

Abstract
Leukodystrophies are a broad class of genetic disorders that result in disruption or destruction of central myelination. Although the mechanisms underlying these disorders are heterogeneous, there are many common symptoms that affect patients irrespective of the genetic diagnosis. The comfort and quality of life of these children is a primary goal that can complement efforts directed at curative therapies. Contained within this report is a systems-based approach to management of complications that result from leukodystrophies. We discuss the initial evaluation, identification of common medical issues, and management options to establish a comprehensive, standardized care approach. We will also address clinical topics relevant to select leukodystrophies, such as gallbladder pathology and adrenal insufficiency. The recommendations within this review rely on existing studies and consensus opinions and underscore the need for future research on evidence-based outcomes to better treat the manifestations of this unique set of genetic disorders.
Keywords: Leukodystrophy, Consensus, Therapy, Care, Outcomes, Prevention
1. Introduction
Leukodystrophies are a heterogeneous collection of genetic disorders that, while individually rare, collectively affect as many as 1 in 7500 individuals [1]. Patients with leukodystrophies, and their families, encounter a wide range of health problems and unique challenges to their care. These patients have a wide variety of issues, ranging from behavioral and sleeping difficulties, to requirements for assisted ventilation, to potential surgical interventions [2,3]. Hospitalizations and related health-care needs account for more than $59 million of health care costs each year in total [4]. For a minority of leukodystrophies, there are curative options, such as hematopoietic stem cell transplantation [5]. However, even in the absence of curative treatment, evidence supports a comprehensive treatment and care plan for all patients with leukodystrophies [6–8]. In addition to significant morbidity, one third of children with a leukodystrophy will succumb to the underlining disease and its complications by the age of 8 years [9].
The general tenets of our approach to the care of children with leukodystrophies are that there are common core symptoms shared among these disorders, that it is important to have a comprehensive approach that encompasses all relevant organ systems and includes the health of the care providers, and, importantly, that all leukodystrophies are treatable.
In this review, we will discuss specific management options for each affected system. We will also address disease-specific concerns, for example the adrenal insufficiency associated with adrenoleukodystrophy and the need for intracranial arteriopathy screening in a subset of patients with Aicardi-Goutières Syndrome. As leukodystrophy-related clinical studies are an area of active need, these recommendations rely primarily on clinical consensus and extrapolated data regarding the management described for other neurologic disorders.
2. Musculoskeletal and skin issues
Dysfunction of the musculoskeletal system is one of the most universal concerns among patients with leukodystrophies. Abnormalities in muscle tone such as spasticity and dystonia can result in secondary medical complications and negatively impact respiratory status, mobility, hygiene, self-care, sleeping patterns, and sexual function. Furthermore, the importance of ambulation for independence, bone and joint health, and emotional well-being cannot be overstated.
2.1. Spasticity
Spasticity is defined as velocity-dependent hypertonia with hyper-reflexia that is typically accompanied by weakness [10]. It occurs as the result of injury to the myelin and/or axons of the primary motor pathways (i.e. corticospinal tracts) of the central nervous system and is, anecdotally, one of the most common symptoms reported in patients with leukodystrophies. Patients often have a combination of tone abnormalities, including truncal hypotonia mixed with appendicular hypertonia, dystonia, and other movement disorders, which may change over time [11]. Of importance, a sudden and persistent change in tone should prompt an in-depth assessment to determine the etiology. An acute increase in tone is commonly the result of an intercurrent illness or pain, although it may also be due to new central nervous system pathology.
Clinical assessment of spasticity should include an evaluation by a physical therapist, ideally employing standardized scoring systems to produce quantifiable metrics that can be tracked longitudinally (Table 1). Although these particular scoring systems have not been validated in children with leukodystrophies, they can be applied with caution in this population. In addition to specific scales to measure the severity of the motor dysfunction, the impact of altered tone on quality of life (QoL) can be assessed using standardized scales such as the Pediatric Quality of Life Inventory (PedsQL) [12]. The PedsQL is a self-reported assessment of health-related QoL. The Vineland Scales of Adaptive Behavior Screener can be useful for children that are young or with relatively low functional levels [13]. An important area of future research should be the validation of these scales within the leukodystrophy population, as these unique disorders are typically progressive (unlike cerebral palsy) and may affect a younger population than was used to design the currently available assessment tools.
Table 1.
Assessment tools for tone abnormalities and movement disorders.
| Hypertonia Assessment Tool (HAT) [27] |
|
| Modified Ashworth Scale (MAS) [14] |
|
| Modified Tardieu Scale (MTS) [15,28] |
|
| Global Dystonia Rating Scale (GDS) [27] |
|
| Movement Disorder Childhood Rating Scale (MD-CRS) [29] |
|
Abnormal tone can cause significant medical complications, many of which may necessitate medical intervention (Table 2). In milder cases of spasticity without significant axial hypotonia, oral medications such as baclofen or diazepam in combination with physical therapy and daily stretching routines are usually sufficient [14–17]. Chemodenervation with botulinum toxin or intramuscular neural lysis with phenol can be useful to target focal areas of spasticity that impede functional tasks (e.g. adductor muscles to facilitate hygiene, gastrocnemius muscles to improve ambulation) [14–18]. The assessment and administration of chemodenervation can be performed by a variety of specialists, but requires an experienced medical provider with specialized training in this technique. Further guidance, as needed, can be provided by physiatry or orthopedics.
Table 2.
Complications of abnormal tone.
|
If these initial medical interventions are not effective, or if oral medications are not suitable or tolerated, consideration can be given to more invasive approaches. Intrathecal baclofen enables the use of higher doses of medication with fewer systemic side effects as compared to oral baclofen, but requires an implanted medical device that poses the added risk of infection or a mechanical failure that can lead to drug withdrawal [14,15,19–21]. In select cases, surgical interventions to lengthen or sever tendons or nerve pathways can facilitate mobility, prevent joint deformity, contractures, and fractures [14,22,24,25]. Consideration should be taken to the underlying risk of the procedure and for the use of anesthesia in this fragile population. Perioperative complications in a study of children with cerebral palsy undergoing scoliosis repair for example correlated with the number of pre-operative medical issues [26].
2.2. Dystonia
Dystonias are hyperkinetic movement disorders characterized by generalized or focal involuntary muscle contractions, frequently resulting in twisting and repetitive movements or abnormal postures [10]. Despite their prevalence in leukodystrophies, dystonias are often under-diagnosed and can worsen with physical discomfort or voluntary movement.
One of the most efficient treatments for generalized dystonia is the anticholinergic medication trihexyphenidyl (Artane), which is typically well tolerated in this patient population [3,17–22]. As trihexyphenidyl can result in constipation, it is important to manage this side effect as clinically indicated. Other options are dopaminergic drugs, such as l-dopa, and the dopamine-depleting drug tetrabenazine [14,30–32]. Oral baclofen and benzodiazepines can also be also effective, but require higher doses than are typically used in the management of spasticity [14,30]. In patients with leukodystrophies, D2 receptors blockers should be avoided because of the risk for tardive movement disorders [33]. Although rarely observed in this population, focal dystonias can be treated with botulinum toxin injection, which is typically preferable to oral medications [14,30,31,34]. More invasive treatments include intrathecal baclofen and, in rare cases, deep brain stimulation (DBS) [14,30,31,34,35].
2.3. Low bone mass/density and fractures
Patients with neurologic disorders are at high risk for low bone mass and fractures due to lack of mobility, decreased sun exposure, and nutritional deficiencies [36]. Additionally, some leukodystrophies carry inherently higher risk for bony complications: vanishing white matter disease (VWM), AARS2-related disorder, and POLR3-related leukodystrophy are associated with endocrine dysfunction and secondary osteoporosis, and patients with cerebrotendinous xanthomatosis are at increased risk for granulomatous lesions of the bones and fractures [2,37,38]. Bone health should be actively monitored in most patients with leukodystrophies, with particular attention paid to patients with steroid exposure, epilepsy, a history of prior fractures, nutritional deficiencies, and those who are non-ambulatory [39–41]. Some anticonvulsants, including phenobarbital, phenytoin, carbamazepine, and valproic acid all have been reported to have detrimental effects on bone mineralization [41]. Long-term use of proton pump inhibitors for reflux can also result in increased bone fractures, possibly through decreased calcium absorption [42].
In general, increased PTH levels can be interpreted as an initial marker of poor calcium intake or absorption. Calcium level alone is not a sufficiently sensitive marker of low calcium reserves. Patients should also have at least yearly monitoring of vitamin D levels (25-OH-D), as poor nutrition and limited sun exposure can contribute to the risk of low bone density for age (Tables 3 and 4).
Table 3.
Vitamin D status definitions as per the Endocrine Society [43].
| Deficiency | 25(OH)D below 20 ng/mL (50 nmol/L) |
| Insufficiency | 21–29 ng/mL (52.5–72.5 nmol/L) |
| Sufficiency | 30–100 ng/mL |
Table 4.
Guidelines for the treatment of Vitamin D deficiency from the Endocrine Society [43].
| Infants and toddlers 0–1 year |
|
| Children 1–18 years |
|
| Adults |
|
| Unique dosing needs for obesity, malabsorption, abnormal vitamin D metabolism (e.g. secondary to medications) |
|
As a complement to laboratory testing, imaging studies can useful in the diagnosis of bony disease. Basal bone density scans, typically dual-energy X-ray absorptiometry (DEXA or DXA, L–spine and Whole Body Less Head) scans are useful screening tools for demineralization in patients who are at risk for fractures [39,40,44]. An initial DEXA screening is recommended around five [5] years of age, as this is the earliest age for which a dataset exists to correct for age, gender, ethnicity, and height. Follow-up imaging would be dependent upon degree of osteopenia/osteoporosis. Standard X-rays are not recommended as a screening tool for bone demineralization, although for patients who are immobilized or have a history of fracture, lateral spine X-rays can be used to screen for vertebral fractures. If contractures or scoliosis prohibit standard bone imaging, a forearm or distal lateral femur scan can be obtained [44]. As with other complex areas of care, a consultation with a bone specialist or endocrinologist is recommended at an early stage of the clinical course.
2.4. Scoliosis and hip dislocation
Although the true prevalence of scoliosis and hip dislocation in this unique patient population is unknown, clinical experience suggests that it is common, particularly in advanced stages of disease [2] (Fig. 1). One epidemiological study on orthopedic and neurologic manifestations in the leukodystrophy population found that scoliosis occurs in 70% and hip dysplasia in 89% of patients [45]. Some individuals may develop a progressive “windswept” appearance as a result of unequal tone in the lower extremities causing asymmetric hip dislocation. Unfortunately, no natural history studies of the leukodystrophy population have been performed to characterize relationship between scoliosis and hip dislocation. Studies in neuromuscular diseases suggest that the scoliosis may accompany or precede pelvic obliquity, although spinal deformity can also be a compensation for pelvic malalignment [46].
Fig. 1.
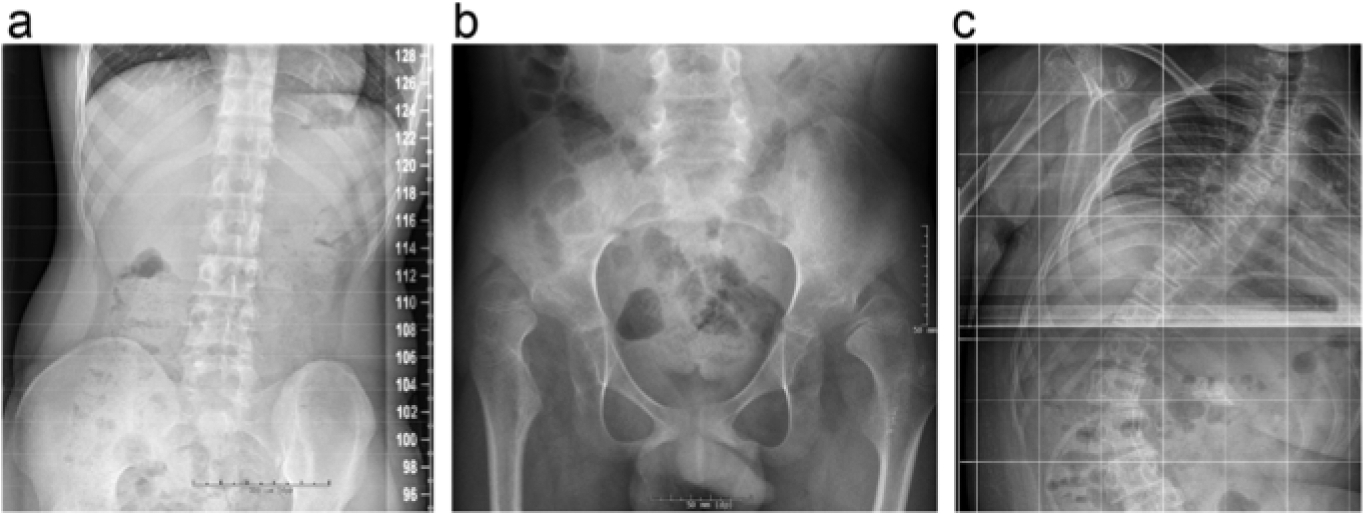
Examples of orthopedic disorders in children with leukodystrophies. Pelvic tilt in a patient with Krabbe disease is shown by hip radiograph (a). Bilateral coxa valga with bilateral dislocated femoral heads is demonstrated in a patient with Pelizaeus-Merzbacher (b). Severe thoracolumbar scoliosis and pelvic tilt is shown in a patient with X-linked Adrenoleukodystrophy (c).
2.4.1. Hip dislocation
Hip issues are common in the leukodystrophy population as a consequence of spasticity, dystonia, and decreased mobility. Dislocation at the hip joints may impair mobility and bone mass. In patients with cerebral palsy, regular hip surveillance programs have been effective in improving long term outcomes and reducing rates of hip dislocation [47] and scoliosis [48]. These programs include physical examinations every six months and regular X-rays starting at 2 years of age and recurring annually until skeletal maturity [73]. With any clinical concerns, patients should have further imaging studies of the hips and/or spine and be referred to specialists in orthopedics, physiatry, and physical therapy. Physiatry and orthopedics can help to guide discussions about appropriate management options, which should take into consideration the overall health of the patient and the family’s goals of care. Not all hip dislocations require surgical intervention. Surgery should be considered if the dislocation is painful, impairs mobility, or poses other risks to the patient’s well-being. Conservative approaches, including adductor releases and tone management, are preferable in patients under five years of age. After six years of age, reconstructive surgery may be considered.
2.4.2. Scoliosis
Scoliosis in patients with leukodystrophies may be progressive and can seriously impact health and quality of life, posing particular risks to respiratory and cardiac function. Progressive scoliosis may diminish lung function, a relationship referred to as “thoracic insufficiency syndrome” [49]. Scoliosis management guidelines for other neurodegenerative disorders recommend a brief spinal exam by clinical observation at each clinic visit [76–78]. If scoliosis is suspected, anterior-posterior and lateral spine X-rays can be obtained, with referral to an orthopedic surgeon as clinically indicated. Based on general recommendations, braces and external frames may be appropriate in milder cases. Spinal orthoses can maximize chest expansion capacity by optimizing seating position, though these will not affect the rate of scoliosis progression [49]. Spinal surgery is often considered if the curve exceeds a Cobb angle of 40–50°[50].
2.5. Ambulation
While some patients with leukodystrophies are able to achieve independent ambulation, most will eventually experience some degree of impairment in their mobility. Preserving and optimizing ambulation or, at a minimum, maintaining weight-bearing exercises, is widely considered important for the overall health and quality of life. Without this, patients may experience secondary complications including contractures and joint dislocation, and impaired bone health.
Patients should be screened for any treatable conditions that may impact ambulation and weight-bearing ability, including abnormal tone or pain (Table 5). Among patients with impaired ambulation, falls potentially pose a serious health risk. During clinical encounters, physical therapists and/or physiatrists can help to assess fall risk and the degree of mobility independence. Although they have only been validated in other patient populations, standardized tools such as the Gross Motor Functional Classification System (GMFCS) can be helpful for these assessments [51,52]. Other simple tools, such as the 10-meter walk test, can be used to monitor ambulation at various stages of disease.
Table 5.
Barriers to ambulation.
|
Physical therapists can help to guide the options for age-appropriate devices to assist mobility and maximize independence. These devices include orthotics, braces, gait trainers, walkers, lifts, and standers. Outpatient physical therapy can also help to preserve motor skills.
2.6. Skin care
Most skin infections are readily amenable to simple prevention strategies, such as daily skin surveys by the primary caregiver. With each clinical evaluation, we recommend a full head-to-toe assessment – with clothes removed – to examine for areas of skin breakdown and pressure, with particular attention placed on areas where orthotic devices, braces or other medical equipment comes in contact with skin and the diaper area for incontinent patients. Families should be educated about skin wounds and provided with educational handouts (Table 6). Non-blanching erythema or indurated skin may indicate an emerging skin abrasion or decubitus ulcer and might necessitate a change in hardware, bed care, or behavioral strategies. Physical therapy can help to review seating and orthotic devices at each visit, as an ill-fitting adaptive device can result in preventable skin breakdown.
Table 6.
Challenges to skin integrity in the leukodystrophy population.
|
2.6.1. Leukodystrophy-specific skin issues
Uniquely, patients with Aicardi-Goutières Syndrome (AGS) have several skin manifestations such as chilblains, which require specialized wound care. Chilblains are necrotic lesions typically found on the hands, feet, elbows, and the pinna of the ears [53]. Children with AGS can also experience acrocyanosis and periungual skin infections. Ichthyosis can be associated with several leukoencephalopathies, including multiple sulfatase deficiency and Sjogren-Larsson Syndrome [54,55].
3. Nutrition, bowel, and urinary tract guidelines
3.1. Hypersalivation
Sialorrhea or excessive drooling is a common problem in children with neurodevelopmental disabilities [56,57]. Importantly, it can be associated with physical, social, and psychological distress. It can result in skin maceration, and in severe cases, secondary respiratory problems. Sialorrhea can be due to a variety of medical issues, including dental problems (gingivitis, dental caries, and malocclusion), gastroesophageal reflux, obstructive sleep apnea, and dysphagia leading to excess pooling of oral secretions [57,58]. Speech-language pathologists and physical therapists can evaluate patients and help to optimize adaptive equipment. First-line interventions to help with excessive drooling include oromotor or behavioral exercises, positioning, replacing medications that stimulate saliva secretion, as well as optimization of constipation, scoliosis, and gastroesophageal reflux [57,59,60].
Medications for hypersalivation, particularly anticholinergic agents, should be used cautiously in this patient population because of thickening of secretions and central sedative effects. Anticholinergics, which include hyoscine (oral/transdermal Scopolamine) and trihexyphenidyl (Artane), decrease mucus secretions [37]. Sublingual 1% atropine ophthalmic solution has also been used with success [61]. Glycopyrrolate successfully reduces the production of saliva with fewer central nervous system side effects and is approved for use in children older than 3 years of age [59]. More intense or invasive treatments for intractable aspiration of saliva, such as, targeted botulinum toxin A injections and salivary gland surgery, can be administered by a trained provider [57,60,62].
3.2. Upper gastrointestinal complications
Swallowing dysfunction, gastroesophageal reflux, and feeding issues are common among individuals with leukodystrophies. Chronic malnutrition and limited fluid intake can have various adverse effects on growth, brain development, immune function, bowel/bladder health, and overall quality of life [63]. Malnutrition in the context of leukodystrophy is often polyfactorial, influenced by dietary intake and basal metabolic needs, as well as non-nutritional factors such as dysphagia and gastroesophageal reflux [63–65]. Patients at highest risk for nutritional issues include those with impaired communication, impaired mobility, and dysphagia, the latter of which is a risk factor for aspiration pneumonia [65–68]. Particular attention should be paid to making sure that the diet is nutritionally complete. This is especially important if food aversions, such as texture, temperature, or consistency preferences, are present due to behavioral or swallowing dysfunction. Even children who gain weight adequately are at risk for micronutrient deficiency if they are too selective in their food choices. If there is any question, a dietician can be consulted.
We encourage each clinical visit to include a comprehensive assessment of feeding-related risk factors, including dental issues, oral/pharyngeal dysphagia, gastroesophageal reflux, and constipation. These regular feeding and nutrition assessments can occur at time of diagnosis and regularly every 3–6 months. The goal of these visits is to optimize oral feeding and continually assess the need for nutritional interventions [64]. Speech-language pathologists, occupational therapists, and physical therapists are equipped to assess oral sensorimotor function, swallowing and feeding-related issues. They can suggest appropriate adaptations, including proper positioning, adjustment of food consistency, pacing of feeding, and equipment [40,42,43,54,55]. It can be particularly helpful for a speech-language therapist or occupational therapist to observe the child eating and evaluate oral-facial structures and function, performance on various food types, efficiency (volume and timing), and oral-pharyngeal status (e.g., bolus cohesion, oral residue, pharyngeal response). The general physical examination should include assessment for malnutrition, as indicated by hydration status, skin color, and subcutaneous fat distribution. The child should also be examined for signs of oromotor dysfunction, coughing, a change in growth parameters, and the efficiency or duration/ease of feeding [40,45,54,56]. Primary caregivers may be able to provide additional information about day-to-day feeding issues at home.
If the clinical bedside speech-language pathology or occupational therapy assessment is suggestive of dysphagia or a risk for aspiration, further instrumental diagnostic studies may be indicated. These include videofluoroscopic swallow study (VFSS), modified barium swallow study (MBS), or fiberoptic endoscopic study examination of swallow (FEES). Instrumental swallow assessment is useful in determining specific areas of concern (e.g., oral, pharyngeal) and safety of swallowing various food consistencies (e.g., aspiration, laryngeal penetration, airway protective reflexes). If these initial studies are unrevealing, the patient can be clinically re-evaluated, typically at 3–6 month intervals. With any significant change in clinical status, the swallow assessment can be repeated sooner. If there is concern for aspiration risk as suggested by coughing, choking, wet or gurgly voice, oromotor dysfunction (e.g., effortful, food escaping lips, food residue in mouth after swallow), difficulty maintaining weight, or general feeding difficulties (e.g., food refusals, emesis), a further investigation may be warranted. With documented aspiration, difficulty maintaining weight, and marked inefficiency of oral nutritional intake, an expedited consultation with gastroenterology or general surgery for consideration of gastrostomy (G-tube) or jejunostomy (J-tube) tube placement may be indicated.
3.2.1. Gastroesophageal reflux
Gastroesophageal reflux (GER) is common among individuals with neurologic dysfunction [7,69]. Pathologic GER can have adverse effects on feeding and sleep, and can cause vomiting, esophagitis, respiratory compromise, dental issues, and malnutrition [70]. Aspiration of oral-pharyngeal matter or secondary aspiration of refluxed pharyngeal-esophageal matter may lead to lung disease and secondary respiratory-related death among children with neurological impairments [71].
Importantly, GER is a clinical diagnosis that does not necessitate confirmatory studies. For children with reflux, speech and physical therapy can help to optimize position and food consistency during feeding [70,72]. Adjunctive medications, such as acid buffering agents, antisecretory agents, and prokinetic agents can be helpful as well [70]. Ranitidine, lansoprazole, and omeprazole are also effective options [7,73]. Of note, proton pump inhibitors can be associated with decreased absorption of calcium, magnesium, iron, and vitamin B12, and therefore patients on long-term treatment with these agents should be monitored for these deficiencies [42]. If medical approaches fail to manage pathologic GER and its secondary medical complications, surgical interventions should be considered. Nissen fundoplication is often offered in conjunction with a gastrostomy or gastrojejunal tube placement [7,70,71]. Evidence supports effectiveness of these procedures for reducing esophagitis, GER, aspiration pneumonia, respiratory illness, failure to thrive, reflux-related hospitalization, and death [71]. As such, local expertise and family preference should be considered in decision-making process.
3.3. Gastrostomy tubes
Gastrostomy tubes are widely considered a safe and effective way to maintain the nutritional needs of neurologically impaired individuals, although no formal studies have directly assessed this important topic in patients with leukodystrophies [74–76]. G-tube placement should be considered in the context of persistently poor weight gain despite optimization of nutrition or in the presence of oromotor dysfunction that impacts safe and efficient swallowing [74,77–79]. Unfortunately, the first encounter with a gastroenterologist and/or general surgeon is often delayed until after a child is no longer orally feeding, losing weight, and/or declining, at which point G-tube placement carries an inherently higher risk [76].
Ideally, the G-tube can be a tool to keep the child healthy, reduce the frequency of hospitalizations, facilitate safe and easy administration of medications, and prevent serious respiratory complications [74]. Children with leukodystrophies may benefit from G-tube placement even as oral feeds continue. Specifically, a G-tube can be employed to deliver unpalatable supplemental nutrition or medications; to provide safe intake of thin liquids when aspiration is observed on only that consistency; and to provide supplemental nutrition when the child does not meet nutritional needs in a timely manner due to inefficiency or rapid fatigue. While there are benefits to tube placement, parents report that children with nasogastric (NG) tubes vomit frequently (particularly in patients younger than 1 year of age) and children with G-tube experience nausea, loss of hunger, as well as skin issues including granulation tissue and irritation [80]. Little evidence exists to support a specific type of feeding tube, however anecdotal clinical evidence indicates greater difficulty transitioning to oral feeds, greater difficulty managing oral secretions, and higher levels of food aversions associated with NG versus G-tube. Also, in the absence of clinically significant gastroesophageal reflux, there is no evidence to guide choice of a Nissen fundoplication with G-tube placement compared to G-tube alone [70,74,75]. Endoscopic placement versus surgical placement should be based on the patient’s overall health status, anesthesia risk, and available resources and expertise.
The decision to place a gastrostomy is often complicated by psychosocial factors, including perceived parental culpability for child’s feeding difficulty, implied deterioration of the child’s condition, impact of the feeding tube on social relationships, child’s enjoyment of feeding (e.g., tastes, smells) and maintaining feeding skills for later oral intake [81]. This many explain why some families may agree to a nasogastric tube, but are hesitant about use of a gastrostomy tube or jejunostomy tube. Additional education, counseling, and time can be helpful to families making these decisions [81]. Furthermore, G-tube placement is often misinterpreted as a permanent step. When addressing interventions, it can be helpful to emphasize “quality of life care” rather than “end of life care” [73]. From the family’s perspective, this language alone contributes to emotional distress and may discourage them to pursue G-tube placement, even though it could significantly improve the family’s quality of life. Encouraging families to meet with a specialist early in the clinical course can work to avoid such misconceptions. This conversation should ideally occur in an outpatient setting during an anticipatory conversation as part of a broader care plan.
3.4. Bowel motility
Impaired bowel motility is a common problem in many patients with neurologic impairment [7,73]. Constipation is an easily treated, yet frequently overlooked source of chronic pain that can significantly impact quality of life and lead to serious secondary complications, such as urinary retention. While there is no standard definition for constipation, it is generally considered as two or fewer bowel movements per week. The diagnosis is made clinically by obtaining a patient history and/or rectal exam. Assessment of fluid intake should also be made at time of examination, as chronic dehydration is a risk factor for constipation. Families can provide additional information on frequency of stooling, as well as appearance based on the Bristol Stool Chart [82]. A plain abdominal X-ray is only indicated if impaction is suspected and is not needed for the diagnosis of constipation.
Education of primary caregivers and implementation of appropriate prevention strategies, such as simple dietary changes, increased hydration, and supplemental dietary fiber or adding enteral formula with fiber if the child is G-tube fed, are the key elements in the management of constipation (Table 7) [83]. If symptoms persist, polyethylene glycol or lactulose can be used as stool softeners [73]. Sodium phosphate enemas are particularly effective at managing constipation in children with limited mobility. Stool stimulants such as senna or bisacodyl can be helpful as well. While these may lead to dependency, priority should be given to optimization of quality of life. Finally, if the treatment of acute constipation necessitates disimpaction, the medical team should consider consulting a gastroenterologist or general surgeon.
Table 7.
Family education on constipation.
|
3.5. Bladder health
Urinary dysfunction is a common and important complication seen in the leukodystrophy population. Dysautonomia, neurogenic bowel and bladder, and constipation are risk factors for secondary urinary dysfunction. Urinary incontinence represents a significant source of embarrassment and stigmatization. From a psychosocial perspective, it is important to destigmatize the issue and shift the focus to improving patient quality of life.
Symptoms may manifest as urinary incontinence, urgency, or retention, all of which increase the risk of a bacterial infection of the bladder and kidney. In non-verbal patients, untreated urinary tract infections (UTIs) may result in serious pain and discomfort, and may contribute to life threatening illness or hospitalization, or seizures [8]. Of note, pediatric patients with neurologic impairment may be either hyperthermic or hypothermic at times of infections. Diagnostic tests for urinary tract infections should be ordered urgently, as delays can increase the severity of UTI and decrease quality of life. This evaluation should include a urinalysis with urine culture, with isolation of more than 100,000 colonies per milliliter considered a reasonable threshold for clinically significant bacteriuria. Urinalysis with urine culture is not appropriate for patients with a history of intermittent or chronic catheterization, as often the bladder is colonized.
It is our general recommendation that a urology consultation should be considered if a female patient presents with two or more UTIs annually, or if a male patient presents with one or more UTIs annually. Consultation can also be helpful in the context of symptoms such as delayed urinary stream, urgency, or secondary enuresis. Renal and bladder ultrasounds with voiding studies may be helpful to assess for neurogenic bladder, and urodynamic studies can help identify issues related to sphincter or bladder control. Prophylactic anti-microbial agents may be used on a case-by-case basis under the guidance of a urologist or infectious disease specialist. Patients taking anti-seizure medications should be monitored closely for urinary dysfunction, as some of these medications can predispose to nephrolithiasis. With bladder retention, consideration should be given to urinary catheterization as guided by urology [7,8]. A delay in catheterization can increase the risk of UTI recurrence and lead to serious infectious complications and potential renal dysfunction.
3.6. Gastrointestinal and urinary health: leukodystrophy-specific guidelines
As gallbladder involvement has been detected in more than half of patients with metachromatic leukodystrophy (MLD), we recommend that patients with MLD are followed by a gastroenterologist with scheduled gallbladder ultrasounds [84,85]. Abdominal computed tomography (CT) can be used if reliable gallbladder ultrasounds are not available [84]. The most common abnormalities include wall thickening and polyps [84]. Gallbladder dysfunction can present as abdominal pain, which can be difficult to distinguish from other issues, such as spasticity. Polyps smaller than 5 mm may be followed annually. As determined by gastroenterology, laparoscopic cholecystectomy should be considered for polyps larger than 5 mm. Gallstones have been reported in patients with cerebrotendinous xanthomatosis as well, although the true incidence of this complication warrants formal study [37,86].
Metachromatic leukodystrophy can also be associated with acid-base disturbances in approximately one quarter of patients [87]. This is likely secondary to renal sulfatide accumulation and can result in a significant metabolic acidosis, particularly in the context of clinical stressors [87]. Patients with Aicardi-Goutières syndrome can sustain inflammatory complications to the kidneys, including a lupus-like glomerulonephritis, and these patients should have regular urinalysis to assess for proteinuria.
4. Respiratory health, sleep, and communication
4.1. Progressive respiratory insufficiency
Respiratory complications such as aspiration pneumonia and community-acquired pneumonia are a common source of serious morbidity and mortality among individuals with neurodegenerative disorders (Table 8) [88]. Importantly, many potentially life-threatening complications are amenable to preventive strategies. Primary respiratory failure may occur in the late stages of some leukodystrophies, particularly those associated with peripheral nerve dysfunction, such as MLD. Bulbar dysfunction, such as seen in Type II Alexander Disease, is associated with central apneas. Finally, dystonic dysphonia, such as that seen in H-ABC and 4H syndrome, and obstructive symptoms seen in many leukodystrophies with hypotonia can result in intermittent obstructive apnea.
Table 8.
Concerning features for respiratory insufficiency and/or aspiration.
|
Dysphagia, as discussed in detail in the upper gastrointestinal complications section, can contribute to acute or chronic lung disease. A clinical swallow assessment by a speech-language or occupational therapist in combination with instrumental diagnostic studies (e.g. VFSS, MBS, or FEES) is helpful in the identification of aspiration risk [38,72]. After the initial evaluation, we recommend that re-assessment recurs every 3–12 months as guided by disease progression or appearance of new signs, symptoms or risk factors (Table 6). More frequent swallow assessments are indicated in high-risk patients, including those with clinical signs of oropharyngeal weakness, oral coordination issues, or sialorrhea, a clinical history of pneumonia, or coughing during meals [31,34,35].
It is important to identify the cause of respiratory insufficiency (e.g. weakness, obstruction, chronic injury, severe scoliosis), as this information can guide the options for intervention. Clinical recommendations and goals of care should encompass the family’s quality of life considerations, as well as cultural and religious beliefs. Consultation with pulmonology, gastroenterology, and/or otolaryngology should be considered early in the disease course to guide preventive strategies, foster therapeutic relationships, and enable anticipatory discussions of future supportive interventions such as G-tube, tracheostomy, and mechanical ventilation. Pulmonary care ideally should be preventative more than reactive.
A comprehensive interventional strategy for respiratory insufficiency should include infection prevention, airway maintenance, and mechanical support. Infection prevention includes annual influenza vaccination, positioning and feeding modifications, regular hand washing, and avoidance of sick contacts when possible [8]. Palivizumab, which targets respiratory syncytial virus (RSV), may have a role in select cases. Key airway maintenance strategies include repositioning, ambulation, physical therapy, as well as targeted interventions such as chest physical therapy, vest therapy, or cough-assist devices [7]. Some patients may benefit from regular use of a suction aspirator machine at home. Additional medical and surgical options are available to treat persistent sialorrhea, as discussed above. Salivary Botox injections may help to reduce aspiration pneumonia risk, although this option has not yet been formally studied [56].
Because of progressive respiratory insufficiency, a discussion regarding the possibility of using mechanical ventilation will be required in most cases. Available options, which should be considered in consultation with a qualified pulmonologist, include invasive mechanical ventilation, continuous positive airway pressure (CPAP), bilevel positive airway pressures (BiPAPs), or supplemental oxygen. In contrast to neuromuscular disorders, primary respiratory failure in the leukodystrophy population typically occurs in the context of severe cognitive impairment. The family, patient, and leukodystrophy provider should have anticipatory discussions of mechanical ventilation in the context of ongoing goals of care discussions [73].
4.2. Communication
Maintaining communication between the affected individual and caregivers is among the most important and underappreciated goals of a comprehensive care strategy [68,89,90]. Simplistically, a language disorder is an impairment in the comprehension and/or the use of spoken, written, or symbol systems involving form (e.g. grammar, syntax), content (e.g. vocabulary and meaning) and functional use (e.g. pragmatics) of language [91]. The array of language impairments in an individual patient with leukodystrophy depends largely on the specific regions of the brain impacted by abnormal myelin development, abnormal myelin homeostasis, and secondary neuronal injury. The progressive loss of language in children with leukodystrophies is an area needing further formal study.
Children with leukodystrophies, like others affected by neurological impairments, often present with neuromotor speech disorders (i.e., dysarthria, apraxia of speech) [69,89,92]. Dysarthria is used to describe a group of speech disorders that are caused by abnormal strength, speed, range, steadiness, tone or accuracy of speech movements [93]. Features of dysarthria include spastic, flaccid, hypokinetic, hyperkinetic, or ataxic speech. Unlike dysarthria, apraxia of speech is caused by difficulty planning or programming commands that direct speech movements in sequence [93]. A child with neuromotor speech impairments, including dysarthria or apraxia, has difficulty with sound production, and this is often accompanied by reduced intelligibility and comprehensibility of speech. As a result, these children are isolated socially, exhibit maladaptive behaviors for communication, and have limited communication partners.
Speech-language pathology (SLP) evaluation should be considered for any patient with communication difficulties or bulbar neuromotor features. A comprehensive exam will assess motor speech function, language skills (verbal and written expression and comprehension), communication effectiveness, and evaluation for communication tools [68,89,90,92,94–96]. In order to best guide the individual family, the speech-language pathologist should ask primary caregivers how they communicate with the child. This often serves as a good introduction to a broader “goals of care” conversation, but also provides valuable insight into the communication abilities of the patient at home.
A comprehensive augmentative and alternative communication (AAC) evaluation should be consider for any child whose current methods of communication are not effective in meeting the child’s daily communication needs [97]. For some children, AAC may be used in specific situations identified as problematic, while others may use AAC as a primary means of communication. During the AAC assessment, the family and child work with a SLP to identify appropriate symbolic representations (e.g., photos, symbols, text), message types (e.g., full utterance, word-by-word, spelling), voice output (e.g., recorded digitized, synthesized speech), technology level (e.g., no tech, low tech, high tech), and access (e.g., direct with hand or eye gaze, indirect scanning) options [89,90]. In order to meet a patient’s individual communication needs, cognition, language, vision, hearing, and physical skills should all be considered. It is important to adapt to a child’s evolving communication needs, which include new communication environments and changes in physical, cognitive, and language skills associated with typical development as well as disease progression.
The value of communication cannot be overstated; the difference between complete absence of communication and the simple ability to communicate “yes” or “no” represents an extreme change in quality of life. Perhaps the most extreme version of this scenario is poignantly illustrated by accounts of individuals suffering from “locked-in-syndrome” where the body and mind continue to experience the pains and pleasures of the physical world but are unable to communicate [98].
4.3. Sleep
Sleep dysregulation, characterized by recurring episodes of difficulty initiating and/or maintaining sleep, is a common feature in the leukodystrophy population that can negatively affect quality of life of both patients and caregivers [99]. While the specific incidence of sleep disorders in the leukodystrophy population is unknown, sleep disorders occur in over half of all children with severe multi-system disability [100]. Common sleep problems include difficulty with sleep onset or maintenance, sleep-related breathing disorders, abnormal circadian rhythms, and hypersomnolence [100]. Obstructive and central sleep apneas are also common in children with leukodystrophies and may necessitate specialist care and interventions. Untreated obstructive sleep apnea (OSA) can result in a variety of medical issues, including excessive daytime sleepiness, headaches, and even serious secondary cardiac complications. Neurologic irritability, such as that seen in early onset disorders such as infantile Krabbe and Aicardi-Goutières Syndrome, can in some cases severely impact sleep as well. Sleep dysfunction can also be the result of other medical issues, including gastroesophageal reflux, pain, and spasticity that interrupt sleep. Each of these causes should be managed accordingly.
An important first step in managing sleep disorders is optimizing sleep hygiene, with an emphasis on a consistent sleep schedule, avoiding screen time 1–2 h prior to bedtime, and minimizing unnecessary medical interventions at night [99]. Primary caregivers can record a sleep diary to help characterize the patient’s sleep patterns and identify problem areas. While there are no FDA-approved medications for the treatment of insomnia in children, off-label options include clonidine, tricyclic antidepressants, and benzodiazepines [99,100]. In clinical practice, melatonin is often used to help with sleep initiation. Referral to a sleep medicine provider should be considered, particularly if OSA is suspected.
5. Neurologic issues
Many patients with a leukodystrophy experience a wide range of neurologic complaints, including pain, irritability, and cognitive impairment, all of which may negatively impact the quality of life of the patient and primary caregivers. The degree of cognitive impairment is largely dictated by the extent of the neural networks affected and the severity of the underlying injury. Even within a family, each affected child may demonstrate variable rates of cognitive decline. As the typical course of a leukodystrophy is progressive, school and home accommodations should be continually re-assessed and adjusted to the child’s evolving needs. In addition to the medical team, social workers can help families navigate these complex issues.
5.1. Pain
Pain, irritability, and sleep are central to maintaining quality of life, but are often under-recognized and undertreated [37]. Validated pain assessment tools are categorized by age and cognitive ability [73]. In very young children, the CRIES (Cry, Requires O2, Increased Vital Signs, Expression, Sleeplessness) or FLACC (Face, Legs, Activity, Cry, Consolability) scales can be used. Older children may be able to use numerical or illustrated pain scales, such as the Wong-Baker FACES® Pain Rating Scale, to describe pain levels. A child with an AAC system for communication may use alternative methods for describing discomfort or pain, such as a gesture dictionary or photographic body representations.
After the child is determined to be in pain, the caregivers and clinical team should investigate common triggers for discomfort. The list of potential occult causes is extensive, but includes dental abscesses, bowel obstruction and constipation, pancreatitis, bone fractures, acute joint dislocation, tone issues, respiratory compromise, skin breakdown, and urinary tract infections [101]. In addition, current medications should be reviewed for agents that may be contributing to pain or worsen existing neurologic symptoms [101]. Peripheral neuropathies are common in select leukodystrophies, such as Krabbe (globoid cell) and MLD, and can result in neuromuscular dysfunction as well as discomfort.
The World Health Organization has published guidelines for pain management for pediatric oncology, which can be adapted to children with chronic neurologic disease [73,102]. According to these guidelines, the basic tenets of pediatric pain management include [73]: treating by ladder (standardized escalation plan), treating by clock (scheduled timing), treating by mouth (using the least invasive route possible), and treating by child (individualized therapies). Gabapentin can be particularly helpful for managing neuroirritability and neuropathic pain, though this approach has not been formally studied in the leukodystrophy population [7,101]. Benzodiazepines and neuroleptics may also be effective for managing agitation [101].
5.2. Seizures
Not just a manifestation of late-stage neurodegeneration, seizures affect almost 50% of patients with leukodystrophies [9]. In rare cases, seizures may be the presenting symptom, such as in Alexander Disease [90]. Epilepsy is common feature of Krabbe disease, megalencephalic leukoencephalopathy (MLC), sialic acid storage disorders, peroxisomal disorders, and L-2-hydroxyglutaric aciduria. Epilepsy is defined as two or more unprovoked seizures, a single seizure with a high risk for a second seizure, or the presence of a known epilepsy syndrome [103,104]. Seizure mimics are common and should be considered during the evaluation of any potential epileptic event (Table 9) [105].
Table 9.
Common seizure mimics.
|
A careful clinical history will allow providers to determine whether a child has had a clinical seizure [104], and clinical history alone is often sufficient for the diagnosis of epilepsy [101]. Features such as focality at onset, duration, and context are important in the evaluation of seizures. A routine electroencephalogram (EEG) may help establish a formal diagnosis of seizures and guide subsequent medication selection, and video EEG monitoring can be useful to distinguish between seizures and non-epileptic events. Following a clinical seizure, we recommend that patients are referred to a neurologist, who will guide the need for and selection of prophylactic medication [104]. There is no evidence to support the use of anti-seizure medications prior to the onset of clinical seizures, as these medications do not prevent the development of epilepsy and the majority of leukodystrophy patients will not develop seizures [92]. As such, the first step after resolution of a seizure should be an assessment for provoking factors, such as fever, electrolyte dysregulation, medication withdrawal, and infection. Urinary tract infections are an especially common risk factor for seizure in the leukodystrophy population because of underlying issues with urinary retention and voiding [8]. For provoked seizures, the patient is unlikely to require a daily prophylactic anticonvulsant medication. Regardless of daily prevention needs, abortive anti-seizure options, such as rectal diazepam and buccal or intranasal midazolam, can be provided for patients with prolonged or clustered seizures [101].
5.3. Autonomic nervous system dysfunction
Autonomic dysfunction affects many patients with leukodystrophies and, given the autonomic nervous system’s role in maintaining global homeostasis, can result in diverse symptomatology (Table 10) [100,106,107]. Autonomic symptoms are a particularly prominent feature of several leukodystrophies, including later-onset (type II) Alexander Disease and adult-onset autosomal dominant leukodystrophy (ADLD) [108,109].
Table 10.
| System | Examples of dysfunction | Potential evaluation and management |
|---|---|---|
| Genital-urinary system |
|
|
| Gastrointestinal system |
|
|
| Cardiac and vascular systems |
|
|
| Sudomotor function |
|
|
| Ophthalmology |
|
|
| Pulmonary |
|
|
| Neurology |
|
|
Autonomic dysfunction can be inherent to the neurodegenerative process or can be triggered acutely by pain or infection. The acute onset of a cluster of dysautonomic symptoms is referred to as an “autonomic storm”. Potential triggers should be evaluated in patients who are at risk for autonomic dysfunction. Medications that may be helpful in the preventative management of dysautonomia include gabapentin, cyproheptadine, baclofen, beta-blockers, and clonidine [100]. For acute attacks, diphenhydramine, acetaminophen, or ibuprofen can be useful.
5.4. Additional neurologic considerations
Several leukodystrophies are associated with significant behavior issues, including inattention, irritability, hyperactivity, and aggression. Infantile Krabbe disease classically is characterized by hyperirritability. Although not evaluated by controlled clinical studies, gabapentin is typically chosen as the first line medication given its safety profile. It is reasonable to start at 15–20 mg/kg/d divided 2–3 times daily and to escalate as needed to 60 mg/kg/d. Non-validated alternatives include prega-balin, topiramate, tricyclic antidepressants, and valproic acid. In refractory cases, benzodiazepines can be used with caution. Many adult presentations of leukodystrophies, including MLD, Krabbe, Alexander Disease, and Hereditary Diffuse Leukodystrophy with Spheroids, can be associated with significant psychiatric symptoms.
The cerebral demyelinating lesions associated with X-linked Adrenoleukodystrophy (X-ALD) may manifest symptoms that mimic of attention-deficit hyperactivity disorder. This can be immediately differentiated through the imaging findings. The presence of brain lesions may qualify boys with X-ALD for bone marrow transplantation, but only if detected at an early stage. Patients with more advanced cerebral X-ALD may experience significant behavior disturbances. Clinical experience suggests that risperidone and valproic acid may be helpful mood and behavior stabilizers in this population.
Certain leukodystrophies can result in significant peripheral nerve involvement and pain, and may include Krabbe, MLD, AARS2 related leukodystrophy, Polyglucosan body disease and Pelizaeus-Merzbacher disease, among others. Since the signs of peripheral neuropathy may be overshadowed by central neurologic signs, and patients may be unable to communicate the symptoms of neuropathy, careful attention should be given to detect the presence of peripheral nerve involvement. Eliciting symptoms of dysesthesia should be included in routine neurologic examinations, in particular for early onset disorders such as infantile Krabbe and late infantile MLD. When peripheral neuropathy is identified, consideration of appropriate treatment to manage these sometimes-painful symptoms should be given utmost attention.
After genetic diagnosis, most patients with leukodystrophies do not require subsequent imaging outside of clinical trials. Exceptions include patients affected by l-2-hydroxyglutaric aciduria who are at increased risk for brain malignancy and therefore necessitate serial brain imaging. Patients with SAMHD1-associated Aicardi-Goutières Syndrome (AGS) are at increased risk for cerebrovascular accidents, and may benefit from annual brain MRI and MR angiograms. The evidence behind the frequency of testing remains to be established through future formal studies.
6. Endocrine guidelines
6.1. Adrenal insufficiency
Up to 90% of male patients affected by X-ALD will ultimately develop adrenal insufficiency at any point between infancy and adulthood [94,95]. Adrenal insufficiency should be considered in any patient with malaise and fatigue and signs including hypotension, hyponatremia, hyperpigmentation, weight loss, vomiting, and poor growth. A simple and reliable screening test, very long chain fatty acids (VLCFA), can help to diagnose X-ALD and should be considered for all boys with suspected adrenal insufficiency. Although potentially fatal, adrenal insufficiency can be treated with corticosteroid supplementation. Once present, adrenal insufficiency in X-ALD is usually chronic, requiring daily oral steroid administration with increased dosing at times of clinical stress.
Adrenal insufficiency is assessed through laboratory measurements of adrenocorticotropic hormone (ACTH) and morning cortisol levels. We recommend that patients with X-ALD should be screened for adrenal insufficiency every 6 months beginning at the time of diagnosis, although the frequency of this testing has yet to be validated. Additional testing can be considered in the context of illness requiring any escalation of medical care. An endocrinologist may also recommend additional studies such as aldosterone, plasma renin activity, and ACTH stimulation tests when biochemical values are equivocal. Boys undergoing stem cell transplantation for X-ALD should be closely monitored for adrenal function. It is important to provide families with a letter of instruction on steroid stress dosing and testing to be given to outside providers and emergency rooms.
6.2. Ovarian failure
Primary ovarian failure may arise as a complication of a number of leukodystrophies, including AARS2-related diseases, as well as some mitochondrial and chromosomal (ex.18q-) disorders. Females diagnosed with vanishing white mater (VWM) disease may experience primary or secondary ovarian failure. These patients may need screening measurements of estradiol, luteinizing hormone (LH), and follicular stimulating hormone (FSH) as guided by endocrinology or gynecology.
6.3. Other leukodystrophy-specific endocrine issues
Patients with hypomyelination, hypogonadotropic hypogonadism and hypodontia (4H syndrome) typically experience a variety of endocrine abnormalities, including hypogonadotropic hypogonadism, hypothyroidism, and, less commonly, growth hormone deficiency [111]. Although there is no clinical evidence, we recommend that patients be screened at diagnosis and then annually for growth failure. Additionally, testing of testosterone, luteinizing hormone, and follicular stimulating hormone levels should be considered at the age of expected puberty (by 13 years old for females and by 14 years old for males). A slow or flat rate of growth as plotted on a growth chart may be indicative of growth hormone failure. Thyroid stimulating hormone (TSH) and T4 can be used as marker of hypothyroidism, with hormone replacement therapy administered as needed. For cases of hypogonadotropic hypogonadism, the benefits and risks of sex steroid replacement should be discussed thoroughly with the patient and family. Hypothyroidism has also been reported in patients with cerebrotendinous xanthomatosis [37,112] and in patients with Aicardi-Goutières syndrome and these patient populations should be specifically screened for this condition.
6.4. Post-transplantation endocrine considerations
Patients who have undergone therapeutic stem cell transplantation are at increased risk for endocrinopathy secondary to medication, irradiation, and the transplant itself. These patients should be considered for referral to endocrinology for regular growth evaluation or for growth hormone screening if growth failure is observed. These patients also may benefit from regular monitoring of thyroid function, with clinical follow-up for thyroid nodules. After transplant, patients are also at risk for gonadal failure, which can present as delayed puberty. Post-transplant osteoporosis should also be assessed and treated as clinically indicated.
7. Additional system-specific concerns
7.1. Autoimmune disorders
In general, autoimmune disorders are overrepresented in patients with Aicardi-Goutières Syndrome (AGS) and include thyroid dysfunction, systemic lupus erythematosus (SLE), celiac disease, and gastrointestinal inflammatory diseases [113]. Patients affected by AGS may benefit from annual testing of TSH levels, as well as clinical screening for other autoimmune diseases. With any concerns, we recommend an evaluation by endocrinology or rheumatology.
7.2. Cardiac issues
Some leukodystrophies are known to be associated with cardiac issues, in addition to dysautonomia, and require regular visits with cardiology. These most notably include Aicardi-Goutières Syndrome, 18q-syndrome, infantile sialic acid storage disorders, and fucosidosis. Mitochondrial leukoencephalopathies can also be associated with cardiomyopathy and cardiac rhythm abnormalities. Due to the underlying abnormalities in cholesterol metabolism, cerebrotendinous xanthomatosis may be associated with accelerated coronary heart disease and hypertrophy of the atrial septum [37,114]. Additionally, respiratory complications such as obstructive sleep apnea can cause secondary cardiac abnormalities. If suspected, prompt referral to a cardiologist is recommended.
7.3. Ophthalmologic issues
Regular ophthalmological monitoring can prevent many of the ocular complications associated with leukodystrophies. Potential problems range from glaucoma, which is observed in patients with Aicardi-Goutières Syndrome (AGS) and peroxisomal disorders, to the ocular malformations found in oculodentodigital dysplasia (ODDD) (Table 11) [113]. Progressive myopia is a common manifestation of 4H or POLR3-related leukodystrophy and requires regular ophthalmological assessments. Optic atrophy is a common feature in a number of leukodystrophies. Eye movement abnormalities, including the nystagmus common to many hypomyelinating leukodystrophies including Pelizaeus-Merzbacher disease, can impair function and academic activities such as reading. Macular degeneration with perifoveal crystalline inclusions is common in Sjogren-Larsson Syndrome, a disorder of lipid metabolism. With any ocular concerns, we recommend evaluation with ophthalmology, and vision services as appropriate. Finally, facial weakness and decreased blinking can result in significant risk of ocular dryness with subsequent injury to the cornea. Careful observation and questioning to elicit this complication should be included in general health maintenance. Where appropriate, ocular lubricants should be used.
Table 11.
Ophthalmologic complications of leukodystrophies.
| Leukodystrophy | Potential issues |
|---|---|
| 4H or POLR3-related leukodystrophy [111] |
|
| Aicardi-Goutieres Syndrome (AGS) [113] |
|
| Peroxisome biogenesis disorders [113] |
|
| Oculodentodigital dysplasia (ODDD) [113] |
|
| Cerebroretinal microangiopathy with calcifications and cysts disease (CRMCC, coats plus syndrome) |
|
| Leukoencephalopathy, brain calcifications, and cysts disease (LCC) |
|
| Retinal vasculopathy with cerebral leukodystrophy (RVCL) |
|
| Sjogren-Larsson Syndrome (SLS) |
|
| Hypomyelination and congenital cataracts (HCC) |
|
| Pelizaeus-Merzbacher disease |
|
| Cerebrotendinous xanthomatosis [37] |
|
7.4. Dental guidelines
In addition to the regular dental care needed for all patients, children affected by select leukodystrophies require regular dental evaluations, preferably by specialists who are aware of the disease-specific considerations. Patients affected by Cockayne syndrome have predisposition to develop cavities. 4H syndrome, or POLR3-related leukodystrophy, is associated with variety of teeth abnormalities, including delayed eruption of teeth or natal teeth. Patients with Aicardi-Goutières syndrome and cerebrotendinous xanthomatosis are at increased risk for tooth loss. Strong consideration should be given to finding a specialist in pediatric special-needs dentistry.
8. Coordination of care
8.1. Biopsychosocial assessment
The biopsychosocial assessment should paint a picture of “where the family is coming from” and will begin to set a “compass” to guide care [73].This might include questions to address the medical condition of the patient and the patient’s and/or family’s understanding of the diagnosis. In order to fully care for a child, it is important to understand living arrangements of the patient, the accessibility of those living arrangements based on patient’s degree of disability, and the access to transportation services. Additional considerations include the access to support programs, the family’s composition and challenges, the support system for the parents and family, and parental health and coping mechanisms. A social worker often guides these discussions and allows the team to start to building trust with the family and positions the team in a better position to help as important end-of-life decisions need to be discussed with the family. The ultimate goal is to develop or adopt a standardized intake form that can be used across GLIA and leukodystrophy care network clinical sites. This form would address clinical, research, and quality of life elements. The clinical team of physicians and social work should have evolving discussions about goals of care with the family.
8.2. Clinical care plan
As demonstrated by the extensive and complex medical issues out-lined in this article, leukodystrophy clinical care and encounters can be overwhelming for families and challenging to medical and allied health care providers. To help families understand the process, a member of the care team should help to outline a longitudinal clinical care plan: starting at diagnosis and continuing over the course of their medical care. This will improve the relationship between family and clinical care team by increasing transparency and communication. In writing, basic disease information and a summary of the key areas of clinical focus should also be provided to families. The handout should list each provider and their specific recommendations for follow up visits, studies, and medications (Table 12).
Table 12.
Example clinical care plan.
| Today your child saw the following providers | |
|---|---|
| Neurology (Dr. X, contact information) | Prescribes X medication (dosage and schedule) for seizures |
| Prescribes X medication (dosage and schedule) for behavioral issues | |
| Recommends X study to be completed | |
| Physiatry (Dr. X, contact information) | Prescribes X medication (dosage and schedule) for spasticity |
| Physical Therapy (X, contact information) | Recommends the following exercises and stretches |
| Nutrition (X, contact information) | Recommends the following changes to your child’s diet |
In order to facilitate communication, “red flags” that may warrant consultation with a specialty care center should be detailed, such as the symptoms of adrenal failure in boys with adrenoleukodystrophy. Additionally, to empower families, the documents should also include space for patients/families to independently keep track of their local care team visits, medication changes, lab results, and medical treatments. All patients with leukodystrophies should have the opportunity to be involved in clinical research, including natural history studies.
8.3. Transitions in care
There are several key transitions of care that occur during the life of patients with leukodystrophies: inpatient versus outpatient; pediatric to adult care; home to group care, insurance coverage changes, changes in access to services; and end of life care. Conversations on transitions of care should be started early to familiarize the family with the concepts and the idea that with most patients with leukodystrophies have evolving needs. Earlier access to palliative care can guide the families with difficult decision-making and work to set the “compass”. As a whole, our health care systems need to dedicate more effort to educating families in order to overcome longstanding stigmas associated with palliative care and help distinguish it from hospice care. The primary goal of palliative care is the treatment of suffering and improvement of quality of life [73]. Hospice can be a valuable resource for end of life care [73].
8.4. Strengthening family supports
There are many aspects of quality of life relevant to both patients and families that should be addressed in addition to the clinical visit [73]. This can include access to care, billing and insurance issues, continuing education, and family counseling. Social workers, essential members of the core team, can help families to access resources outside of their immediate support group. Database-linked surveys could identify key quality of life issues not fully addressed during clinical encounters. This can be in turn used to determine, revise, and optimize resources and approaches used to support the family at home and between visits. Existing metrics for assessing quality of life, including PedsQL and other questionnaires have not yet been validated in this population.
Leukodystrophy centers should develop resource-categorized central web resources, such as the Global Leukodystrophy Initiative (GLIA) and National Organization for Rare Disorders (NORD) websites that will provide educational materials and links to leukodystrophy clinics, research centers, and disease-specific advocacy groups, such as MLD Foundation, Foundation to Fight H-ABC, PMD Foundation, etc. In general, online resources are able to quickly report on relevant innovations, changes in care strategies and resources, to communicate about research and therapeutic opportunities, and allow a more active role as parents, clients, and patient advocates.
9. Conclusions
Our goal with this document is to provide a framework to address the multi-faceted needs of patients with leukodystrophies, maximizing their quality of life. A dedicated leukodystrophy center is only one part of the important network of providers and may not be accessible to all patients and their families. A primary goal is to create a wider network of physicians who are qualified to accommodate patients who do not have regular access to sites within formal leukodystrophy care centers. Efforts are underway to develop a system that allows more experienced specialists to train providers beyond the geographical reach of the clinical sites and have specialized physicians actively communicate with local physicians and help them access the resources they need.
The ultimate goal of care for a patient with leukodystrophy is to enhance both the quality and duration of life. While a definitive diagnosis may inform disease-specific therapies and research eligibility, and represents a major milestone in the patient’s clinical odyssey, the lack of a diagnosis should not preclude comprehensive preventative and symptomatic care. All patients deserve, at a minimum, a comprehensive prevention and symptom management plan. Delivery of such care requires the involvement of a multidisciplinary team, ideally in the context of a dedicated leukodystrophy center, working in collaboration with local pediatricians and health care providers. As guided by the patient’s changing needs, the team may include geneticists and genetic counselors, neurologists, complex care pediatricians, pulmonologists and respiratory therapists, gastroenterologists, speech therapists, endocrinologists, physiatrists, orthopedic surgeons and physical/occupational therapists, specialists in palliative and hospice care, and social workers.
9.1. About the Global Leukodystrophy Initiative
The Global Leukodystrophy Initiative (GLIA), a consortium of leukodystrophy experts and patient advocates, was founded in 2013 with the aim of standardizing guidelines for the diagnosis and management of leukodystrophies. For more information, or to join our organization, please visit us at theglia.org or contact the GLIA Coordinator, Omar Sherbini, at (215) 590–3068.
Funding sources
The GLIA consensus meeting was funded in part by a grant from the Departments of Neurology and Genetics at Children’s National Medical Center and the members of the Leukodystrophy Alliance. Additional funding includes, LB: supported by the National Institutes of Health (NIH) National Center for Advancing Translational Sciences (Award Number UL1TR001876); GB: Research Scholar Junior 1 award from the Fonds de Recherche du Québec en Santé (FRQS) (2012–2016) and the New Investigator Salary Award from the Canadian Institutes for Health Research (2017–2022); JLB: Supported by the PCMC Foundation, NIH DP2 MH100008, March of Dimes Foundation research grant, and the Vanishing White Matter Foundation; SHE: Supported by funding from the Hunter’s Hope Foundation for Development of Clinical Practice Guidelines (CPGs); JLF: Supported by funding from the National Institutes of Health, National Institute of Child Health and Human Development via Children’s National Medical Center Child Health Research Career Development Award (2K12HD001399-11) (salary and research support); EM: Endocrine Exploratory Research Fund, Division of Endocrinology and Diabetes, Children’s Hospital of Philadelphia; WR: Funded in part by the Sterol and Isoprenoid Consortium (U54HD061939) which is part of Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research (ORDR), NCATS, in collaboration with the Eunice Kennedy Shriver National Institute of Child Health and Human Development; KVH: Supported by grants from the Lucile Packard Foundation (salary support) and the Child Neurology Foundation (research and salary support); AV: Supported by grants from the National Institutes of Health, National Institute of Neurologic Disorders and Stroke (1K08NS060695) and the Myelin Disorders Bioregistry Project.
Conflicts of interest
As members of GLIA, we participate in clinical and research programs dedicated to the care of leukodystrophy patients. HA is serving as Principal Investigator for an X-linked Adrenoleukodystrophy (X-ALD) therapeutic trial sponsored by BlueBirdBio, Inc.; DS is President and Chair of the Board at the MLD Foundation; DH is a member of GLIA and is the President and Chair of the Board of the PMD Foundation, and the Leukodystrophy Alliance. AF is a paid member of the Drug Monitoring and Safety Committee for BlueBirdBio, Inc., TEVA Pharmaceuticals, Stealth Pharmaceuticals, and is a paid consultant for Vertex Pharmaceuticals, Ambry Genetics, and Aevi Genomics. Otherwise, there are no reports of conflicts of interest.
Abbreviations:
- 4H
hypomyelination, hypogonadotropic hypogonadism and hypodontia syndrome
- AAC
augmentative and alternative communication
- ACTH
adrenocorticotropic hormone
- ADLD
adult-onset autosomal dominant leukodystrophy
- AGS
Aicardi-Goutières Syndrome
- AxD
Alexander Disease
- BiPAP
bilevel positive airway pressure
- CPAP
Continuous Positive Airway Pressure
- CRIES
Cry, Requires O2, Increased Vital Signs, Expression, Sleeplessness Scale
- CT
computed tomography
- CTX
cerebrotendinous xanthomatosis
- DBS
deep brain stimulation
- DEXA or DXA
dual-energy X-ray absorptiometry
- EEG
electroencephalogram
- FEES
fiberoptic endoscopic evaluation of swallow
- FLACC
Face, Legs, Activity, Cry, Consolability Scale
- FSH
follicular stimulating hormone
- G-tube
gastrostomy tube
- GER
gastroesophageal reflux
- GJ-tube
gastrojejunostomy tube
- GLIA
Global Leukodystrophy Initiative
- GMFCS
Gross Motor Function Classification System
- LH
luteinizing hormone
- MBS
modified barium swallow study
- MLC
megalencephalic leukoencephalopathy
- MLD
metachromatic leukodystrophy
- MRI
Magnetic Resonance Imaging
- ODDD
oculodentodigital dysplasia
- OSA
obstructive sleep apnea
- PedsQL
Pediatric Quality of Life Inventory
- PTH
Parathyroid Hormone
- QoL
quality of life
- SLE
systemic lupus erythematosus
- SLP
speech-language pathology
- UTI
urinary tract infection
- VWM
vanishing white matter disease
- X-ALD
X-linked Adrenoleukodystrophy
Footnotes
Uncited reference
[23]
References
- [1].Gordon HB, Letsou A, Bonkowsky JL, The leukodystrophies, Semin. Neurol 34(3) (2014) 312–320. [DOI] [PubMed] [Google Scholar]
- [2].Van Haren K, Bonkowsky JL, Bernard G, Murphy JL, Pizzino A, Helman G, et al. , Consensus statement on preventive and symptomatic care of leukodystrophy patients, Mol. Genet. Metab 114 (4) (2015) 516–526. [DOI] [PubMed] [Google Scholar]
- [3].Eichler FS, Cox TM, Crombez E, Dali CI, Kohlschutter A, Metachromatic Leukodystrophy: an assessment of disease burden, J. Child Neurol 31 (13) (2016) 1457–1463. [DOI] [PubMed] [Google Scholar]
- [4].Brimley CJ, Lopez J, van Haren K, Wilkes J, Sheng X, Nelson C, et al. , National variation in costs and mortality for leukodystrophy patients in US children’s hospitals, Pediatr. Neurol 49 (3) (2013), (156–62.e1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Helman G, Van Haren K, Bonkowsky JL, Bernard G, Pizzino A, Braverman N, et al. , Disease specific therapies in leukodystrophies and leukoencephalopathies, Mol. Genet. Metab 114 (4) (2015) 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nelson C, Mundorff MB, Korgenski EK, Brimley CJ, Srivastava R, Bonkowsky JL, Determinants of health care use in a population-based leukodystrophy cohort, J. Pediatr 162 (3) (2013), (624–8.e1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Escolar ML, West T, Dallavecchia A, Poe MD, LaPoint K, Clinical management of Krabbe disease, J. Neurosci. Res 94 (11) (2016) 1118–1125. [DOI] [PubMed] [Google Scholar]
- [8].Anderson HM, Wilkes J, Korgenski EK, Pulsipher MA, Blaschke AJ, Hersh AL, et al. , Preventable infections in children with leukodystrophy, Ann. Clin. Transl. Neurol 1 (5) (2014) 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bonkowsky JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava R, The burden of inherited leukodystrophies in children, Neurology 75 (8) (2010) 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW, Classification and definition of disorders causing hypertonia in childhood, Pediatrics 111 (1) (2003) e89–97. [DOI] [PubMed] [Google Scholar]
- [11].Costello DJ, Eichler AF, Eichler FS, Leukodystrophies: classification, diagnosis, and treatment, Neurologist 15 (6) (2009) 319–328. [DOI] [PubMed] [Google Scholar]
- [12].Varni JW, Seid M, Rode CA, The PedsQL: measurement model for the pediatric quality of life inventory, Med. Care 37 (2) (1999) 126–139. [DOI] [PubMed] [Google Scholar]
- [13].Berg AT, Smith SN, Frobish D, Beckerman B, Levy SR, Testa FM, et al. , Longitudinal assessment of adaptive behavior in infants and young children with newly diagnosed epilepsy: influences of etiology, syndrome, and seizure control, Pediatrics 114 (3) (2004) 645–650. [DOI] [PubMed] [Google Scholar]
- [14].Vadivelu S, Stratton A, Pierce W, Pediatric tone management, Phys. Med. Rehabil. Clin. N. Am 26 (1) (2015) 69–78. [DOI] [PubMed] [Google Scholar]
- [15].Delgado MR, Hirtz D, Aisen M, Ashwal S, Fehlings DL, McLaughlin J, et al. , Practice parameter: pharmacologic treatment of spasticity in children and adolescents with cerebral palsy (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Neurology 74 (4) (2010) 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Papavasiliou AS, Management of motor problems in cerebral palsy: a critical update for the clinician, Eur. J. Paediatr. Neurol 13 (5) (2009) 387–396. [DOI] [PubMed] [Google Scholar]
- [17].Ronan S, Gold JT, Nonoperative management of spasticity in children, Childs Nerv. Syst 23 (9) (2007) 943–956. [DOI] [PubMed] [Google Scholar]
- [18].Simpson DM, Gracies JM, Graham HK, Miyasaki JM, Naumann M, Russ-man B, et al. , Assessment: botulinum neurotoxin for the treatment of spasticity (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology, Neurology 70 (19) (2008) 1691–1698. [DOI] [PubMed] [Google Scholar]
- [19].Motta F, Antonello CE, Analysis of complications in 430 consecutive pediatric patients treated with intrathecal baclofen therapy: 14-year experience, J. Neurosurg. Pediatr 13 (3) (2014) 301–306. [DOI] [PubMed] [Google Scholar]
- [20].Albright AL, Ferson SS, Intrathecal baclofen therapy in children, Neurosurg. Focus 21 (2) (2006), e3. [DOI] [PubMed] [Google Scholar]
- [21].Hoving MA, van Raak EP, Spincemaille GH, Palmans LJ, Becher JG, Vles JS, Efficacy of intrathecal baclofen therapy in children with intractable spastic cerebral palsy: a randomised controlled trial, Eur. J. Paediatr. Neurol 13 (3) (2009) 240–246. [DOI] [PubMed] [Google Scholar]
- [22].Abbott R, Neurosurgical management of abnormal muscle tone in childhood, Pediatr. Clin. N. Am 51 (2) (2004) 457–475. [DOI] [PubMed] [Google Scholar]
- [23].Buizer AI, van Schie PE, Bolster EA, van Ouwerkerk WJ, Strijers RL, van de Pol LA, et al. , Effect of selective dorsal rhizotomy on daily care and comfort in non-walking children and adolescents with severe spasticity, Eur. J. Paediatr. Neurol 21 (2) (2017) 350–357. [DOI] [PubMed] [Google Scholar]
- [24].McLaughlin J, Bjornson K, Temkin N, Steinbok P, Wright V, Reiner A, et al. , Selective dorsal rhizotomy: meta-analysis of three randomized controlled trials, Dev. Med. Child Neurol 44 (1) (2002) 17–25. [DOI] [PubMed] [Google Scholar]
- [25].Thomason P, Baker R, Dodd K, Taylor N, Selber P, Wolfe R, et al. , Single-event multilevel surgery in children with spastic diplegia: a pilot randomized controlled trial, J. Bone Joint Surg. Am 93 (5) (2011) 451–460. [DOI] [PubMed] [Google Scholar]
- [26].Bendon AA, George KA, Patel D, Perioperative complications and outcomes in children with cerebral palsy undergoing scoliosis surgery, Paediatr. Anaesth 26(10) (2016) 970–975. [DOI] [PubMed] [Google Scholar]
- [27].Pavone L, Burton J, Gaebler-Spira D, Dystonia in childhood: clinical and objective measures and functional implications, J. Child Neurol 28 (3) (2013) 340–350. [DOI] [PubMed] [Google Scholar]
- [28].Haugh AB, Pandyan AD, Johnson GR, A systematic review of the Tardieu Scale for the measurement of spasticity, Disabil. Rehabil 28 (15) (2006) 899–907. [DOI] [PubMed] [Google Scholar]
- [29].Battini R, Sgandurra G, Petacchi E, Guzzetta A, Di Pietro R, Giannini MT, et al. , Movement disorder-childhood rating scale: reliability and validity, Pediatr. Neurol 39 (4) (2008) 259–265. [DOI] [PubMed] [Google Scholar]
- [30].Jankovic J, Medical treatment of dystonia, Mov. Disord 28 (7) (2013) 1001–1012. [DOI] [PubMed] [Google Scholar]
- [31].Lubarr N, Bressman S, Treatment of generalized dystonia, Curr. Treat. Options Neurol 13 (3) (2011) 274–289. [DOI] [PubMed] [Google Scholar]
- [32].Mink JW, Special concerns in defining, studying, and treating dystonia in children, Mov. Disord 28 (7) (2013) 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cardoso F, Jankovic J, Dystonia and dyskinesia, Psychiatr. Clin. North Am 20(4) (1997) 821–838. [DOI] [PubMed] [Google Scholar]
- [34].Deon LL, Gaebler-Spira D, Assessment and treatment of movement disorders in children with cerebral palsy, Orthop. Clin. North Am 41 (4) (2010) 507–517. [DOI] [PubMed] [Google Scholar]
- [35].Albright AL, Barry MJ, Shafton DH, Ferson SS, Intrathecal baclofen for generalized dystonia, Dev. Med. Child Neurol 43 (10) (2001) 652–657. [DOI] [PubMed] [Google Scholar]
- [36].Bishop N, Arundel P, Clark E, Dimitri P, Farr J, Jones G, et al. , Fracture prediction and the definition of osteoporosis in children and adolescents: the ISCD 2013 Pediatric Official Positions, J. Clin. Densitom 17 (2) (2014) 275–280. [DOI] [PubMed] [Google Scholar]
- [37].Federico A, Dotti MT, Gallus GN, Cerebrotendinous xanthomatosis, in: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al. (Eds.), GeneReviews(R), University of Washington, Seattle, Seattle (WA), 1993, (GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved). [Google Scholar]
- [38].Martini G, Mignarri A, Ruvio M, Valenti R, Franci B, Del Puppo M, et al. , Long-term bone density evaluation in cerebrotendinous xanthomatosis: evidence of improvement after chenodeoxycholic acid treatment, Calcif. Tissue Int 92 (3) (2013) 282–286. [DOI] [PubMed] [Google Scholar]
- [39].Bianchi ML, Leonard MB, Bechtold S, Hogler W, Mughal MZ, Schonau E, et al. , Bone health in children and adolescents with chronic diseases that may affect the skeleton: the 2013 ISCD Pediatric Official Positions, J. Clin. Densitom 17 (2) (2014) 281–294. [DOI] [PubMed] [Google Scholar]
- [40].Henderson RC, Berglund LM, May R, Zemel BS, Grossberg RI, Johnson J, et al. , The relationship between fractures and DXA measures of BMD in the distal femur of children and adolescents with cerebral palsy or muscular dystrophy, J. Bone Miner. Res 25 (3) (2010) 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Coppola G, Fortunato D, Mainolfi C, Porcaro F, Roccaro D, Signoriello G, et al. , Bone mineral density in a population of children and adolescents with cerebral palsy and mental retardation with or without epilepsy, Epilepsia 53 (12) (2012) 2172–2177. [DOI] [PubMed] [Google Scholar]
- [42].Ito T, Jensen RT, Association of long-term proton pump inhibitor therapy with bone fractures and effects on absorption of calcium, vitamin B12, iron, and magnesium, Curr. Gastroenterol. Rep 12 (6) (2010) 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. , Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline, J. Clin. Endocrinol. Metab 96 (7) (2011) 1911–1930. [DOI] [PubMed] [Google Scholar]
- [44].Zemel BS, Stallings VA, Leonard MB, Paulhamus DR, Kecskemethy HH, Harcke HT, et al. , Revised pediatric reference data for the lateral distal femur measured by Hologic Discovery/Delphi dual-energy X-ray absorptiometry, J. Clin. Densitom 12 (2) (2009) 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Holmes L, Cornes MJ, Foldi B, Miller F, Dabney K, Clinical epidemiologic characterization of orthopaedic and neurological manifestations in children with leukodystrophies, J. Pediatr. Orthop 31 (5) (2011) 587–593. [DOI] [PubMed] [Google Scholar]
- [46].Patel J, Shapiro F, Simultaneous progression patterns of scoliosis, pelvic obliquity, and hip subluxation/dislocation in non-ambulatory neuromuscular patients: an approach to deformity documentation, J. Child. Orthop 9 (5) (2015) 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hagglund G, Lauge-Pedersen H, Persson Bunke M, Rodby-Bousquet E, Windswept hip deformity in children with cerebral palsy: a population-based prospective follow-up, J. Child. Orthop 10 (4) (2016) 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wynter M, Gibson N, Willoughby KL, Love S, Kentish M, Thomason P, et al. , Australian hip surveillance guidelines for children with cerebral palsy: 5-year review, Dev. Med. Child Neurol 57 (9) (2015) 808–820. [DOI] [PubMed] [Google Scholar]
- [49].Cunin V, Early-onset scoliosis: current treatment, Orthop. Traumatol. Surg. Res 101 (1 Suppl) (2015) S109–18. [DOI] [PubMed] [Google Scholar]
- [50].Downs J, Bergman A, Carter P, Anderson A, Palmer GM, Roye D, et al. , Guidelines for management of scoliosis in Rett syndrome patients based on expert consensus and clinical evidence, Spine 34 (17) (2009) E607–17. [DOI] [PubMed] [Google Scholar]
- [51].Morris C, Bartlett D, Gross motor function classification system: impact and utility, Dev. Med. Child Neurol 46 (1) (2004) 60–65. [DOI] [PubMed] [Google Scholar]
- [52].Kehrer C, Blumenstock G, Raabe C, Krageloh-Mann I, Development and reliability of a classification system for gross motor function in children with metachromatic leucodystrophy, Dev. Med. Child Neurol 53 (2) (2011) 156–160. [DOI] [PubMed] [Google Scholar]
- [53].Kolivras A, Aeby A, Crow YJ, Rice GI, Sass U, Andre J, Cutaneous histopathological findings of Aicardi-Goutieres syndrome, overlap with chilblain lupus, J. Cutan. Pathol 35 (8) (2008) 774–778. [DOI] [PubMed] [Google Scholar]
- [54].Renaud DL, Clinical approach to leukoencephalopathies, Semin. Neurol 32 (1) (2012) 29–33. [DOI] [PubMed] [Google Scholar]
- [55].Rizzo WB, S’Aulis D, Jennings MA, Crumrine DA, Williams ML, Elias PM, Ichthyosis in Sjogren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion, Arch. Dermatol. Res 302 (6) (2010) 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Faria J, Harb J, Hilton A, Yacobucci D, Pizzuto M, Salivary botulinum toxin injection may reduce aspiration pneumonia in neurologically impaired children, Int. J. Pediatr. Otorhinolaryngol 79 (12) (2015) 2124–2128. [DOI] [PubMed] [Google Scholar]
- [57].Montgomery J, McCusker S, Lang K, Grosse S, Mace A, Lumley R, et al. , Managing children with sialorrhoea (drooling): experience from the first 301 children in our saliva control clinic, Int. J. Pediatr. Otorhinolaryngol 85 (2016) 33–39. [DOI] [PubMed] [Google Scholar]
- [58].Erasmus CE, van Hulst K, Rotteveel JJ, Willemsen MA, Jongerius PH, Clinical practice: swallowing problems in cerebral palsy, Eur. J. Pediatr 171 (3) (2012) 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Eiland LS, Glycopyrrolate for chronic drooling in children, Clin. Ther 34 (4) (2012) 735–742. [DOI] [PubMed] [Google Scholar]
- [60].Blommaert D, van Hulst K, Hoogen FJ, Erasmus CE, Wortmann SB, Diagnosis and management of drooling in children with progressive dystonia: a case series of patients with MEGDEL syndrome, J. Child Neurol 31 (10) (2016) 1220–1226. [DOI] [PubMed] [Google Scholar]
- [61].Sharma A, Ramaswamy S, Dahl E, Dewan V, Intraoral application of atropine sulfate ophthalmic solution for clozapine-induced sialorrhea, Ann. Pharmacother 38 (9) (2004) 1538. [DOI] [PubMed] [Google Scholar]
- [62].Lungren MP, Halula S, Coyne S, Sidell D, Racadio JM, Patel MN, Ultrasound-guided botulinum toxin type a salivary gland injection in children for refractory sialorrhea: 10-year experience at a large tertiary children’s hospital, Pediatr. Neurol 54 (2016) 70–75. [DOI] [PubMed] [Google Scholar]
- [63].Penagini F, Mameli C, Fabiano V, Brunetti D, Dilillo D, Zuccotti GV, Dietary intakes and nutritional issues in neurologically impaired children, Nutrients 7(11) (2015) 9400–9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rempel G, The importance of good nutrition in children with cerebral palsy, Phys. Med. Rehabil. Clin. N. Am 26 (1) (2015) 39–56. [DOI] [PubMed] [Google Scholar]
- [65].Morgan AT, Dodrill P, Ward EC, Interventions for oropharyngeal dysphagia in children with neurological impairment, Cochrane Database Syst. Rev 10 (2012) Cd009456. [DOI] [PubMed] [Google Scholar]
- [66].Ashford J, McCabe D, Wheeler-Hegland K, Frymark T, Mullen R, Musson N, et al. , Evidence-based systematic review: oropharyngeal dysphagia behavioral treatments. Part III—impact of dysphagia treatments on populations with neurological disorders, J. Rehabil. Res. Dev 46 (2) (2009) 195–204. [PubMed] [Google Scholar]
- [67].Frymark T, Schooling T, Mullen R, Wheeler-Hegland K, Ashford J, McCabe D, et al. , Evidence-based systematic review: oropharyngeal dysphagia behavioral treatments. Part I—background and methodology, J. Rehabil. Res. Dev 46 (2) (2009) 175–183. [PubMed] [Google Scholar]
- [68].Schlosser RW, Wendt O, Effects of augmentative and alternative communication intervention on speech production in children with autism: a systematic review, Am. J. Speech Lang. Pathol 17 (3) (2008) 212–230. [DOI] [PubMed] [Google Scholar]
- [69].Aisen ML, Kerkovich D, Mast J, Mulroy S, Wren TA, Kay RM, et al. , Cerebral palsy: clinical care and neurological rehabilitation, Lancet Neurol 10 (9) (2011) 844–852. [DOI] [PubMed] [Google Scholar]
- [70].Czinn SJ, Blanchard S, Gastroesophageal reflux disease in neonates and infants: when and how to treat, Paediatr. Drugs 15 (1) (2013) 19–27. [DOI] [PubMed] [Google Scholar]
- [71].Stone B, Hester G, Jackson D, Richardson T, Hall M, Gouripeddi R, et al. , Effectiveness of fundoplication or gastrojejunal feeding in children with neurologic impairment, Hosp. Pediatr 7 (3) (2017) 140–148. [DOI] [PubMed] [Google Scholar]
- [72].Jadcherla SR, Peng J, Moore R, Saavedra J, Shepherd E, Fernandez S, et al. , Impact of personalized feeding program in 100 NICU infants: pathophysiology-based approach for better outcomes, J. Pediatr. Gastroenterol. Nutr 54 (1) (2012) 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Klick JC, Hauer J, Pediatric palliative care, Curr. Probl. Pediatr. Adolesc. Health Care 40 (6) (2010) 120–151. [DOI] [PubMed] [Google Scholar]
- [74].Cook S, Hooper V, Nasser R, Larsen D, Effect of gastrostomy on growth in children with neurodevelopmental disabilities, Can. J. Diet. Pract. Res 66 (1) (2005) 19–24. [DOI] [PubMed] [Google Scholar]
- [75].Livingston MH, Shawyer AC, Rosenbaum PL, Jones SA, Walton JM, Fundoplication and gastrostomy versus percutaneous gastrojejunostomy for gastroesophageal reflux in children with neurologic impairment: a systematic review and meta-analysis, J. Pediatr. Surg 50 (5) (2015) 707–714. [DOI] [PubMed] [Google Scholar]
- [76].McGreevy CM, Pentakota SR, Mohamed O, Sigler K, Mosenthal AC, Berlin A, Gastrostomy tube placement: an opportunity for establishing patient-centered goals of care, Surgery (2016). [DOI] [PubMed] [Google Scholar]
- [77].Leonard H, Ravikumara M, Baikie G, Naseem N, Ellaway C, Percy A, et al. , Assessment and management of nutrition and growth in Rett syndrome, J. Pediatr. Gastroenterol. Nutr 57 (4) (2013) 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Slattery J, Morgan A, Douglas J, Early sucking and swallowing problems as predictors of neurodevelopmental outcome in children with neonatal brain injury: a systematic review, Dev. Med. Child Neurol 54 (9) (2012) 796–806. [DOI] [PubMed] [Google Scholar]
- [79].Wheeler-Hegland K, Ashford J, Frymark T, McCabe D, Mullen R, Musson N, et al. , Evidence-based systematic review: oropharyngeal dysphagia behavioral treatments. Part II—impact of dysphagia treatment on normal swallow function, J. Rehabil. Res. Dev 46 (2) (2009) 185–194. [PubMed] [Google Scholar]
- [80].Pahsini K, Marinschek S, Khan Z, Dunitz-Scheer M, Scheer PJ, Unintended adverse effects of enteral nutrition support: parental perspective, J. Pediatr. Gastroenterol. Nutr 62 (1) (2016) 169–173. [DOI] [PubMed] [Google Scholar]
- [81].Craig GM, Scambler G, Spitz L, Why parents of children with neurodevelopmental disabilities requiring gastrostomy feeding need more support, Dev. Med. Child Neurol 45 (3) (2003) 183–188. [DOI] [PubMed] [Google Scholar]
- [82].Amarenco G, Bristol Stool Chart: prospective and monocentric study of “stools introspection” in healthy subjects, Prog. Urol 24 (11) (2014) 708–713. [DOI] [PubMed] [Google Scholar]
- [83].Araujo LA, Silva LR, Mendes FA, Digestive tract neural control and gastrointestinal disorders in cerebral palsy, J. Pediatr 88 (6) (2012) 455–464. [DOI] [PubMed] [Google Scholar]
- [84].Kim J, Sun Z, Ezekian B, Schooler GR, Prasad VK, Kurtzberg J, et al. , Gallbladder abnormalities in children with metachromatic leukodystrophy, J. Surg. Res 208 (2017) 187–191. [DOI] [PubMed] [Google Scholar]
- [85].McFadden K, Ranganathan S, Pathology of the gallbladder in a child with metachromatic leukodystrophy, Pediatr. Dev. Pathol 18 (3) (2015) 228–230. [DOI] [PubMed] [Google Scholar]
- [86].Canelas HM, Quintao EC, Scaff M, Vasconcelos KS, Brotto MW, Cerebrotendinous xanthomatosis: clinical and laboratory study of 2 cases, Acta Neurol. Scand 67 (5) (1983) 305–311. [DOI] [PubMed] [Google Scholar]
- [87].Lorioli L, Cicalese MP, Silvani P, Assanelli A, Salvo I, Mandelli A, et al. , Abnormalities of acid-base balance and predisposition to metabolic acidosis in Metachromatic Leukodystrophy patients, Mol. Genet. Metab 115 (1) (2015) 48–52. [DOI] [PubMed] [Google Scholar]
- [88].Bach JR, Baird JS, Plosky D, Navado J, Weaver B, Spinal muscular atrophy type 1: management and outcomes, Pediatr. Pulmonol 34 (1) (2002) 16–22. [DOI] [PubMed] [Google Scholar]
- [89].Ball LJ, Fager S, Fried-Oken M, Augmentative and alternative communication for people with progressive neuromuscular disease, Phys. Med. Rehabil. Clin. N. Am 23 (3) (2012) 689–699. [DOI] [PubMed] [Google Scholar]
- [90].Millar DC, Light JC, Schlosser RW, The impact of augmentative and alternative communication intervention on the speech production of individuals with developmental disabilities: a research review, J. Speech Lang. Hear. Res 49 (2) (2006) 248–264. [DOI] [PubMed] [Google Scholar]
- [91].Bishop DV, Snowling MJ, Thompson PA, Greenhalgh T, Phase 2 of CATALISE: a multinational and multidisciplinary Delphi consensus study of problems with language development: terminology, J. Child Psychol. Psychiatry (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Brady NC, Bruce S, Goldman A, Erickson K, Mineo B, Ogletree BT, et al. , Communication services and supports for individuals with severe disabilities: guidance for assessment and intervention, Am. J. Intellect. Dev. Disabil 121 (2) (2016) 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Duffy JR, Motor Speech Disorders: Substrates, Differential Diagnosis, and Management, 2nd ed., Elsevier Mosby, St. Louis, MO, 2005. [Google Scholar]
- [94].Ko ML, McConachie H, Jolleff N, Outcome of recommendations for augmentative communication in children, Child Care Health Dev. 24 (3) (1998) 195–205. [DOI] [PubMed] [Google Scholar]
- [95].Murray E, McCabe P, Ballard KJ, A systematic review of treatment outcomes for children with childhood apraxia of speech, Am. J. Speech Lang. Pathol 23 (3) (2014) 486–504. [DOI] [PubMed] [Google Scholar]
- [96].Boyle J, McCartney E, Forbes J, O’Hare A, A randomised controlled trial and economic evaluation of direct versus indirect and individual versus group modes of speech and language therapy for children with primary language impairment, Health Technol. Assess. (Winch. Eng.) 11 (25) (2007) 1–139, (iii–iv, xi–xii). [DOI] [PubMed] [Google Scholar]
- [97].Beukelman DRMP, Augmentative and Alternative Communication: Supporting Children and Adults With Complex Communication Needs, 4th ed., Paul H Brookes Publishing Co, Baltimore, MD, 2013. [Google Scholar]
- [98].Haan J, Locked-in: the syndrome as depicted in literature, Prog. Brain Res 206 (2013) 19–34. [DOI] [PubMed] [Google Scholar]
- [99].Malow BA, Byars K, Johnson K, Weiss S, Bernal P, Goldman SE, et al. , A practice pathway for the identification, evaluation, and management of insomnia in children and adolescents with autism spectrum disorders, Pediatrics 130 (Suppl. 2) (2012) S106–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Schwantes S, O’Brien HW, Pediatric palliative care for children with complex chronic medical conditions, Pediatr. Clin. N. Am 61 (4) (2014) 797–821. [DOI] [PubMed] [Google Scholar]
- [101].Wusthoff CJ, Shellhaas RA, Licht DJ, Management of common neurologic symptoms in pediatric palliative care: seizures, agitation, and spasticity, Pediatr. Clin. N. Am 54 (5) (2007) 709–733, (xi). [DOI] [PubMed] [Google Scholar]
- [102].McGrath PA, Development of the World Health Organization guidelines on cancer pain relief and palliative care in children, J. Pain Symptom Manag 12 (2) (1996) 87–92. [DOI] [PubMed] [Google Scholar]
- [103].Fisher RS, Redefining epilepsy, Curr. Opin. Neurol 28 (2) (2015) 130–135. [DOI] [PubMed] [Google Scholar]
- [104].Bergey GK, Management of a first seizure, Continuum (Minneapolis, Minn) 22 (1 Epilepsy) (2016) 38–50. [DOI] [PubMed] [Google Scholar]
- [105].Smith PE, Epilepsy: mimics, borderland and chameleons, Pract. Neurol 12 (5) (2012) 299–307. [DOI] [PubMed] [Google Scholar]
- [106].Axelrod FB, Genetic autonomic disorders, Semin. Pediatr. Neurol 20 (1) (2013) 3–11. [DOI] [PubMed] [Google Scholar]
- [107].Axelrod FB, Chelimsky GG, Weese-Mayer DE, Pediatric autonomic disorders, Pediatrics 118 (1) (2006) 309–321. [DOI] [PubMed] [Google Scholar]
- [108].Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E, et al. , GFAP mutations, age at onset, and clinical subtypes in Alexander disease, Neurology 77 (13) (2011) 1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Terlizzi R, Calandra-Buonaura G, Zanigni S, Barletta G, Capellari S, Guaraldi P, et al. , A longitudinal study of a family with adult-onset autosomal dominant leukodystrophy: clinical, autonomic and neuropsychological findings, Auton. Neurosci 195 (2016) 20–26. [DOI] [PubMed] [Google Scholar]
- [110].Nahhas N, Sabet Rasekh P, Vanderver A, Padiath Q, Autosomal dominant leukodystrophy with autonomic disease, in: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al. (Eds.), GeneReviews(R), University of Washington, Seattle, Seattle (WA), 1993, (GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved). [PubMed] [Google Scholar]
- [111].Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bu-giani M, et al. , Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations, Neurology 83 (21) (2014) 1898–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Idouji K, Kuriyama M, Fujiyama J, Osame M, Hoshita T, Hypothyroidism with increased serum levels of cholestanol and bile alcohol—analogous symptoms to cerebrotendinous xanthomatosis, Rinsho Shinkeigaku 31 (4) (1991) 402–406. [PubMed] [Google Scholar]
- [113].Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. , Clinical and molecular phenotype of Aicardi-Goutieres syndrome, Am. J. Hum. Genet 81 (4) (2007) 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Valdivielso P, Calandra S, Duran JC, Garuti R, Herrera E, Gonzalez P, Coronary heart disease in a patient with cerebrotendinous xanthomatosis, J. Intern. Med 255 (6) (2004) 680–683. [DOI] [PubMed] [Google Scholar]