrogen receptor [ER]–positive), SKOV 3 (ER‐positive), and OVCAR 5 (ER‐negative) cells were stripped of hormone for 48 hours prior to seeding. Cell culture media contained hormone‐stripped FBS (which contains epidermal growth factor [EGF]) to maintain RAS/mitogen‐activated protein kinase (MAPK) signaling. Cells were treated with 10 nM 17β‐estradiol, 10 nM tamoxifen, and 20 nM trametinib in various combinations, as indicated, for 48 hours. Cell proliferation was measured by MTT assay. Data are representative of eight biological replicates. Histograms represent means, and error bars represent SD. Statistical significance is indicated and determined by one‐way ANOVA with post hoc Tukey's test. The results show that MEK inhibition in the presence of estrogen depletion was associated with significantly decreased growth in all three cell lines. However, MEK inhibition in the presence of estrogen had no effect among ER‐positive cell lines (OVCAR 3 and SKOV 3). On the contrary, MEK inhibition alone was sufficient to reduce the cell proliferation in the ER‐negative cell line (OVCAR 5). *p < .05. (B): OVCAR 3 cells (ER‐positive) were grown in hormone‐stripped cell culture media containing FBS (which contains EGF) for 48 hours prior to seeding. Cells were treated with 10 nM 17β‐estradiol, 10 nM tamoxifen, 20 nM trametinib, and 20 nM erlotinib in various combination, as indicated, for 48 hours. Cell proliferation was measured by MTT assay. Data are representative of eight biological replicates. Histograms represent means, and error bars represent SD. Statistical significance is indicated and determined by one‐way ANOVA with post hoc Tukey's test. The results show that MEK inhibition by trametinib in the presence of estrogen depletion was associated with decreased proliferation; however, if tamoxifen and MEK inhibition were given simultaneously, there was no decrease in proliferation (presumably because tamoxifen may have a partial agonist effect on ER). Similarly, erlotinib (to reduce RAS‐induced MAPK signaling) in combination with estrogen depletion led to decreased cell proliferation (Fig. 2). *p < .0001. (C): OVCAR 3 cells (ER‐positive) were grown in hormone‐stripped cell culture media containing FBS (which contains EGF) for 48 hours and then treated with 10 nM 17β‐estradiol, 10 nM tamoxifen, 20 nM trametinib, and 20 nM erlotinib in various combinations, as indicated, for 48 hours. Whole cell lysates were collected and analyzed by Western blot. Combination of trametinib or erlotinib along with estrogen depletion successfully reduced the phosphorylation of ERK, AKT, and ER. The same effects were not observed with tamoxifen alone or for tamoxifen in combination with trametinib (Fig. 2). Abbreviations: E2, estradiol; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; MEKi, MEK inhibitor; pAKT, phospho‐AKT; pER, phospho‐ER, pERK, phosphor‐ERK; TMX, tamoxifen.
We then used trametinib, tamoxifen, estrogen, and erlotinib to deconstruct the signaling axis in OVCAR 3 cells. Cells grown in estrogen grew at comparable levels to cells grown with estrogen and tamoxifen (Fig. 3B). Furthermore, the estrogen and tamoxifen combination and single‐agent tamoxifen treatment did not show any reduction in ERK phosphorylation, ER phosphorylation, or AKT phosphorylation by Western blot (Fig. 3C). We propose that this occurs because tamoxifen can activate the membrane estrogen receptor (MER) to signal through AKT and phosphorylate ER (Fig. 2, Scenario 2). When cells were treated with trametinib, tamoxifen, and estrogen in combination, we observed no reduction in cell growth (Fig. 3B). However, significant reduction in phospho‐ERK signal was observed by Western blot, yet phospho‐ER and phospho‐AKT levels were unchanged (Fig. 3C). We propose that proliferation is maintained because MER can still promote growth signaling through AKT, and in turn, AKT phosphorylates ER to promote growth (Fig. 2, Scenario 3). Lastly, when cells were treated with trametinib or erlotinib in the absence of estrogen and tamoxifen, cellular growth was inhibited (Fig. 3B). Moreover, we observed a drastic reduction in phospho‐AKT, phospho‐ERK, and phospho‐ER by Western blot (Fig. 3C). We hypothesize that the growth inhibition occurs when ER activity is reduced because of upstream AKT and ERK being inhibited (Fig. 2, Scenarios 4 and 5).
Discussion
Herein, we unravel the biologic basis for an exceptional response to trametinib and letrozole that was unexpected in the context of prior rapid disease progression on trametinib and tamoxifen and on letrozole alone. Hence, two different therapeutic regimens that each targeted both the ER and MEK pathways produced remarkably different clinical effects. There are important lessons that emerge from consideration of this case: (a) the oncology dogma against revisiting prior failed drugs (in this case, trametinib and letrozole had each been given before) may not always hold true [19], even to the extent that two drugs that each failed were successful when given together; (b) drugs targeting the same receptor through different mechanisms may have disparate biologic impact that can be elucidated by bedside to bench interrogation; and (c) combination therapy affecting the estrogen receptor and MEK pathways may be beneficial in advanced cancer.
Antiestrogen is standard of care for ER‐positive breast cancer and may also be important in other malignancies [20]. In the context of excess mitogen‐activated protein kinase (MAPK) signaling (KRAS mutation in this case), the precise choice of antiestrogen therapy could be critical, as observed in both our patient and in our in vitro assays. The classic pathway of estrogen action is dependent on ERα and ERβ. Tamoxifen can act as an agonist in some tissues and as an antagonist in other tissues [16], providing one possible source for diverging behaviors between treatment with tamoxifen and letrozole. In addition, nonclassical mechanisms of steroid action may be dependent on MERs. The MER is a cell surface receptor that can activate the RAS and PI3K pathways [21]. We present evidence that proliferation in an in vitro context analogous to our patient's tumor relies upon MER‐mediated ERK and AKT signaling, which promotes phosphorylated nuclear ER. Inhibiting both MER (by estrogen depletion) and ERK pathways is necessary to halt proliferation and may justify the use of aromatase inhibitors rather than tamoxifen when MAPK activity is elevated. Of note, our study uses EGFR activation to activate RAS and its downstream signaling partners in order to phenocopy the effects of a KRAS mutation. However, EGFR can also have downstream consequences beyond those mediated by active RAS. Thus, although our experimental observations are consistent with our hypotheses and follow from consideration of described mechanisms of these drugs and biomarkers, alternative hypotheses could also be presented and could not be ruled out with the present study. Future studies could investigate patient‐derived xenografts from individuals with similar molecular characteristics to determine if they or their xenografts respond similarly. Furthermore, there are limited data describing the association between estrogen receptor positivity and KRAS mutation among LGSOC; this also requires further investigation.
In summary, we report a woman with aggressive ER‐positive, KRAS‐mutated LGSOC who achieved a remarkable response to combination therapy with the MEK inhibitor trametinib and aromatase inhibitor letrozole, even though the tumor had progressed on a combination of a PI3K inhibitor and different MEK inhibitor, as well as on trametinib itself and the selective estrogen receptor modulator tamoxifen, and on letrozole alone. The molecular mechanisms underlying this exceptional response were elucidated by in vitro modeling that demonstrated that the known partial agonist/antagonist mechanism of tamoxifen compared with the estrogen depletion of letrozole was likely the critical feature that explained the different responses to the combination of each agent with an MEK inhibitor. It has previously been suggested that customized combination therapy may be a key to achieving tumor control when there are important genomic co‐alterations [9, 10], especially when certain gene product pathways, such as KRAS or PI3K, are activated [22]. Our current observations indicate that drugs with subtly different mechanisms of action targeting the same receptor may have markedly different anticancer activity when used in combinations.
Author Contributions
Conception/design: Shumei Kato, Thomas McFall, Edward Stites, Razelle Kurzrock
Provision of study material or patients: Edward Stites, Razelle Kurzrock
Data analysis and interpretation: Shumei Kato, Thomas McFall, Kenta Takahashi, Kasey Bamel
Manuscript writing: Shumei Kato, Thomas McFall, Edward Stites, Razelle Kurzrock
Final approval of manuscript: Shumei Kato, Thomas McFall, Kenta Takahashi, Kasey Bamel, Sadakatsu Ikeda, Ramez N. Eskander, Steven Plaxe, Barbara Parker, Edward Stites, Razelle Kurzrock
Disclosures
Shumei Kato: Foundation Medicine (C/A), Roche (H); Barbara Parker: Pfizer, Novartis, Oncternal, Genentech (RF—institution), Merck (OI), Bioatla (C/A—spouse), EMD Serono (other—travel—spouse); Razelle Kurzrock: Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Konica Minolta (RF), LOXO, Pfizer, X‐Biotech, Actuate Therapeutics, Genentech, NeoMed (C/A), Roche (H), IDbyDNA, CureMatch, Inc. (OI), CureMatch, CureMetrix (SAB). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1. Supporting Information.
Figure S1
Acknowledgments
This work was supported in part by the Joan and Irwin Jacobs Fund and by the National Institutes of Health (grant NIH P30 CA023100 to R.K.) and by NIH K22CA216318 to E.S., NIH T32CA009370 to T.M., and NIH P30CA014195 to E.S.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com.
Disclosures of potential conflicts of interest may be found at the end of this article.
Contributor Information
Shumei Kato, Email: smkato@health.ucsd.edu.
Edward Stites, Email: estites@salk.edu.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Torre LA, Trabert B, DeSantis CE et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018;68:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gershenson DM. Low‐grade serous carcinoma of the ovary or peritoneum. Ann Oncol 2016;27:i45–i49. [DOI] [PubMed] [Google Scholar]
- 4. Bodurka DC, Deavers MT, Tian C et al. Reclassification of serous ovarian carcinoma by a 2‐tier system: A Gynecologic Oncology Group study. Cancer 2012;118:3087–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gershenson DM, Sun CC, Bodurka D et al. Recurrent low‐grade serous ovarian carcinoma is relatively chemoresistant. Gynecol Oncol 2009;114:48–52. [DOI] [PubMed] [Google Scholar]
- 6. Wong KK, Lu KH, Malpica A et al. Significantly greater expression of ER, PR, and ECAD in advanced‐stage low‐grade ovarian serous carcinoma as revealed by immunohistochemical analysis. Int J Gynecol Pathol 2007;26:404–409. [DOI] [PubMed] [Google Scholar]
- 7. Gershenson DM, Sun CC, Iyer RB et al. Hormonal therapy for recurrent low‐grade serous carcinoma of the ovary or peritoneum. Gynecol Oncol 2012;125:661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Farley J, Brady WE, Vathipadiekal V et al. Selumetinib in women with recurrent low‐grade serous carcinoma of the ovary or peritoneum: An open‐label, single‐arm, phase 2 study. Lancet Oncol 2013;14:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sicklick JK, Kato S, Okamura R et al. Molecular profiling of cancer patients enables personalized combination therapy: The I‐PREDICT study. Nat Med 2019;25:744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodon J, Soria JC, Berger R et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat Med 2019;25:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato S, Kim KH, Lim HJ, et al. Real‐world data from a molecular tumor board demonstrates improved outcomes with a precision N‐of‐One strategy. Nat Commun 2020;11:4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Riggs BL, Hartmann LC. Selective estrogen‐receptor modulators – mechanisms of action and application to clinical practice. N Engl J Med 2003;348:618–629. [DOI] [PubMed] [Google Scholar]
- 13. Filardo EJ, Quinn JA, Bland KI et al. Estrogen‐induced activation of Erk‐1 and Erk‐2 requires the G protein‐coupled receptor homolog, GPR30, and occurs via trans‐activation of the epidermal growth factor receptor through release of HB‐EGF. Mol Endocrinol 2000;14:1649–1660. [DOI] [PubMed] [Google Scholar]
- 14. Vendrell JA, Bieche I, Desmetz C et al. Molecular changes associated with the agonist activity of hydroxy‐tamoxifen and the hyper‐response to estradiol in hydroxy‐tamoxifen‐resistant breast cancer cell lines. Endocr Relat Cancer 2005;12:75–92. [DOI] [PubMed] [Google Scholar]
- 15. Likhite VS, Stossi F, Kim K et al. Kinase‐specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol Endocrinol 2006;20:3120–3132. [DOI] [PubMed] [Google Scholar]
- 16. Gallo MA, Kaufman D. Antagonistic and agonistic effects of tamoxifen: Significance in human cancer. Semin Oncol 1997;24:S1‐71–S71‐80. [PubMed] [Google Scholar]
- 17. Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. N Engl J Med 2003;348:2431–2442. [DOI] [PubMed] [Google Scholar]
- 18. Rojas M, Yao S, Lin YZ. Controlling epidermal growth factor (EGF)‐stimulated RAS activation in intact cells by a cell‐permeable peptide mimicking phosphorylated EGF receptor. J Biol Chem 1996;271:27456–27461. [DOI] [PubMed] [Google Scholar]
- 19. Naing A, Kurzrock R. Chemotherapy resistance and retreatment: A dogma revisited. Clin Colorectal Cancer 2010;9:E1–E4. [DOI] [PubMed] [Google Scholar]
- 20. Munoz J, Wheler J, Kurzrock R. Expression of estrogen and progesterone receptors across human malignancies: New therapeutic opportunities. Cancer Metastasis Rev 2015;34:547–561. [DOI] [PubMed] [Google Scholar]
- 21. Soltysik K, Czekaj P. Membrane estrogen receptors ‐ is it an alternative way of estrogen action? J Physiol Pharmacol 2013;64:129–142. [PubMed] [Google Scholar]
- 22. Kato S, Okamura R, Sicklick JK et al. Prognostic implications of RAS alterations in diverse malignancies and impact of targeted therapies. Int J Cancer 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein‐coupled receptor for estrogen. Mol Cell Endocrinol 2007;265‐266:138‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1. Supporting Information.
Figure S1