

ABSTRACT
Retinoblastoma (RB) is commonly-seen cancer in children. The p53 pathway dysfunction, which can lead to elevated MDM2 or MDM4 (p53 antagonists) protein expression, is frequently observed in almost all human cancers, including RB. The present study attempted to investigate the underlying mechanism from the perspective of non-coding RNA regulation. Here, we demonstrated that p53 and miR-129 were positively correlated with each other in RB. miR-129 directly targeted MDM2/4 to inhibit expression, therefore counteracting MDM2/4-mediated p53 signaling suppression and modulating RB cell proliferation and apoptosis. Moreover, p53 could activate the transcription of miR-129 via binding to the miR-129 promoter region, therefore forming a regulatory loop with MDM2/4 to affect RB progression. Altogether, the p53/miR-129/MDM2/4/p53 regulatory loop can modulate RB cell growth. We provide a solid experimental basis for developing novel therapies for RB.
KEYWORDS: Retinoblastoma (RB), p53, miR-129, MDM2/4, feedback loop
Introduction
Retinoblastoma (RB), caused by mutations in gene RB1, is considered the third most commonly seen childhood cancer [1,2]. RB can be diagnosed in early childhood: 2/3 by age two and 95% by age five [3]. Therapy methods need to consider curing the illness and preserving eyesight with minimization of the long-term side effects.
The p53 pathway dysfunction is frequently observed in almost all human cancers, leading to elevated MDM2 or MDM4 (p53 antagonists) protein expression [4–8]. The gene amplification of tumor somatic cells or the germline polymorphism at MDM2 and MDM4 sites may lead to the up-regulation of expression, which activates higher basal expression levels and predisposes individuals to cancer [5–8]. Also, more stable forms of MDM4 could be produced by various splicing variants of MDM4 mRNAs (messenger RNAs), subsequently suppressing p53 [9]. Multiple mechanisms for knocking down p53 via MDM2/MDM4 have been proposed, including alterations within MDM2 transcription regulation, alterations within the stability of mRNAs, and microRNAs (miRNAs)-mediated perturbations within the translation of proteins, or a combination of multiple mechanisms. More importantly, RB expresses wild-type (w-t) p53 [10], and cytogenetic studies have revealed that MDM4 gene gain is observed in about 65% of RBs, which is linked to the increase in the mRNA expression and protein levels. According to the studies, RBs show to be sensitive to MDM2/4 antagonists, and efforts are being made to target these proteins using small molecule antagonists [11,12]. Searching for more regulatory mechanisms of this important tumor suppressor pathway may provide more RB treatment directions.
As widely recognized, miRNAs target mRNAs to modulate gene expression, which leads to inhibition of transcription or transcriptional instability while reducing protein expression, and we have recently checked their effect on RB [13]. MDM2/4-mediated p53 signaling deregulation could be modulated by miRNAs [14]. Since MDM2/4 expression is commonly upregulated in RB, we searched online microarray analyses for miRNAs that might target MDM2/4 to regulate their expression negatively. Of deregulated miRNAs in RB, miR-129-3p (fold change = −363.3, p < 0.05) and miR-129-5p (fold change = −91.21, p < 0.05) are dramatically reduced in RB tissues and cell lines [15]. Besides, as predicted by lncTar, miR-129 may target MDM2/4; according to an integrative and comprehensive database, ChIP-Atlas, which nearly covers all public ChIP-seq data submitted to the Sequence Read Archives (SRA) within NCBI, DDBJ, or ENA, p53 may activate miR-129 transcription via targeting miR-129 promoter region. Subsequently, we hypothesize that p53, miR-129, and MDM2/4 may form a regulatory loop to modulate RB progression.
Herein, p53 and miR-129 expression and correlation within RB tissue samples and cells, and the dynamic function of p53 and miR-129 upon RB cell proliferation and apoptosis are examined. The putative bindings between miR-129 and MDM2/4 are validated, and the effects of miR-129 and MDM2/4 combination on RB cell and p53 signaling are evaluated. Finally, the predicted binding of p53 to the miR-129 promoter is examined. These data indicate that we present a regulatory loop consisting of p53, miR-129, and MDM2/4 that may modulate RB cell proliferation and apoptosis via MDM2/4-mediated p53 signaling deregulation.
Materials and methods
Clinical tissue samples
Ten cases of normal retinal tissues and thirty cases of RB tissue samples were collected from patients hospitalized at The First Hospital of Hunan University of Chinese Medicine with the approval of the Research Ethics Committee at the hospital and performed in strict accordance with international standards. The informed consent was signed and obtained from each patient enrolled. Tissue samples were stored at −80°C until further use.
Cell lines, cell culture, and cell transfection
A normal retinal cell line, ARPE-19 (ATCC® CRL-2302™), and two RB cell lines, Y79 (ATCC® HTB-18™) and WERI-RB-1 (ATCC® HTB-169™) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI-1640 medium (Catalog No. 30–2001, ATCC) supplemented with 10% FBS and 1% penicillin-streptomycin (Invitrogen) at 37°C in a humidified incubator with 5% CO2.
P53 expression was achieved by transfecting si-p53 (GeneCopoecia, Guangzhou, China). MDM2/4 expression was achieved by the transfection of the MDM2/4 overexpressing vector (GeneCopoecia, Guangzhou, China). The expression of miR-129 was achieved by transfection of miR-129 mimics or miR-129 inhibitor (Genepharma, Shanghai, China) by using Lipofectamine 3000 (Invitrogen). The primers for plasmid construction and siRNA sequence were listed in Table S1.
PCR-based analysis of miRNA and mRNA expression
After extracting the total RNA from target tissues or cells using Trizol reagent (Invitrogen), the total RNA was reverse-transcribed using a miScript Reverse Transcription kit (Qiagen, Germany), taking RNU6B expression as an endogenous control. The mRNA expression was examined using an SYBR Green PCR Master Mix (Qiagen), taking GAPDH expression as an endogenous control. All data were processed, and the relative expression levels were calculated using the 2−ΔΔCT method. The primers were listed in Table S1.
Immunoblotting assays
The protein levels of p53, p21, MDM2, and MDM4 in tissue samples and cells were detected by performing immunoblotting assays following the methods described before [16,17,18]. Total proteins were extracted from target tissues or cells, separated using SDS-PAGE, and then transferred onto PVDF membrane for primary antibody incubation. The following antibodies were used: anti-p53 (ab26, Abcam, Cambridge, CA, USA), anti-p21 (ab109520, Abcam), anti-MDM2 (ab38618, Abcam), anti-MDM4 (ab49993, Abcam) and anti-GAPDH (ab8245, Abcam). After the incubation with these primary antibodies overnight at 4°C, the blots were then incubated with the HRP-conjugated secondary antibody. Signals were visualized using ECL Substrates (Millipore, USA). The gray scale of the blot was analyzed by image J. GAPDH protein level was used as an endogenous control. The MDM2/MDM4 expression was normalized to GAPDH, and the group of NC mimics or NC inhibitor was set as control (set as 1).
Cell viability detected by MTT assay
By following the methods described before [16], the cell viability was detected. After transfection or treatment, MTT (20 μl at a density of 5 mg/ml; Sigma-Aldrich) was added, and another 4-h incubation was conducted. At the end of the incubation, DMSO (200 μl) was added for formazan dissolving. Next, OD values were measured at 490 nm, and the relative cell viability was calculated, taking the non-treated cell viability (control) as 100%.
DNA synthesis capacity detected by EdU using FACS analysis
Cells were cultured in a 6-well plate at a density of 3 × 106 cells/well, transfected or co-transfected. Replace the culture medium in each well with medium added with 1 ml EdU solution (final concentration of 50 μM) and incubate the cells for 2 h. At the end of the incubation, collect the cells, centrifuge the cells at 350 × g for 5 min and discard the supernatant, resuspend the cells with 1 ml PBS, centrifuge the cells at 350 × g for 5 min again, and fix the cells with 1 ml 4% paraformaldehyde for 15 ~ 30 min after discarding the supernatant. After washing with PBS, each well was added with 1 ml 0.5% TritonX-100 and incubated at room temperature for 10 min. After washing with PBS, each well was added with 1× Apollo® staining agent and incubated at room temperature for 10 min in the dark. At the end of the incubation, centrifuge the cells at 600 × g for 10 min and discard the supernatant. Add 3 ml 0.5% TritonX-100, wash three times at room temperature centrifuge the cells at 600 × g for 10 min and discard the supernatant, and resuspend the cells with 500 μl PBS. Flow cytometry was carried out immediately after dyeing.
Cell apoptosis detected by flow cytometer assay
Flow cytometer analysis was performed using the Annexin V-FITC apoptosis detection kit (Keygen, China), following the methods described before [16]. Propidium iodide (PI) was used for the nucleus staining, and the apoptotic rate was detected using the BD Accuri C6 flow cytometer (BD, USA). Ex (the excitation wavelength) = 488 nm. Em (the emission wavelength) = 530 nm.
Luciferase reporter assay
The fragment of MDM2 or MDM4 3’-UTR was amplified by PCR and cloned to the downstream of the Renilla psiCHECK2 vector (Promega, Madison, WI, USA), named wt-MDM2 or MDM4 3’-UTR. For the MDM2 or MDM4 mutant reporter, the study mutated the predicted miR-129 binding site in the seed region of MDM2 or MDM4 was mutated, respectively. These reporter vectors were then co-transfected in HEK293 cells (ATCC, USA) together with miR-129 mimics or miR-129 inhibitor, respectively. Forty-eight hours later, the luciferase activity changes were monitored using the Dual-Luciferase Reporter Assay System (Promega). Renilla luciferase activity was normalized to Firefly luciferase activity for each transfected well.
Regarding p53 binding to the miR-129 promoter, HEK293 cells were co-transfected with p53 overexpression vector andpGL3 luciferase reporter constructs harboring the miR-129 target sequence (named wt). The mutant-type vector contains a mutation in any of the predicted p53 binding sites in the miR-129 promoter (named mut-Site A, mut-Site B, or mut-Site AB). Twenty-four hours later, the luciferase activity changes were monitored using a Dual-Luciferase Assay System (Promega, WI, USA).
Chromatin immunoprecipitation (ChIP)
Target cells (107 cells) were cross-linked using 1% formaldehyde for 10 min and then immunoprecipitated with anti-p53 (ab1101, abcam) using Magnetic Chip kit (Thermo, USA). Briefly, chromatin was digested with micrococcal nuclease, sheared by sonication and then lysates were clarified by centrifugation at 10,000 rpm for 10 min at 4°C. The supernatant was incubated with anti-p53 or IgG (negative control) overnight at 4°C with rotation. After being pulled down with protein G agarose beads, the target protein-DNA complexes were cleaned, released, and eluted for subsequent experiment. The promoter regions in miR-129 harboring the predicted p53 binding sites were amplified using ChIP-PCR primers. The fold-enrichment was calculated following the formula: FE = 2 (IgG CT-Sample CT). The amplification efficiency of p21 promoter was used as a positive control, and nonimmune IgG was used as a negative control. The primers were listed in Table S1.
Statistical analysis
Data were processed using the SPSS Statistics 17.0 software and expressed as means ± SD of at least three independent experiments. The differences were compared by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test or independent sample t-test. A P value of less than 0.05 is considered statistically significant.
Results
Expression and correlation of p53 and miR-129 in tissue samples and cells
First, we detected p53 and miR-129 expression and association within tissues and cells. In 30 cases of RB tissues, p53 and miR-129 expression was dramatically reduced, in comparison with that in normal healthy retinal tissue samples (Figure 1(a-b)). Within tissues, p53 showed to be positively related to miR-129 (Figure 1(c)). Consistently, the protein levels of p53 showed to be significantly decreased within cancerous tissue samples (Figure 1(d-e)). In two RB cells, Y79 and WERI-RB-1, both miR-129 expression and p53 protein levels were downregulated considerably compared to those in a normal cell line, ARPE-19 (Figure 1(f-h)).
Figure 1.
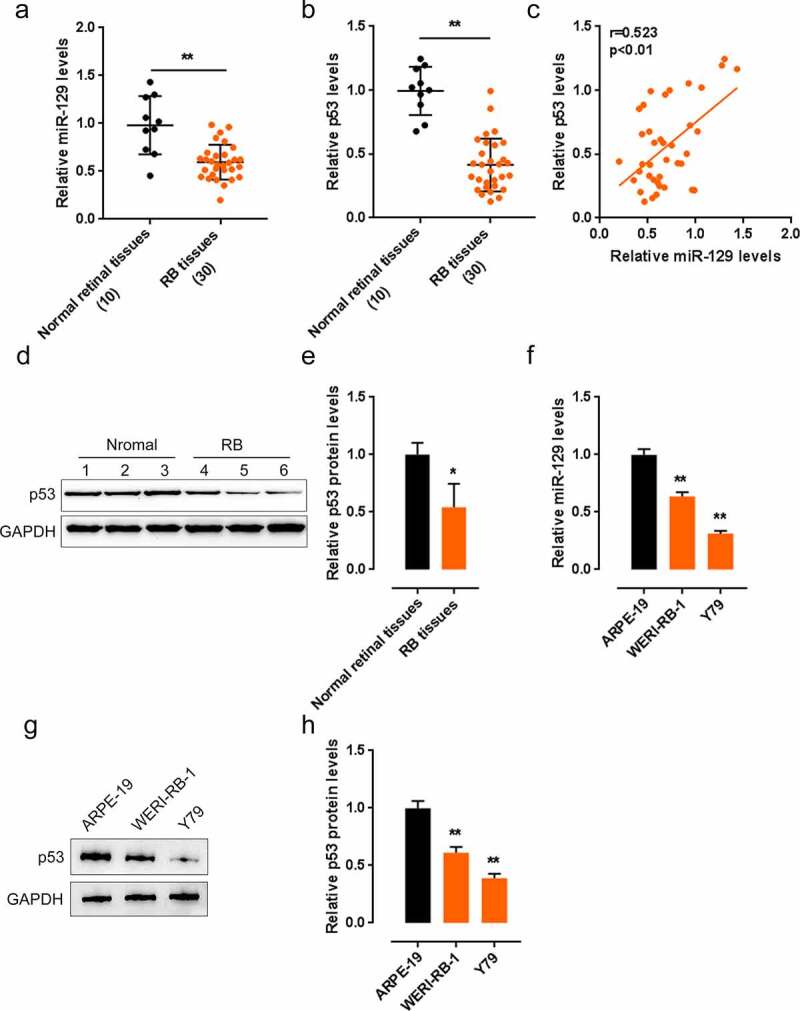
Expression and correlation of p53 and miR-129 in tissue samples and cell lines. (a–b) The expression of p53 and miR-129 in 10 cases of non-cancerous retinal tissues and 30 cases of retinoblastoma (RB) tissues examined by qPCR. (c) The correlation of p53 and miR-129 expression in tissue samples analyzed by Pearson’s correlation analysis. (d–e) The protein levels of p53 in tissue samples determined by Immunoblotting. (f) The expression of p53 and miR-129 in two RB cell lines, Y79 and WERI-RB-1, and a normal cell line, ARPE-19, examined by qPCR. (g–h) The protein levels of p53 in cell lines examined by Immunoblotting
Effect of miR-129 upon p53, p21, and RB cell lines
Since miR-129 and p53 are positively correlated with each other in RB, next, the effect and dynamic of miR-129 and p53 on RB cells were evaluated. QPCR confirmed that the expression of miR-129 was generated within Y79 and WERI-RB-1 cells via transfection of miR-129 mimics/inhibitor (Figure 2(a)). According to the results of Immunoblotting, p53 was knocked down via si-p53 transfection (Figure 2(b)). After that, we co-transfected two RB cell lines with miR-129 mimics as well as si-p53, then evaluated them for p53, p21, Bax, Bcl-2, and cleaved-caspase 3 protein levels. According to Figure 2(b-d), miR-129 overexpression increased the protein levels of p53, p21, Bax, and cleaved-caspase 3 and decreased Bcl-2 protein; p53 knockdown decreased the protein levels of p53, p21, Bax, and cleaved-caspase 3 and increased Bcl-2 protein. p53 knockdown could partially reverse miR-129 overexpression effects on these proteins.
Figure 2.
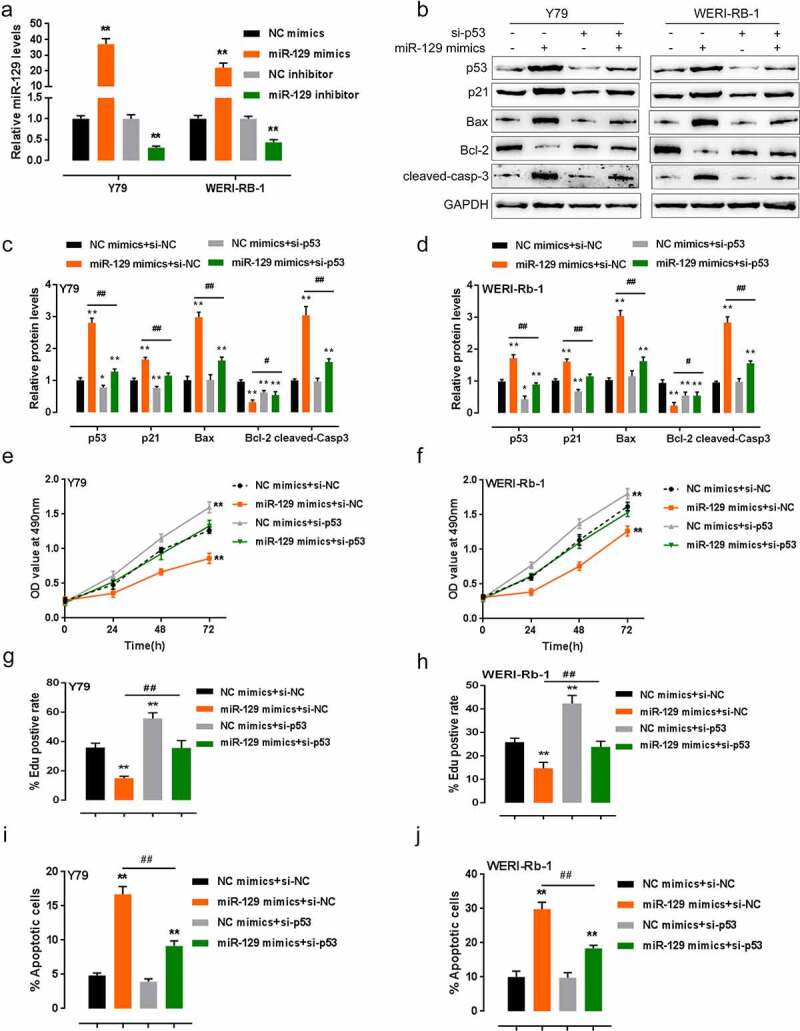
Dynamic effects of p53 and miR-129 on p53, p21, and RB cells. (a) miR-129 expression in two RB cell lines achieved by transfection of miR-129 mimics or miR-129 inhibitor, as confirmed by qPCR. Two RB cell lines were co-transfected with si-p53 and miR-129 mimics, the protein levels of p53, p21, Bax, Bcl-2, and cleaved-caspase 3 were examined by Immunoblotting (b–d); the cell viability of co-transfected cells examined by MTT assays (e–f); the DNA synthesis capacity of co-transfected cells examined by FACS analysis (g–h); the cell apoptosis was examined by Flow cytometry (i–j). The data are presented as mean ± SD of three independent experiments. *P< 0.05, **P< 0.01, compared to control group; #P< 0.05, ##P< 0.01, compared to miR-129 mimics + si-NC (negative control) group
Regarding their cellular functions, miR-129 overexpression significantly inhibited, while p53 knockdown dramatically promoted the cell activity and DNA synthesis ability; p53 knockdown could partially reverse how miR-129 overexpression affected RB cells (Figure 2(e-h)). Consistently, miR-129 overexpression promoted, while p53 knockdown inhibited RB cell apoptosis; p53 knockdown also reversed how miR-129 overexpression affected RB cells (Figure 2(i-j)), the FACS plots were shown in Fig. S1.
miR-129 directly targets MDM2/4 and affects p53 signaling
According to the prediction of lncTar, miR-129 may target the 3’-UTR of MDM2 and MDM4, two essential proteins in RB progression [3,19,20]. Next, we validated the miR-129 regulation of MDM2/4 and the putative binding between miR-129 and MDM2/4. Within Y79 and WERI-RB-1 cell lines, the protein levels of MDM2/4 showed to be considerably downregulated via the overexpression of miR-129, whereas upregulated via the inhibition of miR-129 (Figure 3(a-d)). For luciferase reporter assay, we constructed wild-type and mutant-type luciferase reporter vectors, namely wt-MDM2 3’-UTR/wt-MDM4 3’-UTR and mut-MDM2 3’-UTR/mut-MDM4 3’-UTR, respectively. Mut-MDM2 3’-UTR and mut-MDM4 3’-UTR contained a 6 or 7 bp mutation in the predicted miR-129 binding site (Figure 3(e)). We co-transfected Y79 and WERI-RB-1 cells, respectively, with these vectors and miR-129 mimic or miR-129 inhibitor and examined the luciferase activity. Figure 3(g-h) showed that wt-MDM2/4 3’-UTR luciferase activity could be considerably downregulated via the overexpression of miR-129 while upregulated via the inhibition of miR-129, and mutating the predicted miR-129 binding site could induce a decrease in luciferase activity to near the original value. These findings indicate miR-129 could negatively regulate MDM2/4 via binding to the 3’-UTR of MDM2/4.
Figure 3.
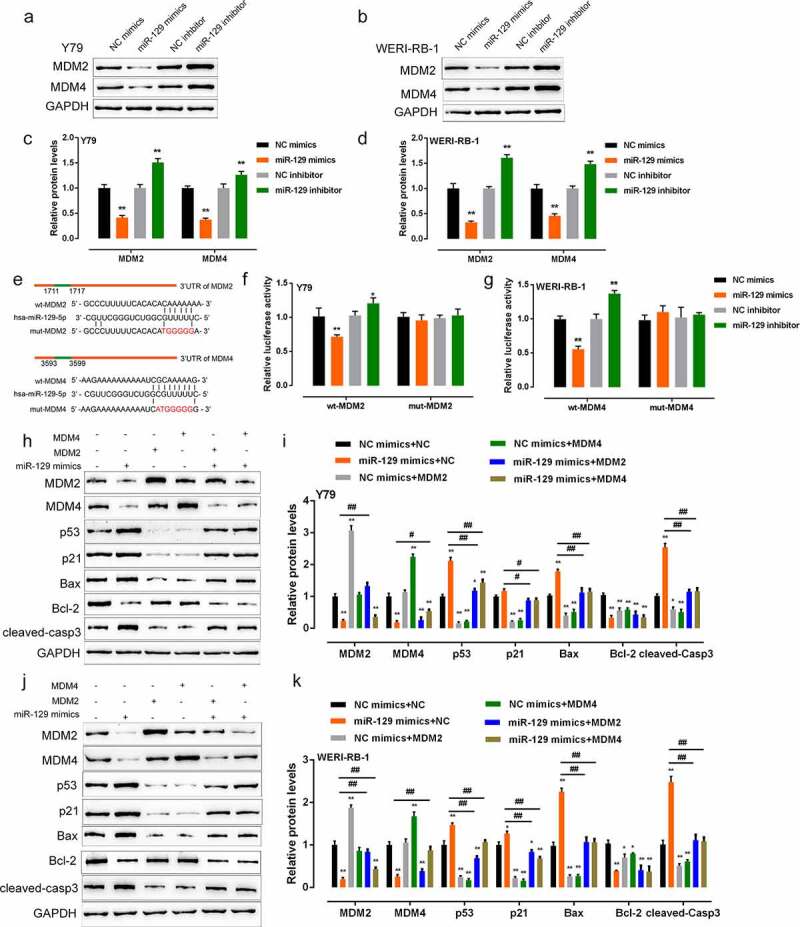
miR-129 directly targets MDM2/4 and affects p53 signaling (a–d) Y79 and WERI-RB-1 cells were transfected with miR-129 mimics or miR-129 inhibitor and examined for the protein levels of MDM2 and MDM4. (e–g) Wild-type or mutant-type MDM2/4 3’-UTR vector containing wild or mutated miR-129 binding site was constructed and co-transfected into Y79 and WERI-RB-1 cells, respectively, with miR-129 mimics or miR-129 inhibitor. The luciferase activity was examined. (h–k) MDM2/4 overexpression in Y79 and WERI-RB-1 cells was achieved by transfection of MDM2/4 overexpressing vector, as confirmed by Immunoblotting. Y79 and WERI-RB-1 cells were co-transfected with miR-129 mimics and MDM2/4 overexpressing vector and examined for the protein levels of MDM2/4, p53, p21, Bax, Bcl-2, and cleaved-caspase 3. The data are presented as mean ± SD of three independent experiments. *P< 0.05, **P< 0.01, compared to control group; #P< 0.05, ##P< 0.01, compared to miR-129 mimics + NC group
As we have mentioned, MDM2/4 serve as antagonists for p53 [3]. Since miR-129 directly binds to MDM2/4, then the dynamic function of miR-129 and MDM2/4 upon p53 signaling is examined. We co-transfected Y79 and WERI-RB-1 cell lines with miR-129 mimics and the overexpressing vector of MDM2/4, then examined them for MDM2, MDM4, p53, p21, Bax, Bcl-2, and cleaved-caspase 3 protein levels. miR-129 overexpression remarkably decreased MDM2, MDM4, and Bcl-2 proteins while increased p53, p21, Bax, and cleaved-caspase 3 proteins, MDM2 or MDM4 overexpression increased MDM2/4 and Bcl-2 while decreased p53, p21, Bax, and cleaved-caspase 3 proteins; the overexpression of MDM2/4 could partially reverse miR-129 overexpression’s effects (Figure 3(h-k)). These findings indicate miR-129 modulates p53 signaling via targeting MDM2/4.
miR-129 modulates RB cell proliferation and apoptosis via MDM2/4
RB expressed wild-type p53 [10], elevated expression of p53 antagonists like MDM2/4 [4–6] may lead to the deregulation of p53 signaling, therefore contributing to RB progression. Thus, we examined the effect of miR-129 and MDM2/4 combination on RB cell proliferation and apoptosis. Figure 4(a-f) showed that the overexpression of miR-129 dramatically inhibited, while MDM2 or MDM4 overexpression significantly promoted RB cell activity as well as DNA synthesis ability; MDM2 or MDM4 overexpression could significantly attenuate the inhibitory effect of miR-129 overexpression on the capacity of RB cells to proliferate. Consistently, miR-129 overexpression promoted, while MDM2 or MDM4 overexpression inhibited RB cell apoptosis; MDM2/4 overexpression could partially reverse how miR-129 overexpression exerted its effect (Figure 4(g-h)). In summary, miR-129 regulates the proliferation and apoptosis of RB cells via its downstream targets MDM2/4.
Figure 4.
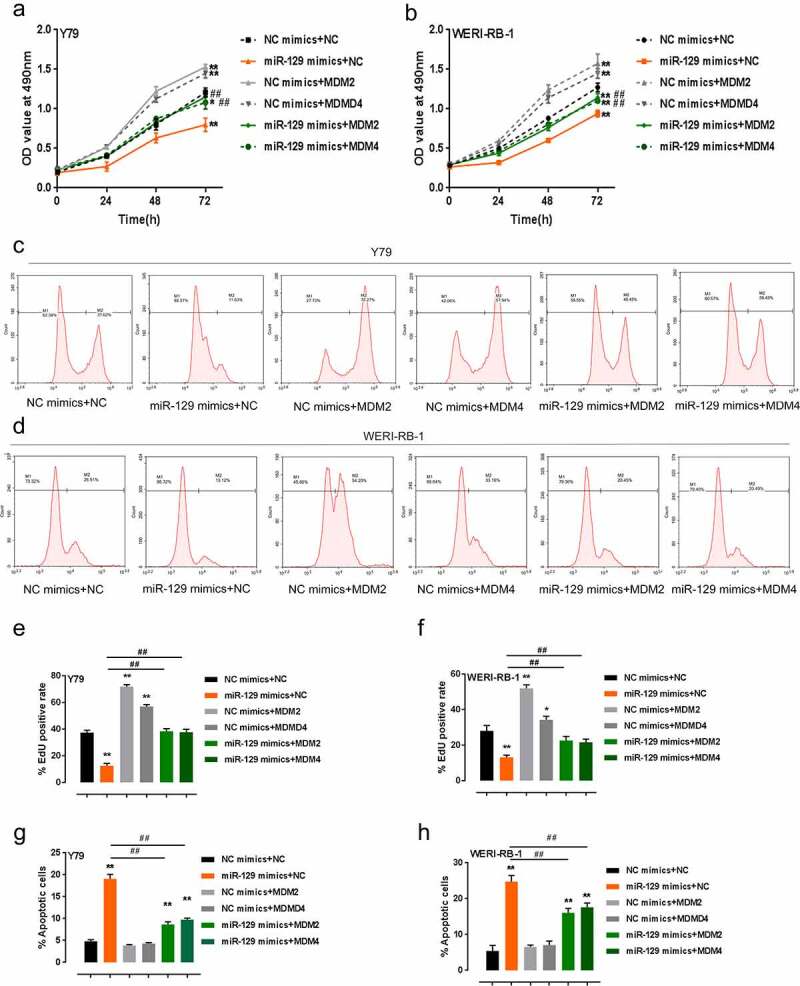
miR-129 modulates RB cell proliferation and apoptosis via MDM2/4 Y79 and WERI-RB-1 cells were co-transfected with miR-129 mimics and MDM2/4 overexpressing vector, cell viability was examined by MTT assay (a–b); DNA synthesis capacity was examined by FACS analysis (c–f); apoptosis of co-transfected RB cells examined by Flow cytometer assay (g–h). The data are presented as mean ± SD of three independent experiments. *P< 0.05, **P< 0.01, compared to control group; #P< 0.05, ##P< 0.01, compared to miR-129 mimics + NC group
P53 activates the transcription of miR-129 via binding to the miR-129 promoter region
It is reported by the present study that p53 and miR-129 are positively correlated with each other in RB. Via an integrative and comprehensive database, ChIP-Atlas, which nearly covers all public ChIP-seq data submitted to the Sequence Read Archives (SRA) within NCBI, DDBJ, or ENA, we found that p53 might bind to the miR-129 promoter to activate its transcription. Subsequently, we validated if p53 bound to the promoter of miR-129. According to Immunoblotting results, the p53 overexpressing vector successfully achieved p53 overexpression in RB cell lines (Figure 5(a-b)). The underlying p53 binding element (BE) within the miR-129 promoter predicted by the Jaspar database is shown in Figure 5(c). We mutated any of the putative p53 binding sites within the miR-129 promoter region to construct a mutant-type BE (Figure 5(c)). The wild-type or mutant-type BE was subcloned; then, the constructs were co-transfected with negative control (NC) or p53 overexpressing vector into HEK293 (Figure 5(d)). Figure 5(d) shows that when the p53 vector was co-transfected with wild-type BE, it significantly promoted the luciferase activity compared with NC; double mutant of site A and B dramatically reduced the luciferase activity both in cells transfected with NC or p53 overexpression vector. Moreover, as confirmed by real-time ChIP assay, in the miR-129 promoter region, the binding level of p53 antibody to miR-129 BE showed to be significantly higher compared with that from IgG within Y79 and WERI-RB-1 cells (Figure 5(e-f)), which means p53 might activate the expression of miR-129 promoter via binding to the miR-129 promoter region. Within both cell lines, p53 knockdown inhibited, while p53 overexpression promoted miR-129 expression and p53 protein levels (Figure 5(g-h)). These findings indicate p53 activates the transcription of miR-129 via binding to the miR-129 promoter region.
Figure 5.
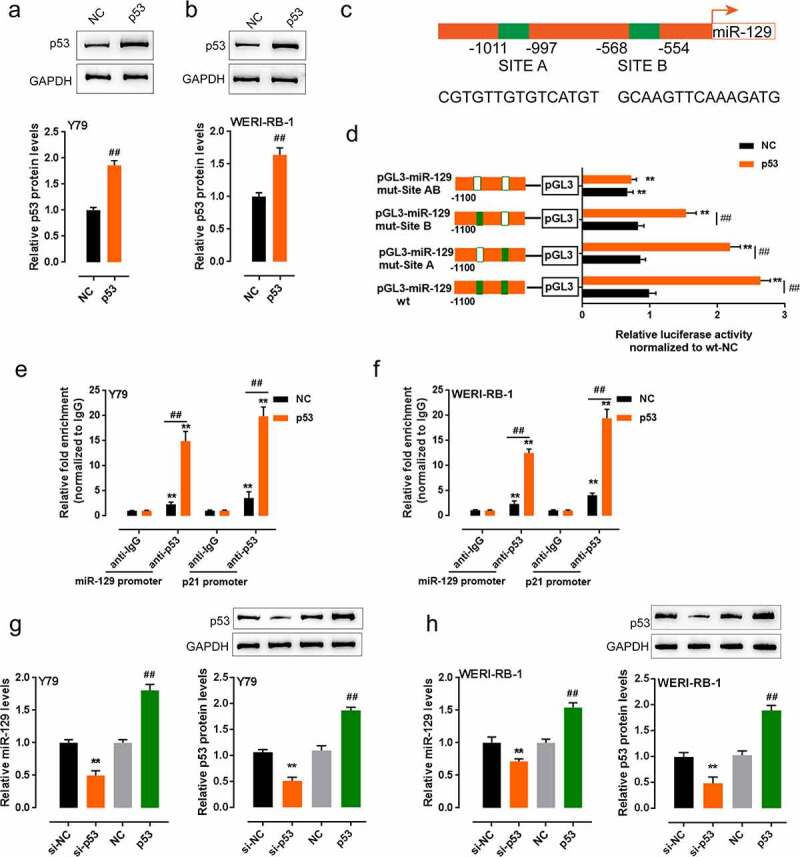
P53 binds to the promoter of miR-129 to activate its transcription. (a–b) p53 overexpression in Y79 and WERI-RB-1 cells achieved by transfection of p53 overexpressing vector, as confirmed by Immunoblotting. (c) A schematic diagram of a potential p53 binding element in the promoter region of the miR-129 gene predicted by Jaspar database. A wt-miR-129 promoter luciferase reporter vector and a mut-miR-129 promoter luciferase reporter vector containing a mutation in any of the predicted p53 binding site in miR-129 promoter were constructed. (d) the indicated luciferase reporter vectors were co-transfected into HEK293 cells with NC (negative control) or p53 overexpressing vector. The luciferase activity was then determined by using dual luciferase assays. (e–f) The level of p53 antibody binding to miR-129 promoter or p21 promoter (positive control) in NC or p53 vector transfected Y79 and WERI-RB-1 cells examined by ChIP assay, compared to IgG. (g–h) Y79 and WERI-RB-1 cells were transfected with si-p53 and examined for miR-129 expression. The data are presented as mean ± SD of three independent experiments. **P < 0.01, compared to pGL3-miR-129 wt, anti-IgG or si-NC group; ##P < 0.01, compared to NC group
Discussion
This study demonstrated that p53 and miR-129 were positively correlated with each other in RB. miR-129 directly targeted MDM2/4 to inhibit expression, therefore counteracting MDM2/4-mediated p53 signaling suppression and modulating RB cell proliferation and apoptosis. Moreover, p53 could activate the transcription of miR-129 via binding to the miR-129 promoter region, therefore forming a regulatory loop with MDM2/4 to affect RB progression.
The tumor suppressor protein p53 could protect the cells from cellular damage and malignant transformation [21]. The p53 gene is closely related to human malignant tumors [22]. The wild-type p53 gene has anticancer effects and inhibits angiogenesis. When the p53 gene is mutated or deleted, wild-type p53-dependent anticancer effects would be lost [23]. As previously reported, here, we find that the expression and protein levels of p53 show to be considerably reduced within RB tissue samples and cells. Meanwhile, miR-129 expression, which was predicted to be activated by p53 via binding to the promoter, is also downregulated in RB tissue samples and cells, and p53 and miR-129 are positively correlated with each other. miR-129 exerts a tumor-suppressive effect on many cancers, like gastric carcinoma [24,25], breast carcinoma [26], bladder cancer [27], and prostate carcinoma [28]; thus, we hypothesize miR-129 could play a tumor-suppressive role within RB. As expected, miR-129 overexpression in RB cell lines inhibits RB cell proliferation and promotes cell apoptosis, while p53 knockdown promotes tumor cell proliferation while inhibits cell apoptosis. Above all, miR-129 overexpression could partially reverse the impacts caused by p53 knockdown.
In RB, we could knock down a complete p53 gene by MDM4 (also known as MDMX) overexpression [12]. Sixty-five percent of human RBs have additional MDM4 copies, while 10% of human RBs have additional MDM2 copies [12]. Although there are structural similarities, MDM4 and MDM2 exert their effects on inhibiting p53 via different mechanisms [29]. As a ubiquitin ligase, MDM2 could suppress the activity of p53 and enable p53 ubiquitination for degradation via binding to the p53 transactivation domain [30]. While MDM4 has no ubiquitous protein-ligase activity; instead, it could effectively block the activity of p53 via binding to its transactivation domain [31]. MDM4 mRNA and protein levels were elevated in RBs with MDM4 gene amplification [12]. MDM4 was negatively related to the level of p53 and p21 proteins, according to what has been revealed by breast tumors with MDMX amplification [31]. Recently, MDM2 has been reported as a targeted gene of miR-129 in glioma cells [32]. In the present study, as predicted by online tools, miR-129 could bind to MDM2/4, consistent with the results from luciferase reporter assay. Via direct binding, miR-129 regulates the expression and protein levels of MDM2/4 negatively. Consistently, MDM2/4 overexpression remarkably downregulates p53, p21, pro-apoptotic Bax and cleaved-caspase 3 protein levels, while miR-129 overexpression exerts an opposite effect on these factors; MDM2/4 overexpression could partially reverse the impacts caused by miR-129 overexpression. Regarding the cellular function, miR-129 overexpression suppresses the proliferation whereas enhances the apoptosis of RB cells, which could be significantly reversed by MDM2/4 overexpression. These findings indicate miR-129 modulate RB cell proliferation and apoptosis via targeting MDM2/4 and MDM2/4-mediated p53 suppression.
As we have revealed, p53 and miR-129 are positively correlated with each other in RB. In cardiomyocytes, p53 modulates prohibition expression via binding to the miR-128 promoter region and subsequently affects apoptosis [33]. In B-cell lymphomas, wt p53 increases miR-124 levels via binding to the miR-124 promoter region, therefore suppressing p65, MYC, and BCL2 [34]. Here, Chip-Atlas database and online tool Jasper also predicted that p53 might bind to the miR-129 promoter, consistent with luciferase reporter and ChIP assays. Via binding to the miR-129 promoter region, p53 also activates the transcription of miR-129, therefore forming a regulatory loop with MDM2/4 to modulate RB cell proliferation and apoptosis.
Based with these findings, we conclude that the p53/miR-129/MDM2/4/p53 regulatory loop can modulate RB cell growth. We provide a solid experimental basis for developing novel therapies for RB.
Supplementary Material
Acknowledgments
This study was supported by Key Laboratory of Hunan Provincial Key Laboratory for Preventing and Treating Five Diseases of Obstetrics and Gynecology (2017TP1018); Key Subject Construction Project of Traditional Chinese Medicine Ophthalmology of the State Administration of Traditional Chinese Medicine (ZK1801YK015) and Science and Technology Plan of Changsha City (K1501014-31, kc1704005).
Funding Statement
Hunan Provincial Key Laboratory Construction Project for Prevention and Treatment of Ophthalmology and Otolaryngology Diseases with Chinese Medicine (2017TP1018) and National Administration of Traditional Chinese Medicine Key Discipline Construction Project of Ophthalmology of Traditional Chinese Medicine (ZK1801YK015).
Authors’ contributions
Xiaolei Yao made a substantial contribution to the conception and design of the work and drafted the manuscript; Hui Shen participated in some experiments; Qinghua Peng analyzed and interpreted the data; Jingsheng Yu revised the work critically for important intellectual content
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
All available data were present in this manuscript.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Watts P. Retinoblastoma: clinical features and current concepts in management. J Indian Med Assoc. 2003;101(8):464–6, 468. [PubMed] [Google Scholar]
- [2].Hurwitz RL, Chévez-Barrios P, Boniuk M, et al. Retinoblastoma: from bench to bedside. Expert Rev Mol Med. 2003;5(1):1–14. [DOI] [PubMed] [Google Scholar]
- [3].McEvoy JD, Dyer MA.. Genetic and epigenetic discoveries in human retinoblastoma. Crit Rev Oncog. 2015;20(3–4):217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci. 2007;120(Pt 3):371–378. [DOI] [PubMed] [Google Scholar]
- [5].Bond GL, Hu W, Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119(5):591–602. [DOI] [PubMed] [Google Scholar]
- [6].Kulkarni DA, Vazquez A, Haffty BG, et al. A polymorphic variant in human MDM4 associates with accelerated age of onset of estrogen receptor negative breast cancer. Carcinogenesis. 2009;30(11):1910–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Atwal GS, Kirchhoff T, Bond EE, et al. Altered tumor formation and evolutionary selection of genetic variants in the human MDM4 oncogene. Proc Natl Acad Sci U S A. 2009;106(25):10236–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wynendaele J, Böhnke A, Leucci E, et al. An illegitimate microRNA target site within the 3′ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010;70(23):9641–9649. [DOI] [PubMed] [Google Scholar]
- [9].Mancini F, Di Conza G, Moretti F. MDM4 (MDMX) and its transcript variants. Curr Genomics. 2009;10(1):42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kato MV, Shimizu T, Ishizaki K, et al. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett. 1996;106(1):75–82. [DOI] [PubMed] [Google Scholar]
- [11].Brennan RC, Federico S, Bradley C, et al. Targeting the p53 pathway in retinoblastoma with subconjunctival Nutlin-3a. Cancer Res. 2011;71(12):4205–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Laurie NA, Donovan SL, Shih C-S, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444(7115):61–66. [DOI] [PubMed] [Google Scholar]
- [13].Theriault BL, Dimaras H, Gallie BL, et al. The genomic landscape of retinoblastoma: a review. Clin Exp Ophthalmol. 2014;42(1):33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].To KH, Pajovic S, Gallie BL, et al. Regulation of p14ARF expression by miR-24: a potential mechanism compromising the p53 response during retinoblastoma development. BMC Cancer. 2012;12(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martin, Martin J, Bryar P, et al. Differentially expressed miRNAs in retinoblastoma. Gene. 2013;512(2):294–299. [DOI] [PubMed] [Google Scholar]
- [16].Li L, Lv G, Wang B, et al. The role of lncRNA XIST/miR-211 axis in modulating the proliferation and apoptosis of osteoarthritis chondrocytes through CXCR4 and MAPK signaling. Biochem Biophys Res Commun. 2018;503(4):2555–2562. [DOI] [PubMed] [Google Scholar]
- [17].Liu Z, Dou C, Yao B, et al. Ftx non coding RNA-derived miR-545 promotes cell proliferation by targeting RIG-I in hepatocellular carcinoma. Oncotarget. 2016;7(18):25350–25365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tu K, Liu Z, Yao B, et al. MicroRNA-519a promotes tumor growth by targeting PTEN/PI3K/AKT signaling in hepatocellular carcinoma. Int J Oncol. 2016;48(3):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Laurie NA, Shih CS, Dyer MA. Targeting MDM2 and MDMX in retinoblastoma. Curr Cancer Drug Targets. 2007;7(7):689–695. [DOI] [PubMed] [Google Scholar]
- [20].Pritchard EM, Dyer MA, Guy RK. Progress in small molecule therapeutics for the treatment of retinoblastoma. Mini Rev Med Chem. 2016;16(6):430–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. [DOI] [PubMed] [Google Scholar]
- [22].Tomkova K, Tomka M, Zajac V. Contribution of p53, p63, and p73 to the developmental diseases and cancer. Neoplasma. 2008;55(3):177–181. [PubMed] [Google Scholar]
- [23].Oki E, Tokunaga E, Nakamura T, et al. Genetic mutual relationship between PTEN and p53 in gastric cancer. Cancer Lett. 2005;227(1):33–38. [DOI] [PubMed] [Google Scholar]
- [24].Liu Q, Jiang J, Fu Y, et al. MiR-129-5p functions as a tumor suppressor in gastric cancer progression through targeting ADAM9. Biomed Pharmacother. 2018;105:420–427. [DOI] [PubMed] [Google Scholar]
- [25].Wang Q, Yu J. MiR-129-5p suppresses gastric cancer cell invasion and proliferation by inhibiting COL1A1. Biochem Cell Biol. 2018;96(1):19–25. [DOI] [PubMed] [Google Scholar]
- [26].Meng R, Fang J, Yu Y, et al. miR-129-5p suppresses breast cancer proliferation by targeting CBX4. Neoplasma. 2018;65(4):572–578. [DOI] [PubMed] [Google Scholar]
- [27].Cao J, Wang Q, Wu G, et al. miR-129-5p inhibits gemcitabine resistance and promotes cell apoptosis of bladder cancer cells by targeting Wnt5a. Int Urol Nephrol. 2018;50(10):1811–1819. [DOI] [PubMed] [Google Scholar]
- [28].Xu S, Ge J, Zhang Z, et al. MiR-129 inhibits cell proliferation and metastasis by targeting ETS1 via PI3K/AKT/mTOR pathway in prostate cancer. Biomed Pharmacother. 2017;96:634–641. [DOI] [PubMed] [Google Scholar]
- [29].Toledo F, Krummel KA, Lee CJ, et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell. 2006;9(4):273–285. [DOI] [PubMed] [Google Scholar]
- [30].Lu F, Chi S-W, Kim D-H, et al. Proteomimetic libraries: design, synthesis, and evaluation of p53-MDM2 interaction inhibitors. J Comb Chem. 2006;8(3):315–325. [DOI] [PubMed] [Google Scholar]
- [31].Danovi D, Meulmeester E, Pasini D, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24(13):5835–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moradimotlagh A, Arefian E, Rezazadeh Valojerdi R, et al. MicroRNA-129 inhibits glioma cell growth by targeting CDK4, CDK6, and MDM2. Mol Ther Nucleic Acids. 2020;19:759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li J, Aung LHH, Long B, et al. miR-23a binds to p53 and enhances its association with miR-128 promoter. Sci Rep. 2015;5(1):16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jeong D, Kim J, Nam J, et al. MicroRNA-124 links p53 to the NF-kappaB pathway in B-cell lymphomas. Leukemia. 2015;29(9):1868–1874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All available data were present in this manuscript.