

Abstract
Recent advances in our understanding of RNA biology have uncovered crucial roles for RNA in multiple disease states, ranging from viral and bacterial infections to cancer and neurological disorders. As a result, multiple laboratories have become interested in developing drug-like small molecules to target RNA. However, this development comes with multiple unique challenges. For example, RNA is inherently dynamic and has limited chemical diversity. In addition, promiscuous RNA-binding ligands are often identified during screening campaigns. This Tutorial Review overviews important considerations and advancements for generating RNA-targeted small molecules, ranging from fundamental chemistry to promising small molecule examples with demonstrated clinical efficacy. Specifically, we begin by exploring RNA functional classes, structural hierarchy, and dynamics. We then discuss fundamental RNA recognition principles along with methods for small molecule screening and RNA structure determination. Finally, we review unique challenges and emerging solutions from both the RNA and small molecule perspectives for generating RNA-targeted ligands before highlighting a selection of the “Greatest Hits” to date. These molecules target RNA in a variety of diseases, including cancer, neurodegeneration, and viral infection, in cellular and animal model systems. Additionally, we explore the recently FDA-approved small molecule regulator of RNA splicing, risdiplam, for treatment of spinal muscular atrophy. Together, this Tutorial Review showcases the fundamental role of chemical and molecular recognition principles in enhancing our understanding of RNA biology and contributing to the rapidly growing number of RNA-targeted probes and therapeutics. In particular, we hope this widely accessible review will serve as inspiration for aspiring small molecule and/or RNA researchers.
Introduction
Around the world, researchers are continuously searching for new drugs to treat a wide variety of diseases. While this process can involve continuing drug development for established disease targets, it can also involve searching for new disease targets that may be “druggable,” particularly for conditions that show drug resistance or that have limited treatment options. Currently, most therapies target proteins,1 and while development of protein-targeted drugs has been crucial in modern medicine, limitations in developing protein-targeted therapeutics in many diseases have inspired some scientists to explore targeting other biomolecules.
The search for new drug targets has led scientists to explore targeting RNA, an interest which has been amplified as a result of the so-called “RNA Revolution,” which included the discovery of important RNA functions in multiple disease states.2 Antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs), which act via complementary base pairing with the target of interest, have contributed greatly in the identification of therapeutically tractable target RNAs and have been successful as therapies in a few cases.3 Small molecule drugs, however, remain the avenue of choice for disease treatment when available, owing to generally more favorable pharmacological properties as well as the ease of tunability via medicinal chemistry approaches. Moreover, their ability to modulate RNA structure has made them complementary tools to sequence-recognizing ASOs.3
Further adding to rising interest in RNA targeting are recent discoveries of ubiquitous functional RNA molecules in human cells. For example, the Encyclopedia of DNA Elements (ENCODE) project, a follow-up study to the Human Genome Project, sought to map the functional regions throughout the genome.4 This project uncovered that while only 1.5% of our genome is ultimately translated into protein, about 70–90% is transcribed to RNA, showing that the vast majority of the genome yields noncoding (ncRNAs) over protein coding messenger RNAs (mRNAs).4 While multiple ncRNA classes were known at the time of the ENCODE project, including micro-RNAs (miRNAs) and long noncoding RNAs (lncRNAs), this study allowed for mapping of previously unidentified lncRNAs, miRNAs, and other ncRNAs in a variety of cell types.4 In addition, viruses and bacteria have long been known to rely on regulatory RNAs, which have served as additional inspiration to explore similar structural elements in humans.3 Motivated by these discoveries, notable progress has been made in small molecule targeting of RNA, both in understanding the underlying principles of these interactions as well as demonstrating clinical efficacy.
This Tutorial will first overview RNA fundamentals to give the reader insight into the myriad functions of RNA and highlight how these functions are driven by structure. We will next discuss established and emerging principles of small molecule recognition of these structures as well as methods for their identification. We will then explore some of the remaining challenges and emerging solutions in the field, before ending with a selection of “Greatest Hits” that are paving the way towards clinical use or have already received approval. Due to length constraints, our focus will remain on direct small molecule–RNA interactions, rather than drugs that indirectly modulate the transcription or promote the degradation of an RNA of interest, though promising work has been accomplished in those areas as well.5 We apologize to the authors of many works that could not be cited due to reference limitations, and we invite the reader to consult the review articles cited herein to learn more about RNA biology and targeting. We hope that this tutorial review serves as an inspiration and resource for both current researchers as well as aspiring scientists and students alike who are unfamiliar with the RNA field to join us in this exciting quest for RNA-targeting small molecule drugs.
Section 1: overview of RNA classes and targetable functions
There are multiple classes of RNA, with the first classification level often involving their coding potential. While mRNAs are translated into protein, it is important to note that not all regions of a coding RNA undergo translation. For example, pre-mRNAs can contain introns that are removed by splicing and also contain untranslated regions (UTRs) that can have functional roles.2 One example of functional regulation via RNA structure in 5′-UTRs, called the riboswitch, is explored below. In contrast, RNAs that do not undergo translation are termed ncRNA. Some of these are essential for the translation process: transfer RNAs (tRNAs) carry the amino acid building blocks for proteins, and ribosomal RNA (rRNA) catalyzes the reactions necessary for polymerizing the amino acids into a peptide chain. Other ncRNA molecules regulate transcription and signaling events and can be further classified by their lengths and functional roles within the cell. Together, this network of coding and ncRNA molecules, while concurrently challenging the traditional view of RNA as a messenger between DNA and proteins, represents an open space for therapeutic exploration and discovery.
This section will review several important examples from RNA classes in both human and microbial cells that represent attractive therapeutic targets for small molecules. Human RNAs are implicated in conditions such as cancer or neurodegenerative diseases, which can be characterized by dysregulation of various, disease-specific ncRNAs. At the same time, therapeutics for various microbial diseases may be attained by targeting unique RNAs within viruses or bacteria. Overall, misexpressed human RNAs may represent an untapped class of potential drug targets, and viral and bacterial RNAs may allow for the development of novel antimicrobial agents.
miRNAs
miRNAs are initially synthesized as larger primary miRNAs (pri-miRNA) that are first processed by the enzyme Drosha in the nucleus, followed by Dicer in the cytosol, to yield a processed, single-stranded miRNA product that is about 17–22 nucleotides in length.2 miRNAs regulate mRNA expression through complementary base pairing, causing mRNA degradation or inhibition of translation. It is important to note that one miRNA can have multiple targets, meaning that dysregulation of one miRNA can result in wide-sweeping changes in gene expression. Indeed, dysregulation of miRNAs is implicated in disease states such as cancer.2
lncRNAs
lncRNAs are defined as RNAs being over 200 nucleotides in length that lack coding function. The aforementioned ENCODE project specifically identified almost 10 000 lncRNA genes,4 many of which were subsequently found to have a variety of cellular functions, such as guiding epigenetic modification complexes or functioning as decoys that divert another biomolecule away from its target.2 For example, the lncRNA X-inactive Specific Transcript (XIST) is involved in X-inactivation for dosage compensation in female mammals.6 Specifically, XIST becomes overexpressed from the X chromosome that will become inactivated, coating the chromosome and recruiting a multitude of proteins including chromatin remodellers such as histone deacetylases and the Polycomb repressive complex 2 (PRC2), thereby promoting gene silencing and chromosome inactivation.6 As with miRNAs, lncRNAs can also be dysregulated in disease. For example, lncRNA Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) plays roles in normal pre-mRNA splicing by altering the localization and phosphorylation status of splicing factors as well as regulating gene expression.7 However, the overexpression of MALAT1 appears to facilitate various cancer processes. As explored below, MALAT1 has a unique structure that allows for its overaccumulation,7 and it has been successfully modulated by small molecules.8,9
Short tandem repeats and repeat expansions
Multiple RNAs contain 1–6 nucleotide repetitive sequences called short tandem repeats. The exact number of these repeats can vary across individuals, and while naturally-occurring, an excess number of repeats called “expansions” in certain genes results in a variety of genetic conditions.10 These repeats can sequester important proteins to prevent them from executing their normal functions or, if translated, create toxic polypeptides. For example, myotonic dystrophy type 1 (DM1), discussed in the “Greatest Hits” section, results from such an expansion.10
Viral and bacterial RNAs
Multiple viruses utilize RNA for their genome instead of DNA, including those that cause HIV (Human Immunodeficiency Virus), influenza, and hepatitis.11 The HIV-1 RNA genome in particular contains several structural elements that have been successfully targeted with small molecules to inhibit viral replication.11 These successes and others serve as inspiration to target a multitude of other and less explored viruses with RNA genomes. Of particular note, coronaviruses, including the pandemic Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) that causes COVID19, are in this category.11
In addition, bacterial systems rely on metabolite interactions with RNA structures called riboswitches to control gene expression.12 Riboswitches are typically found in the UTR of a gene of interest and are able to detect a related metabolite concentration by direct binding. This event induces a conformational change that allows for transcription or translation of the gene to be regulated. Riboswitches have been identified for multiple metabolites, including amino acids, nucleotides, metals, and vitamins.12 Targeting of riboswitches with synthetic small molecules may have antibiotic potential, as they are critical for bacterial survival, and similar structures have not been found in human cells.13
Section 2: RNA structure and properties
Primary RNA structure and backbone
RNA structural architecture is often referred to as consisting of primary, secondary and tertiary structure. The primary structure of RNA is the nucleotide sequence comprised of three building blocks as described in Fig. 1A.14 Specifically, the “glycosidic bond” connects the ribose sugar to one of four aromatic, nitrogen-containing nucleobases: adenine (A) and guanine (G) purines, and cytosine (C) and uracil (U) pyrimidines (Fig. 1A). The negatively charged sugar-phosphate backbone is comprised of phosphodiester and ribose groups, which, owing to the 2′-hydroxyl moiety, induce distinct sugar “puckering” patterns compared to DNA. The backbone structure is highly dependent on the solvent environment, as RNA folding has to overcome electrostatic repulsion from the phosphate groups. This repulsion can be overcome through association of metal ions, such a divalent magnesium or monovalent sodium.14
Fig. 1.
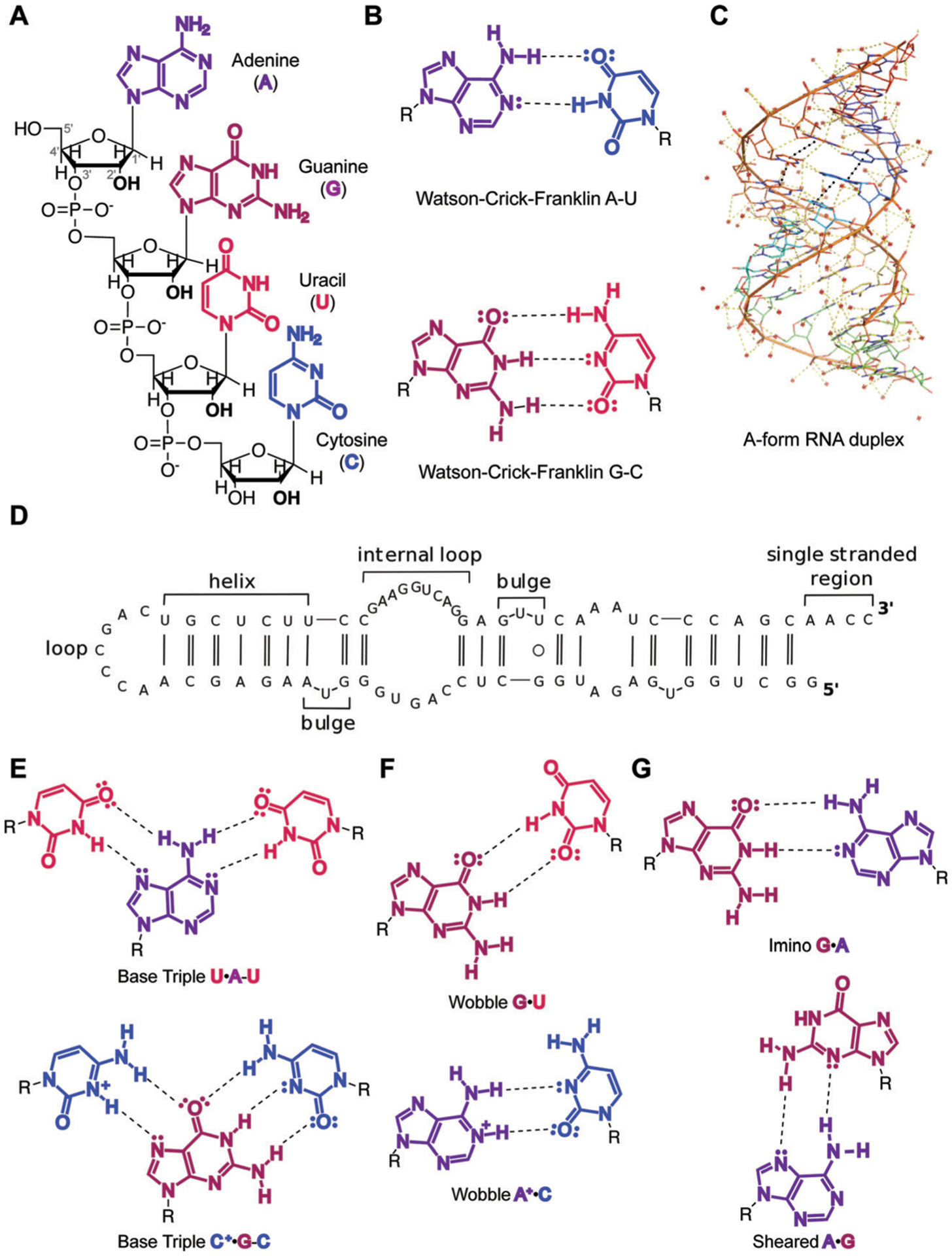
Overview of RNA primary and secondary structure and base pairing patterns. (A) Nucleotide building blocks of RNA primary structure. Each consists of a ribose sugar, containing a 2′ hydroxyl unique to RNA over DNA; phosphodiester groups, which connect the 5′ and 3′ hydroxyl groups of the ribose sugars; and adenine, guanine, uracil and cytosine nucleobases, which define the sequence of an RNA molecule. (B) Canonical Watson–Crick–Franklin hydrogen bonding patterns. Dashed lines represent hydrogen bonds. (C) An A-form RNA duplex secondary structure (PDB ID: 1LNT). Red dots represent metal ions. Yellow dashes indicate electrostatic interactions, including hydrogen bonds. Black dashes indicate stacking interactions. (D) Example of canonical RNA secondary structures as observed in 5′-end of long non-coding RNA (lncRNA) B2-SINE. Single and double solid lines represent A–U and G–C Watson–Crick–Franklin base pairs, respectively, while circles represent wobble base pairs. Adapted from ref. 15 with permission from Elsevier B. V., Copyright 2015. (E) Higher-order base triple interactions. (F) Non-canonical wobble interactions. (G) Purine–purine interactions, including imino and sheared hydrogen bonding patterns.
Base pairing patterns and RNA secondary structure
Secondary structure refers to local folds in the RNA structure created through base pairing, or the interaction between hydrogen bond donor and hydrogen bond acceptor groups on RNA bases. The most common secondary structure in RNA forms through the Watson–Crick–Franklin (WCF) hydrogen bonding pattern, where the canonical base pairs consist of A–U and G–C (Fig. 1B).† RNA base pairing, similar to DNA, allows for the formation of a double helix or duplex. The conformation of the ribose sugar in the RNA backbone leads to the formation of an A-form duplex, distinct from the B-form structure in DNA, and is stabilized through highly favorable and cooperative stacking interactions formed between the planar and polarizable nucleobase surfaces (Fig. 1C, black dashes). At the same time, the increased number of hydrogen bonds results in G–C base pairs having slightly greater stability than A–U.
In addition to duplex regions, nucleotides that do not form hydrogen bonds enable the formation of single-stranded secondary structures unique to RNA. These motifs include apical loops, internal loops, and bulges (Fig. 1D).15 Together, combinations of base paired and single-stranded structural “modules” throughout an RNA provide distinct topologies that may serve as recognition sites by ligands, including proteins and small molecules.
Further adding to the structural complexity of RNA are non-canonical base pairing patterns. The stability of non-canonical base pairs can depend on several factors, including sterics if they require rotation of the glycosidic bond,16 protonation of the bases, and/or direct binding of metals. For example, uridines or protonated cytosines can engage in base pairing with pyrimidines on the other side of the WCF interface. This hydrogen bonding pattern is referred to as Hoogsteen, and it can result in a base triple when combined with the WCF base pair (Fig. 1E). Additionally, uridines can bond with both guanines and protonated adenosines via “wobble” interactions at the WCF face (Fig. 1F). Purines can also interact with each other: guanines can base pair with adenosines through WCF–WCF (imino G–A) or Hoogsteen-sugar edge (sheared G–A) conformations (Fig. 1G). Together, these non-canonical interactions and can provide stabilizing forces for the formation of RNA tertiary structures, which serve as additional recognition sites for natural or synthetic ligands.
RNA tertiary structure
RNA tertiary structures form when secondary structure elements participate in through-space interactions. While these regions may be distant in primary or secondary structure, they come into proximity during RNA folding. The driving force towards formation of these structures is maximization of base stacking, similar to the folding of proteins that is driven by the hydrophobic effect.16
The folding architecture of RNA tertiary structures is largely dependent on coaxial stacking of adjacent helices (the linear alignment of the central axes of two separate helices), which is influenced by the RNA junction (the central connection of adjacent helices) topology.16 A prominent early example came from the structural characterization of the yeast phenylalanine transfer RNA by X-ray crystallography (Fig. 2A).17 In addition to coaxial stacking factors, long-range complementarity in nucleotide sequence enables base pairing interactions that drive the folding of structures such as kissing loops and pseudoknots (Fig. 2B and C).15 Furthermore, consecutive base triples have been documented in MALAT1, forming a unique triple helix structure (Fig. 2D).7 This is believed to confer stability and prevent degradation of the abundant MALAT1 transcript. Lastly, backbone desolvation and charge screening or direct binding by metals can promote RNA tertiary structure formation. For example, guanines are known to form tetrads with each other called G-quartets that are coordinated by potassium ions, which may then stack on top of each other to form highly stable G-quadruplex structures (Fig. 2E) that have been implicated in disease.18 Taken together, these unique tertiary structures offer opportunities for specific recognition by small molecules to enable the interrogation of structure–function relationships and therapeutic potential of these folds.
Fig. 2.
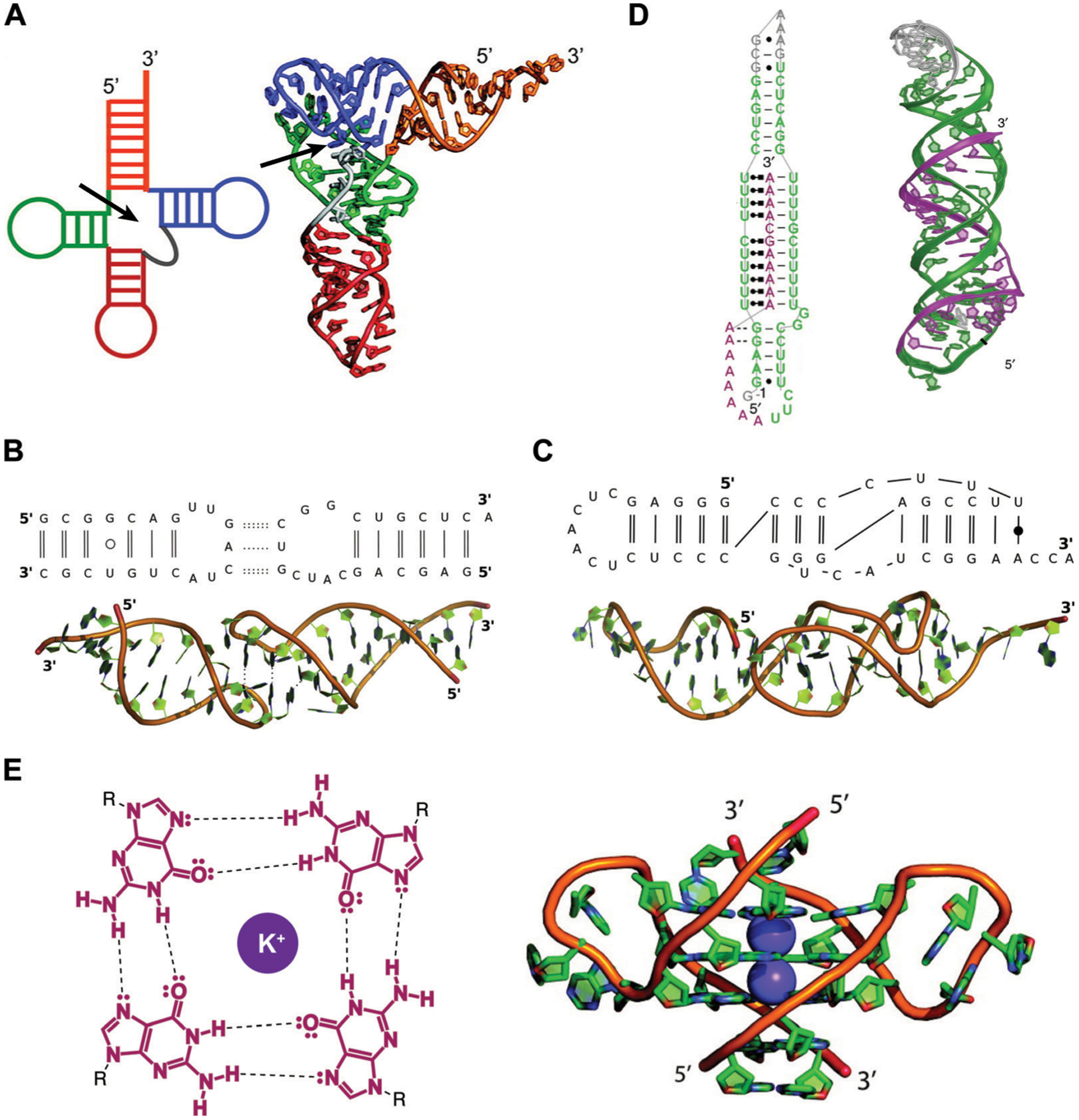
Examples of RNA tertiary structures. (A) Yeast phenylalanine t-RNA (PDB ID: 6TNA). The individual helices arising from the multi-way junction (arrow) that form the cloverleaf-shaped secondary structure (left) were found to stack on each other to determine the L-shaped three-dimensional fold (right). Adapted from ref. 16 with permission from American Chemical Society, Copyright 2011. (B) Example of a kissing loop tertiary interaction formed between substrate and catalytic domain stem-loops of the Neurospora Varkud satellite ribozyme (PDB ID: 2MIO). Dashed lines indicate hydrogen bonding. Wobble and non-WCF base pairs are shown as unfilled and filled circles, respectively. Adapted from ref. 15 with permission from Elsevier B. V., Copyright 2015. (C) Example of a pseudoknot tertiary interaction in the turnip yellow mosaic virus RNA (PDB ID: 1A60). Non-WCF base pair is shown as a filled circle. Adapted from ref. 15 with permission from Elsevier B. V., Copyright 2015. (D) 3′-Triple helix in lncRNA MALAT1 (PDB ID: 4PLX). Uridine-rich stem loop (green) form WCF (simple lines) and Hoogsteen (dot and square lines) base pairs with the A-rich tail (purple). Adapted from ref. 7 with permission from Springer Nature, Copyright 2014. (E) G-quartets are stabilized by potassium cations through interactions with the guanine’s oxygens (left), and stack on top of each other to form G-quadruplex structures (right), as seen in the human telomeric (TERRA) RNA (PDB ID: 3IBK). Dashed lines represent hydrogen bonds, dots represent lone pairs of electrons, and purple circles represent the potassium ion. Adapted from ref. 16 with permission from American Chemical Society, Copyright 2011.
RNA quaternary structure
Diversity of RNA function is often achieved by RNAs interacting with a wide variety of other biomolecules, including DNA, proteins, and other RNAs.2 Such assemblies are referred to as RNA quaternary structures. While all of these interactions can have functional importance, we will focus on RNA:protein interactions, as they represent a class of quaternary structures that are actively being investigated for small molecule regulation.
Understanding the structure of RNA:protein interactions can give further insight into RNA biology and potential targeting of these interfaces with small molecules. For example, many RNA-binding proteins (RBPs) utilize stacking between nucleobases and aromatic amino acids, and/or charge–charge interactions between cationic amino acids and the RNA backbone, to promote binding.14 Stacking and electrostatic interactions appear to be important for RNA recognition from both a protein and small molecule perspective, as discussed below.
In addition, some RBPs are known to have preferential interactions with particular RNA tertiary structures. For example, the aforementioned PRC2 complex appears to interact with a multitude of RNAs. In an effort to explain this promiscuous binding, Cech, Davidovich, and co-workers found that PRC2 preferentially recognized RNAs at G-quadruplex sites.19 Understanding the sequence and structure determinants of RNA–protein interactions will facilitate the discovery of small molecules that modulate these interactions, either by direct disruption or allosteric regulation of these complexes.
It is also important to note that RNAs can form higher order assemblies containing multiple protein and RNA subunits. For example, the spliceosome, an RNA:protein complex that catalyzes the RNA splicing reaction, has been analyzed structurally. Notable work from Yan, Shi, and co-workers20 yielded important insights into the intermolecular interactions between the various subunits of the spliceosome, including the RNA substrate, protein subunits, and small nuclear RNAs (snRNAs), a family of approximately 150 nucleotide RNAs involved in the splicing process.20 Another significant RNA–protein assembly is the ribosome, where structural insight has allowed for understanding of the molecular interactions that take place during translation. For example, the bacterial 70S ribosome, consisting of both the small and large ribosomal subunits and mRNA and tRNA subunits, was analyzed via X-ray crystallography by Ramakrishnan and co-workers.21 The authors observed extensive RNA–RNA and RNA–protein interactions, with many interactions requiring magnesium coordination and other electrostatic interfaces.21
These higher-order assemblies serve as great inspiration for successful RNA targeting in the clinic, as small molecule binders of the bacterial ribosomal RNA and splice sites represent the only FDA-approved RNA-binding drugs to date. Specifically, FDA-approved aminoglycoside antibiotics such as neomycin bind bacterial ribosomal RNA and hinder translation.22 These molecules have found extensive clinical use and have been reviewed elsewhere.22 Risdiplam, the recently approved splicing modifier for treating spinal muscular atrophy (SMA) will be explored below. Together, these advances serve as proof-of-principle for the targeting of RNA-containing complexes.
RNA dynamics
All levels of RNA structure are prone to changes that are regulated by a variety of dynamic cellular cues, such as protein binding and ionic environment. Additionally, post-transcriptional modifications, which are reversible covalent modifications of RNA nucleotides, are also known to modulate RNA structural dynamics.23,24 Advances in structure determination methods continue to provide evidence of diverse conformational landscapes found in RNA, leading to a paradigm shift of considering these molecules as containing structural “ensembles,” i.e. a collection of structures, rather than a single static structure. Importantly, distinct ensemble members are known to differentially regulate functional outcomes and can also provide unique sites for ligand binding and consequent modulation of the conformational equilibria.25
Local changes in RNA dynamics can be induced by post-transcriptional modifications and other regulatory processes and lead to major functional consequences. For example, N6-methyladenosine (m6A) is known to disfavor WCF base pairing due to steric clashes and to simultaneously enhance base-stacking in single-stranded regions, thus altering RNA structure and dynamics.23 The presence of m6A in a hairpin loop at the 5′-end of the aforementioned lncRNA MALAT1 was shown to increase dynamics, thereby enhancing heterogeneous nuclear ribonucleoprotein C (hnRNP C) binding (Fig. 3A).24 In addition, secondary structure rearrangements can affect protein binding. For example, the HIV Trans-Activation Response (TAR) element RNA can sample both ground (GS) and excited state (ES) base-pairings (Fig. 3B), with only the GS able to bind the Cyclin T1–Tat protein complex. The specific biological impact of this sampling is under investigation, but it offers an opportunity for small molecule control of viral transcription and ultimately virion production.26
Fig. 3.
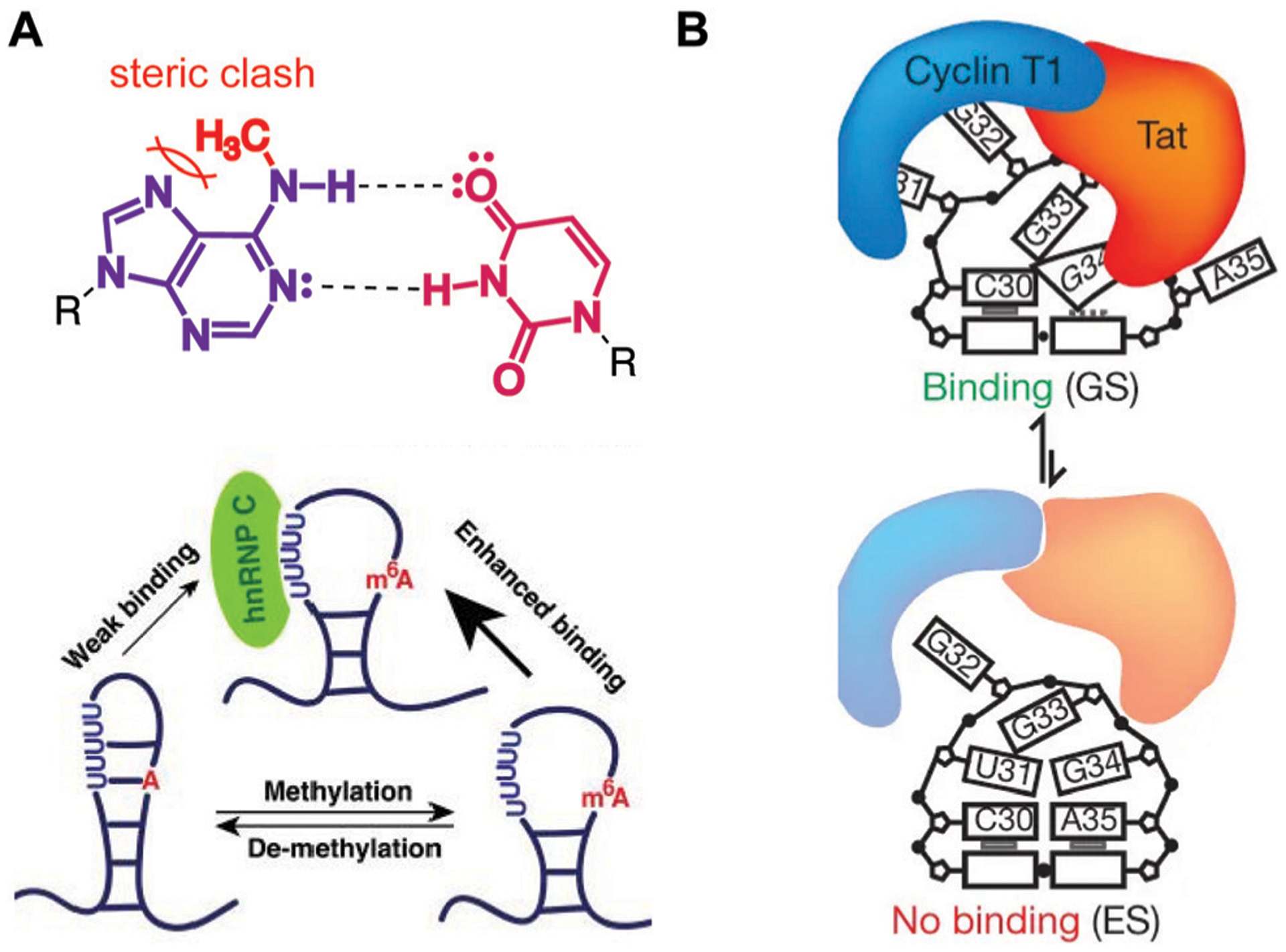
Examples of RNA dynamic regulation and associated functional consequences. (A) N6-methyladenosine (m6A) modification disfavors WCF base pairing (top), promoting single-stranded hairpin formation in MALAT1 and consequent enhanced binding of the heterogeneous nuclear ribonucleoprotein C (hnRNP C, bottom). Adapted from ref. 24 with permission from Springer Nature, Copyright 2015. (B) The ground state (GS) of HIV-1 TAR RNA is more strongly bound by the Cyclin T1-Tat protein complex than the less energetically stable excited state. Adapted from ref. 26 with permission from Springer Nature, Copyright 2012.
Collectively, the folding landscapes and dynamic nature of RNA demonstrate its unique structural architecture and properties compared to other biomolecules. While more similar to DNA than proteins in its building blocks, the dynamic nature of RNA enables this biomolecule to access a broader range of conformations compared to DNA.27 Furthermore, the ensemble nature of a single RNA molecule distinguishes it from proteins, which often form one major thermodynamically stable conformation.28 Indeed, the RNA folding landscape is now widely considered to be more “rugged” than that of proteins, as multiple energetically favorable conformations can exist in equilibrium with each other.28 It is therefore possible that the conformational diversity of RNA molecules compensates for its limited chemical diversity in terms of the four nucleotide building blocks, compared to protein’s twenty amino acids, and that this diversity provides opportunities for unique regulation of this biomolecule by small molecule ligands.
Section 3: principles and methods for studying RNA–small molecule recognition
A unique property of small molecules compared to oligonucleotides is their ability to specifically recognize binding pockets in RNA and thereby modulate a transcript’s function.3 Structural and biophysical characterization of these binding pockets has provided insights into the major driving forces that enable recognition. The following section will highlight several noncovalent interactions important in small molecule–RNA recognition as well as discuss common and emerging methods utilized to detect and characterize binding events.
Fundamental recognition principles and noncovalent interactions
With the increase in RNA–small molecule structure characterization efforts, there are opportunities to analyze these data sets and identify general trends in recognition of RNA by small molecules. The identification of such trends is expected to expedite the discovery of general guidelines for selective RNA targeting as well as clarify differences in recognition of RNA versus proteins (the “traditional” drug targets) by small molecules.
A recent analysis of all RNA–small molecule complexes and comparison to protein–small molecule complexes in the Protein Data Bank (PDB, a depository of solved biomolecular structures) by the Hargrove group, for example, found differences in the types of interactions that are enriched in small molecule recognition with each biomacromolecule.29 For example, the major contributors for RNA recognition by small molecules are stacking and hydrogen-bonding interactions. Stacking interactions are enabled by small molecules with aromatic surfaces. For example, ligands containing extended aromatic ring systems are known to intercalate, i.e. insert themselves between stacked base pairs and strengthen the existing π-interactions. A notable example from this analysis is an acridine-based ligand discovered to stabilize a G-quadruplex structure found in a lncRNA Telomeric Repeat-Containing RNA (TERRA, Fig. 4A).30 Additionally, specific hydrogen bonding can occur between small molecules and nucleotides. In another example from this analysis, the aminoglycoside paromomycin uses its hydroxyl and ether groups to hydrogen bond with an adenine residue in the HIV-1 dimerization initiation site, another important RNA regulatory element (Fig. 4B).31
Fig. 4.
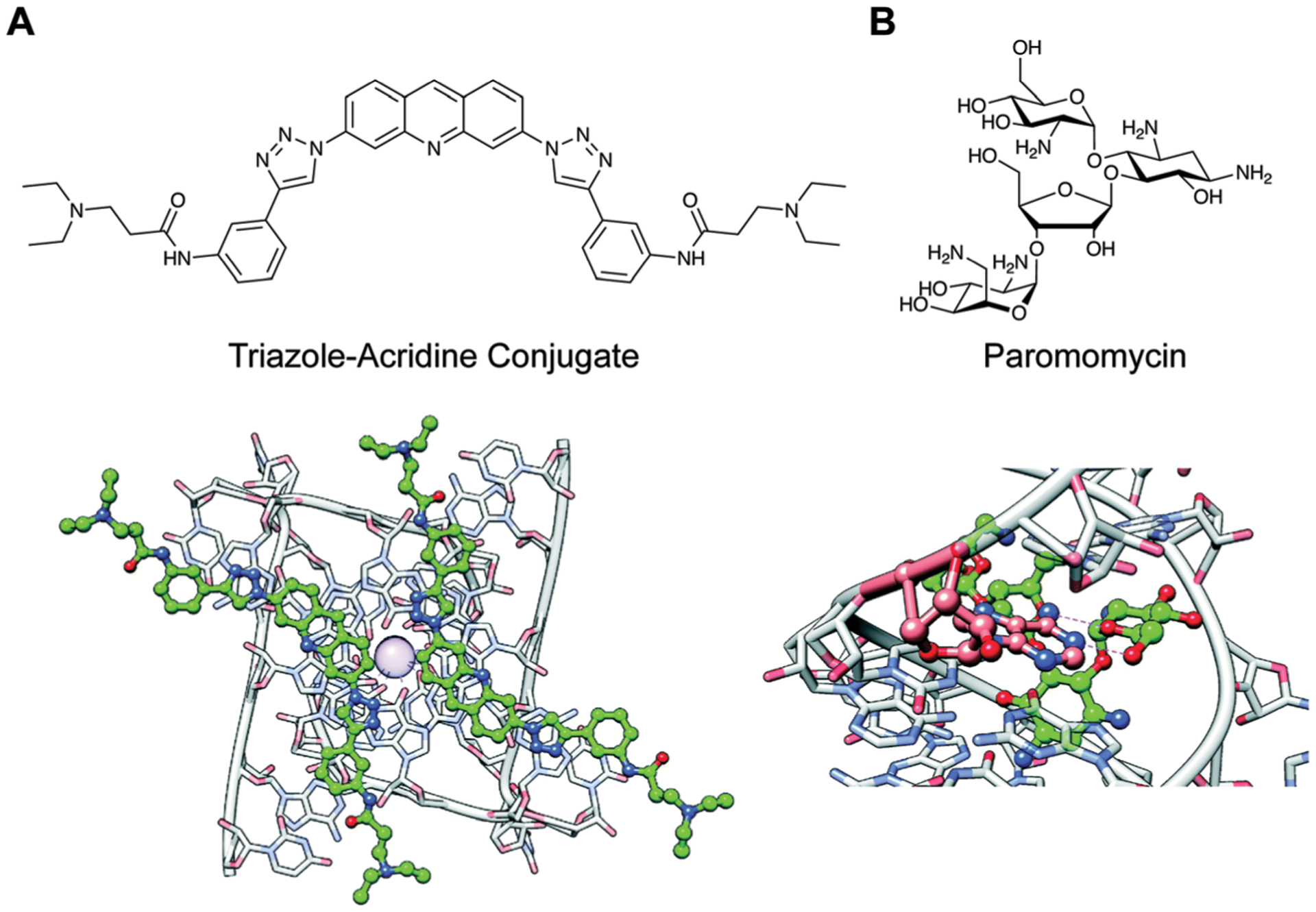
Examples of common noncovalent interactions in RNA–small molecule recognition. Adapted from ref. 29 with permission from The Royal Society of Chemistry, Copyright 2020. (A) Binding of two acridine-based ligands to the Telomeric Repeat-Containing RNA (TERRA) via stacking interactions (PDB ID: 3MIJ). Ball denotes a K+ ion. (B) Hydrogen bonding interactions (dashed lines) between paromomycin and HIV-1 Dimerization Initiation Site (DIS) (PDB ID: 3C44).
In contrast to proteins, interactions were found to be less driven by the hydrophobic effect in RNA, though these effects still play a critical role.29 Complementing this finding is a recent study by Schneekloth and co-workers, in which ligand-bound binding pockets in RNA and proteins from the PDB were compared.32 While these binding pockets were found to have similar volume and buriedness (lack of solvent exposure) parameters, binding pockets in RNA were measured to be much less hydrophobic, consistent with the more polar nature of this biomolecule and suggesting a potential opportunity for specific RNA targeting.32 It is worth noting that the knowledge gained from these studies is currently limited to RNA and proteins that are amenable to structural characterization, and that these principles may be different for more dynamic and disordered recognition regions in both biomolecules. Nevertheless, the identification of differences in both interaction types and hydrophobicity of the binding pockets between these targets has led some to hypothesize, as explored below, that there may be privileged chemical properties of RNA-binding small molecules. As such, this research paves the way towards exploiting distinct RNA-binding properties to rationally design selective ligands for these targets.
Biophysical methods and considerations to detect RNA–small molecule interactions
To detect RNA–small molecule binding events, several in vitro screening methods can be used. This section will review commonly used methods as well as considerations for future studies, including expanding the repertoire of binding techniques and inclusion of functional assays.
Fluorescence-based methods.
Most current methods for identifying small molecule RNA binders rely on the sensitive measure of fluorescence. For example, relative binding affinities can be measured by small molecule titrations with fluorescently-labeled RNA constructs,33 fluorescence indicator displacement (FID) assays,34 or by monitoring changes in intrinsically fluorescent ligands upon RNA titration.8 In FID assays, binding is measured by a small molecule of interest “displacing” an RNA-binding indicator molecule, which causes a fluorescence change in the indicator (Fig. 5A).34 To detect binding events in a high-throughput fashion, fluorescently-labelled RNA constructs can be incubated on small molecule microarrays (SMMs), as demonstrated by Schneekloth and co-workers (Fig. 5B).33 In these assays, small molecules are spotted on a surface via covalent linkages, and the fluorescent RNA is retained on a spot if it is interacting with the molecule.
Fig. 5.
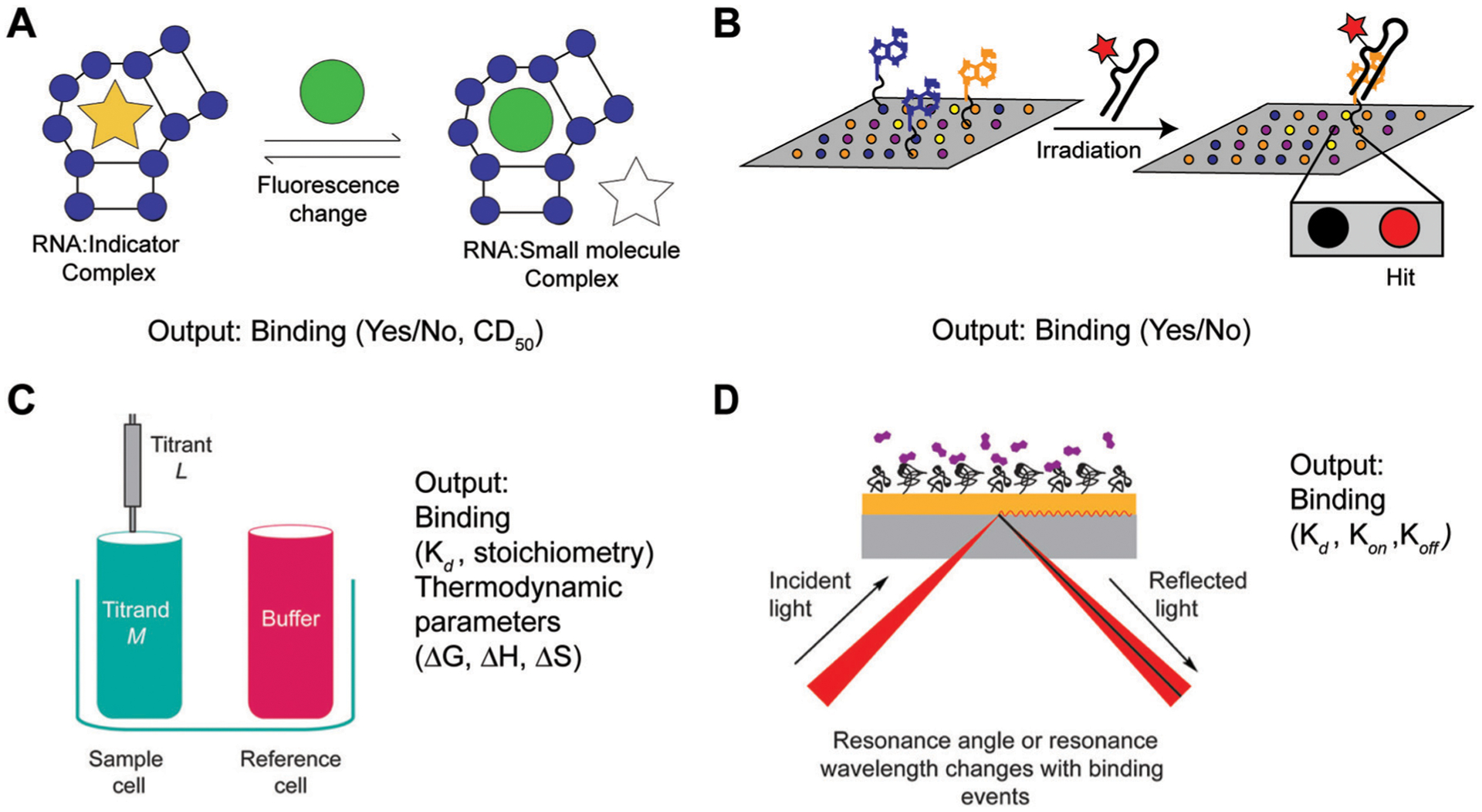
Common screening methods utilized for detecting RNA–small molecule binding. (A) Fluorescence Indicator Displacement (FID) assays detect changes in fluorescence intensity of an indicator as a result of small molecule binding. This method enables detection of binding events if a single small molecule concentration is used, and relative affinities or Competitive Displacement (CD50) values if multiple concentrations are used. Adapted from ref. 34 with permission from Elsevier B.V., Copyright 2019. (B) Small Molecule Microarrays (SMM) enable detection of binding as a result of detecting fluorescent wells upon irradiating a microarray plate. The plate contains immobilized small molecule ligands and is incubated with a fluorescently labeled RNA construct prior to irradiation. (C) Isothermal Titration Calorimetry (ITC) functions through directly measuring changes in heat as a result of small molecule (titrant) binding to the RNA construct (sample cell) relative to buffer (reference cell). As such, this method enables determination of binding affinity (Kd), stoichiometry, as well as Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (ΔS) values. Adapted from ref. 35 with permission from American Chemical Society, Copyright 2019. (D) Surface Plasmon Resonance (SPR) directly measures changes in resonance angle or wavelength of a glass surface containing the immobilized RNA of interest upon addition of a small molecule. This method can be utilized to obtain both the association (kon) and dissociation (koff) constants that comprise binding affinities (Kd), providing insights into the kinetics of a binding event. Adapted from ref. 35 with permission from American Chemical Society, Copyright 2019.
While useful approaches for identifying de novo RNA–small molecule interactions, these methods suffer from several disadvantages that call for careful consideration. FID assays can be biased in detecting ligands with similar binding modes and affinity ranges as the indicator used. Similarly, methods that rely on fluorescently labeled RNA or intrinsically fluorescent ligands may fail to detect binding events that do not result in fluorescence changes, and the reliance of SMMs on ligand immobilization may not accurately represent ligand behavior in solution. Moreover, in in systems that aim to detect disruption of high-affinity binding events, such as inhibition of RNA–protein interactions, fluorescence-based measurements may need to be replaced with more sensitive measures of radiolabeling. Finally, all of these methods can benefit from follow-up studies using other methods.
To gain a deeper understanding of fundamental principles of RNA–small molecule recognition, detailed biophysical studies of such interactions are needed.35 Evaluating thermodynamic and kinetic parameters of these interactions would provide important insights into differential small molecule-based regulation of RNA structural landscapes, and such parameters cannot be directly measured using the aforementioned techniques.
Isothermal titration calorimetry (ITC).
For thermodynamic insights, ITC is most commonly used.35 This method provides a direct measurement of a binding constant (Kd) by reporting the heat change from small molecule addition to the RNA solution, and the resulting curve can be used to calculate all thermodynamic parameters (Fig. 5C). As such, this information can be used to determine enthalpic and entropic contributions in small molecule–RNA binding events.
Surface plasmon resonance (SPR).
For kinetic insights, SPR experiments are typically conducted.35 By immobilizing the RNA on a glass surface and measuring changes in the refraction angle upon small molecule binding, this method outputs kon and koff parameters (Fig. 5D). Understanding if high affinity binding events are driven by fast on-rates or slow off-rates for small molecule–RNA systems, and how these differ for different small molecule and/or RNA classes, will be of significance.
Ideally, binding assays should be complemented with RNA-focused functional assays to fully characterize RNA–small molecule interactions as well as to establish correlations between binding strengths and functional outcomes, which is of rising interest in the field.3 This consideration is also of particular importance given the increase in phenotypic screening campaigns that result in the identification of RNA as targets, as seen recently in studies by Novartis,36 Roche,37 and others. Together, such combined studies of biophysical and functional methods can increase our understanding of requirements for bioactivity of RNA-binding ligands and accelerate the path towards therapeutic drug discovery for these targets.
Structural approaches for analyzing RNA–small molecule interactions
Along with understanding RNA–small molecule interactions through the binding and biophysical assays discussed above, it is also important to gain structural insight into such interactions. Compared to over 100 000 crystal structures and over 10 000 NMR structures for proteins, there are only over 800 crystal structures and over 500 NMR structures of RNAs currently deposited in the PDB. The apparent paucity of known RNA structures is partly the result of several challenges associated with RNA structure determination, including its high negative charge and dynamic nature. Methods for determining RNA structure can be broadly characterized as solid-state or solution-state. This section will focus on four major methods: X-ray crystallography, cryo-electron microscopy (cryo-EM), nuclear magnetic resonance (NMR) spectroscopy, and chemical probing.
Solid-state methods
X-Ray crystallography.
In X-ray crystallography, crystals containing a molecule of interest are analyzed with X-rays, yielding a diffraction pattern that serves as an electron-density map through which the structure of the biomolecule can be modelled. X-Ray crystallography has been a major workhorse for biomolecular structure analysis, with one of the first examples of RNA crystallography in the aforementioned tRNA crystal structure (Fig. 2A).17 Since this work, X-ray crystallography has given crucial insight into RNA apo structures, RNA–RNA interactions, and RNA–protein complexes, most notably the ribosome.21 The aforementioned TERRA lncRNA was crystalized with the acridine mimic shown in Fig. 2E. We will also see the example of Ribocil in the Greatest Hits section.
X-ray derived structures based on crystals generally capture only one conformation of the RNA. Thus, other conformations sampled by the RNA can be missed in these experiments. In addition, as a solid state method, a crystal structure may not be representative of a biologically-relevant structure, as crystal packing may induce non-native conformational changes.38 Finally, the high dynamics and negative charge on RNA can hinder crystal formation, meaning that crystallography is not attainable for many RNAs, in particular large RNAs.
Cryo-EM.
Cryo-EM refers to the rapid incubation of a sample at cryogenic temperatures with subsequent electron microscopy analysis. This rapid cooling results in the formation of vitreous ice, where the water molecules in the solvent do not have time to form a crystal lattice. As a result, the molecule of interest is protected from damage from freezing while maintained in a water-containing environment, and the cold temperatures minimize damage from electron radiation.38 Cryo-EM has allowed for significant understanding of both the ribosome38 and spliceosome.20 Notably, through single-molecule analysis of the electron micrograph, multiple structural “states” can be observed. These states can be used to generate structural ensembles to gain further insight into RNA dynamics, which is not achievable with X-Ray crystallography.38
It is anticipated that further advancements in cryo-EM will facilitate analysis of RNA:ligand complexes even at lower molecular weights. Again, as a solid-state method, it is possible that the solved structures may not necessarily represent the biologically-relevant structures, although circumventing crystal packing may make such artifacts less likely than in crystallography.38
Solution-state methods
NMR spectroscopy.
NMR spectroscopy utilizes magnetic fields to analyze a molecule of interest, wherein individual atoms from the molecule produce chemical shift values related to their identity and surrounding environment. As this method is solution-state, RNA is more likely to assume a biologically-relevant conformation, although there are limitations due to buffer conditions that can be used in NMR. In addition, chemical shift overlap can occur due to the similar chemical functionality of the nucleobases. Thus, NMR analysis is typically limited to shorter RNAs (optimally 50 or fewer nucleotides), unless site-specific radiolabeling is accomplished.39 Notably, Telesnitsky, Summers, and co-workers solved an NMR structure for HIV-1 5′-UTR (155 nucleotides), the longest RNA-only complex analyzed by NMR to date, utilizing site-specific labeling methodologies.40 NMR analysis can also allow for identification of structural ensembles, as was identified by Al-Hashimi and co-workers in a virtual small molecule screen against an RNA element in HIV-1, discussed below.41 Apart from structural information, NMR can also yield information on ligand binding. For example, in water–Ligand Observed via Gradient SpectroscopY (waterLOGSY) magnetization is transferred from the water to the ligand in the presence and absence of an RNA target via through-space interactions called an intermolecular Nuclear Overhauser Effect (NOE).39 Due to the size difference between the RNA–ligand and ligand-only complex, binding can be assessed. We will see another example in the Greatest Hits section where NMR was used to solve the structure of an RNA–small molecule interaction in splicing regulation.
Chemical probing.
Chemical probing refers to incubating RNA with a reactive chemical that forms adducts on nucleobase or sugar groups in a manner that depends on base-pairing or flexibility. There are multiple probing reagents that modify the RNA at distinct positions. For example, SHAPE reagents (selective 2′-hydroxyl acylation analyzed by primer extension) modify the 2′-hydroxyl of the ribose sugar of conformationally flexible nucleotides, generally corresponding to single-stranded regions (Fig. 6A, top).42 Another reagent, dimethylsulfate (DMS) modifies A and C nucleobases at the WCF face, giving insight into base pairing (Fig. 6A, bottom).42 After chemical probing, the RNA is reverse transcribed into copy DNA (cDNA). Modified sites result in either the truncation in the cDNA, where the reverse transcriptase halts, or a mutation that can be detected by deep sequencing in the more recently developed mutational profiling (MaP) approach (Fig. 6B).42 However, it is important to note that reactivity data is used as a restraint in an RNA folding algorithm. Thus, the accuracy of a determined structure is dependent on the validity of the folding algorithm.
Fig. 6.
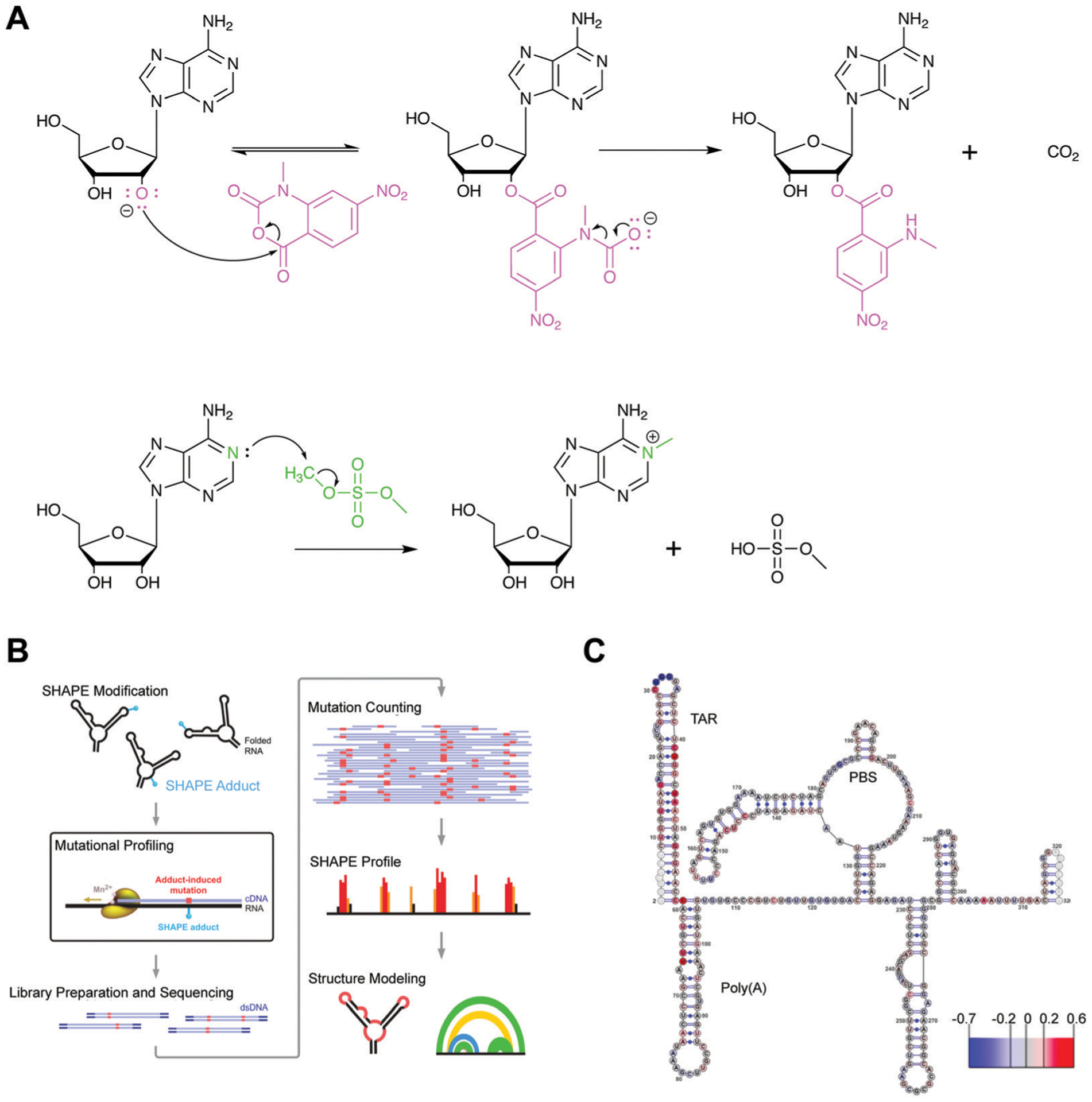
Chemical probing for interrogation of RNA structure. (A) Mechanism for covalent modification by SHAPE reagent 1-methyl-7-nitroisatoic anhydride (1M7, top) and dimethylsulfate (DMS, bottom). In SHAPE probing, deprotonation of the 2′-hydroxyl by a general base facilitates its nucleophilic attack of the anhydride moiety, resulting in the ring opening of 1M7. While this attack is an equilibrium process, the subsequent decarboxylation, generating the final adduct as depicted, is irreversible. In DMS probing, the nitrogen lone pair on A (depicted) or C attack the electrophilic methoxy position, resulting in irreversible covalent modification of the base. (B) Workflow for integration of chemical probing information into a structure model. Reagent modifications are incorporated as mutations during reverse transcription in the presence of manganese, and mutations and detected via sequencing. Reactivities for each nucleotide are incorporated as restraints in a structure model. Adapted from ref. 42 with permission from Springer Nature, Copyright 2014. (C) Chemical probing with HIV TAR following small molecule treatment reveals changes in reactivity for small molecule binding site. Larger scale changes in secondary structure were not observed. Scale shows changes in reactivity between control and small molecule treatment. PBS: primer binding site. Adapted from ref. 33 with permission American Chemical Society, Copyright 2014.
Schneekloth and co-workers33 performed chemical probing in the presence of a small molecule identified utilizing the aforementioned SMM approach against HIV-1 TAR. Probing was carried out with the entire 5′-untranslated region (UTR) of HIV-1, which contains the TAR RNA to validate that the small molecule binding site is maintained even in the presence of a larger RNA construct. The authors noticed that the most significant changes in nucleotide reactivity were observed near the TAR structure, allowing for validation that this site is indeed the small molecule binding site (Fig. 6C).33 Thus, chemical probing represents a potential approach for validating small molecule binding sites and selectivity particularly on large RNAs.
Section 4: challenges and emerging solutions
While the established and emerging methods for studying RNA–small molecule interactions are paving the way towards understanding factors contributing to selective recognition and ultimately clinical advances, many unanswered questions remain in this flourishing field. Both the RNA and small molecule perspectives will be discussed in the following sections.
RNA perspective
Identification of suitable RNA targets.
From the RNA perspective, one major challenge is identifying RNA molecules that are amenable for selective small molecule targeting. Contributing to this challenge is the relative lack of 3D structures for RNA, as highlighted in the previous section. At the same time, a small molecule must identify an RNA of interest in the cell where a large majority (>90%) of RNA consists of rRNA and tRNA. As drug-development efforts extend to capitalizing on RNA’s potential for therapeutic targeting with small molecules, several criteria can be considered when evaluating the suitability of a given RNA.3 One criterion is RNA abundance, as a higher amount of the RNA target in diseased tissue is likely to decrease off-target effects, especially considering that RNAs can have tissue- or disease-specific expression. Additionally, Weeks and co-workers proposed that ideal small molecule recognition sites on RNA would consist of a binding pocket analogous to protein targeting and also have high “information content,” which is defined as structural complexity that makes that particular site unique (e.g. 1 in a 1 000 000 000 from other RNA structures).1
Leveraging RNA conformational landscapes.
RNA dynamics complicates our understanding of small molecule recognition. Dynamics presents challenges for structure-based drug design, as RNA cannot be described as a static structure and a small molecule ligand may only bind one of multiple ensemble members. An emerging strategy to exploit these dynamics is identifying and trapping inactive or non-functional RNA conformations with small molecules. In this case, small molecule-based regulation of RNA function occurs by altering its conformational landscape, as already demonstrated in nature with riboswitch-metabolite binding. Importantly, this strategy is shifting the view that RNA dynamics is an obstacle for selective targeting to leveraging this property towards specific recognition.
In one example, Al-Hashimi and co-workers combined NMR studies and computational simulations to generate an ensemble of representative TAR conformations.41 Small molecules were computationally docked to this set of structures, rather than just one structure, to more fully account for the role of RNA dynamics in small molecule binding. This study uncovered a novel compound, dimethylamiloride (DMA), that showed moderate affinity for TAR.41 In follow-up collaborative studies, the Al-Hashimi and Hargrove groups ultimately found that DMA-169 can stabilize the aforementioned ES conformation of TAR RNA (Fig. 7A), as can argininamide (Fig. 7B). Interestingly, other TAR-binding ligands, such as neomycin (Fig. 7B) do not stabilize this conformation.43 Since the ES conformation is poorly recognized by the Tat protein complex,26 these results provide evidence that a strategy of modulating structural ensembles with synthetic ligands may indeed be used to inhibit RNA function.
Fig. 7.
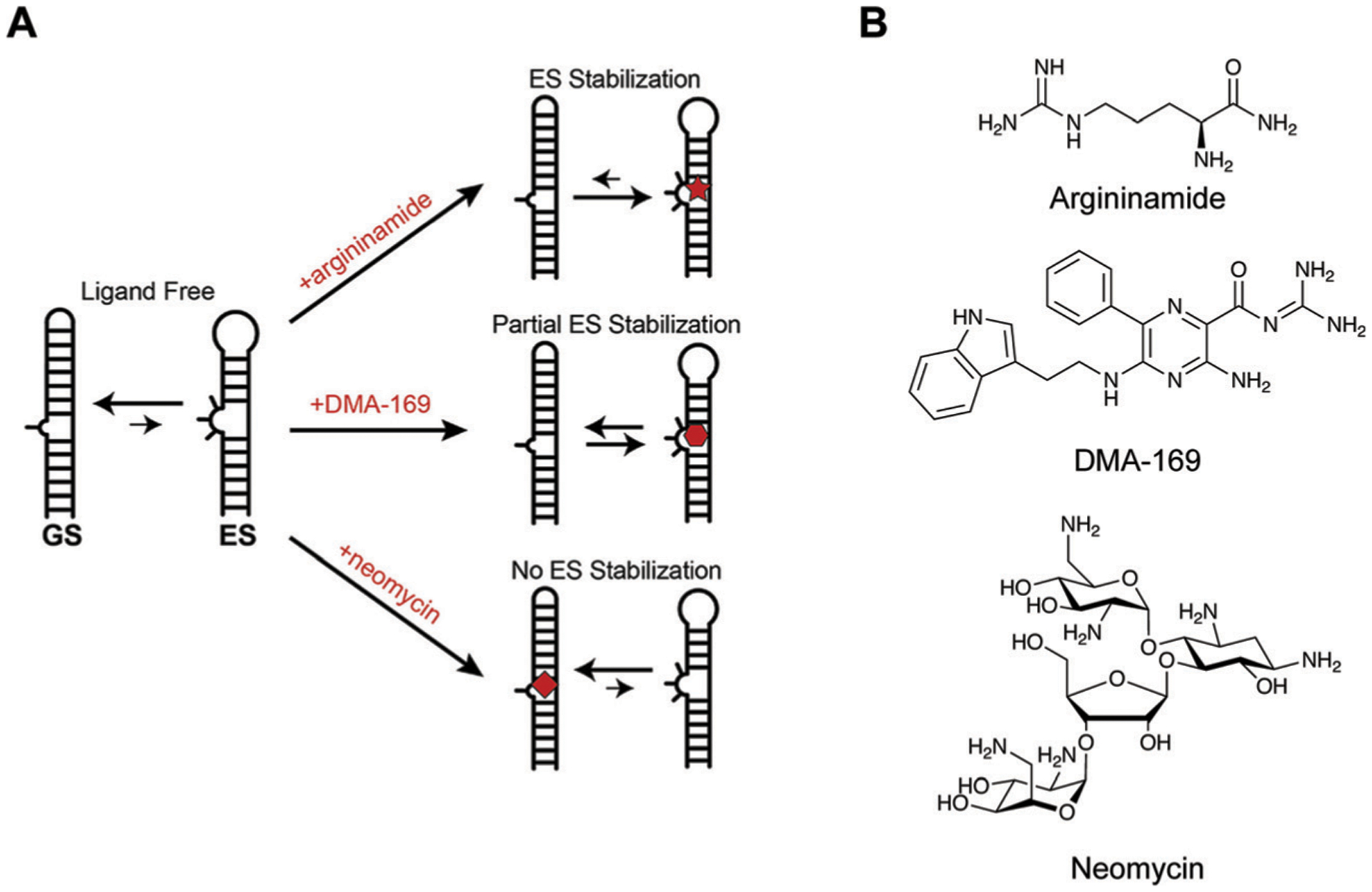
Structural dynamics of HIV-1 TAR are modulated with small molecules. (A) TAR oscillates between a ground state (GS) and excited state (ES) characterized by base flipping and angular rearrangement of the helix. Several ligands, including argininamide and DMA-169, were shown to alter excited state accessibility, whereas other ligands such as neomycin were not. Adapted from ref. 43 with permission from Elsevier, Copyright 2020. (B) The structures of argininamide, DMA-169, and neomycin.
Small molecule perspective
From the small molecule perspective, many have suggested that we are hindered by a limited understanding of the general driving principles behind preferential and selective RNA recognition. Several unanswered questions remain. For example: what is the interplay between affinity and selectivity necessary for favorable binding and functionally-relevant events? What are the physical and chemical properties of molecules that lead to such events?
Insights into this area would aid in busting the myth of RNA being perceived as ‘undruggable’, a notion that largely emerged as a result of repeated identification of promiscuous chemical scaffolds and low hit rates in high-throughput screening campaigns targeted against RNA. One hypothesis is that these initial failures may be the result of using small molecule screening libraries that are enriched in protein-binding chemical scaffolds, as they represent the traditional target for small molecule ligands. As discussed in previous sections, the forces and noncovalent interactions that drive small molecule recognition of RNA and proteins are different, providing support for this hypothesis.
Exploration of RNA-privileged chemical space.
To address the possibility that screening libraries are biased towards proteins, an understanding and validation of an RNA privileged “chemical space” is necessary. In cheminformatics research, chemical space is a concept referring to a multi-dimensional space defined by properties analyzed for a given set of molecules. Molecules that adhere to similar properties will be located in the same region of that space, which enables comparisons of small molecule groups. By quantifying and comparing twenty physicochemical properties of RNA- and protein-binding molecules, as represented by the curated RNA-Targeted BIoactive ligaNd Database (R-BIND) and FDA libraries, respectively, the Hargrove group indeed found that RNA-binding small molecules have different properties from those of protein-binding small molecules (Fig. 8A).44 Similar distinguishing properties for hit RNA-binding ligands from a large Astra Zeneca library were later identified by Disney and co-workers.45 While similar in properties, the hit ligands contained previously unexplored chemical matter in RNA targeting, supporting the notion of chemical space analyses as a tool to identify novel RNA-privileged chemotypes.
Fig. 8.
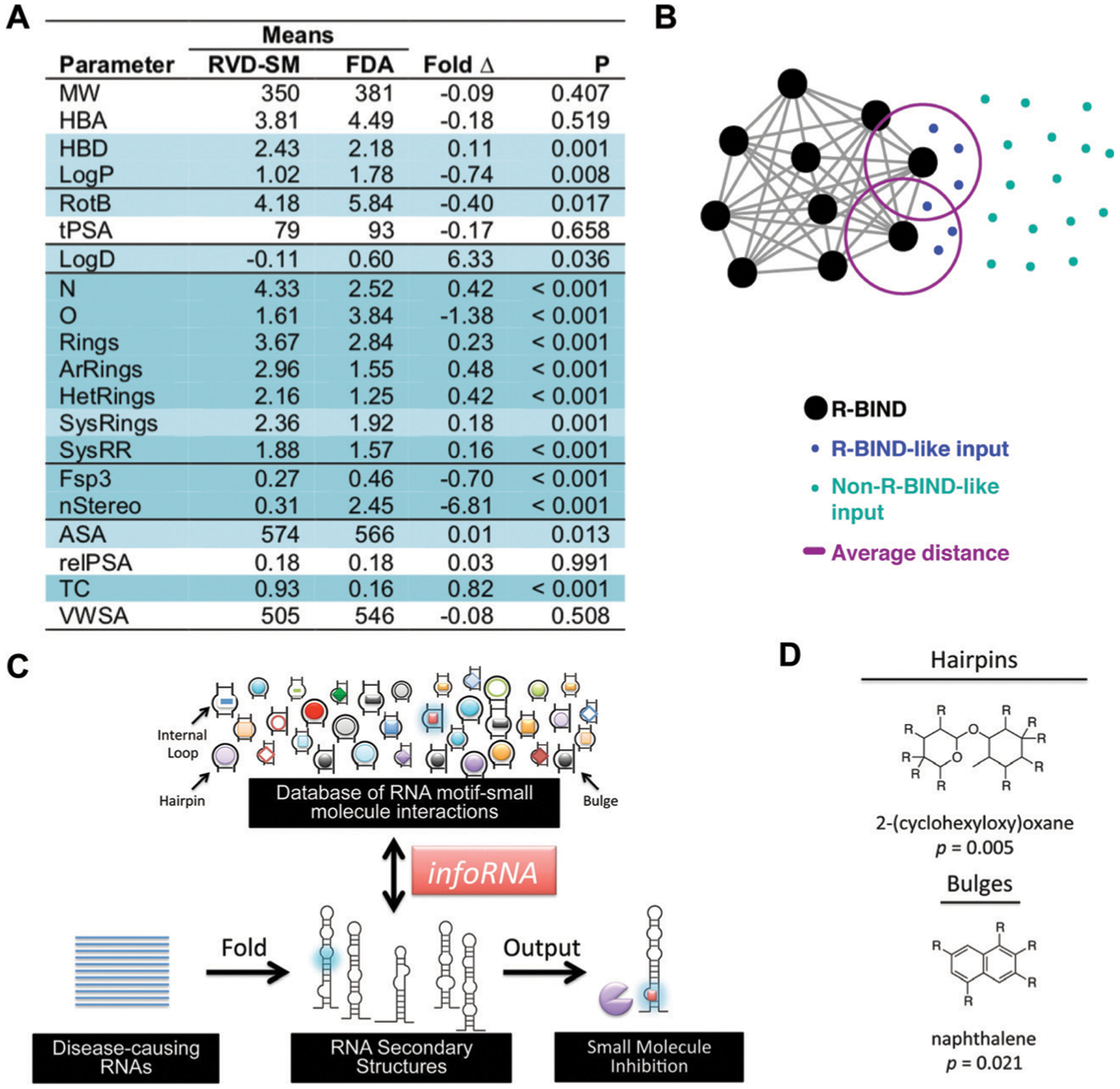
Approaches towards discovering privileged properties and chemotypes of RNA-binding small molecules. (A) Comparison of 20 physicochemical properties of bioactive, RNA-binding small molecules (R-BIND) and FDA ligands (mostly protein-targeted). R-BIND members were found to have increased nitrogen atom count, decreased number of sp3-hybridized carbons, among other properties. Statistically significant differences determined by Mann Whitney U test are indicated by P values of <0.05 in light blue and highly significant differences as <0.001 in dark blue. MW = molecular weight, HBA = hydrogen bond acceptors, HBD = hydrogen bond donors, log P = n-octanol/water partition coefficient, RotB = number of rotatable bonds, tPSA = topological polar surface area, log D = n-octanol/water partition coefficient, N = number of nitrogen atoms, O = number of oxygen atoms, rings = number of rings, ArRings = number of aromatic rings, HetRings = number of heteroatom-containing rings, SysRings = number of ring systems, SysRR = ring complexity, Fsp3 = fraction of sp3-hybridized carbons, nStereo = number of stereocenters, ASA = accessible surface area, RelPSA = relative polar surface area, TC = total charge, VWSA = van der Waals surface area. Adapted from ref. 44 with permission from John Wiley & Sons, Copyright 2017. (B) Nearest Neighbor algorithm utilized to select R-BIND-like molecules as developed by Hargrove and co-workers. Each cheminformatic parameter from (A) is defined as a “dimension”, quantified along its dimension, and the Euclidian distance (smallest distance in space) between each library member is measured and averaged (purple line). The user’s input molecule is considered “R-BIND-like” (turquoise dots) or enriched in RNA-binding properties if it is found to be within the measured average distance to an R-BIND member (blue dots). Adapted from ref. 47 with permission from American Chemical Society, Copyright 2019. (C) Summary of infoRNA platform utilized to select potential binders of an RNA target. By inputting an RNA sequence of interest, infoRNA is mined for similar RNA motifs to output lead small molecules that can be tested and optimized for specific targeting. Adapted from ref. 48 with permission from American Chemical Society, Copyright 2016. (D) Examples of privileged chemotypes for binding of RNA hairpin or bulge secondary structures as discovered by infoRNA mining. Adapted from ref. 48 with permission from American Chemical Society, Copyright 2016.
The finding of a putative “RNA-privileged” chemical space further corroborates the view of differential driving forces for RNA recognition by small molecules and offers the possibility of enriching screening libraries with the newly discovered properties to increase hit rates. Indeed, recent work by Merck demonstrated that building an RNA-focused set of small molecules upon identifying the physicochemical properties that prevent protein binding ultimately increased propensity towards binding an array of RNA targets.46 To make the curation of RNA-privileged screening libraries accessible to the broader scientific community, Hargrove and co-workers recently reported a website interface with a “Nearest Neighbor” algorithm, which identifies how similar a molecule of interest is to R-BIND database members (Fig. 8B).47 Ultimately, these and similar approaches are expected to provide tools for scientists for efficient discovery of novel RNA-binding small molecules.
Towards specific sequence and motif targeting.
Another area of interest has been in identifying properties of small molecules that enable preferential recognition of a specific RNA sequence or structural motif over another. Towards this goal, Disney and co-workers conducted 2-dimensional combinatorial screening (2DCS) by testing a range of secondary structure motifs for binding to a large set of small molecules5 and then curated a database of these interactions called infoRNA to serve as a discovery platform (Fig. 8C).48 Analyses of the database have also led to the identification of privileged chemotypes for targeting specific secondary structure motifs (Fig. 8D). In another example, the aforementioned PDB analysis of RNA–small molecule complexes found an enrichment of cation–π and stacking interactions in purine-rich sites, offering opportunities for rational design.29 Finally, advances in machine learning technologies can enable additional insights. Both the recent finding of enriched non-canonical base pairing interactions in selective small molecule–RNA recognition events,49 as well as the application of pattern recognition methodologies to characterize differential small molecule-based recognition of RNA structures,50 may be exploited for rational design in the future.
Section 5: selection of “Greatest Hits” to date
In this section, we will cover the recent advances and promising biological examples of small molecule regulation of RNA. We will first explore molecules that have been validated in cellular systems before moving to molecules with evaluation in animal models. We will conclude this section with a recently FDA-approved drug, showcasing the promise and clinical utility of RNA-targeting small molecules.
MALAT1/RNA triple helices
As discussed above, the MALAT1 lncRNA is overexpressed in multiple cancers and drives cancer proliferation. Notably, a triple helix structure appears to facilitate its cellular abundance and overexpression in cancer and other diseases, thereby posing this element as an attractive target for small molecules.7 The Hargrove group generated derivatives of a known nucleic acid scaffold called furamidine and identified DPF (diphenylfuran) p8 (Fig. 9A) as the first high affinity and selective molecule for the triple helix.8 Since this work, Baird, Le Grice, and co-workers identified two MALAT1 triple helix binders, SM5 (Fig. 9B) and SM16, through the aforementioned SMM platform. These molecules were shown to modulate triple helix dynamics in vitro as well as reduce MALAT1 transcript abundance and branching in breast cancer organoids (Fig. 9C).9 More recent work by the Hargrove group found that DPFp8 derivatives were triple helix stabilizers and can prevent in vitro exonucleolytic degradation, making these molecules complementary to SM5/SM16 ligands.51 Together, these probes have strong potential for future mechanistic studies of the triple helix structure, as well as spatiotemporal regulation of MALAT1 transcript abundance.51
Fig. 9.
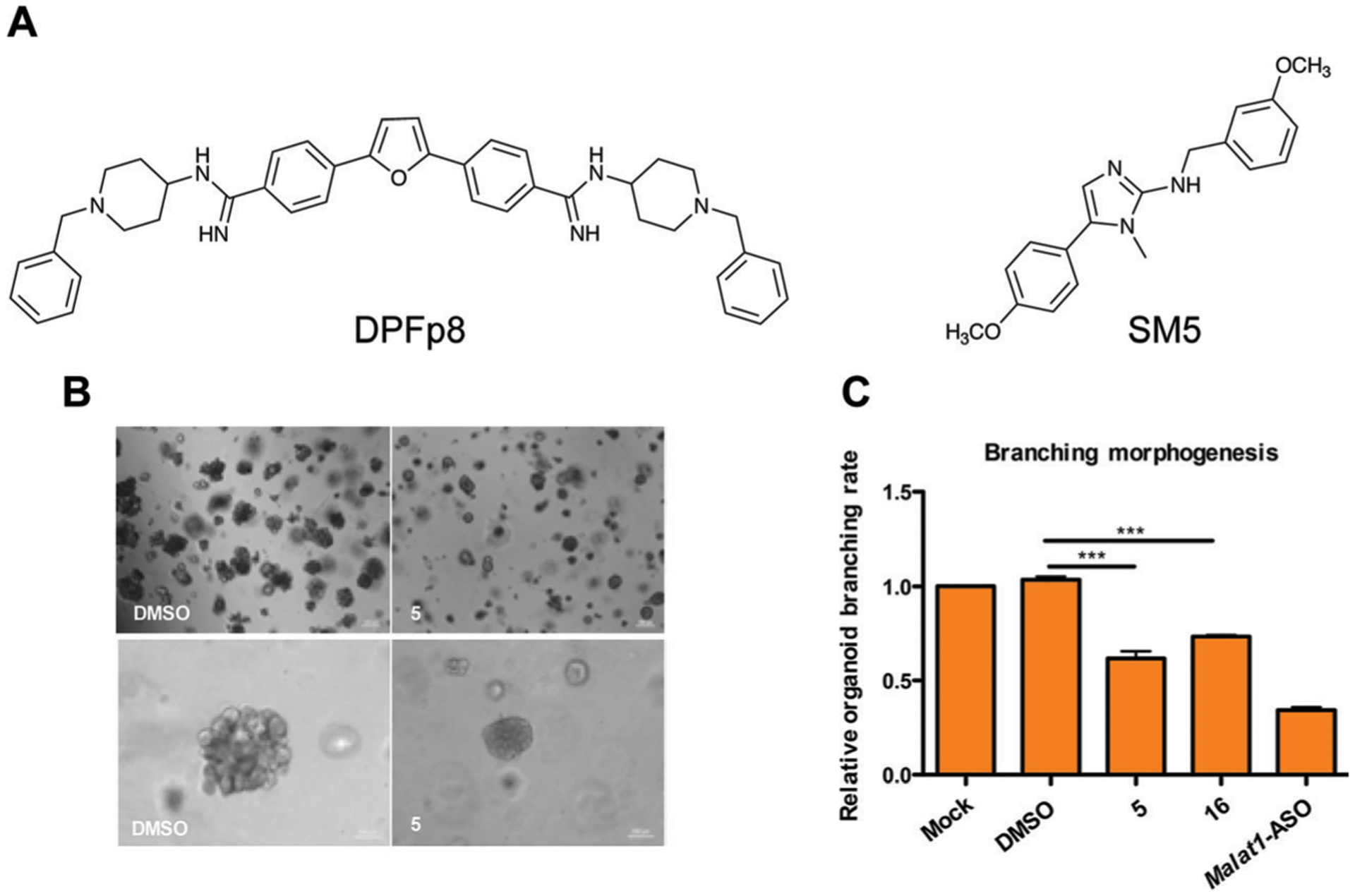
Small molecule targeting of the MALAT1 triple helix. (A) Structures of DPFp8 and SM5, which selectively bind the MALAT1 triple helix over controls. (B) Treatment of breast cancer organoids with SM5 significantly reduces organoid size (upper panel and lower panel differ by magnification). Adapted from ref. 9 with permission from American Chemical Society, Copyright 2019. (C) SM5 and SM16 (not shown) significantly reduce branching morphogenesis of breast cancer organoids. Adapted from ref. 9 with permission from American Chemical Society, Copyright 2019. ASO: antisense oligonucleotide.
RNA repeat expansions
Repeat expansions are promising drug targets as they have a large number of potential identical binding sites for a small molecule. In a notable example, Disney and co-workers10 targeted a CUG expansions in myotonic dystrophy type 1 (DM1). This condition results from hundreds to thousands of CUG expansions in the dystrophia myotonica protein kinase (DMPK) 3′-UTR, whereas a healthy individual typically has less than 30 expansions. These repeats sequester the Muscleblind-like 1 (MBNL1) splicing regulator, which in turn causes mRNA splicing abnormalities.10 Using the aforementioned infoRNA database, the authors generated a multivalent molecule, 2H-K4NMeS (Fig. 10A), which rescued splicing defects (Fig. 10B) and reduced the number of CUG:MBNL1 foci within the cell (Fig. 10C).10
Fig. 10.
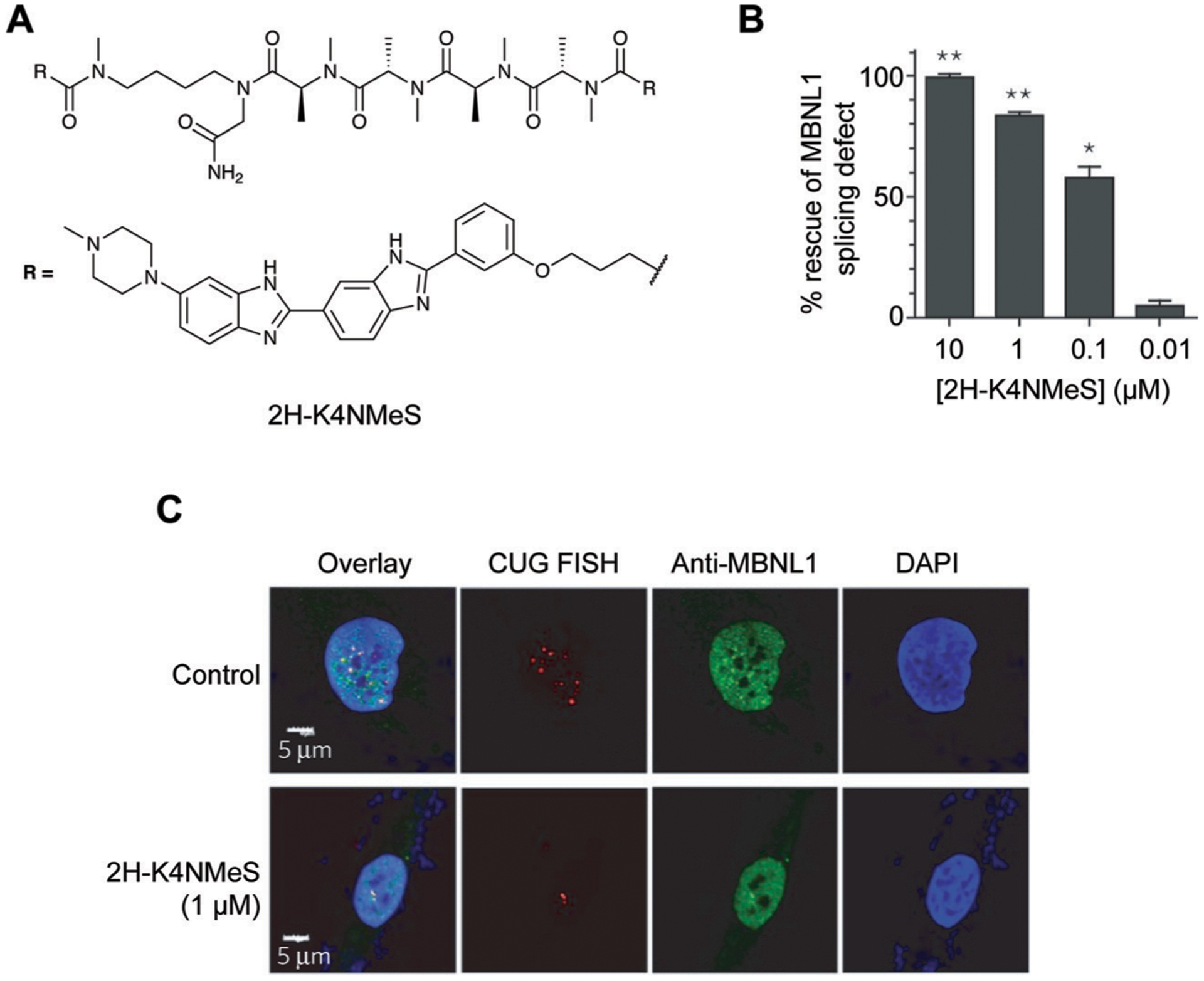
Targeting CUG repeat expansions with a multivalent ligand in DM1. (A) Structure of 2H-K4NMeS. (B) 2H-K4NMeS treatment reduces CUG:MBNL1 foci. RNA was detected with Fluorescence In Situ Hybridization (FISH), where fluorescently-labeled oligonucleotides complementary to an RNA of interest are incubated in cells to show their subcellular localization. Nuclei are labeled with DAPI (4′,6-diamidino-2-phenylindole). Adapted from ref. 10 with permission from Springer Nature, Copyright 2016. MBNL1 was detected using immunofluorescence, where a fluorescently-labeled antibody for MBNL1 was incubated in cells to show its subcellular localization. White regions in the “Overlay” show where FISH, MBNL1, and DAPI signals overlap. (C) Treatment with 2H-K4NMeS shows dose-dependent rescue of MBNL1 splicing defect. Adapted from ref. 10 with permission from Springer Nature, Copyright 2016.
Enterovirus 71 internal ribosomal entry site
As noted above, there is growing interest in small molecule targeting of viral RNAs.11 A recent contribution by Brewer, Hargrove, Tolbert, and co-workers targets the internal ribosomal entry site (IRES) of Enterovirus 71 (EV-71), one of the main agents of hand, foot, and mouth disease.52 The EV-71 IRES relies on a structure, called stem-loop 2 (SLII), that interacts with human RBPs, including hnRNP A1, which promotes viral translation, and AUF1, which suppresses translation. By screening an amiloride-based library, the authors discovered one molecule, DMA-135 (Fig. 11A), that exhibited strong binding for SLII and caused a significant perturbation in the SLII structure (Fig. 11B). DMA-135 significantly decreased viral translation in a dose dependent manner (Fig. 11C), significantly increased the interaction between SLII and AUF1 in cells (Fig. 11D), and significantly reduced viral titer in a dose-dependent manner.52 This study supports the use of small molecules for allosteric regulation of RNA structure and stabilization of RNA–protein interactions.
Fig. 11.
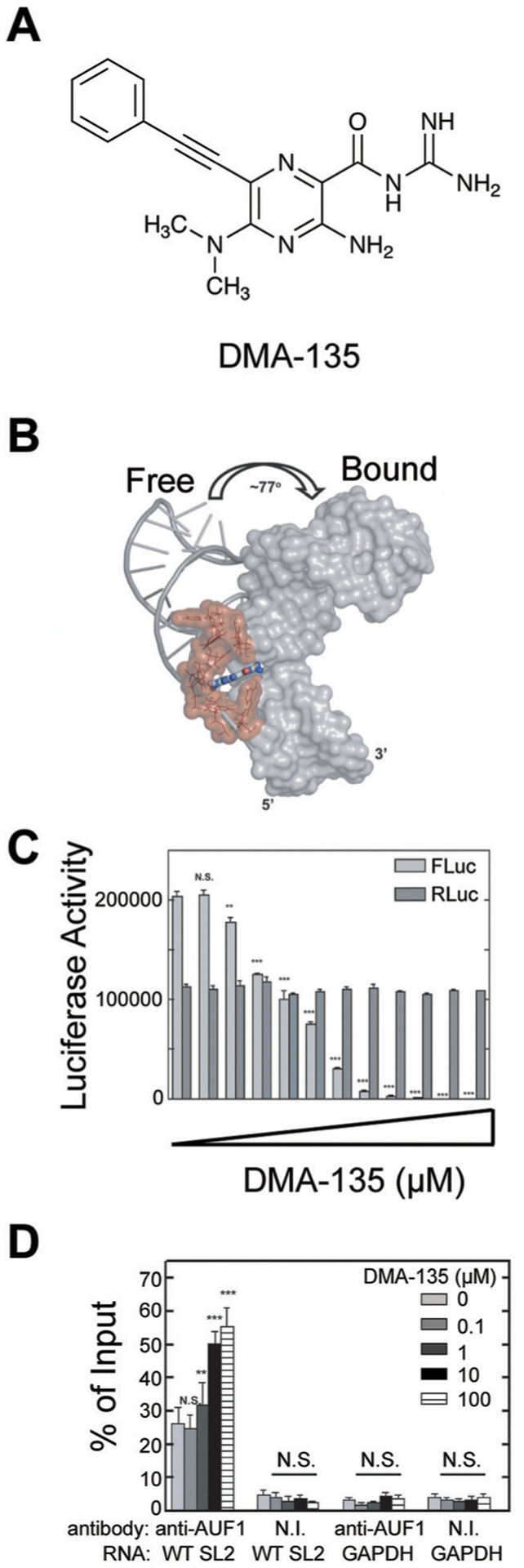
Small molecule allosteric modulation of EV71 SLII. (A) The structure of DMA-135. (B) DMA-135 results in a 77° interhelical shift in SLII as shown by NMR. Adapted from ref. 52 with permission from Springer Nature, Copyright 2020. (C) A luciferase-based reporter assay shows reduction of FLuc translation from 5′ UTR upon DMA-135 treatment as compared to RLuc control. Adapted from ref. 52 with permission from Springer Nature, Copyright 2020. (D) AUF1 protein was isolated via immunoprecipitation and bound RNA was detected via qPCR. Dose-dependent increase in bound SL2 RNA was observed upon DMA-135 treatment as compared to GAPDH control RNA. N. I. non-immune antibody. Adapted from ref. 52 from Springer Nature, 2020 under Creative Commons CC-BY 4.0 License.
RNA G-quadruplexes in cancer and neurological disorders
G-quadruplexes are found in the mRNAs of several oncogenes and can reduce translation. As a result, the stabilization of these structures via small molecules is an attractive therapeutic goal for the treatment of cancer.53 Multiple examples of small molecules targeting G-quadruplexes have been documented, including for the mRNA for NRAS and KRAS,53 which are implicated in multiple cancers but have not been successfully inhibited in the clinic.3
Targeting G-quadruplexes has also been explored in neurological disorders.3 For example, a GGGGCC repeat expansion in C9orf72 is implicated in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).54 These repeats can generate nuclear RNA foci that bind and antagonize the healthy function of other RNA-binding proteins by sequestering them as well as be translated into toxic, cytoplasmic dipeptide repeats. Both species have been hypothesized to contribute to the pathology of ALS and FTD. Neidle, Patani, Fratta, Isaacs, and co-workers identified three ligands that selectively stabilized RNA G-quadruplexes over those in DNA. Through favoring G-quadruplex formation, these ligands were hypothesized to reduce dipeptide translation and were shown to significantly reduce RNA foci in patient-derived cells.54 One molecule, DB1273 (Fig. 12A), significantly reduced RNA foci (Fig. 12B) and dipeptide levels both in cells and Drosophila models, where it also increased fly survival.54
Fig. 12.
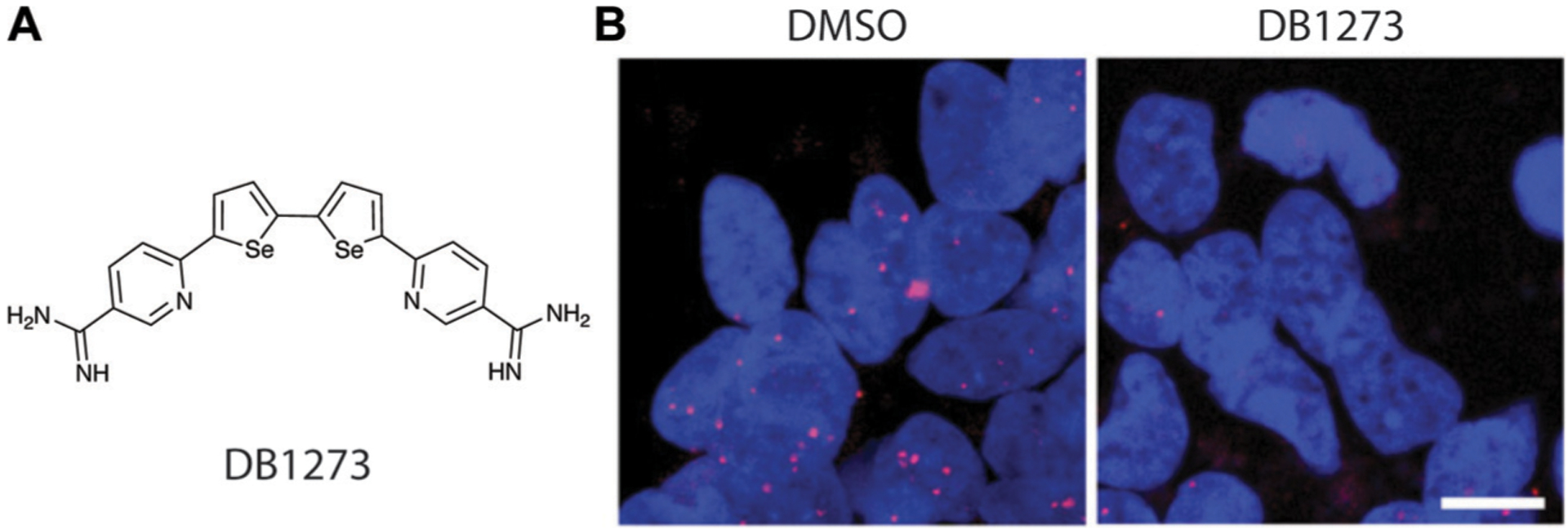
Small molecule disruption of foci containing G-quadruplex GGGGCC repeat expansions in ALS/FTD. (A) Structure of DB1273. (B) DB1273 significantly reduces GGGGCC RNA foci (red) in cellular nuclei (blue). Adapted from ref. 54, Copyright 2017, John Wiley and Sons in accordance to Creative Commons CC BY 4.0 License.
The riboflavin riboswitch
As discussed above, riboswitches are found in bacterial systems and are a naturally-occurring example of RNA–small molecule recognition. As a result, targeting these systems may facilitate antimicrobial development. Merck identified Ribocil, which binds to the bacterial riboflavin riboswitch, thereby deactivating riboflavin synthesis genes and resulting in bacterial death.13 The authors evaluated binding affinities for two stereoisomers of Ribocil, termed Ribocil-A and Ribocil-B. Ribocil-B (Fig. 13A) was found to have significantly a higher binding affinity than Ribocil-A, with a Kd of 6.6 nM and over 10 000 nM, respectively. These changes in binding affinity were also reflected in biological assays, where much lower concentrations of Ribocil-B were needed to inhibit bacterial growth as compared to Ribocil-A.
Fig. 13.
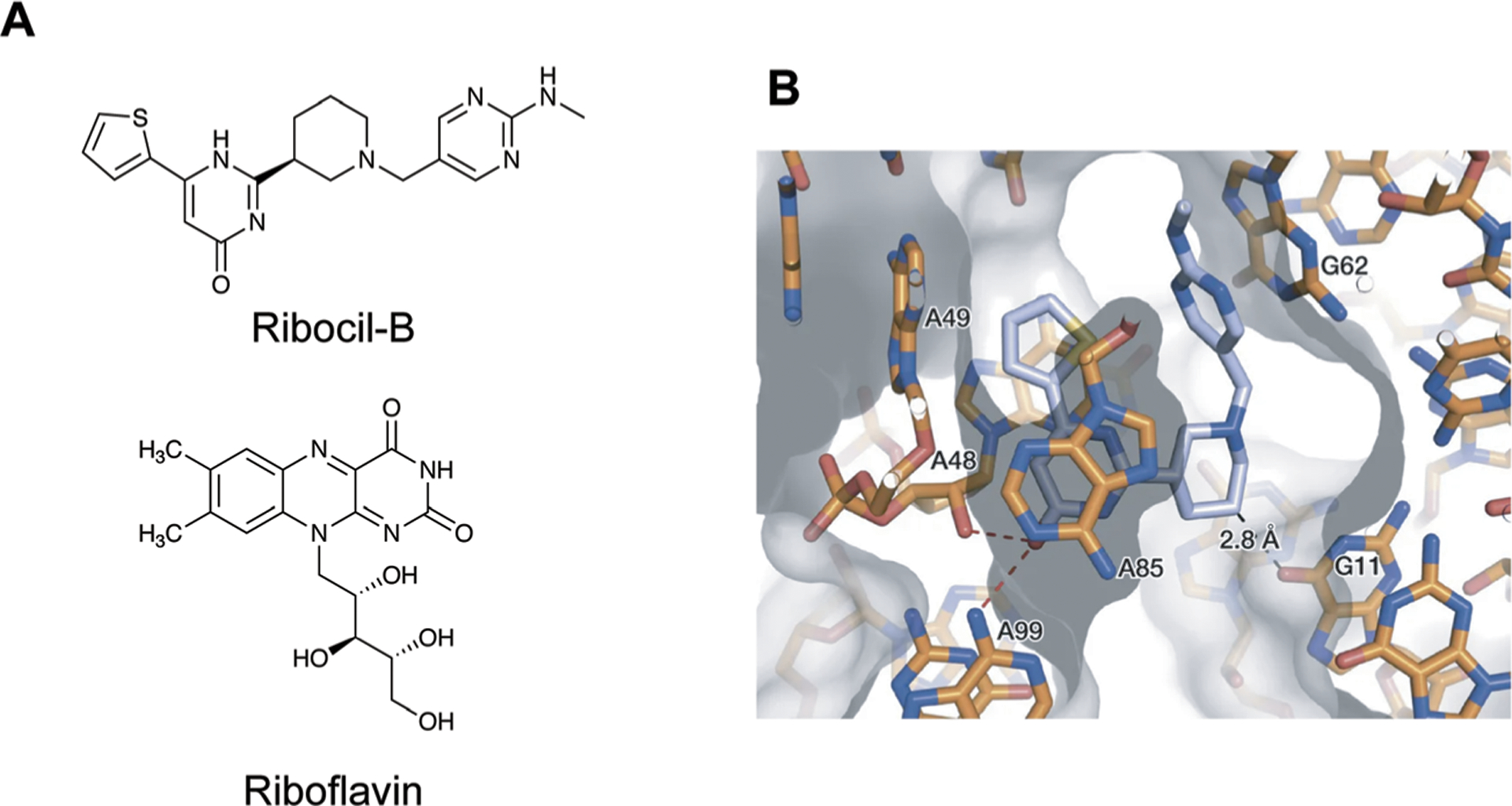
Small molecule targeting of the riboflavin riboswitch. (A) The structures of Ribocil-B and Riboflavin. (B) Crystal structure of Ribocil-B (blue sticks) in complex with riboflavin riboswitch. Ribocil binding is stabilized by stacking interactions with A48 and A85 and hydrogen bonding between the oxygen and A48 and A99. Additional stacking interactions and a methyl hydrogen bond are observed on the other face of the molecule. Adapted from ref. 13 with permission from Springer Nature, Copyright 2015.
Mechanistically, Ribocil-B functions as a riboflavin mimic, causing an equivalent conformational change in the riboswitch to deactivate the riboflavin synthesis pathway by occupying the same binding site. Interestingly, Ribocil is not structurally similar to riboflavin, yet is able to occupy this site through similar noncovalent interactions, including stacking interactions and hydrogen bonds, that mediate this recognition (Fig. 13B).13 Although Ribocil induced rapid development of resistant strains through mutation of the riboflavin aptamer, another derivative, Ribocil-C, successfully reduced systemic E. coli infection in a mouse model.13 Combined, insights from these studies showcase RNA’s ability to discriminate between minute changes in small molecule structure, in this case stereochemistry, despite more limited chemical functionality. This work also highlights the potential for targeting this biomolecule with small molecules that are structurally distinct from the natural ligand.
RNA splicing in spinal muscular atrophy
As RNAs can have multiple splice isoforms, one avenue within RNA-based drug design is to identify molecules that alter splicing to mitigate disease states.55 For example, spinal muscular atrophy (SMA) is the result of loss-of-function in a gene called survival of motor neuron 1 (SMN1), resulting in loss of muscle function and early death.36 Humans have a paralog gene called SMN2, but the resulting transcript from this gene lacks the seventh exon, critical for protein stability, and thus does not rescue loss of SMN1 function. Drugs that could favor Exon 7 inclusion in SMN2 are a promising treatment option (Fig. 14A).36
Fig. 14.
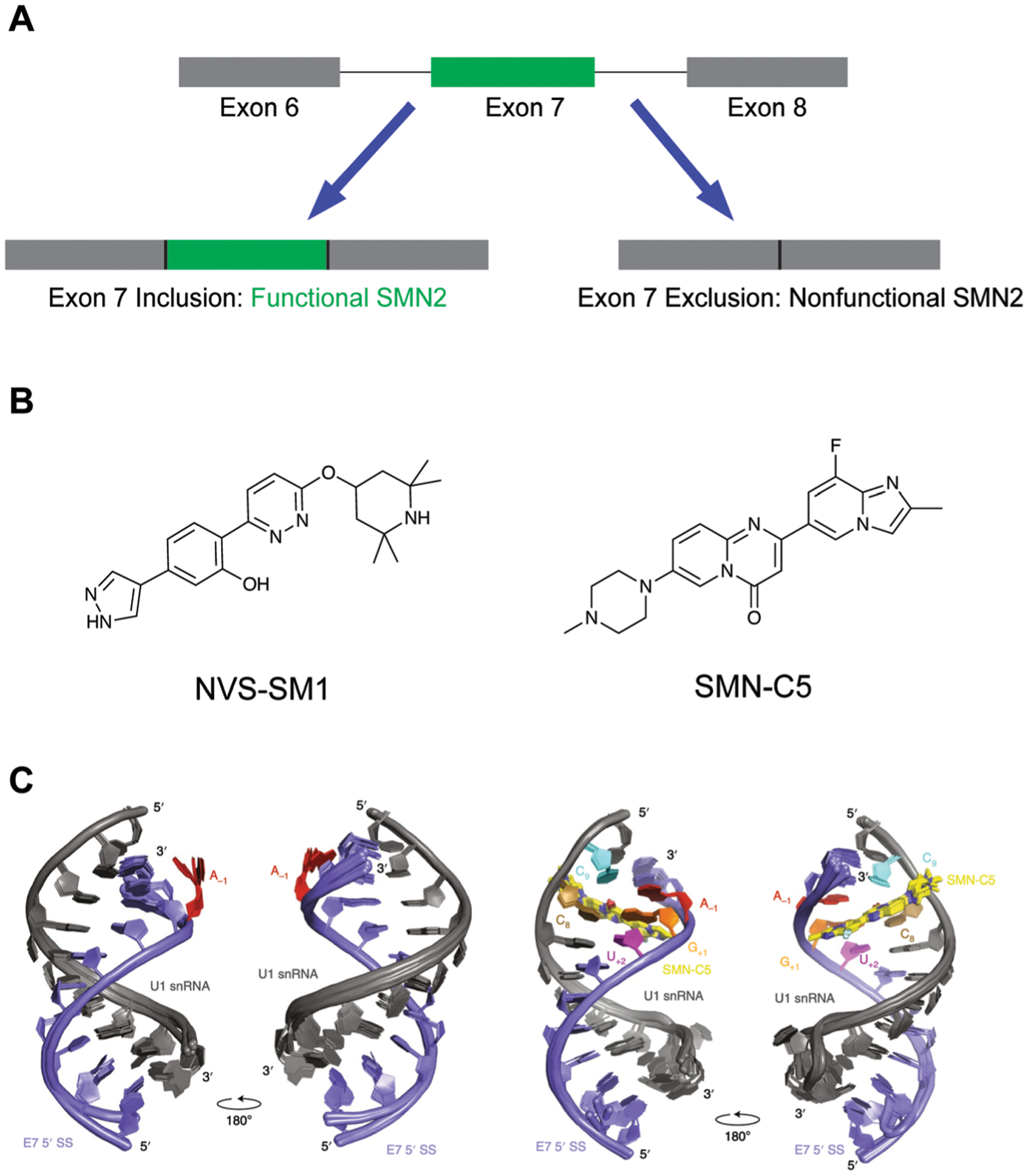
Small molecule modulation of splicing in SMA. (A) Schematic of SMN2 splicing. Inclusion of Exon 7 results in a functional SMN2 protein. (B) Structures of two documented SMN2 splicing modulators. (C) SMN-C5 (yellow) stabilizes an adenine residue (red) between Exon 7 and a spliceosomal U1 small nuclear ribonucleoprotein (snRNP), facilitating Exon 7 inclusion. SS: splice site. Adapted from ref. 37 with permission from Springer Nature, Copyright 2019.
Two promising molecules, NVS-SM1 by Novartis36 (Fig. 14B, left) and SMN-C5 by Roche37 (Fig. 14B, right), were identified through phenotypic screens to favor Exon 7 inclusion through interacting with the RNA components of the splicing machinery. In particular, SMN-C5 was found to interact with the U1 snRNP complex, and an NMR structure for the apo- and SMN-C5-bound U1 snRNA and Exon 7 splice site was solved by Campagne, Allain, and co-workers via NMR (Fig. 14C).37 It was found that SMN-C5 binds the major groove of the splice site, and electrostatic interactions between SMN-C5 and a U1 snRNA phosphate group facilitates binding. Stabilization of an adenine residue via hydrogen bonding maintains the adenine within the duplex, as compared to being flipped out as seen in the apo structure. As a result, this increased stabilization enhances Exon 7 stability and promotes splicing. In addition, this splice site structure is unique from other spice sites, thereby facilitating selectivity.37
Excitingly, an SMN-C5 derivative called risdiplam (Evrysdi™) by Roche/Genentech in collaboration with PTC therapeutics recently received FDA approval for the treatment of SMA.56 Risdiplam improved patient outcomes in both infantile and later-onset SMA in two separate clinical trials and represents the first orally-available drug for SMA.56 The approval of risdiplam showcases the promise of small molecule targeting of RNA and higher-order RNA assemblies, and also how structural biology studies can give greater insight into the mode of action of these drugs. It is certain that the success of risdiplam will spur interest into RNA targeting and modulation of splicing in a variety of diseases.
Outlook
The field of RNA-targeted drug discovery is rapidly progressing. Daily discoveries of RNA functions and structures, as well as privileged small molecule scaffolds, are accelerating the development of RNA-based drugs into the clinic. Small molecule approaches have great potential for therapeutic development as well as functioning in complement with ASOs and siRNAs for laboratory research. Identifying small molecule–RNA recognition principles, from both the RNA perspective and the small molecule perspective, will continue to facilitate such development. The importance of understanding fundamental recognition for these processes is elegantly illustrated with risdiplam, where the elucidation of its mechanism of action on a molecular level serves to explain its clinical success. In a larger sense, it is clear that drug development is moving past a protein-only mindset and that RNA’s rich structure and function make it an excellent therapeutic target to be further explored.
Several additional lines of research can be envisioned to progress the field. For example, more work is needed to parse the sequence and structure dependencies of small molecule–RNA interactions. The possibility of using molecules to selectivity interact with unique RNA structure or sequences of interest, such as protein-binding motifs, is an exciting avenue for future work. As seen in the EV-71 example above, this work may also include searching for modulators of RNA–protein interactions via allosteric regulation as opposed to competition for a given binding site. In addition, research is needed to understand to what extent currently approved protein-targeting therapeutics may bind RNA, either as part of their mechanism of action or as part of off-target effects. Similarly, natural products are currently underexplored for RNA recognition, and the often complex structures of many natural products may demonstrate novel selectivity or biological activity.
Given the unique chemical properties and modes of recognition that appear to be enriched in RNA-binding small molecules, it is possible that these structures may impact cell uptake and bioavailability. Perhaps, for example, certain transporters preferentially interact with RNA-binding small molecules on account of these unique structures or propensity for cationic chemotypes. Future work should explore these uptake mechanisms to aid in generating molecules with high bioavailability. This may be particularly relevant for targeting RNA that are enriched in certain cellular compartments.
Finally, it is most clear that continued basic understanding of RNA biology will be critical towards developing these novel therapeutics. RNA has been found to be core to a wide variety of cellular processes. Every day, new discoveries in chemistry and RNA biology are overturning what we thought we knew and allowing for ever-growing progress.2 One particularly exciting area of expanding research is the study of RNA’s role in liquid–liquid phase separation that drives the formation of membraneless cellular compartments, several of which have important disease implications.57 The possibility of modulating this dynamic cellular process with small molecules is a fascinating direction that will require cross-talk between the phase separation and RNA–small molecule targeting field.
If even just the last few years are anything to guide us, it is clear that the future in the RNA-targeting field will bring incredible advances. New research, coming out almost daily, is unlocking the clues of how to capitalize on RNA to change the course of human health. We greatly look forward to the upcoming years in RNA-targeting and all the treatments and promise it will bring.
Key learning points.
The reader will learn about emerging cellular functions mediated by RNA structure and the potential for small molecule-based modulation of those functions.
The reader will learn about the unique physicochemical and structural properties of RNA that differentiate it from other biomacromolecules.
The reader will learn about the noncovalent interactions and driving forces behind small molecule–RNA recognition.
The reader will learn about common methods used to characterize RNA–small molecule interactions as well as associated challenges and emerging solutions in the field.
The reader will learn about pioneering and recent successful examples of small molecule–RNA targeting, including risdiplam, which has received clinical approval.
Acknowledgements
The authors would like to thank the members of the Hargrove Lab for their invaluable feedback on this manuscript. The authors acknowledge financial support from Duke University, the National Science Foundation (CAREER 1750375), the U.S. National Institutes of General Medicine (R35 GM124785) and the Prostate Cancer Foundation. JPF was further supported by the Duke University Department of Biochemistry fellowship and Duke University School of Medicine scholarship funding.
Biographies

James P. Falese is a Biochemistry graduate student at Duke University School of Medicine. He received his BS in Biochemistry and Molecular Biology from Wake Forest University in 2018. His research focuses on RNA structure and protein interactions in cancer. James also enjoys mentoring and hiking.

Anita Donlic was born in Bosnia and Herzegovina. She obtained her BS in Chemistry at Methodist University in 2014 as a Davis United World College Scholar and a Methodist University Presidential Scholar. During her PhD at Duke University under the guidance of Prof. Amanda Hargrove, she developed the first small molecules to target the triple helix of the long non-coding RNA MALAT1 and was awarded a Joe Taylor Adams fellowship. In 2020, she began postdoctoral studies with Prof. Clifford Brangwynne at Princeton University, where she’s engineering approaches to study and perturb RNA-based membraneless organelles. She enjoys mentoring and science outreach.

Amanda E. Hargrove is an Associate Professor of Chemistry at Duke University. Prof. Hargrove earned her PhD in Organic Chemistry from the University of Texas at Austin followed by a postdoctoral fellowship at Caltech. Her laboratory at Duke works to understand the fundamental drivers of selective small molecule:RNA recognition and to use this knowledge to functionally modulate viral and oncogenic RNA structures. Her passions outside the lab include developing course-based undergraduate research experiences, working toward equity in chemistry at the departmental and national level, and watching old movies with her awesome family.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
We note that these canonical base-pairing patterns have been historically referred to as Watson–Crick and that our usage of the term Watson–Crick–Franklin acknowledges the critical yet traditionally underrecognized contributions of Dr Rosalind Franklin in X-ray crystallography to solving the structure of DNA. It is our hope to further the acceptance of this term and encourage its broader adaptation by using it in this tutorial.
References
- 1.Warner KD, Hajdin CE and Weeks KM, Nat. Rev. Drug Discovery, 2018, 17, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cech TR and Steitz JA, Cell, 2014, 157, 77–94. [DOI] [PubMed] [Google Scholar]
- 3.Donlic A and Hargrove AE, Wiley Interdiscip. Rev.: RNA, 2018, 9, e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The ENCODE Project Consortium, Nature, 2012, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costales MG, Childs-Disney JL, Haniff HS and Disney MD, J. Med. Chem, 2020, 63, 8880–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, Lander ES, Hess S, Plath K and Guttman M, Nature, 2015, 521, 232–236 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA and Steitz JA, Nat. Struct. Mol. Biol, 2014, 21, 633–640 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donlic A, Morgan BS, Xu JL, Liu A, Roble C Jr. and Hargrove AE, Angew. Chem., Int. Ed, 2018, 57, 13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abulwerdi FA, Xu W, Ageeli AA, Yonkunas MJ, Arun G, Nam H, Schneekloth JS Jr., Dayie TK, Spector D, Baird N and Le Grice SFJ, ACS Chem. Biol, 2019, 14, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R and Disney MD, Nat. Chem. Biol, 2017, 13, 188–193 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hermann T, Wiley Interdiscip. Rev.: RNA, 2016, 7, 726–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandal M and Breaker RR, Nat. Rev. Mol. Cell Biol, 2004, 5, 451–463. [DOI] [PubMed] [Google Scholar]
- 13.Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, Malinverni JC, Mayhood T, Villafania A, Nahvi A, Murgolo N, Barbieri CM, Mann PA, Carr D, Xia E, Zuck P, Riley D, Painter RE, Walker SS, Sherborne B, de Jesus R, Pan W, Plotkin MA, Wu J, Rindgen D, Cummings J, Garlisi CG, Zhang R, Sheth PR, Gill CJ, Tang H and Roemer T, Nature, 2015, 526, 672–677. [DOI] [PubMed] [Google Scholar]
- 14.Morgan BS and Hargrove AE, Synthetic Receptors for Biomolecules: Design Principles and Applications, The Royal Society of Chemistry, 2015, pp. 253–325. [Google Scholar]
- 15.Blythe A, Fox A and Bond C, Biochim. Biophys. Acta, Gene Regul. Mech, 2015, 1859, 46–58. [Google Scholar]
- 16.Butcher SE and Pyle AM, Acc. Chem. Res, 2011, 44, 1302–1311. [DOI] [PubMed] [Google Scholar]
- 17.Sussman JL, Holbrook SR, Warrant RW, Church GM and Kim SH, J. Mol. Biol, 1978, 123, 607–630. [DOI] [PubMed] [Google Scholar]
- 18.Collie GW, Haider SM, Neidle S and Parkinson GN, Nucleic Acids Res, 2010, 38, 5569–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Goodrich KJ, Gooding AR, Naeem H, Archer S, Paucek RD, Youmans DT, Cech TR and Davidovich C, Mol. Cell, 2017, 65, 1056–1067 and references therein. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Yan C, Hang J, Finci LI, Lei J and Shi Y, Cell, 2017, 169, 918–929. [DOI] [PubMed] [Google Scholar]
- 21.Selmer M, Dunham CM, Murphy IV FV, Weixlbaumer A, Petry S, Kelley AC, Weir JR and Ramakrishnan V, Science, 2006, 313, 1935–1942. [DOI] [PubMed] [Google Scholar]
- 22.Arya DP, Aminoglycoside antibiotics: from chemical biology to drug discovery, John Wiley and Sons, Hoboken, New Jersey, 2007. [Google Scholar]
- 23.Roost C, Lynch SR, Batista PJ, Qu K, Chang HY and Kool ET, J. Am. Chem. Soc, 2015, 137, 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu N, Dai Q, Zheng G, He C, Parisien M and Pan T, Nature, 2015, 518, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganser LR, Kelly ML, Herschlag D and Al-Hashimi HM, Nat. Rev. Mol. Cell Biol, 2019, 20, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dethoff EA, Petzold K, Chugh J, Casiano-Negroni A and Al-Hashimi HM, Nature, 2012, 491, 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belmont P, Constant J-F and Demeunynck M, Chem. Soc. Rev, 2001, 30, 70–81. [Google Scholar]
- 28.Chen S-J and Dill KA, Proc. Natl. Acad. Sci. U. S. A, 2000, 97, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Padroni G, Patwardhan NN, Schapira M and Hargrove AE, RSC Med. Chem, 2020, 11, 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collie GW, Sparapani S, Parkinson GN and Neidle S, J. Am. Chem. Soc, 2011, 133, 2721–2728. [DOI] [PubMed] [Google Scholar]
- 31.Vicens Q and Westhof E, Structure, 2001, 9, 647–658. [DOI] [PubMed] [Google Scholar]
- 32.Hewitt WM, Calabrese DR and Schneekloth JS Jr., Bioorg. Med. Chem, 2019, 27, 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sztuba-Solinska J, Shenoy SR, Gareiss P, Krumpe LRH, Le Grice SFJ, O’Keefe BR and Schneekloth JS Jr., J. Am. Chem. Soc, 2014, 136, 8402–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wicks SL and Hargrove AE, Methods, 2019, 167, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juru AU, Patwardhan NN and Hargrove AE, ACS Chem. Biol, 2019, 14, 824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, Beibel M, Renaud NA, Smith TM, Salcius M, Shi X, Hild M, Servais R, Jain M, Deng L, Bullock C, McLellan M, Schuierer S, Murphy L, Blommers MJ, Blaustein C, Berenshteyn F, Lacoste A, Thomas JR, Roma G, Michaud GA, Tseng BS, Porter JA, Myer VE, Tallarico JA, Hamann LG, Curtis D, Fishman MC, Dietrich WF, Dales NA and Sivasankaran R, Nat. Chem. Biol, 2015, 11, 511–517. [DOI] [PubMed] [Google Scholar]
- 37.Campagne S, Boigner S, Rüdisser S, Moursy A, Gillioz L, Knörlein A, Hall J, Ratni H, Cléry A and Allain FH, Nat. Chem. Biol, 2019, 15, 1191–1198 and references therein. [DOI] [PubMed] [Google Scholar]
- 38.Brown A and Shao S, Curr. Opin. Struct. Biol, 2018, 52, 1–7. [DOI] [PubMed] [Google Scholar]
- 39.Thompson RD, Baisden JT and Zhang Q, Methods, 2019, 167, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keane SC, Heng X, Lu K, Kharytonchyk S, Ramakrishnan V, Carter G, Barton S, Hosic A, Florwick A, Santos J, Bolden NC, McCowin S, Case DA, Johnson B, Salemi M, Telesnitsky A and Summers MF, Science, 2015, 348, 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM and Al-Hashimi HM, Nat. Chem. Biol, 2011, 7, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siegfried NA, Busan S, Rice GM, Nelson JA and Weeks KM, Nat. Methods, 2014, 11, 959–965 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganser LR, Kelly ML, Patwardhan NN, Hargrove AE and Al-Hashimi HM, J. Mol. Biol, 2020, 432, 1297–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan BS, Forte JE, Culver RN, Zhang Y and Hargrove AE, Angew. Chem., Int. Ed, 2017, 56, 13498–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haniff HS, Knerr L, Liu X, Crynen G, Boström J, Abegg D, Adibekian A, Lekah E, Wang KW, Cameron MD, Yildirim I, Lemurell M and Disney M, Nat. Chem, 2020, 12, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizvi NF, Santa Maria JP Jr., Nahvi A, Klappenbach J, Klein DJ, Curran PJ, Richards MP, Chamberlin C, Saradjian P, Burchard J, Aguilar R, Lee JT, Dandliker PJ, Smith GF, Kutchukian P and Nickbarg EB, SLAS Discovery, 2019, 384–396. [DOI] [PubMed] [Google Scholar]
- 47.Morgan BS, Sanaba BG, Donlic A, Karloff DB, Forte JE, Zhang Y and Hargrove AE, ACS Chem. Biol, 2019, 14, 2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M and Childs-Disney JL, ACS Chem. Biol, 2016, 11, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oliver C, Mallett V, Gendron RS, Reinharz V, Hamilton WL, Moitessier N and Waldispühl J, Nucleic Acids Res, 2020, 48, 7690–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eubanks CS, Forte JE, Kapral GJ and Hargrove AE, J. Am. Chem. Soc, 2017, 139, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donlic A, Zafferani M, Padroni G, Puri M and Hargrove AE, Nucleic Acids Res, 2020, 48, 7653–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davila-Calderon J, Patwardhan N, Chiu L, Sugarman A, Cai Z, Penutmutchu SR, Li M, Brewer G, Hargrove AE and Tolbert BS, Nat. Commun, 2020, 11, 4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bugaut A, Rodriguez R, Kumari S, Hsu STD and Balasubramanian S, Org. Biomol. Chem, 2010, 8, 2771–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert-Jaramillo J, Abdelkarim S, Clayton EL, Clarke M, Konrad MT, Nicoll AJ, Mitchell JS, Calvo A, Chio A, Houlden H, Polke JM, Ismail MA, Stephens CE, Vo T, Farahat AA, Wilson WD, Boykin DW, Zetterberg H, Partridge L, Wray S, Parkinson G, Neidle S, Patani R, Fratta P and Isaacs AM, EMBO Mol. Med, 2018, 10, 22–31 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Havens MA, Duelli DM and Hastings ML, Wiley Interdiscip. Rev.: RNA, 2013, 4, 247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.FDA.gov, 2020, FDA Approves Oral Treatment for Spinal Muscular Atrophy, Retrieved from https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
- 57.Shin Y and Brangwynne CP, Science, 2017, 357, eaaf4382. [DOI] [PubMed] [Google Scholar]