

ABSTRACT
The tumor microenvironment (TME) within mucosal neoplastic tissue in oral cancer (ORCA) is greatly influenced by tumor-infiltrating lymphocytes (TILs). Here, a clustering method was performed using CIBERSORT profiles of ORCA data that were filtered from the publicly accessible data of patients with head and neck cancer in The Cancer Genome Atlas (TCGA) using hierarchical clustering where patients were regrouped into binary risk groups based on the clustering-measuring scores and survival patterns associated with individual groups. Based on this analysis, clinically reasonable differences were identified in 16 out of 22 TIL fractions between groups. A deep neural network classifier was trained using the TIL fraction patterns. This internally validated classifier was used on another individual ORCA dataset from the International Cancer Genome Consortium data portal, and patient survival patterns were precisely predicted. Seven common differentially expressed genes between the two risk groups were obtained. This new approach confirms the importance of TILs in the TME and provides a direction for the use of a novel deep-learning approach for cancer prognosis.
KEYWORDS: Head and neck cancer, oral cancer, cibersort, tumor-infiltrating lymphocytes, tumor microenvironment, the cancer genome atlas, international cancer genome consortium, deep learning
Introduction
Head and neck cancer (HNSC) is currently garnering much attention; approximately 53,260 patients have been newly diagnosed with HNSC in 2020 – thus far in the United States – and HNSC-associated estimated deaths ranks 10th among the major malignant cancer types in the U.S.1 Although the number of patients accounts for only 2.9% of the newly reported cancer cases, the incidence rate of HNSC is 11.4% and the mortality rate is 2.5% over a period of 5 years, from 2013 to 2017, and the 5-year relative survival rate is only 66.2% from 2010 to 2016.2 Therefore, the development of new HNSC biomarkers is important to overcome the lack of research data as a result of the small number of patients.
Over the past several years, many studies have been conducted to develop a novel HNSC biomarker that predicts prognosis.3–8 However, limitations of the current potential biomarkers make clinical application very challenging.9 The importance of tumor-infiltrating lymphocyte (TIL) information has previously been reported,10,11 as TIL levels identify high-risk groups of patients with oral tongue squamous cell carcinoma.12 In particular, balanced levels of CD8 + T cells and regulatory T cells (Tregs) directly affect the survival rate of patients with oral cancer (ORCA).13 In addition, the elevated abundance of cancer-associated fibroblasts is highly correlated with patient survival.14
In TIL-focused analysis, determining increases in gene expression levels via differentially expressed gene (DEG) analysis is eventually used to identify individual potential gene biomarkers. In this case, within the analysis results, it is possible that there may be elevated false-potential biomarker genes that are not significantly related to the immune system. Thus, broad identification of general immune microenvironments may be more effective than relying on the identification of individual levels of biomarker genes when analyzing immune-related survival rates.15,16 Thus, CIBERSORT, a popular TIL prediction method, was selected. Through the LM22 signature matrix, CIBERSORT provided the most diverse predictions of abundance levels across TIL subsets among the tumor microenvironment (TME) deconvolution tools that were currently available. This was consistent with our purpose of identifying the “cell type-wide” immune cell landscape. Using the support regression vector-based machine learning method, Newman et al. have demonstrated that CIBERSORT effectively resolves cell subtypes with similar gene expression patterns via benchmarking analysis.17 CIBERSORT analysis for various cancer types has enabled the development of novel biomarkers,18–20 confirming the importance of focusing on immune cell fractions within the TME.
Several existing studies have identified survival patterns by analyzing gene expression profiles using deep learning,21–23 but deep learning studies that reveal TIL-specific patterns using secondary information, such as CIBERSORT, have not been published thus far. Therefore, in this study, deep learning was suggested as an alternative strategy for identifying biomarkers that may provide details about survival patterns. In this strategy, heterogeneous TME information, RNA expression data of selected ORCA subgroups in the HNSC cohort of The Cancer Genome Atlas (TCGA), was fed into a deep neural network (DNN) classifier coupled to CIBERSORT.
Materials and Methods
RNA expression data and derived immunotype-predicted data preprocessing
The RNA-Sequencing (RNA-Seq) ORCA datasets were downloaded for training candidates and validation. The RNA expression and clinical data for head and neck squamous cell carcinoma were downloaded from the Broad Institute Genome Data Analysis Center Firehose database (https://gdac.broadinstitute.org/). Another ORCA dataset was downloaded from the International Cancer Genome Consortium (ICGC) data portal (https://dcc.icgc.org/). Detailed patient information after data preprocessing is provided in Table 1.
Table 1.
Clinical characteristics of the cohort of patients with head and neck cancer in The Cancer Genome Atlas for which the oral cancer data were filtered
| Total |
Low-risk group |
High-risk group |
||
|---|---|---|---|---|
| Number (Percentage) | ||||
| Age (years) | 0–39 | 4(2.3) | 3(4.1) | 1(1.0) |
| 40–49 | 20(11.6) | 8(10.8) | 12(12.1) | |
| 50–59 | 58(33.5) | 27(36.5) | 31(31.3) | |
| 60–69 | 42(24.3) | 14(18.9) | 28(28.3) | |
| 70–79 | 34(19.7) | 15(20.3) | 19(19.2) | |
| 80+ | 15(8.7) | 7(9.5) | 8(8.1) | |
| Sex | Male | 124(71.7) | 53(71.6) | 71(71.7) |
| Female | 49(28.3) | 21(28.4) | 28(28.3) | |
| N stage | N0 | 75(43.4) | 34(45.9) | 41(41.4) |
| N1 | 28(16.2) | 8(10.8) | 20(20.2) | |
| N2 | 62(35.8) | 29(39.2) | 33(33.3) | |
| N3 | 2(1.2) | 0(0.0) | 2(2.0) | |
| NX | 6(3.5) | 3(4.1) | 3(3.0) | |
| T stage | T1 | 14(8.1) | 10(13.5) | 4(4.0) |
| T2 | 56(32.4) | 29(39.2) | 27(27.3) | |
| T3 | 33(19.1) | 15(20.3) | 18(18.2) | |
| T4 | 66(38.2) | 17(23.0) | 49(49.5) | |
| TX | 4(2.3) | 3(4.1) | 1(1.0) | |
| M stage | M0 | 166(96.0) | 68(91.9) | 98(99.0) |
| M1 | 2(1.2) | 2(2.7) | 0(0.0) | |
| MX | 5(2.9) | 4(5.4) | 1(1.0) | |
The immune cell fractions in both TCGA and ICGC datasets were predicted via CIBERSORT using the LM22 signature matrix with a 100× permutation count without applying quantile normalization, as directed on the website. After running CIBERSORT, 203 out of the 566 samples with a CIBERSORT p-value > 0.05 were removed. Another 77 samples from sites of the hypopharynx, larynx, and oropharynx that obviously did not belong to ORCA were removed. A total of 113 samples from tongue sites (tongue base and oral tongue) were also removed, and the clinical prognosis was different from the validation cohort, gingivobuccal cancer.24,25 The remaining 173 samples were obtained for further analysis. A detailed flowchart of the pipeline is shown in Figure 1(a).
Figure 1.
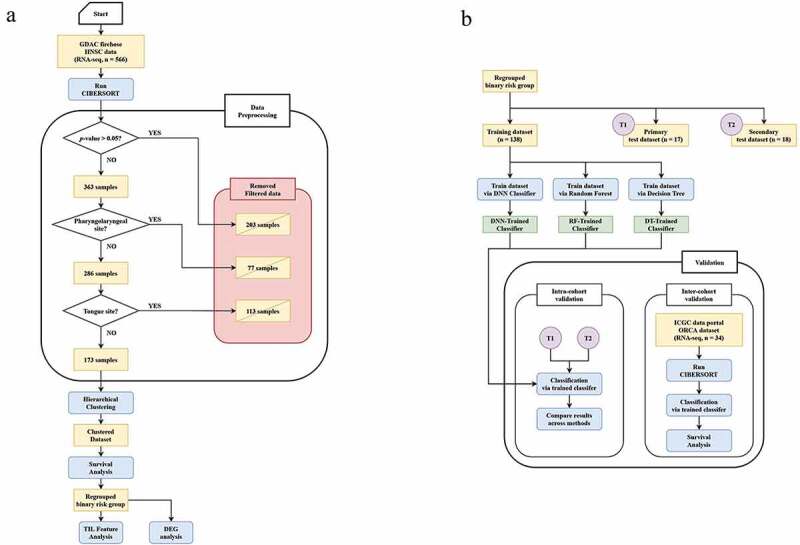
(a) Pipeline flowchart depicting the data preprocessing step. (b) Pipeline flowchart for processing the classifier establishment step, including the validation process using a deep neural network (DNN) classifier. GDAC, Genome Data Analysis Center; HNSC, head and neck cancer; RNA-seq, RNA sequencing; TIL, tumor-infiltrating lymphocyte; DEG, differentially expressed gene; DNN, deep neural network; RF, random forests; DT, decision tree; ICGC, International Cancer Genome Consortium; ORCA, oral cancer
Statistical analysis
K-means clustering and hierarchical clustering were performed using the scikit-learn Python package (version 0.22.1). Consensus clustering was performed using the ConsensusClusterPlus R package (version 1.50.0). Significant differences in each LM22 fraction were compared using the Mann‒Whitney U test. Survival analysis was performed using the lifeline Python package (version 0.24.2). Significance between survival curves was analyzed using the log-rank test. The t-test boxplot visualization was performed using the Statannot Python package (version 0.2.2).
DEG analysis
DEG analysis between TCGA risk groups was performed using the R limma package (version 3.42.2) .26 P-values < 0.05 and |log fold change | > 1.2 threshold were applied to the result.
Survival prediction by deep learning classification
Deep learning classification was performed using a DNN classifier in the TensorFlow module (version 1.14.0) in Python and included 2000 steps with a 7 × 7 hidden layer. The hidden unit size (7 × 7) was diversified by square multiplication from 2 to 30, determining the optimal value with the highest accuracy. Loss was calculated using softmax cross entropy, Adagrad for optimizer, and Relu for the activation function. Accuracy was autonomously calculated using the internal evaluation function. A detailed flowchart of the pipeline is shown in Figure 1(b).
Analysis environment
The overall analysis was performed using Python (version 3.7.9; Python Software Foundation, Wilmington, DE, USA) and R (version 3.6.1; The R Foundation, Vienna, Austria) software. Any other version of Python/R packages of interest may be checked in an established conda environment within the provided docker image.
Results
Survival analysis of clustered CIBERSORT results from TCGA data
To obtain unsupervised classified results, the CIBERSORT results were subjected to various clustering analyses. The most important step in clustering was to determine the appropriate clustering method and optimal k-value. In this study, three clustering methods were considered for classification during candidate training: Hierarchical, K-means, and consensus clustering. These clustering methods were all incorporated into the “intracohort validation” process, and the results were calculated for each k-value via mutual information (MI), normalized MI (NMI), and adjusted MI (AMI) methods. To concisely depict only the valid analysis results, the hierarchical clustering method was utilized with a k-value = 3 that exhibited every measuring value (Table 2).
Table 2..
Mutual information (MI), Normalized MI (NMI) and Adjusted MI (AMI) scores of potential clustering methods. The most acceptable scores among variated k values across each clustering/measuring method are highlighted.
| Consensus (k = 3) |
Hierarchical (k = 2) |
Hierarchical (k = 3) |
Hierarchical (k = 4) |
K-means (k = 2) |
K-means (k = 3) |
|
|---|---|---|---|---|---|---|
| MI | 0.423562 | 0.405244 | 0.687885 | 1.037712 | 0.426107 | 0.686712 |
| NMI | 0.414230 | 0.592863 | 0.654062 | 0.818148 | 0.674453 | 0.642146 |
| AMI | 0.400079 | 0.589346 | 0.645963 | 0.809483 | 0.671388 | 0.633973 |
A Kaplan‒Meier (K-M) graph was plotted to depict overall survival information in the clinical data for each defined cluster. Cluster 2 (green) exhibited a better prognosis compared to clusters 1 and 3 (Figure 2(a)). The two groups (cluster 1, red and cluster 3, green) did not exhibit meaningful differences between them (p = .88740); therefore, the two groups were combined into a single high-risk group (Figure 2(b)).
Figure 2.
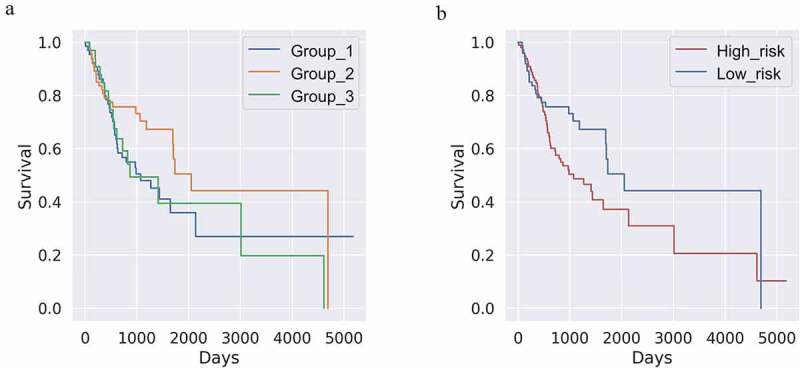
(a) Kaplan‒Meier (k-m) plot of K-means clustering after cell-type identification by estimating relative subsets of RNA transcripts (k = 3 and n = 173). The yellow line (class 3) shows a distinct favorable survival pattern. (p-value = 0.26592) (b) K-M plot of Figure 2a regrouped by binary risk group. Groups corresponding to the blue and green lines in Figure 2a are merged into one high-risk group. (p-value = 0.01441)
Differences in the TIL fraction between the high- and low-risk groups
The Mann‒Whitney U test was used to determine the differences between the high- and low-risk groups in the TCGA ORCA dataset based on the abundance of each of the 22 TIL subsets (Figure 3). As shown in the boxplots, the counts of naïve and memory B cells, CD8 + T cells, activated memory CD4 + T cells, follicular helper T cells, Tregs, resting natural killer (NK) cells, monocytes, M1 macrophages, resting dendritic cells, and resting mast cells were significantly increased in the low-risk group, whereas a significant increase in the naïve CD4 + T cell, gamma delta T cell, M0 macrophage, activated mast cell, and eosinophil TIL counts were observed in the high-risk group. No difference was observed between the two groups with respect to plasma cell, resting memory CD4 + T cell, activated NK cell, M2 macrophage, activated dendritic cell, and neutrophil counts.
Figure 3.
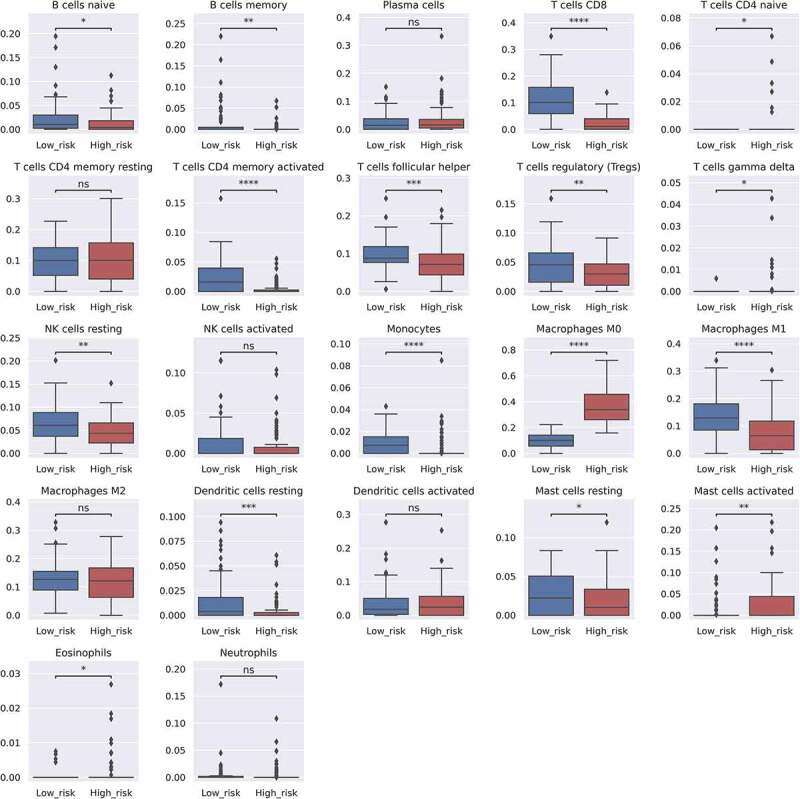
Bar plots indicating the differences in the estimated LM22 fraction between the high and low survival risk groups. Each p-value is written above the bar plots (NS: p > .05, *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001, and ****: p ≤ 0.0001). Y-axis indicates predicted fraction level of each cell subtype
Common DEGs between high- and low-risk groups
To explore prominent potential biomarker genes, DEG analysis was performed between each predicted risk group in both TCGA and ICGC patients, confirming seven common DEGs in total. Small proline-rich protein 3 (SPRR3) was upregulated in the low-risk group, while collagen type XI alpha 1 chain (COL11A1), collagen type X alpha 1 chain (COL10A1), matrix metallopeptidase 11 (MMP11), matrix metallopeptidase 13 (MMP13), collagen triple helix repeat containing 1 (CTHRC1), and ring finger protein 128 (RNF128) showed significant upregulation in the high-risk group.
Survival prediction based on the classified CIBERSORT results
To verify if the survival rate of other public ORCA cohorts might be predicted based on the patterns observed in the current data group, a deep learning model using a DNN classifier was established. To validate the classifier model’s accuracy, two strategies were employed, i.e., “intracohort validation” and “intercohort validation.”
In the first step, 80% of the samples (n = 138) in the TCGA ORCA dataset were used as inputs to train the DNN classifier. The remaining samples were divided into two to perform validation tests. The accuracy for the former test group was 100%, whereas for the latter, it was 94.4% (n = 17 for group 1, n = 18 for group 2), demonstrating a significant survival group prediction level of 97.2%, on average. A time-dependent graph depicting the changes in the loss function/internal accuracy of the training set and accuracies in individual datasets is shown in Figure 4. Because the classifier predicted the survival pattern of the sample group based on the CIBERSORT results, the classifier was used to analyze a completely different RNA-Seq ORCA dataset.
Figure 4.
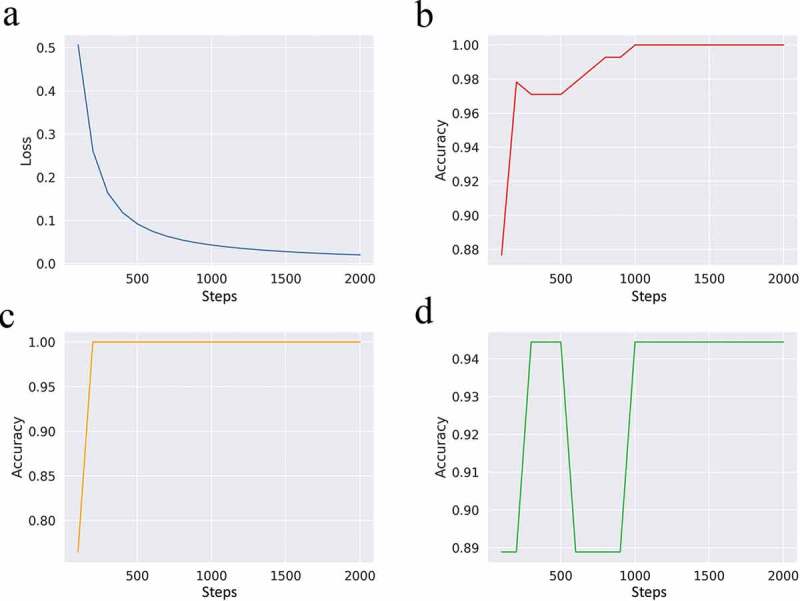
Scalar visualization of the established deep neural network (DNN) classifier model over steps in the loss function (a) and accuracy with the training datasets (b), primary test set (c), and secondary test set (d)
There are some concerns that the remarkable performance of the classifier might be attributed to overfitting, which is always associated with endeavors aimed at achieving maximum accuracy. Thus, the performance of the classifier must be validated using cohorts from completely different batches. The obtained ICGC ORCA RNA-Seq dataset was classified using a previously established classifier. The differential survival patterns between samples from the predicted high-risk (n = 6) and low-risk groups (n = 28) were analyzed using the K-M method. As shown in Figure 5, the survival of the predicted high-risk group was significantly lower than that of the predicted low-risk group (p = .00685). Detailed patient information for each predicted group is provided in Table 3.
Figure 5.
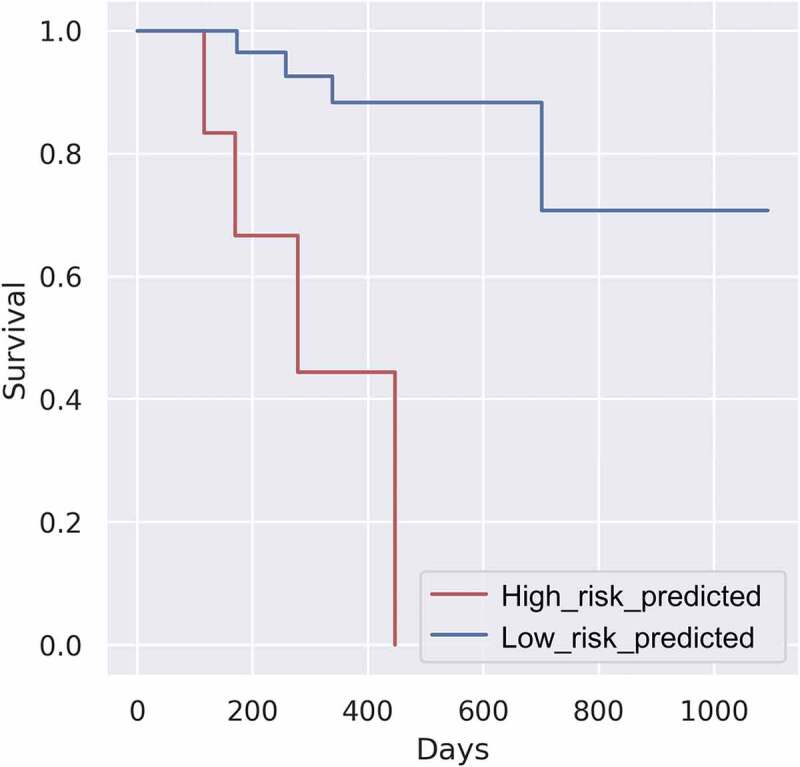
Kaplan‒Meier survival plot of the predicted International Cancer Genome Consortium oral cancer dataset. (p-value: 0.00685)
Table 3.
Clinical characteristics of the risk-group predicted cohort of patients with oral cancer in the International Cancer Genome Consortium data
| Total |
Low-risk group |
High-risk group |
||
|---|---|---|---|---|
| Number (Percentage) | ||||
| Age (years) | 0–39 | 7(20.5) | 7(25.0) | 0(0.0) |
| 40–49 | 10(29.4) | 8(28.6) | 2(33.3) | |
| 50–59 | 11(32.4) | 10(35.7) | 1(16.7) | |
| 60–69 | 5(14.7) | 3(10.7) | 2(33.3) | |
| 70–79 | 1(2.9) | 0(0.0) | 1(16.7) | |
| 80+ | 0(0.0) | 0(0.0) | 0(0.0) | |
| Sex | Male | 28(82.4) | 22(78.6) | 6(100.0) |
| Female | 6(17.6) | 6(21.4) | 0(0.0) | |
| N stage | N0 | 8(23.5) | 8(28.6) | 0(0.0) |
| N1 | 17(50.0) | 12(42.9) | 5(83.3) | |
| N2 | 9(26.5) | 8(28.6) | 1(16.7) | |
| N3 | 0(0.0) | 0(0.0) | 0(0.0) | |
| NX | 0(0.0) | 0(0.0) | 0(0.0) | |
| T stage | T1 | 0(0.0) | 0(0.0) | 0(0.0) |
| T2 | 0(0.0) | 0(0.0) | 0(0.0) | |
| T3 | 2(5.9) | 2(7.1) | 0(0.0) | |
| T4 | 32(94.1) | 26(92.9) | 6(100.0) | |
| TX | 0(0.0) | 0(0.0) | 0(0.0) | |
| M stage | M0 | 34(100.0) | 28(100.0) | 6(100.0) |
| M1 | 0(0.0) | 0(0.0) | 0(0.0) | |
| MX | 0(0.0) | 0(0.0) | 0(0.0) | |
To validate the classifier’s performance over similar modeling methods, the validation result was compared to other results of the same training data using two methods: random forests and decision tree methods over the same pipeline. The accuracy of the random forest method was nearly 94.1% on average between the two intracohort validation datasets, but the established classifier with the method did not show any significant survival rates between the predicted risk groups in intercohort validation. In contrast, the decision tree method significantly predicted survival rates between the two groups in external validation, but the internal validation result had the worst accuracy across methods, scoring only 82.4% on average. All K-M plots, receiver operating characteristic curve plots, and corresponding area under the curve scores as validation results of the three methods over the pipeline are provided in Supplemental Figures 4 and 5.
Discussion
The analysis of hidden patterns within gene expression data is a tremendous strategy to attaining an in-depth understanding of functional genomics. However, the complexity of biological networks and the large number of genes make data analysis very difficult; thus, some clustering algorithms help derive useful information by identifying patterns in gene expression data.27 Based on this idea, by clustering the ORCA CIBERSORT results, which accurately estimate immune composition, we aimed to achieve a more immune-specific and noise-free clustering efficiency. In addition, to examine whether the results could be validly analyzed, the immunological characteristics of each high- and low-risk group were determined by comparing the estimated immune cell fraction identified using CIBERSORT in each group.
The TME is an important indicator of the clinical and prognostic factors of cancer. Bin Liang et al. have identified the effects of 22 immune cell subsets in patients with HNSC, and used their characteristics to reveal clinical relevance and define independent prognosis factors in advance.28 Furthermore, in this study, the actual risk groups were predicted by classifying the signature patterns of TIL fractions in patients with HNSC by combining them with survival information. As a result of this effort, a few patients with HNSC, whose survival rates were extremely low, were successfully identified.
The purpose of this study was to identify the clearest differences between the two risk groups and to introduce these data patterns into a classifier, so as to identify the classification method/optimal k-value that resulted in the most distant and significant clusters. Therefore, there was a significant difference between this approach and other clustering measurement methods, such as MI, NMI, and AMI. The measured score plays an important role in determining the optimal method/k-value based on survival patterns, but it cannot serve as the sole evidence. For example, in the overall measuring scores of the hierarchical clustering method as shown in Table 2, the score with a k-value = 4 (MI = 1.037712, NMI = 0.818148, AMI = 0.809483) was significantly higher compared to that of a k-value = 3 (MI = 0.687885, NMI = 0.654062, AMI = 0.645963), but in the K-M analysis, the p-value associated with the former condition was poor (k = 3, p = .1359 and k = 4, p = .2524). The survival plot based on K-M analysis is shown in Supplemental Figure 1–3.
Differences in LM22 subtypes between the risk groups were also investigated. Many immune cell subtypes that enhance immunity were decreased in the high-risk group. Focusing on T-cell subgroups, the high-risk group showed decreased counts of activated memory CD4 + T cell fractions. Given that the prediction level of the naïve CD4 + T cell fraction showed an opposing regulation pattern, these data suggested that CD4 + T cell differentiation affected survival rates. Although further investigations on memory CD4 + T cells are required, several studies on cell development have revealed that memory CD4 + T cell development in tumors is crucial for various immunotherapeutic treatments, such as immune blockade therapy, in the context of enhancing the effectiveness of the anti-tumor response.29 In addition, activated memory CD4 + T cell-induced activation of CD8 + T cells increases the direct kill rate of cancer, which is illustrated in the same context as a dramatic difference in the current cell type data. The counts of follicular helper T cells, which have been reported to play important roles in various cancer microenvironments,30–32 are also decreased in the high-risk group. A high fraction level of gamma/delta T cells has been found in patients with HNSC compared to that of normal patients.33 The current corresponding result also indicated that the elevated gamma/delta T cell fraction might affect the low survival rates of the predicted high-risk patient group.
Increased monocyte levels enhance macrophage polarization into M1 macrophages, which produce proinflammatory cytokines and reactive oxygen/nitrogen species that are crucial for host defense and tumor cell killing.34 Chronic inflammation abnormalities and the accompanying oxidative stress lead to the development of various diseases, such as cancer.35,36 In this context, the current predictions of monocyte and macrophage phenotype abundance revealed some interesting results. Specifically, although the M2 macrophage fraction did not show differences between the two groups, the M0 macrophage fraction counts were significantly increased in the high-risk group, while those of the M1 macrophages and monocytes exhibited a proportionately opposite regulation pattern, suggesting that the proinflammatory deactivation caused by significantly decreased macrophage polarization played a crucial role in determining the survival rate in the high-risk group. The resting and activated mast cell fractions also showed an opposite regulation pattern. Mast cell accumulation in tumor tissue is either beneficial or detrimental to tumors; although further understanding of the key roles of mast cells in cancer is required, several studies have summarized the correlation between mast cells and cancer.37
In particular, it is interesting to note that this result is somewhat different from the survival analysis results based on Tregs and M0 macrophages reported by Bin Liang et al.28 Both studies showed low survival rates in the groups containing low M0 macrophage counts, but the current study showed a fraction level with an opposite pattern in the Treg counts between risk groups. This indicated that the classifier we established focused on the difference in the cell subtype count with a more significant effect on survival among the 22 input channels, considering the overall actual TIL composition within malignant tissues. It also suggests that analyzing the survival patterns using individual factors, such as single immune-cell types, may be problematic because the TIL composition in the TME is heterogeneous.
Eosinophil activity affects tumors in various ways due to their immunobiological characteristics; they affect anti-tumor responses due to their destructive features, but also promote tumor proliferation by inhibiting Th1 responses or increasing Th2 responses.38 Although tumor-associated eosinophils are generally observed in hematological solid tumors with a favorable prognosis, 39 the eosinophil elevation levels in this study show an opposite tendency. Further studies on the roles of eosinophils in ORCA would contribute to better comprehension of the results. Additionally, both naïve and memory B cells had downregulated fractions in high-risk patients. This result suggests potential hazards of B cell deficiency, since the role of tumor-infiltrating B cells within the TME remains controversial40 and is a challenge that requires further investigation.
Toruner et al. have reported significant downregulation of SPRR3 in oral squamous cell carcinoma compared to that in normal tissues,41 in two individual studies.42,43 The current results were identical to that research. COL11A1 and COL10A1, which express circulating extracellular-matrix (ECM)-related proteins, are significantly elevated in breast cancer, gastric cancer, and pancreatic cancer.44 Although further study is required to apply this result to ORCA, the current study enhances the hypothesis of the role of the two genes in various types of cancer. Pal et al. have reported that MMP11 and MMP13 are stimulated by the tumor-specific ECM protein thrombospondin 1 (THBS1), whose expression is highly elevated in both cultured human ORCA cell lines and their co-cultivated mouse fibroblast cells. The expression level of CTHRC1 is highly correlated with metastasis of ORCA cells.45 Downregulation of RNF128 expression is correlated with poor prognosis in various types of malignancies.46,47 However, in the current study, RNF128 was upregulated in the high-risk group compared to that of the low-risk group. The Cox proportional-hazards model was used to acquire common DEGs, but no significant coefficient with survival was found among the genes included. In light of these results, determining the cancer survival prognosis with a single biomarker gene might overlook the importance of a cohort comprising the TME.
The benefits gained from the clustering of existing gene expression data and analysis methods are well established.48 However, the ability to achieve useful results by clustering secondarily analyzed expression data (in this case, CIBERSORT) has been questionable. In this study, evidence was provided on the benefits of clustering CIBERSORT data that contained information on tumor-infiltrating immune cell subsets. Further study and understanding of these cell subsets in in vivo environments in various cancers are needed.
Due to the lack of a public RNA-Seq ORCA dataset, the intercohort validation was performed using only a single cohort. In addition, there were some existing challenges that overcame the skewed intercohort validation result. In spite of efforts to establish binary risk group models with simultaneous sample-balanced, survival-distinct, and significant features, predicted high-risk patients were only 17.6% of the total patients, even though most of them were at the T4 stage.
Conclusion
Despite efforts to develop novel biomarkers for HNSC, more research is needed. To establish an accurate survival information-specific predictive model, the public RNA-Seq ORCA dataset was virtually dissected and transformed into TIL-specific data using CIBERSORT. When these data were fed into the DNN classifier, it successfully predicted survival patterns of the predicted risk groups in an independent ICGC ORCA cohort.
Through this study, a novel approach based on deep learning is suggested that has potential application in various types of cancer.
Supplementary Material
Funding Statement
This work was supported by the National Research Foundation of Korea [NRF-2018R1C1B6005304; NRF-2018R1A5A2023879; NRF-2020R1C1C1003741].
Author notes
Yeongjoo Kim and Ji Wan Kang contributed equally to this study.
Availability of data and material
The datasets used or analyzed during the current study are available from the corresponding author upon reasonable request.
Code availability
Code that support the findings of this study are available in Github.
(URL: https://github.com/asdoper0630/ORCA, DOI: 10.5281/zenodo.4553322).
Ethics approval
All procedures described fulfilled the requirements of internal or national ethics committees from the author-affiliated institutions or those of the cited references.
Authors’ contributions
All authors contributed to the study conception and design. Data collection/preprocessing and analysis were performed by Yeongjoo Kim and Ji Wan Kang. Yeongjoo Kim and Ji Wan Kang contributed equally to the first draft of the manuscript, and all authors commented on previous versions of the manuscript. All authors have read and approved the final manuscript.
Disclosure of interest
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics. Cancer J Clin. 2020;70(1):7–9 [DOI] [PubMed] [Google Scholar]
- 2.Howlader N, Noone A, Krapcho M, et al. SEER cancer statistics review. National Cancer Institute. 1975;2008 [Google Scholar]
- 3.Wilson G, Grover R, Richman P, et al. Bcl-2 expression correlates with favourable outcome in head and neck cancer treated by accelerated radiotherapy. Anticancer Res. 1996;16(4C):2403–2408 [PubMed]
- 4.Shen Y, Liu J, Zhang L, et al. Identification of Potential Biomarkers and Survival Analysis for Head and Neck Squamous Cell Carcinoma Using Bioinformatics Strategy: A Study Based on TCGA and GEO Datasets. BioMed Research International. 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang Y, Feng G, Zhong S, et al. An Inflammation-Immunity Classifier of 11 Chemokines for Prediction of Overall Survival in Head and Neck Squamous Cell Carcinoma. Medical science monitor: international medical journal of experimental and clinical research. 2019;25:4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngan H-L, Liu Y, Fong AY, et al. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Science Alliance. 2020;3(6):e201900545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon EJ, Ha M, Jang JY, et al. Identification and Complete Validation of Prognostic Gene Signatures for Human Papillomavirus-Associated Cancers: Integrated Approach Covering Different Anatomical Locations. Journal of Virology. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pak K, Oh S-O, Goh TS, et al. A user-friendly, web-based integrative tool (ESurv) for survival analysis: development and validation study. Journal of medical Internet research. 2020;22(5):e16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santosh ABR, Jones T, Harvey J. A review on oral cancer biomarkers: Understanding the past and learning from the present. Journal of cancer research and therapeutics. 2016;12(2):486. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen N, Bellile E, Thomas D, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head & neck. 2016;38(7):1074–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huo M, Zhang Y, Chen Z, et al. Tumor microenvironment characterization in head and neck cancer identifies prognostic and immunotherapeutically relevant gene signatures. Scientific Reports. 2020;10(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heikkinen I, Bello IO, Wahab A, et al. Assessment of tumor-infiltrating lymphocytes predicts the behavior of early-stage oral tongue cancer. The American journal of surgical pathology. 2019;43(10):1392–1396 [DOI] [PubMed] [Google Scholar]
- 13.Watanabe Y, Katou F, Ohtani H, et al. Tumor-infiltrating lymphocytes, particularly the balance between CD8+ T cells and CCR4+ regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology. 2010;109(5):744–752 [DOI] [PubMed] [Google Scholar]
- 14.Graizel D, Zlotogorski‐Hurvitz A, Tsesis I, et al. Oral cancer‐associated fibroblasts predict poor survival: Systematic review and meta‐analysis. Oral diseases. 2020;26(4):733–744 [DOI] [PubMed] [Google Scholar]
- 15.Gnjatic S, Bronte V, Brunet LR, et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. Journal for immunotherapy of cancer. 2017;5(1):1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Zhou H, Zheng J, et al. Identification of immune-related prognostic biomarkers based on the tumor microenvironment in 20 malignant tumor types with poor prognosis. Frontiers in oncology. 2020;10:1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Khodadoust MS, Liu CL, et al. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Systems Biology. Springer; 2018. p. 243–259 [DOI] [PMC free article] [PubMed]
- 18.Zhou R, Zhang J, Zeng D, et al. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I–III colon cancer. Cancer Immunology, Immunotherapy. 2019;68(3):433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Zhang S, Yan J. IRF1 association with tumor immune microenvironment and use as a diagnostic biomarker for colorectal cancer recurrence. Oncology letters. 2020;19(3):1759–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue Y, Tong L, LiuAnwei Liu F, et al. Tumor‑infiltrating M2 macrophages driven by specific genomic alterations are associated with prognosis in bladder cancer. Oncology reports. 2019;42(2):581–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urda D, Montes-Torres J, Moreno F, et al, editors. Deep learning to analyze RNA-seq gene expression data. International work-conference on artificial neural networks; 2017: Springer
- 22.Chen Y, Li Y, Narayan R, et al. Gene expression inference with deep learning. Bioinformatics. 2016;32(12):1832–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van IJzendoorn DG, Szuhai K, Briaire-de Bruijn IH, et al. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS computational biology. 2019;15(2):e1006826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla S, Pranay A, D'Cruz AK, et al. Immunoproteomics reveals that cancer of the tongue and the gingivobuccal complex exhibit differential autoantibody response. Cancer Biomarkers. 2009;5(3):127–135 [DOI] [PubMed] [Google Scholar]
- 25.Nair S, Singh B, Pawar PV, et al. Squamous cell carcinoma of tongue and buccal mucosa: clinico-pathologically different entities. European Archives of Oto-Rhino-Laryngology. 2016;273(11):3921–3928 [DOI] [PubMed] [Google Scholar]
- 26.Smyth GK. Limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and Bioconductor. Springer; 2005. p. 397–420 [Google Scholar]
- 27.Datta S, Datta S. Evaluation of clustering algorithms for gene expression data. BMC bioinformatics. 2006;7(S4):S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang B, Tao Y, Wang T. Profiles of immune cell infiltration in head and neck squamous carcinoma. Bioscience reports. 2020;40(2):BSR20192724. [DOI] [PMC free article] [PubMed]
- 29.Hope JL, Stairiker CJ, Bae E-A, et al. Striking a balance–cellular and molecular drivers of memory T cell development and responses to chronic stimulation. Frontiers in immunology. 2019;10:1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu-Trantien C, Loi S, Garaud S, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. The Journal of clinical investigation. 2013;123(7):2873–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amé-Thomas P, Le Priol J, Yssel H, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia. 2012;26(5):1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu F, Zhang H, Chen J, et al. Immune signature of T follicular helper cells predicts clinical prognostic and therapeutic impact in lung squamous cell carcinoma. International immunopharmacology. 2020;81:105932. [DOI] [PubMed] [Google Scholar]
- 33.Bas M, Bier H, Schirlau K, et al. Gamma–delta T-cells in patients with squamous cell carcinoma of the head and neck. Oral oncology. 2006;42(7):691–697 [DOI] [PubMed] [Google Scholar]
- 34.Laskin DLJCRIT. chemical toxicity: a battle of forces. Macrophages and Inflammatory Mediators. 2009;22:1376–1385.Laskin DLJCrit. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. 2009;22(8):1376–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shacter E, Weitzman SA, Chronic inflammation and cancer. 2002. [PubMed]
- 36.Ohshima H, Tazawa H, Sylla BS, et al. Prevention of human cancer by modulation of chronic inflammatory processes. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;591(1–2):110–122 [DOI] [PubMed] [Google Scholar]
- 37.Maciel TT, Moura IC, Hermine O. The role of mast cells in cancers. F1000prime reports. 2015;7. [DOI] [PMC free article] [PubMed]
- 38.정일엽, Hypereosinophilia-associated Diseases and the Therapeutic Agents in Development. Hanyang Medical Reviews, 2013. 33.
- 39.Gatault S, Legrand F, Delbeke M, et al. Involvement of eosinophils in the anti-tumor response. Cancer Immunology, Immunotherapy. 2012;61(9):1527–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wouters MC, Nelson BH. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clinical Cancer Research. 2018;24(24):6125–6135. doi: 10.1158/1078-0432.CCR-18-1481. [DOI] [PubMed] [Google Scholar]
- 41.Toruner GA, Ulger C, Alkan M, et al. Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer genetics and cytogenetics. 2004;154(1):27–35 [DOI] [PubMed] [Google Scholar]
- 42.Zucchini C, Biolchi A, Strippoli P, et al. Expression profile of epidermal differentiation complex genes in normal and anal cancer cells. International journal of oncology. 2001;19(6):1133–1141 [DOI] [PubMed] [Google Scholar]
- 43.Chen B-S, Wang M-R, Cai Y, et al. Decreased expression of SPRR3 in Chinese human oesophageal cancer. Carcinogenesis. 2000;21(12):2147–2150 [DOI] [PubMed] [Google Scholar]
- 44.Kim H, Watkinson J, Varadan V, et al. Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC medical genomics. 2010;3(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu G, Sengupta PK, Jamal B, et al. N-glycosylation induces the CTHRC1 protein and drives oral cancer cell migration. Journal of Biological Chemistry. 2013;288(28):20217–20227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee Y-Y, Wang C-T, Huang SK-H, et al. Downregulation of RNF128 predicts progression and poor prognosis in patients with urothelial carcinoma of the upper tract and urinary bladder. Journal of Cancer. 2016;7(15):2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei C-Y, Zhu M-X, Yang Y-W, et al. Downregulation of RNF128 activates Wnt/β-catenin signaling to induce cellular EMT and stemness via CD44 and CTTN ubiquitination in melanoma. Journal of hematology & oncology. 2019;12(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’haeseleer P. How does gene expression clustering work? Nat Biotechnol. 2005;23(12):1499–1501. doi: 10.1038/nbt1205-1499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used or analyzed during the current study are available from the corresponding author upon reasonable request.