

Abstract
Why layers II/III of entorhinal cortex (EC) deteriorate in advance of other regions during the earliest stages of Alzheimer's disease is poorly understood. Failure of retrograde trophic support from synapses to cell bodies is a common cause of neuronal atrophy, and we accordingly tested for early-life deterioration in projections of rodent layer II EC neurons. Using electrophysiology and quantitative imaging, changes in EC terminals during young adulthood were evaluated in male rats and mice. Field excitatory postsynaptic potentials, input/output curves, and frequency following capacity by lateral perforant path (LPP) projections from lateral EC to dentate gyrus were unchanged from 3 to 8–10 months of age. In contrast, the unusual presynaptic form of long-term potentiation (LTP) expressed by the LPP was profoundly impaired by 8 months in rats and mice. This impairment was accompanied by a reduction in the spine to terminal endocannabinoid signaling needed for LPP-LTP induction and was offset by an agent that enhances signaling. There was a pronounced age-related increase in synaptophysin within LPP terminals, an effect suggestive of incipient pathology. Relatedly, presynaptic levels of TrkB—receptors mediating retrograde trophic signaling—were reduced in the LPP terminal field. LTP and TrkB content were also reduced in the medial perforant path of 8- to 10-month-old rats. As predicted, performance on an LPP-dependent episodic memory task declined by late adulthood. We propose that memory-related synaptic plasticity in EC projections is unusually sensitive to aging, which predisposes EC neurons to pathogenesis later in life.
SIGNIFICANCE STATEMENT Neurons within human superficial entorhinal cortex are particularly vulnerable to effects of aging and Alzheimer's disease, although why this is the case is not understood. Here we report that perforant path projections from layer II entorhinal cortex to the dentate gyrus exhibit rapid aging in rodents, including reduced synaptic plasticity and abnormal protein content by 8–10 months of age. Moreover, there was a substantial decline in the performance of an episodic memory task that depends on entorhinal cortical projections at the same ages. Overall, the results suggest that the loss of plasticity and related trophic signaling predispose the entorhinal neurons to functional decline in relatively young adulthood.
Keywords: aging, entorhinal cortex, lateral perforant path, long-term potentiation, memory, TrkB
Introduction
The groundbreaking studies of Van Hoesen et al. (1991) and Braak and Braak (1991) showed that Alzheimer's disease (AD)-related pathology (cell loss, tangles) initially appears in temporal lobe structures. Among those, superficial layers (II, III) of entorhinal cortex (EC), and particular hippocampal subfields, are the first to deteriorate. Pathology has been proposed to “spread” from these first affected zones along projection pathways to other regions (Khan et al., 2014; Llorens-Martin et al., 2014). Degeneration of lateral EC (LEC) neurons in humans may be presaged by lesser impairments, as suggested by imaging studies showing that activity in LEC is depressed in healthy older individuals relative to young adults during episodic learning, an effect associated with poorer retention (Reagh et al., 2018). These findings accord with the hypothesis that early LEC deterioration is a precipitating condition for, rather than a consequence of, AD. They also raise questions about the causes of pre-AD impairments and whether such disturbances occur in other mammals. Available evidence from rodents, monkeys, and humans indicates that neuron loss is not characteristic of the aged entorhinal cortex in the absence of disease. The clear implication is that the observed functional problems are because of cell biological disturbances that affect neuronal operations but do not threaten neural viability (Gómez-Isla et al., 1996; Gazzaley et al., 1997; Merrill et al., 2000; Rapp et al., 2002).
Related to the above, layer II EC neurons express high levels of reelin, a glycoprotein implicated in development and plasticity. Reelin levels are altered in AD (Botella-López et al., 2006; Chin et al., 2007; Doehner and Knuesel, 2010; Herring et al., 2012) and are reduced in LEC of 24-month-old rats but only in cases with impaired learning (Stranahan et al., 2011). In 12–month-old rats, layer II LEC neurons also have dense deposits of the protease cathepsin D, suggesting lysosomal dysfunction, and are reduced in size relative to young adults (Bi et al., 2000). Neurophysiological studies of projections from EC layer II to the dentate gyrus (DG) granule cells provide further evidence for age-related changes in rodents. Barnes et al. (2000) established that long-term potentiation (LTP) of contacts between medial EC (MEC) and the DG is substantially reduced in 24-month-old rats versus young adults. Notably, baseline responses were subnormal in the old animals, an observation pointing to the possibility that the plasticity impairment was secondary to synapse loss. Subsequent experiments identified a significant reduction in the magnitude of LTP in LEC–DG synapses in 12- to 24-month-old rodents (Froc et al., 2003). Paired-pulse facilitation (PPF), a conventional measure of transmitter mobilization, was also reduced in the older rats.
Overall, available data indicate that layer II EC connections within the DG deteriorate during the second year of life in rodents. Anatomical and functional disturbances are evident throughout telencephalon during this period, and there is evidence for dendritic atrophy in cortex (Dickstein et al., 2013; Samson and Barnes, 2013) and hippocampus (Lauterborn et al., 2016) by middle age. It is accordingly possible that the unusual AD vulnerability of EC and select hippocampal subfields is because of an interaction between generalized effects of aging and a heightened regional sensitivity to AD-specific factors. Alternatively, AD-related deterioration could reflect an unusually early arrival at a pathology threshold because of a faster than normal rate of aging.
If accelerated aging is characteristic of mammalian layer II LEC, then we would expect to find defects in LEC innervation of the DG relatively early in rodent adulthood. This possibility is also of interest regarding the hypothesis that decreased synaptic plasticity negatively affects retrograde trophic support for the parent neurons, resulting in gradual atrophy (Harrington and Ginty, 2013; Ito and Enomoto, 2016). Accordingly, the present studies evaluated the status of LEC–DG synapses at different stages of young adulthood and the encoding of an aspect of episodic memory that is dependent on these connections.
Materials and Methods
Animals.
Studies used male C57BL/6J mice and Long–Evans rats (Charles River) tested at 3, 6, or 8–10 months of age. Animals were group housed with access to food and water ad libitum and maintained on a 12 h light/dark cycle; all studies were conducted during the light phase of the cycle. All procedures were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine, and were in accordance with the National Institutes of Health Guideline for the Care and Use of Laboratory Animals.
Hippocampal slice electrophysiology.
Hippocampal slices (350 µm) were prepared from the middle third of hippocampi of rats and mice as described previously (Wang et al., 2016b); the mean number of slices per animal across all experiments was two. Slices were cut using a McIlwain tissue chopper for rat (Ted Pella) and a vibrating tissue slicer (model VT1000S, Leica) for mouse, and collected into oxygenated ice-cold artificial CSF (aCSF) with high magnesium as follows (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 5 MgSO4, 26 NaHCO3, and 10 dextrose. Immediately after cutting, slices were transferred into an interface recording chamber with constant perfusion of oxygenated aCSF (31 ± 1°C; 95% O2/5% CO2) as follows (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 1.5 MgSO4, 26 NaHCO3, 2.5 CaCl2, and 10 dextrose. Experiments were initiated ∼1.5 h later and were run for up to 4 h. Field EPSPs (fEPSPs) were elicited using a bipolar stimulating electrode (twisted 65 µm formvar-insulated nichrome wire; catalog #762000, A-M Systems) and recorded with a glass pipette (2 m NaCl, 2–3 MΩ). Baseline stimulation was applied at 0.05 Hz with intensity adjusted to elicit 40–50% of the maximum population spike-free fEPSP.
For LTP analyses, recording and stimulating electrodes were placed visually ∼500–600 μm apart from each other, in either the outer third or middle third of DG molecular layer (suprapyramidal blade) for stimulation of the lateral perforant path (LPP) or medial perforant path (MPP), respectively. Electrode placement in each field was verified using paired-pulse stimulation, which elicits response facilitation in the LPP and response depression in the MPP (Dahl and Sarvey, 1989; Christie and Abraham, 1994; Wang et al., 2016b). After 20 min of baseline recordings, potentiation was induced using a single 1 s train of high-frequency stimulation (100 Hz; HFS) for the LPP. Because previous studies from our group found that three brief (0.5 s) trains of 100 Hz stimulation with the trains separated by 20 s was optimal to elicit MPP LTP (mppLTP), this stimulation protocol was used for this pathway (Trieu et al., 2015; Wang et al., 2016b). For mppLTP only, the aCSF included 1 μm picrotoxin that was infused for 40 min before stimulation (Hanse and Gustafsson, 1992). In all cases, responses to 0.05 Hz stimulation were collected for at least 60 min after HFS, and the level of LTP was assessed as the percentage change in mean fEPSP slope during the last 5 min of post-HFS recording relative to the mean fEPSP slope during the last 5 min of baseline (pre-HFS) recordings.
For frequency following analysis, 10 pulses of 10, 20, or 40 Hz stimulation were primarily applied in ascending (10 Hz first) order, and some cases were performed in descending (40 Hz first) order, which did not show a difference in response. There was a minimum of 4 min between trains, and the fEPSP did not potentiate or depress in response to any of the trains. Therefore, all cases were included. For PPF analyses, two pulses (50 ms interstimulus interval) were applied before onset of physostigmine infusion, and again 60 min after the onset of physostigmine infusion. Slope of the paired-pulse responses were measured, and the percentage PPF was calculated as the ratio of the second to the first pulse × 100 (Colgin et al., 2003).
All traces were collected using NACGather 2.0 (Theta Burst). Drugs were introduced via syringe pump to a second infusion line to achieve bath concentrations of 10 μm physostigmine (catalog #0622, Tocris Bioscience), 1 μm JZL184 (catalog #3836, Tocris Bioscience), and <0.001% dimethylsulfoxide (catalog #D5879, Sigma-Aldrich; Wang et al., 2016b, 2018b).
Fluorescence deconvolution tomography.
Rat hippocampal slices (N = 12–15 slices/group from at least three animals per group) and brains (N = 5/group for rats and N = 6/group for mice) were used. Slices were immersion fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2, cryoprotected in 20% sucrose, and subsectioned at 25 µm on a freezing microtome, and then sections were slide mounted, with the first one to two sections discarded to avoid regions of tissue damage (Rex et al., 2009; Wang et al., 2018a). Brains were fresh frozen, cryostat sectioned (25 µm), and then fixed with 4% paraformaldehyde/0.1 m phosphate buffer for 20 min at 4°C. In all cases, tissue was collected from the middle third of the hippocampus. All microscope slide-mounted tissue was processed for dual immunofluorescence as described previously (Chen et al., 2010; Seese et al., 2012; Wang et al., 2018b; Lauterborn et al., 2020). For each experiment, tissue from the age groups to be compared was processed together. Primary antisera included rabbit anti-phosphorylated FAK (1:1000; catalog #ab39967, Abcam), anti-GluN2A (1:25,000; catalog #AB1555, EMD-Millipore), and anti-TrkB (1:2000; catalog #07–225, EMD-Millipore); guinea pig anti-synaptophysin (1:2500; catalog #101004, SySy Systems); and mouse anti-PSD-95 (1:1000; catalog #MA1-045, Thermo Fisher Scientific). Secondary antibodies were goat anti-rabbit Alexa Fluor 594, goat anti-guinea pig Alexa Fluor 488, and goat anti-mouse Alexa Fluor 488 (1:1000 each; Thermo Fisher Scientific).
Digital image z-stacks (105 × 135 µm) were collected with 0.2 µm steps over a depth of 2 µm from the internal (suprapyramidal) blade of the DG middle and outer molecular layers for MPP and LPP fields, respectively. The hippocampal fissure was used to align the long edge of the camera field (hence, the sample box) over the mediolateral dimension of the molecular layer (see Fig. 4A). For rats, it was then possible to image the outer 100 µm of the molecular layer, immediately subjacent to the fissure, which corresponds to lamina (the outer molecular layer) known from numerous neuroanatomical studies to be densely innervated by the LPP (Witter, 1993). For measures in the rat MPP, the sample box was placed 100 μm away from the fissure in the middle of the molecular layer. The molecular layer of mice is significantly thinner than that of rats but contains the same three sublamina (inner, middle, outer molecular layers). Accordingly, for mouse studies we imaged across the depth of the molecular layer and then partitioned the digital images along the granule cell-to-fissure axis to collect measurements for the LPP and MPP in the outer and middle third of the molecular layer, respectively, as described previously (Wang et al., 2018a); in prior studies, these procedures detected significant differences between MPP and LPP for synaptic proteins (Wang et al., 2018a).
Figure 4.

Early aging alters presynaptic markers in the LPP terminal zone. A, Section through hippocampus shows the two zones in the DG outer molecular layer (OML) used for analysis of synaptic proteins. B, Deconvolved immunofluorescence images show the localization of p-FAK (red) and vesicular protein synaptophysin (SYN; green) in rat. Arrows point to double-labeled puncta. Scale bar, 10 µm. C, FDT analysis of the density of p-FAK immunoreactivity (ir) colocalized with synaptophysin. Measurements were made for >100,000 individually reconstructed, double-labeled (2×) synapses per slice. Values for each slice were plotted as the percentage of 2× synapses (y-axis) fitting into an ascending series of labeling-density bins (x-axis). The resultant curves for individual slices were then averaged for a group and plotted as mean ± SEM values. There was a small but significant left shift in the curve for the 8-month-old group (N = 12) relative to that for the 3-month-old group (N = 15). Scatter plot summarizes the percentage of 2× synapses with high-density p-FAK-ir (right of vertical dotted line; *p < 0.05). D, Same analysis as in C but for the density of synaptophysin-ir: there was a pronounced rightward skew in the plot for 8-month-old rats (toward greater synaptic densities) versus the 3-month-old rats, and a large increase in the proportion of terminals with dense synaptophysin-ir (scatter plot; ***p < 0.001). E, Deconvolved images show dual immunofluorescence for TrkB (red) and synaptophysin (green) in mouse. Arrows point to 2× puncta. Scale bars: 4 µm; inset, 1 µm. F, Analysis of the density of TrkB-ir in LPP terminals in mice: there was a pronounced left shift in the density frequency distribution for the 8-month-old group relative to the 3-month-old group (N = 6/group), resulting in a large reduction in the proportion of terminals with dense TrkB-ir (scatter plot; ***p < 0.001).
The z-stacks were processed for iterative deconvolution using Volocity 4.0 software (PerkinElmer). Fluorescence deconvolution tomography (FDT; Chen et al., 2010; Seese et al., 2012, 2013) was used to reconstruct in three dimensions (3D) the 28,350 µm3 sample field from each z-stack, and to quantify numbers and measure immunolabeling intensities of single- and double-labeled elements that were both detected across multiple intensity thresholds and within the size and eccentricity constraints of synapses. Elements were considered to be double labeled if there was any overlap in the fields labeled with the two fluorophores, as assessed in 3D (Chen et al., 2010). Past studies showed that the technique produces a clear separation between proteins in presynaptic versus postsynaptic compartments (Wang et al., 2018b). Approximately 30,000 synaptic profiles were assessed for each 3D sample field. Measures were collected from a minimum of four tissue sections from each hippocampal slice, and from five tissue sections for each brain, to determine the mean number of labeled elements for each region of interest per hippocampal slice or brain. These mean values were then averaged to determine the group mean ± SEM values presented in the text and illustrations.
Behavior.
The serial odor task was conducted using C57BL/6J mice as described and illustrated in previously published work (Wang et al., 2018a; Cox et al., 2019) with slight modification. Animals were not handled before the task; mice were exposed to odor cues and tested for retention on the same day. Briefly, each mouse was put into a testing chamber (Plexiglas, 30 × 25 × 21.5 cm) containing two cups without odors and was allowed to explore for 5 min. The initial cups were then replaced with two cups containing identical odor A, and the animal was allowed to explore for 3 min. The cups were then removed, and the animal was allowed to explore the empty chamber for 3 min. Following this, the mouse was presented with cups containing identical odor pairs B:B and C:C using the same exposure and interinterval timing before the final testing phase, during which the animal was presented with odors A and D (in separate cups; see Fig. 7 within the main text). For all sessions, cups were placed in the same locations, and behavior was videotaped using an overhead recorder. Scorers (blind to group) measured the time that each animal spent exploring the two cups as the number of seconds spent sniffing the odor with their nose within 2 cm of, and directed toward, the odor hole in the cup lid.
Figure 7.

LPP-dependent episodic memory task performance declines from early to late phases of young adulthood in mouse. A, Mice were tested on two paradigms. The serial odors task entailed sampling a sequence of identical odor pairs (A:A, B:B, C:C) followed by a retention trial with a novel odor paired with a previously sampled one (A:D). In the two-odor control task, an identical odor pair (A:A) was followed by a retention trial pairing odor A with a novel odor (A:D). The delay between the first pair and the A:D retention test was the same in the two paradigms. B, The 3-month-old mice (N = 12) preferentially explored the novel versus the familiar odor in the serial odors task (****p< 0.0001, paired Student's t test), whereas 8- to 10-month-old mice (N = 11) did not (p = 0.471, paired Student's t test; n.s., not significant). C, Group measures for the discrimination index calculated from data presented in B (**p = 0.0013; unpaired t test). D, Total time sampling cues in the serial odor retention test did not differ between groups (p = 0.897). E, In the two-odor control task, mice in both age groups spent a greater proportion of sampling time exploring the novel cue (***p ≤ 0.0002, both groups). F, Control saliency tests demonstrated that both odors A and D were equally sampled by mice of both age groups (p = 0.4205 and p = 0.5377 for 3- and 8- to 10-month-old groups, respectively).
The two-odor control task used the same testing chamber as did the serial odor task. For the two-odor task, the mice were placed in the chamber with the empty cups (no odor) for 5 min and then with the initial, identical odor pair for 3 min; odors used in this task were not the same as used in the serial odor task. After removal of the first scented cups, the animals remained in the empty chamber for 15 min (the amount of time covered by exposure to odors B:B and C:C in the serial odor task). At the conclusion of the 15 min, cups containing test odors (familiar odor A and novel odor D) were placed in the chamber, and animals were allowed to explore for 3 min. As for the serial task, behavior was video recorded and scored (for time spent exploring the different odor cups) by observers blind to group.
For both serial odor and two-odor tasks, animals were excluded from analyses if they failed to explore each cup in each session for a minimum of 1 s or if they escaped the chamber; testing was counterbalanced by odor location and group.
The following odorants were used: (+)-limonene (1:4000; catalog #L-2129, Sigma-Aldrich), cyclohexyl ethyl acetate (1.97:4000; catalog #0–7725, International Flavors & Fragrances), Citronellal 96% (1.5:4000; catalog #L15753, Alfa Aesar), octyl aldehyde 99% (1.5:4000; catalog #12948–1000, Acros Organics), anisole 99% (0.85:4000; catalog #153920050, Acros Organics), and 1-pentanol 99% (1.36:4000; catalog #160600250, Acros Organics). All odorants were diluted with mineral oil.
To assess preferential exploration of the novel or familiar odors during testing (both tasks), a discrimination index was calculated as the difference between the time spent sampling the novel odor and the time spent sampling the familiar odor, divided by the total amount of time spent sampling the two odors, and multiplied by 100.
Experimental design and statistical analyses.
For electrophysiological studies, results from individual hippocampal slices are reported. Slices were obtained from the following numbers of animals: five or more animals/group for LPP studies, four animals/group for MPP studies, eight or more animals/group for physostigmine baseline studies, three animals/group for physostigmine paired pulse studies, and three or more animals/group for JZL184 studies. For statistical evaluation, two-tailed unpaired Student's t test was used to compare mean data for two groups, and one-way ANOVA was used for comparisons among three groups, followed by the Bonferroni post hoc test. Input/output curves were assessed by linear regression. For immunofluorescence studies, hippocampal slices (N = 12–15 slices/group) were obtained from at least three animals per age group. The separate immunofluorescence studies of brains were conducted on five rats/group and six mice/group. A two-tailed unpaired Student's t test was used to compare mean data for two groups, and a two-way repeated-measures (RM) ANOVA was used to compare differences in the intensity–frequency curves. For statistical analysis of behavioral measures, we evaluated 11–12 mice/group following exclusion of those animals that failed to explore (as described above). A two-tailed unpaired Student's t test or, for unequal variances, a Mann–Whitney test was used to compare mean data for the two groups. All statistics were conducted using GraphPad Prism, and p ≤ 0.05 was considered to be significant.
Results
Relating rodent and human ages is recognized to be difficult. Based on multiple measures, The Jackson Laboratory suggests that mouse ages 3–6 months correspond to young adulthood and human ages 20–30 years (C. Hagan, unpublished observations; https://www.jax.org/news-and-insights/jax-blog/2017/november/when-are-mice-considered-old#); 10–14 months is middle age (e.g., onset of declines in motor skills; Ernyey et al., 2019) and is approximately equivalent to 38–47 years in humans; and 18–24 months in mice can be considered old and comparable to 56–69 years in humans (Flurkey et al., 2007). Based on these values, and literature on rats and mice (Sengupta, 2013; Dutta and Sengupta, 2016), we considered 10 months to be the beginning of middle age. The above age boundaries are somewhat arbitrary with certain late developmental processes likely continuing into young adulthood. Nonetheless, this period can reasonably be considered as a relatively stable segment of life in which rodents engage in the various aspects of adult life without the neurologic/behavioral issues that characterize the onset of middle age (Ernyey et al., 2019). Accordingly, the present analysis of early aging focused on 3 versus 8- to 10-month-old rats and mice; for some electrophysiological measures, a 6-month-old mouse group was included to evaluate an intervening age.
LPP transmission and LTP
The LPP fEPSPs recorded from the DG outer molecular layer did not differ in size or waveform in slices from mice (Fig. 1A) or rats (Fig. 1B) that were between 3 and 8–10 months of age. Input/output curves (number of axons stimulated vs fEPSP amplitude) were also comparable across age groups (Fig. 1C,D). Note that the curves tended to be more variable in mice than rats. We suspect that this reflects the greater difficulty in reproducing electrode placements in the smaller mouse dentate gyrus. Next, we tested for age effects on LPP synaptic responses to input frequencies (10, 20, and 40 Hz) that routinely occur during behavior in rats (Grossberg, 2009; Colgin, 2016). The three patterns were applied sequentially in either an ascending (10 Hz first) or descending (40 Hz first) order with a minimum of 4 min between trains. As expected, 10 pulses did not produce short-term potentiation during the post-train period. The 10 Hz trains applied to 3-month-old slices produced an initial facilitation of responses that progressively decreased over subsequent stimulation pulses (Fig. 2A). Facilitation was greater for the amplitudes than the slopes of the fEPSPs. The same pattern was obtained in tests of 8-month-old slices with no evidence for a loss of response enhancement; indeed, frequency facilitation tended to be greater in the older cases (amplitude: F(9,189) = 2.838, p = 0.0037; slope: F(9,189) = 1.39, p = 0.123). The 20 Hz trains produced a similar set of results except that the within-train decay of frequency facilitation was more pronounced (Fig. 2A). Here again, the curve for the 8-month-old group was slightly displaced upward from that for the younger rats; but the difference with age was significant (slope: F(9,189) = 2.204; p = 0.0235; amplitude: F(9,189) = 2.09, p = 0.032). The steady decline of facilitation during the train was dramatic at 40 Hz with responses falling below baseline by pulse 6, an effect suggestive of transmitter depletion. There was no evidence for an age-related impairment in the response to 40 Hz stimulation.
Figure 1.

Waveforms of LPP fEPSPs are comparable in early and later adulthood. A, B, Responses from individual slices were normalized to their peak amplitude and then averaged across all slices within a group. At left, mean ± SEM traces for the different age groups were superimposed for mouse (3-month-old, N = 8; 6-month-old, N = 9; 10-month-old, N = 7; A) and rat (3-month-old, N = 7; 8-month-old, N = 7; B); there were no obvious differences between 3-month-old and older rodents. Calibration: (in B) A, B, 50% peak fEPSP amplitude, 10 ms. At right, graphs show the initial slopes, and decay time constants (tau) did not differ between young and older mice (A; slope: p = 0.431; decay tau: p = 0.999, one-way ANOVA; 3-month-old, N = 7; 6-month-old, N = 8; 10-month-old, N = 7) and rats (B; slope: p = 0.159, t test; decay tau: p = 0.5406; 3-month-old, N = 7; 8-month-old, N = 7). C, D, Input/output (fiber volley vs fEPSP amplitude) curves were comparable across ages in mice (C; 3-month-old, N = 5; 6-month-old, N = 6, 10-month-old, N = 5) and rats (D; 3-month-old, N = 7; 8-month-old, N = 8); traces for the different age groups are shown at right. Calibration: (in D) C, D, 1 mV, 10 ms.
Figure 2.
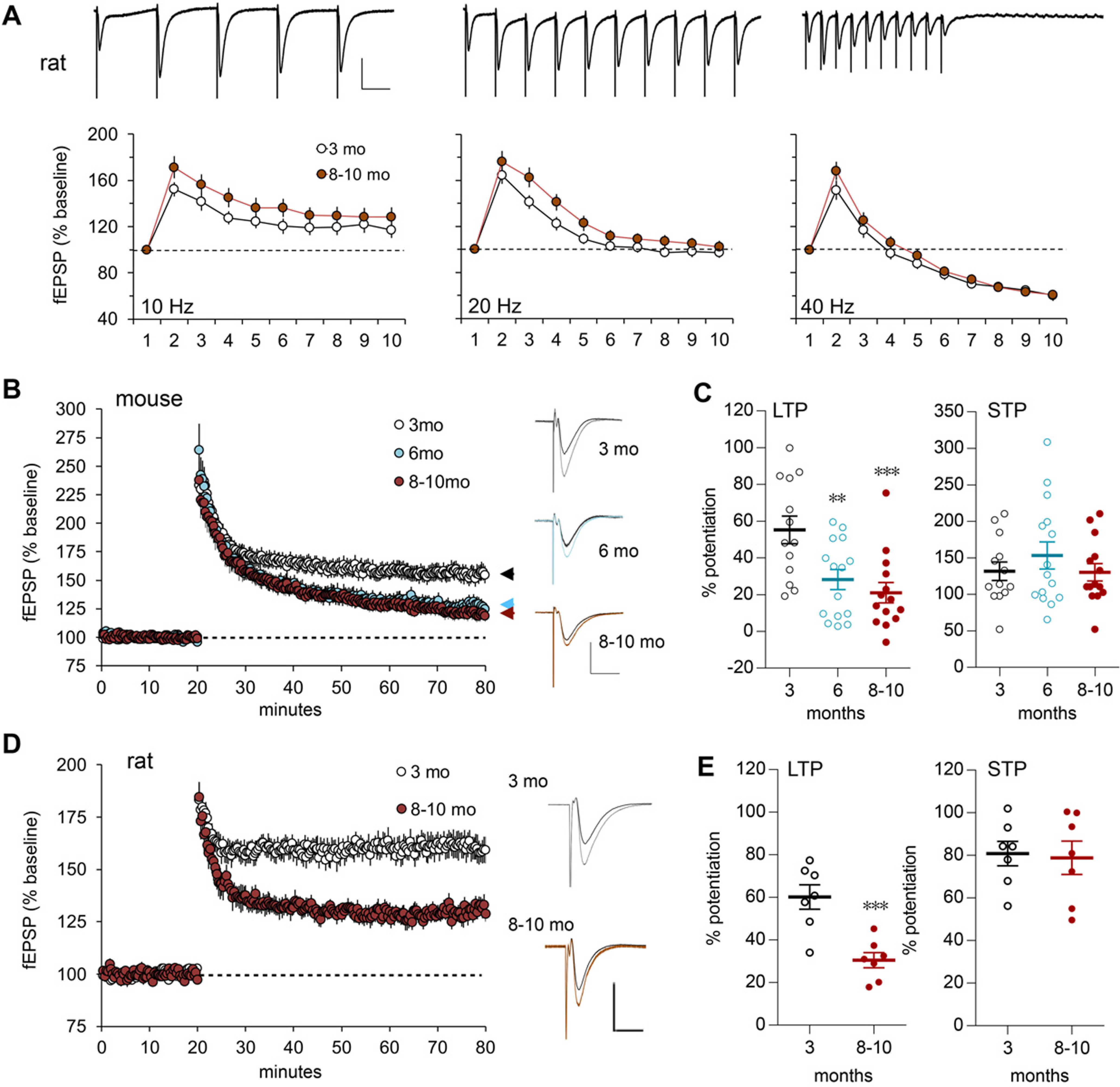
LTP but not frequency facilitation is impaired in the LPP of older adults. A, A train of 10 stimulation pulses was delivered to the LPP at 10, 20, or 40 Hz, and LPP fEPSPs were recorded in the rat DG outer molecular layer. Representative traces above each graph (calibration: 1 mV, 50 ms) show the first five responses for 10 Hz and all responses for 20 and 40 Hz. For 10 and 40 Hz, within-train fEPSP slope facilitation was comparable in 8- to 10-month-old and 3-month-old rats (10 Hz: p = 0.123, F(9,189) = 1.39; 40 Hz: p = 0.055, F(9,189) = 1.897; RM ANOVA). For 20 Hz, facilitation was greater in 8- to 10-month-old versus 3-month-old old rats (p = 0.024, F(9,189) = 2.204; 3-month-old, N = 11; 8- to 10-month-old, N = 12 for all frequencies). B, Following HFS (100 Hz, 1 s) in mice, LPP potentiation (assessed by initial fEPSP slope) was greater and more stable in 3-month-old versus 6- and 8- to 10-month-old mice; representative traces at right show superimposed baseline (darker line) with post-HFS waveforms (calibration: 1 mV, 10 ms; 3-month-old, N = 5; 6-month-old, N = 6; 8- to 10-month-old, N = 7). C, Mean percentage of potentiation over baseline, during the last 5 min of recording after HFS for LTP or during the first 2 min post-HFS for STP, shows that LTP was lower in the two older mouse groups, whereas STP was comparable across groups (for LTP: F(2,39) = 8.283, p = 0.001; **p < 0.01 and ***p < 0.001 vs 3-month-old, post hoc Bonferroni; STP: n.s.). D, lppLTP was greater in 3- versus 8- to 10-month-old rats (calibration: 1 mV, 10 ms; 3-month-old, N = 7; 8- to 10-month-old, N = 7); superimposed traces also show the difference in LTP size with age. E, The mean percentage of LTP at 55–60 min post-HFS was approximately twofold greater in 3- versus 8- to 10-month-old rats (***p = 0.0009, t test), whereas STP was equivalent between groups.
Collectively, these results indicate that baseline synaptic transmission does not deteriorate over the course of young adulthood.
Effects of age on LPP LTP were evaluated in additional animals. Potentiation was substantially reduced in 6- and 8- to 10-month-old versus 3-month-old mice (Fig. 2B). The percentage of potentiation at 60 min post-HFS was 55.3 ± 7.5% at 3 months, but 28.2 ± 5.4% and 21.0 ± 5.5% at 6 and 8–10 months, respectively (one-way ANOVA: F(2,39) = 8.283; p < 0.001; 3-month-old vs older groups; each p < 0.01, Bonferroni post hoc test). In contrast, short-term potentiation (STP), measured over the first minute post-HFS, was comparable across age groups (Fig. 2C). In this system, STP is blocked by NMDAR antagonists (Hanse and Gustafsson, 1992; Wang et al., 2016b); thus, the absence of group differences for STP suggests that initial events producing potentiation were intact across ages tested here, and that age effects on LTP reflect a failure in stabilization.
Age effects on synaptic plasticity were similar for rats. STP was comparable at 3 and 8–10 months of age, but potentiation then decayed toward baseline in the older rats so that the percentage potentiation at 60 min was strikingly smaller in the older group (Fig. 2D,E; p < 0.001, t test). The mouse and rat data constitute the first evidence that a pronounced but selective loss of synaptic plasticity occurs in layer II EC projections during the first 6–8 months of life in rodents.
Early aging disrupts endocannabinoid-triggering mechanisms for LPP-LTP
The LPP uses a form of LTP (lppLTP) wherein potentiation is initiated postsynaptically but expressed presynaptically via enhanced transmitter release. The endocannabinoid 2-arachidonoylglycerol (2-AG) acts as the requisite retrograde messenger (Wang et al., 2016b; 2018b). We tested the possibility that early aging disrupts the endocannabinoid signaling required for lppLTP using brief infusions of physostigmine, a compound that increases constitutive cholinergic signaling by blocking acetylcholine breakdown. Prior work showed that acetylcholine activates the postsynaptic synthetic enzyme (diacylglycerol lipase α) that produces 2-AG and consequently increases 2-AG concentrations in the DG (Wang et al., 2018b). 2-AG acts via presynaptic cannabinoid receptor 1 (CB1R) to depress glutamate release in the LPP and MPP (Wang et al., 2016b, 2018b). As previously described, physostigmine caused a rapid decrease (21.53 ± 1.6%) in the size of fEPSPs elicited by single-pulse LPP stimulation in 3-month-old rats; this effect of physostigmine was substantially reduced in 8-month-old rats (12.07 ± 1.1%; p < 0.0001, t test; Fig. 3A,B). As expected for a decrease in transmitter release, PPF was increased by physostigmine infusion: absent other treatment, PPF was comparable between groups (Fig. 3C), but physostigmine increased PPF to a greater degree above this level in 3- versus 8-month-old rats (Fig. 3D). These are the first results indicating that postsynaptic mobilization of endocannabinoid signaling deteriorates in the LPP before middle age, and that changes in 2-AG production likely contribute to the age-related loss of lppLTP. We next tested whether enhancing 2-AG levels restores lppLTP in the older animals. The compound JZL184 inhibits monoacylglycerol lipase, the enzyme primarily responsible for the breakdown of released 2-AG (Long et al., 2009). JZL184 thus increases 2-AG levels in the DG and, as described previously, nearly doubles lppLTP in 3-month-old rodents (Wang et al., 2016b). JZL184 caused a more modest increase in the percentage of LTP in 8- to 10-month-old rats (48.2 ± 4.1% JZL184 vs 25.5 ± 4.1% vehicle; p = 0.01) without an effect on STP (Fig. 3E,F). The failure of JZL184 to produce supranormal lppLTP aligns with the conclusion that the links between high-frequency activation and the postsynaptic signalosome for 2-AG production and release deteriorate over the course of young adulthood.
Figure 3.

Age-dependent effects of treatments that influence endocannabinoid signaling on synaptic responses and lppLTP in rat. A, Physostigmine (30 μm, gray bar) reduced the slopes of fEPSPs elicited by single-pulse LPP stimulation to a greater extent in slices from 3- versus 8-month-old rats. Traces show representative fEPSPs pre-infusion (dark line) and postinfusion (3-month-old, N = 14; 8-month-old, N = 18; calibration: 1 mV, 10 ms). B, The percentage of reduction in fEPSP slopes (from data in A) at the 60 min time point (****p < 0.0001 vs 3-month-old, t test). C, D, PPF of fEPSP slopes (50 ms interpulse interval) was comparable between age groups before physostigmine administration (C; p = 0.736) but was greater after physostigmine administration in slices from 3-month-old versus 8-month-old rats (D; **p = 0.0071; 3-month-old, N = 6; 8-month-old, N = 9); representative traces shown at left. Calibration: 1 mV, 10 ms. E, JZL184 (JZL; 1 μm in slice bath) enhanced lppLTP in 8- to 10-month-old rats. Above, representative traces from before (dark line) and after HFS. Calibration: 1 mV, 10 ms [vehicle (veh), N = 5; JZL184, N = 10]. F, Graphs summarize, from E, the percentage of LTP and the percentage of STP at 55–60 and 0–2 min after HFS, respectively (**p = 0.01 vs veh, t test).
Age-related changes in LPP terminals
There was no evidence for an age-related reduction in the number of LPP terminals as assessed by counts of synapse-sized puncta immunolabeled for the presynaptic marker synaptophysin in the DG outer molecular layer; indeed, values were slightly higher for 8-month-old versus 3-month-old rats (17,486 ± 349 vs 16,456 ± 251/sample field; p < 0.03). Using dual immunolabeling and FDT, we tested for changes in density of phosphorylated (activated) focal adhesion kinase (p-FAK) colocalized with synaptophysin. FAK is an integrin-associated tyrosine kinase and the activation of integrin-FAK signaling is essential for producing lppLTP (Wang et al., 2018b). FDT creates 3D reconstructions for each of tens of thousands of immunolabeled synapses and measures their density and size within fields of interest. Strong immunostaining for both p-FAK and synaptophysin was present in the DG outer molecular layer of 3- and 8-month-old rats (Fig. 4A–D). Analyses of presynaptic p-FAK concentrations were performed by constructing curves relating the percentage of all immunolabeled elements to an ascending series of immunostaining density bins. Plots for p-FAK colocalized with synaptophysin were slightly but reliably left shifted at 8 months of age relative to 3 months of age (F(16,400) = 4.549, p < 0.0001, two-way RM ANOVA), indicating a modest loss of the active enzyme with age (Fig. 4C). This effect, combined with the more dramatic change in endocannabinoid signaling, provides a plausible explanation for the pronounced loss of plasticity in the 8-month-old group.
A similar analysis of synaptophysin immunoreactivity produced a surprising result: the density frequency distribution for the 8-month-old group was skewed more greatly to the right, toward higher immunolabeling densities, relative to the curve for 3-month-old animals (two-way RM ANOVA, p < 0.0001; Fig. 4D). Synaptophysin is one of the most abundant presynaptic proteins; although its functions are poorly understood, they are generally assumed to involve transmitter vesicle mobilization (Valtorta et al., 2004; Igarashi, 2015; Kokotos et al., 2019). The elevated per-synapse synaptophysin levels are consistent with the tendency toward increased frequency facilitation found in the 8-month-old group but are also suggestive of changes in the trafficking in LPP projections.
Age-related changes in presynaptic TrkB in the LPP
The neurotrophins and their receptors play a critical role in retrograde trophic signaling in the peripheral nervous system. Ligand binding results in receptor internalization at terminals followed by the formation of complex structures that are transported back to the soma, resulting in prosurvival genomic events (Harrington and Ginty, 2013). We accordingly used FDT to test whether the density of the BDNF receptor TrkB, colocalized with synaptophysin in LPP terminals, changes during the course of young adult life and obtained evidence for a marked reduction between 3 and 8 months of age. The TrkB immunolabeling frequency distribution was strongly left shifted, toward lower per-synapse densities, in 8- versus 3-month-old mice (F(10,100) = 9.505, p < 0.0001), and the percentage of contacts with high concentrations of TrkB was substantially reduced at 8 months (p < 0.001, t test; Fig. 4E,F). These results provide additional evidence that presynaptic protein trafficking in the LPP becomes disturbed during early aging and that this extends to key elements of trophic signaling.
Early-onset aging in the medial perforant path
We tested whether the loss of plasticity in layer II EC synapses also occurs in projections from MEC to the DG in rats. These fibers course in the MPP and terminate in the DG middle molecular layer immediately subjacent to the LPP. Mechanisms underlying mppLTP are not well understood, but evidence indicates that induction and expression both occur in the postsynaptic compartment (Harney et al., 2008) and do not involve endocannabinoids (Wang et al., 2016b). Previous studies found that infusions of GABAAR antagonists facilitate robust and reliable mppLTP (Hanse and Gustafsson, 1992; Wang et al., 2016b; Schreurs et al., 2017). In our preparations, three brief (0.5 s) trains of 100 Hz stimulation separated by 20 s combined with infusion of GABAAR blocker picrotoxin (1 μm) proved optimal for inducing mppLTP (Trieu et al., 2015; Wang et al., 2016b); picrotoxin at this low level caused no evident changes in fEPSPs and did not produce epileptiform activity. As with the LPP, there were no detectable age-related changes in MPP synaptic responses or input/output curves (Fig. 5A,B). Repetitive MPP stimulation produces an unusual depression effect (Trieu et al., 2015), and it was accordingly not possible to test frequency following. LTP was clearly reduced in the MPP in 8- versus 3-month olds (30.8 ± 6.8% vs 78.9 ± 9.8% at 60 min post-HFS; p = 0.0005, t test; Fig. 5C,D). In contrast with the LPP, the magnitude of initial MPP potentiation was also lower in the older rats. STP was 128.9 ± 9.7% and 76.4 ± 9.2% for young and older animals, respectively (p = 0.001, t test; Fig. 5D).
Figure 5.

Age-related changes in MPP potentiation in rats. A, With single-pulse stimulation the averaged decay tau and initial fEPSP slope were comparable for 3-month-old (N = 10) and 8-month-old (N = 12) rats (slope, p = 0.3542; decay tau, p = 0.4678; t test). B, The MPP input–output curve was not different between 3-month-old (N = 6) and 8-month-old (N = 12) rats. C, Following HFS, both initial and enduring MPP potentiation were smaller in slices from 8-month-old versus 3-month-old rats. Representative traces show baseline (dark line) and post-HFS responses. Calibration: 1 mV, 10 ms. D, Scatter plots show that for the MPP, the mean percentage of potentiation during the last 5 min of recording (LTP) and the 2 min post-HFS interval (STP) were both significantly lower for slices from 8-month-old versus 3-month-old rats (***p < 0.001; data from C). C, D: 3-month-old, N = 10; 8-month-old, N = 12.
Prior work showed that STP in the MPP is completely blocked by NMDAR antagonist APV (Colino and Malenka, 1993). It is thus possible that the lower STP in 8-month-old animals reflects a failure in the number or operation of NMDARs. We tested the former possibility by evaluating levels of the GluN2A subunit colocalized with postsynaptic marker PSD-95 in the DG middle molecular layer. The GluN2A immunolabeling density frequency distribution was only slightly right shifted in 8- versus 3-month-old rats (F(15,120) = 1.895, p = 0.0299), and there was no age effect on the numbers of synapses with dense GluN2A immunoreactivity (Fig. 6A), suggesting that decreases in synaptic NMDAR densities are not responsible for the age-related loss of potentiation. Disrupted signaling from postsynaptic NMDARs to rapid mechanisms for enhancing fEPSPs is a more likely explanation.
Figure 6.

MPP presynaptic protein levels are altered with age. A, FDT analysis (plotted as in Fig. 4) of the density of postsynaptic GluN2A immunoreactivity (ir) for synapses double labeled (2×) for postsynaptic marker PSD-95 in the DG middle molecular layer (MML) of 3-month-old (N = 5) and 8-month-old (N = 5) rats; scatter plot shows that the percentages of 2× synapses with dense GluN2A-ir (≥95 U, x-axis) did not differ between age groups (p = 0.328, unpaired t test). B, FDT analysis in rats shows a marked right shift in the per-terminal density of synaptophysin-ir in the MPP, the predominant excitatory afferent to the MML; scatter plot shows the increase in the proportion of synapses with dense immunolabeling at 8 months of age versus 3 months of age (**p < 0.01). C, In CA1 stratum radiatum, the per-terminal density of synaptophysin-ir did not differ between age groups. B, C, 3-month-old, N = 15; 8-month-old, N = 12 rat hippocampal slices as assessed in Fig. 4B,D. D, Analysis of the density of presynaptic TrkB-ir in the MPP terminal field; there was a pronounced left shift in density frequency distribution for the 8-month-old group relative to the 3-month-old group (N = 6 mice/group), resulting in a large reduction in terminals with dense TrkB-ir (scatter plot; **p < 0.01).
As described for the LPP, the plasticity failure in the MPP was accompanied by both an increase in synaptophysin levels and a reduction in presynaptic TrkB levels. The density frequency distribution for synaptophysin immunolabeling in the middle molecular layer was strongly right shifted in older versus younger rats (p < 0.0001; Fig. 6B). Notably, this effect appeared to be pathway specific as per-synapse synaptophysin levels did not differ between 3- and 8-month-old rats for projections from CA3 to the apical dendrites of CA1b (Fig. 6C). This suggests that the marked changes seen in the medial and lateral perforant path are not reflections of brain-wide effects of aging. Finally, the percentage of synaptophysin-positive contacts with high concentrations of TrkB in the MPP was also substantially reduced in 8- versus 3-month-old mice (p = 0.0014, Student's t test; Fig. 6D).
Changes in LPP-dependent episodic memory during early adulthood
Activity in LEC is depressed in older individuals relative to young adults during the processing of the semantic or “what” component of episodic memory (Reagh et al., 2018), which is consistent with reports that the elderly are impaired in this type of learning (Samson and Barnes, 2013; Gonzalez-Escamilla et al., 2018). When the deficits first appear is unknown, but evidence suggests performance declines by middle age (Kinugawa et al., 2013). We tested whether declines in the “what” aspect of episodic learning appear during the same period of early adulthood in which lppLTP deficits emerge. These tests were based on recently described protocols for separately measuring the “what”, “where,” and “when” elements of episodic memory in rodents. Those experiments showed that transient chemogenetic silencing of the LPP entirely eliminates encoding of the “what” element and, as expected for episodic learning, this is accompanied by impairments in “where” and “when” encoding (Wang et al., 2018a; Cox et al., 2019). In the present study, mice were presented with a sequence of three different odors (A:A to C:C), separated by 3 min, and then tested whether they recognized that a test odor was not part of the sampled series (Wang et al., 2018a; Cox et al., 2019; Fig. 7A, serial odors test). Importantly, as in human episodic learning, this paradigm does not require prior training or explicit rewards. Previous work established that 3-month-old mice prefer the novel odor D in the test trial versus the previously sampled odor A, and that acquisition is entirely blocked by transient silencing of the LPP (Cox et al., 2019). We replicated the control result for 3-month-old mice but found a marked retention deficit in the 8- to 10-month-old group (p = 0.0013; Fig. 7B,C). Notably, the age-related effect was not because of group differences in sampling time: the total time exploring the cues during the retention test did not differ between the 8- and 3-month-old groups (p = 0.897; Fig. 7D).
Importantly, mice at both 3- and 8-months of age performed similarly in a two-odor test in which they sampled an identical odor pair (A:A) and were subsequently presented with this cue and a novel odor (A:D; Fig. 7A). Both age groups preferentially explored the novel odor D in this task (Fig. 7E). Finally, we verified that odors A and D were equally salient to mice in both age groups (Fig. 7F). Thus, the rapid aging impairment was evident when the older mice were required to remember cues that were part of a prior sequence and was not accompanied by difficulties in detecting or discriminating odors per se.
Discussion
The present results provide the first evidence that synapses formed by layer II entorhinal cortex neurons deteriorate surprisingly early during adulthood in rodents. The effects were selective and not accompanied by the perturbations to basic synaptic operations previously described for the perforant path of aged rodents (Barnes et al., 2000; Froc et al., 2003). Specifically, there were no age-related losses of synapses, or deficits in baseline synaptic transmission or transient response facilitation by repetitive activation. Nonetheless, there was a clear reduction in LTP at 8 months of age in the medial and lateral perforant paths of rats and mice. Relatedly, there was also a substantial loss of endocannabinoid signaling, which involves retrograde communication from spines to terminals, from early to later stages of young adulthood. Specifically, physostigmine, which increases concentrations of constitutively released acetylcholine and thereby enhances 2-AG production, had a lesser effect on LPP-initiated fEPSPs and paired-pulse facilitation in the DG outer molecular layer in 8- versus 3-month-old mice. Past work showed that the effects of physostigmine on LPP responses are completely blocked by an antagonist of CB1R, a protein concentrated on LPP terminals (Wang et al., 2018b). While it is likely that elevated levels of extracellular acetylcholine will have effects on granule cells, it is reasonable to conclude that the physiological assay used here samples endocannabinoid actions on release from LPP terminals.
Studies have described decreases in endocannabinoids with aging (Piyanova et al., 2015) although data for changes during young adulthood are lacking. Nevertheless, the finding provides a plausible explanation for the early loss of lppLTP because endocannabinoid signaling initiated by high-frequency afferent stimulation is essential for producing stable potentiation in the LPP (Wang et al., 2016b, 2018b). Notably, blockade or knockdown of the presynaptic CB1R does not disrupt short-term potentiation of LPP responses, and this STP effect was similarly unaffected by early aging. Thus, our findings support the conclusion that aging has a negative effect on one or more steps in the endocannabinoid mechanism and thereby depresses lppLTP. Multiple lines of evidence suggest that consolidation of this unusual form of plasticity involves atypical signaling by CB1R leading to cytoskeletal changes within LPP terminals (Wang et al., 2018b). Whether age-related disturbances to these presynaptic mechanisms contribute to the loss of LPP plasticity is an interesting question for future research.
Prior studies showed that mppLTP does not rely on endocannabinoid signaling (Wang et al., 2018b) and is induced postsynaptically (Harney et al., 2006). Pharmacological experiments further indicate that mppLTP expression involves the movement of glutamate receptors into the synapse (Harney et al., 2008) and so, in contrast to lppLTP, would also be postsynaptic. A clue about mechanisms is suggested by our finding that MPP STP is negatively affected by early aging. This rapid potentiation is NMDAR dependent but we found no evidence for reductions in synaptic NMDAR levels between early and later stages of young adulthood. These results point to a partial age-related failure in linkages between the NMDARs and the substrates for short- and long-term synaptic modifications.
Because mppLTP appears to be induced and expressed postsynaptically, it is reasonable to hypothesize that the loss of this plasticity reflects age effects on the DG granule cells. By this argument, these neurons—which are atypical in many respects—would be unusually sensitive to aging during early adulthood, resulting in disturbances to the NMDAR-dependent signaling that initiates STP and LTP in the MPP. Unusually rapid granule cell aging could also account for the reductions in lppLTP because generation of the endocannabinoid trigger required for this presynaptic potentiation occurs postsynaptically. In this scenario, generalized changes to the granule cells would include factors that affect the LPP (endocannabinoid production) but not the MPP and vice versa (activity-driven transfer of glutamate receptors into synapses in the MPP but not LPP). Related to this argument, there is evidence that the DG is particularly sensitive to the effects of aging in humans (Dillon et al., 2017) and rodents (Small et al., 2004; Burger, 2010).
While disturbances to the granule cells provide a unifying hypothesis for the observed losses of two very different forms of LTP, we also found that aging is associated with accumulations of the presynaptic marker synaptophysin in both LPP and MPP. This could be an early sign of deterioration because aberrant synaptophysin concentrations are observed with axonal degeneration (Hilbe et al., 2005; Gudi et al., 2017). The question then arises of whether this presynaptic effect is a contributor to, or a consequence of, the decline in LTP. The latter idea accords with the widely discussed hypothesis that plasticity contributes importantly to the maintenance of synaptic junctions (Gogolla et al., 2007). Whatever their causes, perturbations to perforant path terminals could disrupt the flow of retrograde trophic support. Consonant with this, we found that early aging is associated with significant decreases in presynaptic levels of TrkB (Minichiello, 2009; Yoshii and Constantine-Paton, 2010) colocalized with synaptophysin in both LPP and MPP terminal fields; notably, we cannot exclude an age-related effect on postsynaptic TrkB levels in the middle molecular layer that could affect the BDNF/TrkB-dependent LTP in this system (Panja et al., 2014). Neurotrophin receptors (e.g., TrkA, TrkB) expressed on terminals in various systems are internalized on ligand (NGF, BDNF) binding, resulting in the formation of endosomes that are transported back to their parent cell bodies to promote neuronal health and survival (von Bartheld et al., 1996; Ito and Enomoto, 2016; Wang et al., 2016a; Kononenko et al., 2017). From this perspective, the unusual vulnerability of layer II entorhinal neurons could reflect reductions in terminal TrkB levels and thus a decline in neuroprotective, retrograde tropic support. Possibly related to this are results showing that heterozygous deletion of a downstream effector for TrkB causes neuronal pyknosis that is especially pronounced in superficial layers of entorhinal cortex and in frontal cortex (Duffy et al., 2011). It will accordingly be of interest to test for retrograde transport of BDNF in the perforant path and to determine whether this process undergoes predicted decreases during young adulthood in rodents. Relatedly, recent work showed that BDNF/TrkB traffics locally via endosome/autophagosome complexes (amphisomes) in the mossy fiber boutons formed by axons of DG granule cells and that this mechanism serves to promote transmitter release (Andres-Alonso et al., 2019). The occurrence of such events in the LPP could contribute to the presynaptic form of LTP expressed by that pathway, with age-related losses in TrkB being a factor in the decline of plasticity. Finally, there is the question of whether restoration of lppLTP with agents that increase endocannabinoid signaling, as reported here, also rescues TrkB levels on LPP terminals.
Our formulation of the problem of entorhinal vulnerability assumes a certain degree of linearity between events occurring at different stages of adult life: reduced synaptic plasticity gradually decreases trophic support, causing a still slower disruption of processes required for the maintenance of neuronal viability. Alternatively, it is possible that perturbations at different stages of aging arise somewhat independently with the ultimate outcome being determined by a combination of, or interaction between, these negative events. Tests for independence between temporally distinct correlates of entorhinal aging will require means for selectively manipulating those correlates. Absent such data, suggestions about the relationship between problems that appear during young adulthood and late-in-life outcomes will be speculative.
In addition to being a potential contributor to later atrophy of the LEC, early impairments to LPP plasticity are likely to have negative effects on the encoding and processing of memory that depends on the flow of information from cortex to hippocampus. The present studies confirmed this prediction. It is widely accepted that hippocampus is critical for the organization of serial, everyday events into episodes minimally involving the identity of encountered cues (what), their spatial relationships (where), and the order in which they occurred (when; Tulving, 1972). Recent studies established that rodents acquire these three features and, like humans, do so without training or explicit rewards, and that blocking activity in the LEC projection to the DG eliminates all three episodic elements (Wang et al., 2018a). As described here, 8- to 10-month-old mice were impaired in memory tasks involving a sequence of odors (episodic what) but had no difficulty retaining the identity of single cues presented outside of a series. The magnitude of the deficit is surprising but consistent with reports of modest declines in episodic memory by middle age in humans (Kinugawa et al., 2013; Ankudowich et al., 2016, 2017). The serial odors test has an uncertain relationship to other forms of learning in rodents, and additional experiments are needed to determine whether the latter are significantly affected by the degree of LTP impairment found in the present studies. It will be of interest to determine whether the JZL184 treatment that partially restored lppLTP also rescues performance in the serial odors paradigm in 8- to 10-month-old mice. A result of this type was obtained in a mouse model of Fragile-X Syndrome, an animal in which LPP plasticity and learning of sequential odors are profoundly depressed (Wang et al., 2018a).
Footnotes
This work was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant HD-089491 to G.L. and C.M.G.; and National Institute on Drug Abuse Grant DA-04118 to C.M.G. A.A.L. was supported by National Institute of Mental Health Training Grant T32-MH-119049-01. J.Q. was supported by the National Science Foundation Graduate Research Fellowship under Grant DGE-1839285.
The authors declare no competing financial interests.
References
- Andres-Alonso M, Ammar MR, Butnaru I, Gomes GM, Acuña Sanhueza G, Raman R, Yuanxiang PAn, Borgmeyer M, Lopez-Rojas J, Raza SA, Brice N, Hausrat TJ, Macharadze T, Diaz-Gonzalez S, Carlton M, Failla AV, Stork O, Schweizer M, Gundelfinger ED, Kneussel M, et al. (2019) SIPA1L2 controls trafficking and local signaling of TrkB-containing amphisomes at presynaptic terminals. Nat Commun 10:5448. 10.1038/s41467-019-13224-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankudowich E, Pasvanis S, Rajah MN (2016) Changes in the modulation of brain activity during context encoding vs. context retrieval across the adult lifespan. Neuroimage 139:103–113. 10.1016/j.neuroimage.2016.06.022 [DOI] [PubMed] [Google Scholar]
- Ankudowich E, Pasvanis S, Rajah MN (2017) Changes in the correlation between spatial and temporal source memory performance and BOLD activity across the adult lifespan. Cortex 91:234–249. 10.1016/j.cortex.2017.01.006 [DOI] [PubMed] [Google Scholar]
- Barnes CA, Rao G, Houston FP (2000) LTP induction threshold change in old rats at the perforant path–granule cell synapse. Neurobiol Aging 21:613–620. 10.1016/s0197-4580(00)00163-9 [DOI] [PubMed] [Google Scholar]
- Bi X, Yong AP, Zhou J, Gall CM, Lynch G (2000) Regionally selective changes in brain lysosomes occur in the transition from young adulthood to middle age in rats. Neuroscience 97:395–404. 10.1016/s0306-4522(00)00021-x [DOI] [PubMed] [Google Scholar]
- Botella-López A, Burgaya F, Gavín R, García-Ayllón MS, Gómez-Tortosa E, Peña-Casanova J, Ureña JM, Del Río JA, Blesa R, Soriano E, Sáez-Valero J (2006) Reelin expression and glycosylation patterns are altered in Alzheimer's disease. Proc Natl Acad Sci U S A 103:5573–5578. 10.1073/pnas.0601279103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Burger C (2010) Region-specific genetic alterations in the aging hippocampus: implications for cognitive aging. Front Aging Neurosci 2:140. 10.3389/fnagi.2010.00140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, Lauterborn JC (2010) Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci 30:10977–10984. 10.1523/JNEUROSCI.1077-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Massaro CM, Palop JJ, Thwin MT, Yu GQ, Bien-Ly N, Bender A, Mucke L (2007) Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer's disease. J Neurosci 27:2727–2733. 10.1523/JNEUROSCI.3758-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Abraham WC (1994) Differential regulation of paired-pulse plasticity following LTP in the dentate gyrus. Neuroreport 5:385–388. 10.1097/00001756-199401120-00003 [DOI] [PubMed] [Google Scholar]
- Colgin LL (2016) Rhythms of the hippocampal network. Nat Rev Neurosci 17:239–249. 10.1038/nrn.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Kramár EA, Gall CM, Lynch G (2003) Septal modulation of excitatory transmission in hippocampus. J Neurophysiol 90:2358–2366. 10.1152/jn.00262.2003 [DOI] [PubMed] [Google Scholar]
- Colino A, Malenka RC (1993) Mechanisms underlying induction of long-term potentiation in rat medial and lateral perforant paths in vitro. J Neurophysiol 69:1150–1159. 10.1152/jn.1993.69.4.1150 [DOI] [PubMed] [Google Scholar]
- Cox BM, Cox CD, Gunn BG, Le AA, Inshishian VC, Gall CM, Lynch G (2019) Acquisition of temporal order requires an intact CA3 commissural/associational (C/A) feedback system in mice. Commun Biol 2:251. 10.1038/s42003-019-0494-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl D, Sarvey JM (1989) Norepinephrine induces pathway-specific long-lasting potentiation and depression in the hippocampal dentate gyrus. Proc Natl Acad Sci U S A 86:4776–4780. 10.1073/pnas.86.12.4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein DL, Weaver CM, Luebke JI, Hof PR (2013) Dendritic spine changes associated with normal aging. Neuroscience 251:21–32. 10.1016/j.neuroscience.2012.09.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SE, Tsivos D, Knight M, McCann B, Pennington C, Shiel AI, Conway ME, Newson MA, Kauppinen RA, Coulthard EJ (2017) The impact of ageing reveals distinct roles for human dentate gyrus and CA3 in pattern separation and object recognition memory. Sci Rep 7:14069. 10.1038/s41598-017-13853-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehner J, Knuesel I (2010) Reelin-mediated signaling during normal and pathological forms of aging. Aging Dis 1:12–29. [PMC free article] [PubMed] [Google Scholar]
- Duffy AM, Schaner MJ, Wu SH, Staniszewski A, Kumar A, Arévalo JC, Arancio O, Chao MV, Scharfman HE (2011) A selective role for ARMS/Kidins220 scaffold protein in spatial memory and trophic support of entorhinal and frontal cortical neurons. Exp Neurol 229:409–420. 10.1016/j.expneurol.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S, Sengupta P (2016) Men and mice: relating their ages. Life Sci 152:244–248. 10.1016/j.lfs.2015.10.025 [DOI] [PubMed] [Google Scholar]
- Ernyey AJ, Grohmann Pereira T, Kozma K, Kouhnavardi S, Kassai F, Gyertyán I (2019) Following of aging process in a new motor skill learning model, "pot jumping" in rats. Geroscience 41:309–319. 10.1007/s11357-019-00073-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Currer JM, Harrison DE (2007) Mouse models in aging research. In: The mouse in biomedical research, Ed 2 (Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, eds), pp 637–672. New York: Elsevier. [Google Scholar]
- Froc DJ, Eadie B, Li AM, Wodtke K, Tse M, Christie BR (2003) Reduced synaptic plasticity in the lateral perforant path input to the dentate gyrus of aged C57BL/6 mice. J Neurophysiol 90:32–38. 10.1152/jn.00105.2003 [DOI] [PubMed] [Google Scholar]
- Gazzaley AH, Thakker MM, Hof PR, Morrison JH (1997) Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neurobiol Aging 18:549–553. 10.1016/s0197-4580(97)00112-7 [DOI] [PubMed] [Google Scholar]
- Gogolla N, Galimberti I, Caroni P (2007) Structural plasticity of axon terminals in the adult. Curr Opin Neurobiol 17:516–524. 10.1016/j.conb.2007.09.002 [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16:4491–4500. 10.1523/JNEUROSCI.16-14-04491.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Escamilla G, Muthuraman M, Chirumamilla VC, Vogt J, Groppa S (2018) Brain networks reorganization during maturation and healthy aging-emphases for resilience. Front Psychiatry 9:601. 10.3389/fpsyt.2018.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossberg S (2009) Beta oscillations and hippocampal place cell learning during exploration of novel environments. Hippocampus 19:881–885. 10.1002/hipo.20602 [DOI] [PubMed] [Google Scholar]
- Gudi V, Gai L, Herder V, Tejedor LS, Kipp M, Amor S, Sühs KW, Hansmann F, Beineke A, Baumgärtner W, Stangel M, Skripuletz T (2017) Synaptophysin is a reliable marker for axonal damage. J Neuropathol Exp Neurol 76:109–125. 10.1093/jnen/nlw114 [DOI] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B (1992) Postsynaptic, but not presynaptic, activity controls the early time course of long-term potentiation in the dentate gyrus. J Neurosci 12:3226–3240. 10.1523/JNEUROSCI.12-08-03226.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney SC, Rowan M, Anwyl R (2006) Long-term depression of NMDA receptor-mediated synaptic transmission is dependent on activation of metabotropic glutamate receptors and is altered to long-term potentiation by low intracellular calcium buffering. J Neurosci 26:1128–1132. 10.1523/JNEUROSCI.2753-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harney SC, Jane DE, Anwyl R (2008) Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J Neurosci 28:11685–11694. 10.1523/JNEUROSCI.3035-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, Ginty DD (2013) Long-distance retrograde neurotrophic factor signalling in neurons. Nat Rev Neurosci 14:177–187. 10.1038/nrn3253 [DOI] [PubMed] [Google Scholar]
- Herring A, Donath A, Steiner KM, Widera MP, Hamzehian S, Kanakis D, Kölble K, ElAli A, Hermann DM, Paulus W, Keyvani K (2012) Reelin depletion is an early phenomenon of Alzheimer's pathology. J Alzheimers Dis 30:963–979. 10.3233/JAD-2012-112069 [DOI] [PubMed] [Google Scholar]
- Hilbe M, Guscetti F, Wunderlin S, Ehrensperger F (2005) Synaptophysin: an immunohistochemical marker for animal dysautonomias. J Comp Pathol 132:223–227. 10.1016/j.jcpa.2004.07.002 [DOI] [PubMed] [Google Scholar]
- Igarashi KM (2015) Plasticity in oscillatory coupling between hippocampus and cortex. Curr Opin Neurobiol 35:163–168. 10.1016/j.conb.2015.09.005 [DOI] [PubMed] [Google Scholar]
- Ito K, Enomoto H (2016) Retrograde transport of neurotrophic factor signaling: implications in neuronal development and pathogenesis. J Biochem 160:77–85. 10.1093/jb/mvw037 [DOI] [PubMed] [Google Scholar]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, Mayeux R, Duff KE, Small SA (2014) Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci 17:304–311. 10.1038/nn.3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinugawa K, Schumm S, Pollina M, Depre M, Jungbluth C, Doulazmi M, Sebban C, Zlomuzica A, Pietrowsky R, Pause B, Mariani J, Dere E (2013) Aging-related episodic memory decline: are emotions the key? Front Behav Neurosci 7:2. 10.3389/fnbeh.2013.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokotos AC, Harper CB, Marland JRK, Smillie KJ, Cousin MA, Gordon SL (2019) Synaptophysin sustains presynaptic performance by preserving vesicular synaptobrevin-II levels. J Neurochem 151:28–37. 10.1111/jnc.14797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko NL, Claßen GA, Kuijpers M, Puchkov D, Maritzen T, Tempes A, Malik AR, Skalecka A, Bera S, Jaworski J, Haucke V (2017) Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat Commun 8:14819. 10.1038/ncomms14819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Palmer LC, Jia Y, Pham DT, Hou B, Wang W, Trieu BH, Cox CD, Kantorovich S, Gall CM, Lynch G (2016) Chronic ampakine treatments stimulate dendritic growth and promote learning in middle-aged rats. J Neurosci 36:1636–1646. 10.1523/JNEUROSCI.3157-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Cox CD, Chan SW, Vanderklish PW, Lynch G, Gall CM (2020) Synaptic actin stabilization protein loss in Down syndrome and Alzheimer disease. Brain Pathol 30:319–331. 10.1111/bpa.12779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Martin M, Blazquez-Llorca L, Benavides-Piccione R, Rabano A, Hernandez F, Avila J, DeFelipe J (2014) Selective alterations of neurons and circuits related to early memory loss in Alzheimer's disease. Front Neuroanat 8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF (2009) Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol 16:744–753. 10.1016/j.chembiol.2009.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill DA, Roberts JA, Tuszynski MH (2000) Conservation of neuron number and size in entorhinal cortex layers II, III, and V/VI of aged primates. J Comp Neurol 422:396–401. [DOI] [PubMed] [Google Scholar]
- Minichiello L (2009) TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10:850–860. 10.1038/nrn2738 [DOI] [PubMed] [Google Scholar]
- Panja D, Kenney J, D'Andrea L, Zalfa F, Vedeler A, Wibrand K, Fukunaga R, Bagni C, Proud C, Bramham C (2014) Two-stage translational control of dentate gyrus LTP consolidation is mediated by sustained BDNF-TrkB signaling to MNK. Cell Rep 9:1430–1445. 10.1016/j.celrep.2014.10.016 [DOI] [PubMed] [Google Scholar]
- Piyanova A, Lomazzo E, Bindila L, Lerner R, Albayram O, Ruhl T, Lutz B, Zimmer A, Bilkei-Gorzo A (2015) Age-related changes in the endocannabinoid system in the mouse hippocampus. Mech Ageing Dev 150:55–64. 10.1016/j.mad.2015.08.005 [DOI] [PubMed] [Google Scholar]
- Rapp PR, Deroche PS, Mao Y, Burwell RD (2002) Neuron number in the parahippocampal region is preserved in aged rats with spatial learning deficits. Cereb Cortex 12:1171–1179. 10.1093/cercor/12.11.1171 [DOI] [PubMed] [Google Scholar]
- Reagh ZM, Noche JA, Tustison NJ, Delisle D, Murray EA, Yassa MA (2018) Functional imbalance of anterolateral entorhinal cortex and hippocampal dentate/CA3 underlies age-related object pattern separation deficits. Neuron 97:1187–1198.e4. 10.1016/j.neuron.2018.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G (2009) Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol 186:85–97. 10.1083/jcb.200901084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson RD, Barnes CA (2013) Impact of aging brain circuits on cognition. Eur J Neurosci 37:1903–1915. 10.1111/ejn.12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreurs A, Sabanov V, Balschun D (2017) Distinct properties of long-term potentiation in the dentate gyrus along the dorsoventral axis: influence of age and inhibition. Sci Rep 7:5157. 10.1038/s41598-017-05358-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Babayan AH, Katz AM, Cox CD, Lauterborn JC, Lynch G, Gall CM (2012) LTP induction translocates cortactin at distant synapses in wild-type but not Fmr1 knock-out mice. J Neurosci 32:7403–7413. 10.1523/JNEUROSCI.0968-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Chen LY, Cox CD, Schulz D, Babayan AH, Bunney WE, Henn FA, Gall CM, Lynch G (2013) Synaptic abnormalities in the infralimbic cortex of a model of congenital depression. J Neurosci 33:13441–13448. 10.1523/JNEUROSCI.2434-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P (2013) The laboratory rat: relating its age with human's. Int J Prev Med 4:624–630. [PMC free article] [PubMed] [Google Scholar]
- Small SA, Chawla MK, Buonocore M, Rapp PR, Barnes CA (2004) Imaging correlates of brain function in monkeys and rats isolates a hippocampal subregion differentially vulnerable to aging. Proc Natl Acad Sci U S A 101:7181–7186. 10.1073/pnas.0400285101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Haberman RP, Gallagher M (2011) Cognitive decline is associated with reduced reelin expression in the entorhinal cortex of aged rats. Cereb Cortex 21:392–400. 10.1093/cercor/bhq106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu BH, Kramár EA, Cox CD, Jia Y, Wang W, Gall CM, Lynch G (2015) Pronounced differences in signal processing and synaptic plasticity between piriform-hippocampal network stages: a prominent role for adenosine. J Physiol 593:2889–2907. 10.1113/JP270398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulving E (1972) Episodic and semantic memory. In: Organization of memory (Tulving E, Donaldson W, eds), pp 381–403. New York: Academic. [Google Scholar]
- Valtorta F, Pennuto M, Bonanomi D, Benfenati F (2004) Synaptophysin: leading actor or walk-on role in synaptic vesicle exocytosis. Bioessays 26:445–453. 10.1002/bies.20012 [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Hyman BT, Damasio AR (1991) Entorhinal cortex pathology in Alzheimer's disease. Hippocampus 1:1–8. 10.1002/hipo.450010102 [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Williams R, Lefcort F, Clary DO, Reichardt LF, Bothwell M (1996) Retrograde transport of neurotrophins from the eye to the brain in chick embryos: roles of the p75NTR and trkB receptors. J Neurosci 16:2995–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Martin S, Nguyen TH, Harper CB, Gormal RS, Martínez-Mármol R, Karunanithi S, Coulson EJ, Glass NR, Cooper-White JJ, van Swinderen B, Meunier FA (2016a) Corrigendum: flux of signalling endosomes undergoing axonal retrograde transport is encoded by presynaptic activity and TrkB. Nat Commun 7:13768. 10.1038/ncomms13768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Trieu BH, Palmer LC, Jia Y, Pham DT, Jung KM, Karsten CA, Merrill CB, Mackie K, Gall CM, Piomelli D, Lynch G (2016b) A primary cortical input to hippocampus expresses a pathway-specific and endocannabinoid-dependent form of long-term potentiation. eNeuro 3:ENEURO.0160-16.2016. 10.1523/ENEURO.0160-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Cox BM, Jia Y, Le AA, Cox CD, Jung KM, Hou B, Piomelli D, Gall CM, Lynch G (2018a) Treating a novel plasticity defect rescues episodic memory in Fragile X model mice. Mol Psychiatry 23:1798–1806. 10.1038/mp.2017.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Jia Y, Pham DT, Palmer LC, Jung KM, Cox CD, Rumbaugh G, Piomelli D, Gall CM, Lynch G (2018b) Atypical endocannabinoid signaling initiates a new form of memory-related plasticity at a cortical input to hippocampus. Cereb Cortex 28:2253–2266. 10.1093/cercor/bhx126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP (1993) Organization of the entorhinal-hippocampal system: a review of current anatomical data. Hippocampus 3 Spec No:33–44. [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M (2010) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70:304–322. 10.1002/dneu.20765 [DOI] [PMC free article] [PubMed] [Google Scholar]