

Abstract
Synaptic and neuronal loss are major neuropathological characteristics of Parkinson's disease. Misfolded protein aggregates in the form of Lewy bodies, comprised mainly of α-synuclein (αSyn), are associated with disease progression, and have also been linked to other neurodegenerative diseases, including Lewy body dementia, Alzheimer's disease, and frontotemporal dementia. However, the effects of αSyn and its mechanism of synaptic damage remain incompletely understood. Here, we show that αSyn oligomers induce Ca2+-dependent release of glutamate from astrocytes obtained from male and female mice, and that mice overexpressing αSyn manifest increased tonic release of glutamate in vivo. In turn, this extracellular glutamate activates glutamate receptors, including extrasynaptic NMDARs (eNMDARs), on neurons both in culture and in hippocampal slices of αSyn-overexpressing mice. Additionally, in patch-clamp recording from outside-out patches, we found that oligomerized αSyn can directly activate eNMDARs. In organotypic slices, oligomeric αSyn induces eNMDAR-mediated synaptic loss, which can be reversed by the drug NitroSynapsin. When we expose human induced pluripotent stem cell-derived cerebrocortical neurons to αSyn, we find similar effects. Importantly, the improved NMDAR antagonist NitroSynapsin, which selectively inhibits extrasynaptic over physiological synaptic NMDAR activity, protects synapses from oligomeric αSyn-induced damage in our model systems, thus meriting further study for its therapeutic potential.
SIGNIFICANCE STATEMENT Loss of synaptic function and ensuing neuronal loss are associated with disease progression in Parkinson's disease (PD), Lewy body dementia (LBD), and other neurodegenerative diseases. However, the mechanism of synaptic damage remains incompletely understood. α-Synuclein (αSyn) misfolds in PD/LBD, forming Lewy bodies and contributing to disease pathogenesis. Here, we found that misfolded/oligomeric αSyn releases excessive astrocytic glutamate, in turn activating neuronal extrasynaptic NMDA receptors (eNMDARs), thereby contributing to synaptic damage. Additionally, αSyn oligomers directly activate eNMDARs, further contributing to damage. While the FDA-approved drug memantine has been reported to offer some benefit in PD/LBD (Hershey and Coleman-Jackson, 2019), we find that the improved eNMDAR antagonist NitroSynapsin ameliorates αSyn-induced synaptic spine loss, providing potential disease-modifying intervention in PD/LBD.
Keywords: α-synuclein oligomers, astrocytic glutamate, extrasynaptic NMDARs, synaptic damage
Introduction
Intracellular protein aggregates composed of α-synuclein (αSyn, encoded by the SNCA gene) accumulate as Lewy bodies and Lewy neurites in neurons in several neurodegenerative disorders, particularly in Parkinson's disease (PD) and Lewy body dementia (LBD), but also in frontotemporal dementia (FTD) and Alzheimer's disease (AD) (Spillantini et al., 1997; Goedert, 2001; Irwin and Hurtig, 2018; Bassil et al., 2020). PD/LBD and other synucleinopathies are associated with synaptic damage and neuronal loss that affects not only dopaminergic neurons in the substantia nigra but also cerebrocortical and other neuronal subtypes. Yet Lewy body pathology is not well correlated with cell death or clinical symptoms (Schulz-Schaeffer, 2010). Evidence suggests that αSyn aggregate-related synaptic dysfunction or dendritic synapse loss contributes to PD/LBD pathogenesis (Schulz-Schaeffer, 2010). However, the precise mechanism by which αSyn oligomers/aggregates cause synaptic loss and subsequent neuronal cell death is not yet clear.
Soluble αSyn aggregates have been shown to exert pleiotropic pathologic effects on multiple cell types, including neurons, astrocytes, and microglia, and on their organelles, such as mitochondria (Guardia-Laguarta et al., 2014; Delenclos et al., 2019; Sulzer and Edwards, 2019; Bras et al., 2020). Larger aggregates and preformed fibrils of αSyn have been shown to result in decreased expression of synaptic proteins and eventually neuronal cell death (Volpicelli-Daley et al., 2011). Other groups have shown that αSyn fibrils propagate and result in neurotoxicity; moreover, different fibril strains have been associated with cell type- and brain region-specific damage (Peelaerts et al., 2015; Lau et al., 2020). One reported effect on neurons of αSyn oligomers, but not monomers or fibrils, involves increased NMDAR and calcium-permeable AMPAR activation (Diogenes et al., 2012). Additionally, large αSyn oligomers alter both presynaptic and postsynaptic mechanisms of AMPAR-mediated synaptic transmission. This increased excitation, mediated by both NMDARs and AMPARs, may contribute to neuronal cell death in synucleinopathies, by altering intracellular calcium homeostasis (Huls et al., 2011). However, whether αSyn oligomers/fibrils interact with glutamate receptors directly, indirectly, or both remains unclear.
Previously, we and others have shown that another misfolded protein, amyloid-β (Aβ) oligomers, induce an increase in astrocytic Ca2+ levels with consequent glutamate release, which in turn activates primarily extrasynaptic NMDARs (eNMDARs) on neurons, contributing to synapse loss (Pirttimaki et al., 2013, 2017; Talantova et al., 2013); decreased reuptake of glutamate into astrocytes may also participate in this process (Matos et al., 2008; Li et al., 2009, 2011; Booth et al., 2017). Here, we hypothesized that oligomeric αSyn, which is known to interact with cell membranes to increase intracellular calcium levels (Danzer et al., 2007; Kim et al., 2009), could act similarly on astrocytes to induce glutamate release. This hypothesis seemed particularly appealing given recent findings that αSyn contains an apolipoprotein repeat motif (KTKEGV) that enables it to form an amphipathic α-helix in the presence of membranes composed of negatively charged lipids (Tosatto et al., 2012; Ghiglieri et al., 2018), driving the association with lipid membranes and the acquisition of the helical secondary structure. In this manner, αSyn induces curvature and causes remodeling of lipid membranes (Iyer and Claessens, 2019). αSyn oligomers can thus form pores in the cellular membranes that display ion channel-like activity with well-defined conductance levels, resulting in abnormal calcium influx (Danzer et al., 2007; Kim et al., 2009).
Along these lines, we describe an oligomeric αSyn-mediated cascade of events on astrocytes and neurons. Oligomeric αSyn induces a rise in astrocytic Ca2+ that triggers glutamate release, which in turn activates neuronal eNMDARs to contribute to synapse loss. Additionally, we found evidence for direct activation by oligomeric αSyn of eNMDAR-associated ion channels in outside-out patches of neuronal membranes. Finally, we show that the novel drug NitroSynapsin, known to effect pharmacological blockade of eNMDARs while relatively sparing physiological synaptic NMDAR activity, abrogated αSyn-mediated synaptic loss. These results suggest a dual mechanism of synaptic damage because of oligomeric αSyn: indirectly, by increasing glutamate release from astrocytes with resultant increased eNMDAR activity; and directly, by activating eNMDAR-associated ion channel activity on neurons.
Materials and Methods
Mouse astrocyte cultures
Mice used for astrocyte cultures were housed and maintained in the animal facility at the Scintillon Institute or The Scripps Research Institute, and all experiments complied with protocols approved by the Institute Animal Care Committee. Primary astroglial cultures were prepared as previously described (Liu et al., 2006). The brains from 2- to 3-d-old C57BL/6 mouse pups of both genders were rapidly harvested, and the meninges were removed. Brains were dissociated using mechanical means (pipettes and scissors) and enzymatic dissociation (0.25% trypsin for 30 min at 37°C). The cells were then filtered through a 70 µm cell strainer and cultured in DMEM:F12 medium supplemented with 10% FBS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Two weeks after initial isolation, cells were dissociated with Accutase (Stem Cell Technologies, #07920), and the suspension was seeded onto glass coverslips coated with 20 µg/cm2 poly-l-ornithine (Sigma Millipore, #P4638) and 2 μg/cm2 laminin (Trevigen, #3400-010-01).
Human astrocyte cultures
Primary human astrocytes were purchased from a commercial source and were cultured using reagents provided by the supplier (normal human astrocytes; Lonza).
Rat cerebrocortical neuronal cultures
Sprague Dawley rat embryos of both genders were killed on day E18.5 to harvest cortical cells for generation of cultures, as previously described (Talantova et al., 2013). Briefly, cortices were dissected and dissociated in 0.25% trypsin and DNase for 30 min in 37°C. Cell debris were allowed to pellet, and cell suspension was plated on glass coverslips coated with poly-d-lysine. After 17-21 DIV, cerebrocortical neurons in these cultures were used for electrophysiological recordings. Cultured cortical neurons at that stage are known to express different types of glutamate receptors and form functional synapses (Talantova et al., 2013).
Human induced pluripotent stem cell (hiPSC)-derived cerebrocortical neurons
The use of specific hiPSC lines was approved by the institutional review boards of the Scintillon Institute and The Scripps Research Institute, and the University of California, San Diego, School of Medicine. hiPSCs (Coriell, GM02036) were routinely cultured and maintained in our laboratory using a protocol described previously (Ghatak et al., 2019) with some modifications. Briefly, pluripotent cells were plated on Matrigel-coated plates (Corning, #354248) and cultured using mTeSR1 (Stem Cell Technologies, #05850), with medium changed daily. The colonies were manually passaged weekly, using the StemPro EZPassage Disposable Stem Cell Passaging Tool (Thermo Fisher Scientific, #23181010). Neuronal differentiation was performed as previously described (Ghatak et al., 2019), with minor changes. In brief, differentiation was induced by the dual SMAD inhibition protocol, using exposure to 2 μm of each of dorsomorphin (bone morphogenetic protein inhibitor, Tocris Bioscience, #3093), A83-01 (Activin/Nodal inhibitor, Tocris Bioscience, #2939), and PNU 74654 (Wnt/β-catenin inhibitor, Tocris Bioscience, #3534) for 6 d in DMEM/F12 medium supplemented with 20% KO Serum Replacement (Invitrogen). The cells were then scraped and allowed to form floating neurospheres, which were maintained in DMEM/F12 medium supplemented with N2 and B27 (Invitrogen) and 20 ng ml−1 of basic FGF. The neurospheres were subsequently seeded on p-ornithine/laminin-coated dishes to form a monolayer of human neural progenitor cells containing rosettes, which were manually picked and expanded. Human neural progenitor cells were characterized and were found to be all SOX2/Nestin-positive. For terminal differentiation, human neural progenitor cells were treated with 100 nm compound E (EMD Millipore) in the same medium for 2 d, and then maintained in DMEM/F12 medium supplemented with N2 and B27, BDNF (20 ng ml−1) and GDNF (20 ng ml−1). Electrophysiological analysis was performed starting 5 weeks after the initiation of terminal differentiation.
SuperGluSnFR glutamate-sensing probe
SuperGluSnFR, a fluorescence resonance energy transfer (FRET) sensor of glutamate levels, was prepared and used as previously described (Hires et al., 2008; Talantova et al., 2013). To generate the glutamate sensor probe on the surface of cells, HEK-293T cells were trypsinized, seeded at 1 × 105 cells per well, and cotransfected with both SuperGluSnFR and neuroligin plasmids (1 μg each) using Lipofectamine 2000 (Invitrogen). To detect release of glutamate from astrocytes, primary astrocytes were plated on top of HEK-293 cells expressing SuperGluSnFR and the cell-adhesion protein neuroligin, which increases cell-cell apposition (Scheiffele et al., 2000). FRET microscopy was used to monitor glutamate release from pure astrocyte cultures using this astrocyte-HEK cell sensor assay system (Pasti et al., 2001; Talantova et al., 2013). Under these conditions, we detected changes in glutamate concentration within a range of 300 nm to 100 μm, as we have previously detailed (Talantova et al., 2013). Experiments were performed on an inverted microscope (Axiovert 100 M; Carl Zeiss) equipped for epifluorescence microscopy, and images were deconvolved using SlideBook software (Intelligent Imaging Innovations). Light was delivered to the sample through a 63× oil immersion objective (1.4 NA). Cells were superfused continuously at room temperature (22°C) with standard medium containing the following (in mm): NaCl, 146; KCl, 2.5; NaOH, 4; CaCl2, 1; d-glucose, 20; sucrose, 20; HEPES, 10; adjusted to pH 7.4. Drugs and reagents were added using a local microperfusion system (Warner Instruments). Cells were epi-illuminated alternately at 434 and 514 nm, and emitted light was collected at 527 nm for YFP and 476 nm for CFP. The peak CFP/YFP ratio was divided by the baseline CFP/YFP ratio and plotted after baseline normalization to 1. Images were obtained every second after a 400 ms stimulus. A 2 × 2 binning method was used to improve signal-to-noise ratio and minimize photobleaching.
iGluSnFR glutamate-sensing probe
To verify our results with the SuperGluSnFR, we used a second-generation glutamate-sensing probe, iGluSnFR, whose fluorescent emission directly reflects glutamate concentration. For these experiments, primary mouse astrocytes were electroporated (Amaxa Electroporator, Lonza) using program A-023, with iGluSnFR plasmid (Marvin et al., 2013) and plated at 200,000/well on glass coverslips; 48 h later, the cells were imaged (Axiovert 100 M; Carl Zeiss) using a 63× oil objective (1.4 NA) for a duration of 5 min. Cells were superfused continuously at room temperature (22°C) with standard medium containing the following (in mm): NaCl, 146; KCl, 2.5; NaOH, 4; CaCl2, 1; d-glucose, 20; sucrose, 20; HEPES, 10; adjusted to pH 7.4. Drugs and reagents were added using a local microperfusion system (Warner Instruments). Various reagents, including glutamate (100 μm), αSyn monomers (1 μm), αSyn oligomers (750 nM), or tetanus toxin (100 nm), were applied and iGluSnFR intensity was measured. Images were analyzed using Fiji software. Images were obtained every second for the duration of the experiment.
Patch-clamp electrophysiology
Whole-cell recordings were performed with the following internal solution (in mm): K-gluconate, 120; KCl, 5; MgCl2, 2; HEPES, 10; EGTA; 10; Mg-ATP, 4; pH 7.4 and mOsm 290. The external solution was comprised of Ca2+- and Mg2+-free HBSS (Invitrogen) with added CaCl2, 2 mm; HEPES, 10 mm; glycine, 20 μm; TTX, 1 μm (Tocris Bioscience); pH 7.4. Recordings were performed using a Multiclamp 700B amplifier (Molecular Devices) at a data sampling rate of 2 kHz with a Digidata 1440A (Molecular Devices) in gap-free mode and analyzed by Clampfit version 10.6-11.02 software (Molecular Devices) and Origin 2019b (OriginLab). Oligomeric recombinant α-Syn (Anaspec, catalog #AS-55555, 500 nm), NMDA (Sigma Millipore, 100 μm), NMDAR antagonists [APV, (Tocris Bioscience, 50 μm), memantine hydrochloride (Sigma Millipore, 10 μm), or NitroSynapsin (also known as NitroMemantine YQW-036 or NMI-6979, EuMentis Therapeutics 10 μm)], or an AMPAR antagonist (NBQX, Tocris Bioscience, 10 μm) was added to the external bath solution from frozen stocks and applied to the recoded cells via a gravity-flow bath perfusion pencil system (Autom8, AutoMate Scientific).
Acute hippocampal slice preparation
Slices were prepared from 13- to 14-week-old WT mice. Brains were removed under isoflurane anesthesia and placed in ice-cold buffer consisting of (in mm): sucrose, 246; KCl, 2; NaH2PO4, 1.25; NaHCO3, 26; d-glucose, 10; MgSO4, 2; CaCl2, 0.5; Na- l-ascorbate, 1; pH 7.4, and equilibrated with 95% O2 and 5% CO2. We then cut 350-μm-thick hippocampal slices with a vibratome (VT1000S; Leica Microsystems), and allowed slices to recover at room temperature for at least 1 h before recording in aCSF consisting of the following (in mm): NaCl, 124, KCl, 3; NaH2PO4, 1.25; NaHCO3, 26; d-glucose, 10; MgCl2, 0.5-1; CaCl2, 2; pH 7.4, equilibrated in 95% O2 and 5% CO2. Recordings were obtained at room temperature from neurons lying superficial in the slice within 1-6 h of preparation. To record tonic basal glutamatergic currents in hippocampal slices, CA1 neurons were held at −70 mV in nominally magnesium-free aCSF. After obtaining a stable recording, the NMDAR antagonist 3-[(R)−2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP, Tocris Bioscience, 100 μm) and the AMPAR antagonist CNQX (Tocris Bioscience, 50 μm) were perfused into the slices by bath application. Because of its better tissue penetration, CPP was generally used rather than APV. The mean glutamatergic basal current amplitude was measured by the addition of these specific antagonists.
Golgi staining and quantification
Brains from 7-d-old mice were dissected out under isoflurane anesthesia, and 300-μm-thick cortical slices were collected in ice-cold dissection buffer containing the following (in mm): sucrose, 212; KCl, 3; MgCl2, 5; CaCl2, 0.5; NaH2PO4, 1; NaHCO3, 26; d-glucose, 10. Dissection buffer was bubbled with 95% O2/5% CO2 to maintain pH 7.4. Slices were transferred onto a Millicell cell-culture insert (Millipore) and placed in medium 1 containing Eagle's MEM [50% (v/v)], horse serum [25% (v/v)], Earle's Balanced Salt Solution [18% (v/v)], d-glucose, 32-35 mm; HEPES, 25 mm; glutamine, 2 mm; amphotericin B, 2.5 μg/ml; and streptomycin sulfate, 100 μg/ml]. After 24 h, the culture medium was changed to medium 2 of similar composition but containing a lower concentration of horse serum [5% (v/v)]. Thereafter, every 2-3 d, half of the culture medium was replaced with fresh medium. Slices were exposed to monomeric or oligomerized αSyn peptide (containing 750 nM of oligomers) and NitroSynapsin (10 μm) for 7 d, which was replenished with each medium replacement. After 7 d, the slices were fixed and stained using a sliceGolgi Kit (catalog #003760, Bioenno Tech) according to the manufacturer's protocol. In brief, slices were fixed in the aldehyde fixative provided by the manufacturer overnight at 4°C, washed, and immersed in impregnation solution for 7 d. The slices were then imaged under a Nikon C2 microscope with 60× objective. Six distinct fields of secondary or tertiary dendrites were randomly selected for each imaged neuron and analyzed in a masked fashion using Nikon Elements analysis software to calculate dendritic spine density.
Oligomerized αSyn preparation
Endotoxin-free recombinant human α-Syn protein was purchased commercially (Anaspec, #AS-55555-1000). The protein was dissolved in HPLC-grade water (Sigma Millipore, #270733) at a concentration of 200 μm. Monomers were immediately aliquoted and frozen at −80°C for subsequent use. For oligomerization, samples were placed in a thermomixer and shaken continuously at 1400 rpm for 6 d at 37°C, centrifuged at 1000 × g, and the pellet aliquoted and frozen at −80°C for subsequent use.
αSyn characterization by dynamic light scattering (DLS)
αSyn samples, prepared as above, were removed from storage at −80°C, thawed gradually on ice, and gently resuspended. A 10 μl aliquot was then removed and monitored at room temperature on a Zetasizer Nano DLS instrument (Malvern Instruments) in disposable microcuvettes with a 5 s acquisition time. Zetasizer analysis software was used for data analysis and size distribution measurement.
αSyn characterization by atomic force microscopy (AFM)
αSyn samples (200 μm of the stock solution prepared as above) were removed from storage at −80°C, thawed gradually on ice, gently resuspended, and diluted 1:10 in HPLC-grade water to 20 μm concentration (0.2892 mg/ml). The resulting solution was adsorbed to a surface of freshly cleaved mica for 1 min. The liquid was then wicked off the surface of the mica with filter paper. Salt and unbound material were removed by washing twice with HPLC-grade water that was immediately wicked off the surface of the mica with filter paper. Samples were then dried overnight at room temperature. Images were taken on a MultiMode Atomic Force Microscope with a Nanoscope IIIa controller (Bruker) and FESP silicon probes (Bruker) in tapping mode. Images were processed with NanoScope Analysis version 1.50r3 (Bruker).
In vivo brain microdialysis
Microdialysis was performed as previously described (Talantova et al., 2013) on 3- to 4-month-old Thy1-α-synuclein mice (Rockenstein et al., 2002). At this age, transgenic mice show early signs of neuronal cell loss in the neocortex and hippocampus (CA3 region). They also show early signs of neuroinflammation and motor impairment. A microdialysis probe was stereotactically implanted into the right hippocampus under isoflurane anesthesia using the following coordinates (in millimeters from bregma): AP = −2.0, ML = −1.3, DV = −2.0. Dialysis probes were perfused continuously using a syringe pump (PHD 22/2000; Harvard Apparatus) with aCSF composed of the following (in mm): NaCl, 149; KCl, 2.8; CaCl2, 1.2; MgCl2, 1.2; d-glucose, 5.4; ascorbic acid, 0.25; pH 7.2-7.4. An 18 h recovery period was allowed before dialysate collection; during this period, aCSF was perfused at a constant rate of 0.15 μl/min, and the perfusion speed was increased to 0.6 μl/min the next day. A 2 h presampling period was allowed before dialysate was sampled for analysis. Six baseline dialysate samples were collected in polypropylene tubes at 10 min intervals over a 60 min period. Glutamate content was analyzed using liquid chromatography coupled with tandem mass spectrometry, as previously reported (Talantova et al., 2013). Briefly, dialysate samples (2 μl each) were supplanted with 5.5 μl of 100 mm boric acid and then were reacted with 2% (v/v) benzoyl chloride (2.5 μl), quenched with 2% (v/v) formic acid (2.5 μl), and supplemented with 13C6-benzoyl chloride-labeled internal standard (2.5 μl). Derivatized samples (12 μl each) were separated by HPLC (Agilent Technologies, 1260) on a Zorbax rapid-resolution high-definition column (2.1 mm internal diameter × 50 mm in length, 1.8 μm particle size; 50°C) using 10 mm ammonium formic acid [0.15% acid (wt/vol)] for mobile phase A and acetonitrile for mobile phase B as follows: 0% B (0 min), 0% B (3 min), 65% B (7 min), 65% B (8.50 min), 0% B (8.51 min), 0% B (13.25 min). Eluents were analyzed for the constituents of interest using tandem mass spectrometry (Agilent Technologies, 6460 with JetStream ion source). Quantification was performed by isotope dilution using the following standard curve: 250, 500, 1000, and 2000 nm.
Intracellular Ca2+ imaging
Calcium ion measurements were performed as previously described (Talantova et al., 2013). Primary rat cerebrocortical neurons were loaded with Fura-2 AM (4 μm) in imaging buffer containing the following (in mm): NaCl, 146; KCl, 2.5; NaOH, 4; CaCl2, 1; d-glucose, 20; sucrose, 20; HEPES, 10; adjusted to pH 7.4. After loading, the cells were washed twice with imaging buffer. For light stimulation and emission, we used a liquid light guide connected to an ultra-high-speed wavelength switcher fitted with a 175 W xenon arc lamp (λ DG4; Sutter Instrument) and an optimized Fura-2 filter set for obtaining the 350/380 nm excitation ratio (for glass rather than quartz objectives).
Images were collected using a CCD camera mounted on a Carl Zeiss Axiovert 35 microscope and analyzed with SlideBook software. Fluorescence values were calculated as change in fluorescence ratios relative to baseline fluorescence intensity (ΔF/F0). For each experiment, values were calculated by dividing the change in peak fluorescence (ΔF) over resting baseline fluorescence (F0).
Statistical analysis
For testing statistical significance of multiple comparisons, an ANOVA was used with Bonferroni post hoc test. When two groups were compared, a Student's t test was used. p values < 0.05 were considered statistically significant. In general, comparisons of samples were performed in a masked fashion in each experiment.
Results
αSyn oligomers increase glutamate release from astrocytes
We first generated and characterized our preparations of αSyn oligomers by DLS (Fig. 1A). We found that, after the oligomerization process (see Materials and Methods), the solution contained 75% oligomers (defined as soluble aggregates >100 nm in diameter) and larger oligomers/fibrils, as can also be seen in AFM images (Fig. 1B).
Figure 1.

Characterization of αSyn oligomers. A, DLS characterization of αSyn monomer and oligomer preparations shows an increase in size distribution, indicating oligomer formation, shown as percentage of mass and apparent hydrodynamic diameter. B, Representative AFM analysis of αSyn monomer and oligomer preparations shows different αSyn strains/sizes in the oligomer preparation, including larger fibrils. Scale bars are located below each image.
To study whether αSyn oligomers could induce a local increase in glutamate concentration contiguous to the astrocytes, we used two fluorescent glutamate-reporter platforms. First, we found that the iGluSnFR glutamate sensor system (Marvin et al., 2013) detected local changes in glutamate concentration near the astrocyte surface after exposure to αSyn oligomers (but not monomers), potentially reflecting increased glutamate release or decreased glutamate uptake. iGluSnFR is a sensitive probe that detects glutamate at low micromolar levels. The increase in glutamate in this system was because of vesicular release because it was significantly blocked by tetanus toxin (F(2,44) = 7.082, p = 0.022 by ANOVA; monomers vs oligomers t(44) = 3.488, p = 0.0025 and oligomers vs tetanus toxin t(44) = 2.649, p = 0.0224 with Bonferroni post hoc test) (Fig. 2A,B), which is known to block Ca2+-dependent vesicular release (McMahon et al., 1992).
Figure 2.

Oligomeric αSyn induces glutamate release in mouse astrocytes. A, Fluorescent sensor iGluSnFR detected an increase in glutamate following exposure of astrocytes to oligomeric αSyn but not monomers; the increase in glutamate was blocked by tetanus toxin. Dotted line indicates time of addition of αSyn. B, Quantification of glutamate levels measured as area under the curve (n = 7 biological replicates in separate experiments). C, The glutamate sensor SuperGluSnFR, using FRET ratio to monitor glutamate levels after oligomeric αSyn exposure, revealed similar results (n = 7). D, Quantification of microdialysis from mouse brain showing glutamate levels in WT (n = 21) and αSyn transgenic (n = 9) animals. Data are mean ± SEM. *p < 0.05; **p < 0.01; one-way ANOVA with Bonferroni post hoc test for multiple comparisons or a Student's t test for comparison of two groups.
We further confirmed these results using a FRET-based glutamate sensor system (SuperGluSnFR) (Hires et al., 2008; Talantova et al., 2013). To ensure close apposition of this sensor probe (consisting of GluSnFR-transfected HEK-293 cells) to the astrocyte membrane surface, the sensor cells were genetically engineered to coexpress neuroligin (Scheiffele et al., 2000). iGluSnFR and SuperGluSnFR have complementary strengths. Used in conjunction, they provide better insight for the mechanism of glutamate release. While the spatial resolution of the FRET GluSnFR technique is similar to that of iGluSnFR, the former has lower affinity/sensitivity but improved temporal resolution, in the subsecond timescale.
This temporal resolution was important for comparing extrasynaptic glutamate levels with the relatively rapid electrophysiological effects of oligomeric αSyn observed in subsequent patch-clamp recordings. To test the sensitivity of each preparation, a test-pulse of glutamate (30 μm) was applied before αSyn applications (Fig. 2C).
Since astrocytic receptors and their pharmacological properties can change when placed into culture, and to confirm our results in an in vivo setting, we next performed microdialysis measurements of glutamate in a PD/LBD mouse model overexpressing transgenic human αSyn under the Thy1 promoter; in this model system, αSyn is known to oligomerize and accumulate (Rockenstein et al., 2002). As expected, we found that basal glutamate levels were increased compared with WT littermates (t(28) = 2.483, p = 0.0193 by unpaired t test) (Fig. 2D). Our results show that extracellular glutamate accumulates not only in cultured astrocytes but also in living brain in the presence of oligomerized αSyn.
Furthermore, given our tetanus toxin data suggesting that oligomeric αSyn-induced glutamate release from cultured astrocytes was a result of Ca2+-dependent vesicular release, we next examined whether oligomeric αSyn increased Ca2+ influx into astrocytes. For this purpose, we used Fura-2 to detect Ca2+ changes, and we found that αSyn oligomers indeed induced increases in Ca2+ influx into both mouse (Fig. 3A) and human astrocytes (Fig. 3B).
Figure 3.

Oligomeric αSyn increases Ca2+ in mouse and human astrocytes. A, Representative trace of intracellular Ca2+ detected with Fura-2 AM in mouse astrocytes after exposure to oligomeric αSyn. B, αSyn oligomers, but not monomers, induced intracellular Ca2+ increase in human astrocytes (n = 3 independent experiment with 6 replicates each). Data are mean ± SEM.
Increased tonic glutamatergic activity in αSyn transgenic mice
Next, we wanted to assess the potential effect of oligomeric αSyn-induced release of astrocytic glutamate on neurons. For this purpose, we again used transgenic αSyn-overexpressing mice as a model of PD/LBD (Rockenstein et al., 2002). Indeed, compared with WT littermate controls, we found a significant increase in basal inward current in neurons in the CA1 region of hippocampal slices prepared from αSyn transgenic mice in the nominal absence of extracellular Mg2+ (Fig. 4). Pharmacological inhibition of glutamate receptors by the AMPAR antagonist CNQX plus the NMDAR antagonist CPP totally abrogated the increase in basal current, consistent with the notion that the current was mediated by glutamate receptors (t(6) = 2.472, p = 0.0484 by unpaired t test) (Fig. 4). Of interest, the decrease in apparent “noise” in the αSyn transgenic compared with WT slice recordings was because of a decrease in synaptic currents (seen as transient inward deflections of the current trace in Fig. 4). This finding is consistent with the notion that there may have been fewer intact synapses in the αSyn transgenic brains, as we and others have reported previously in slices with oligomeric Aβ overexpression (Talantova et al., 2013). Hence, we next studied the effect of oligomeric αSyn on dendritic spine number.
Figure 4.

Increased tonic glutamatergic current in αSyn transgenic mice. A, Representative whole-cell recording with a patch electrode of tonic glutamatergic currents in CA1 pyramidal neurons from acute hippocampal slices of WT and αSyn transgenic mice at a holding potential of (Vh) = −70 mV. The tonic basal currents were larger in the αSyn transgenic mice versus WT (42 vs 14 pA). B, Quantification of basal tonic current blocked by CPP + CNQX (n = 4 slices each). Data are mean ± SEM. *p < 0.05 (Student's t test).
Oligomerized αSyn triggers dendritic spine loss on neurons
To assess the contribution of excessive glutamatergic activity triggered by αSyn on synapse loss, we next exposed cerebrocortical slices to oligomerized αSyn. These organotypic slices provide an ex vivo system that contains dendritic spines that can be counted under these conditions (Talantova et al., 2013). We found that exposure to αSyn oligomers resulted in the loss of dendritic spines, consistent with the notion that synapses were compromised (F(2,162) = 41.19, p < 0.0001 by ANOVA; control vs αSyn t(162) = 9.053, p < 0.0001 with Bonferroni post hoc test) (Fig. 5). Since we had previously shown that excessive eNMDAR activity could contribute to such synaptic loss (Talantova et al., 2013), we next tested this possibility pharmacologically. We used the novel, dual-allosteric antagonist NitroSynapsin (also known as NitroMemantine YQW-036 or NMI-6979) to block selectively eNMDARs while relatively sparing synaptic NMDARs; this new eNMDAR antagonist was used because, at maximally effective concentrations, as used here, it was previously shown to be more effective than memantine in blocking these channels (Lipton, 2006; Okamoto et al., 2009; Xia et al., 2010; Talantova et al., 2013; Takahashi et al., 2015). Indeed, we found that treatment with NitroSynapsin prevented synaptic spine loss in this paradigm (F(2,162) = 41.19, p < 0.0001 by ANOVA; αSyn vs αSyn + NitroSynapsin t(162) = 3.804, p = 0.0004 with Bonferroni post hoc test) (Fig. 5). Together, these results suggest that events associated with hyperactivation of eNMDARs by oligomeric αSyn may be linked to the subsequent loss of synapses in PD/LBD, and that inhibition of eNMDAR-activated channels may prevent this loss.
Figure 5.

Oligomerized αSyn-evoked eNMDAR activity triggers dendritic spine loss. A, Representative images of dendritic spines labeled by Golgi staining in mouse hippocampal slices. B, Quantification of dendritic spines showing that blockade of eNMDAR activity by NitroSynapsin abrogated dendritic spine loss mediated by oligomerized αSyn. For each treatment, there were two independent experiments; and in each independent experiment, there were two slices. For each slice, at least three neurons were imaged; and spines were counted on six dendrites for each of these neurons. Data are mean ± SEM. **p < 0.01; ****p < 0.0001; one-way ANOVA with Bonferroni post hoc test.
NMDAR antagonists block oligomeric αSyn-induced currents
To investigate the possible direct effect of oligomeric αSyn on neurons, we initially performed whole-cell recordings while applying oligomerized recombinant αSyn preparations to mature rat cerebrocortical neurons. We observed that oligomeric αSyn evoked inward currents in the majority of recorded cells voltage-clamped at −70 mV (6 of 8 neurons). Selective NMDAR antagonists blocked these currents, for example, APV, a glutamate binding-site NMDAR antagonist (Fig. 6A) or NitroSynapsin, a selective eNMDAR antagonist (Fig. 6B). Collectively, these findings are consistent with the notion that oligomerized αSyn-induced currents are mediated by activation of eNMDARs.
Figure 6.
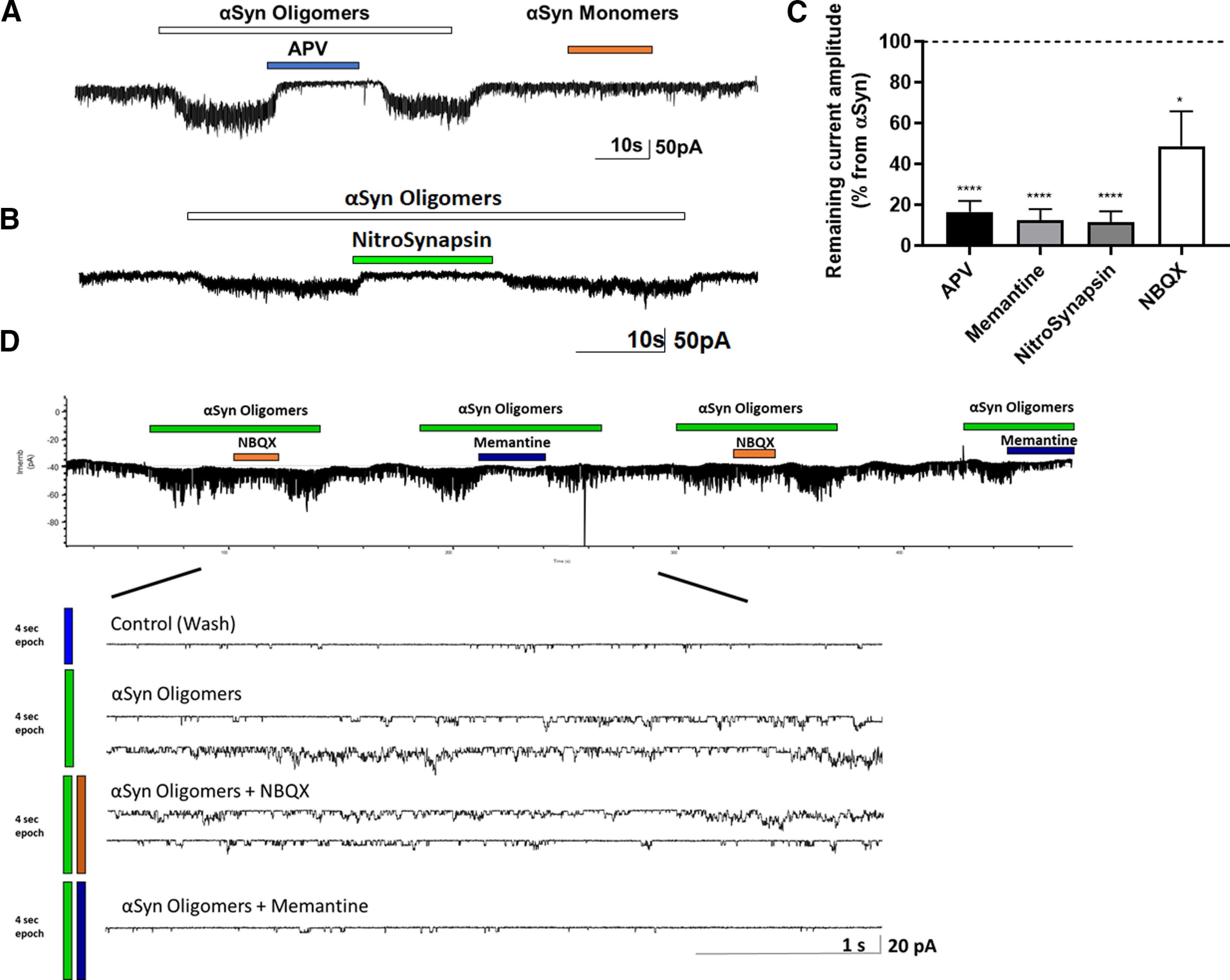
Oligomeric αSyn-induced currents in rat cerebrocortical cultures are blocked by NMDAR antagonists. A, B, Whole-cell recording of oligomeric αSyn-evoked currents. Application of oligomeric, but not monomeric, αSyn-induced tonic currents in rat cerebrocortical neurons. This current was virtually blocked by the specific NMDAR antagonists APV (n = 19 cells), memantine (n = 5 cells), or NitroSynapsin (n = 10 cells). NBQX exerted a minimal effect (n = 5 cells). C, Quantification of the degree of inhibition αSyn oligomer-induced current by the various. Dashed line indicates current level after αSyn oligomers alone. Data are mean ± SEM. *p < 0.05; ****p < 0.0001; paired t test. D, Representative single-channel recording from outside-out patch of αSyn-induced currents (n = 7 patches recorded in 7 independent experiments). Single-channel events were blocked to a greater degree by the eNMDAR antagonist memantine than by the AMPAR antagonist NBQX (Okamoto et al., 2009; Xia et al., 2010; Talantova et al., 2013; Takahashi et al., 2015). Bottom, Same recording on a faster timescale.
Next, we asked whether αSyn might act directly on eNMDARs under these conditions. To begin to approach this question, we pulled outside-out patches from the neuronal somal membrane, lifted the patch pipette away from the layer of the mixed neuronal and glial cells, and applied oligomeric αSyn. We found that αSyn oligomer-induced channel activity was blocked to a greater degree by a maximally effective concentration of the eNMDAR-preferring open-channel blocker memantine than by the AMPAR antagonist NBQX (APV: t(18) = 15.54, p < 0.0001; memantine: t(4) = 16.0, p < 0.0001; NitroSynapsin: t(8) = 16.72, p < 0.0001; NBQX: t(4) = 2.991, p = 0.0403 by paired t test compared with αSyn alone applied without antagonist) (Fig. 6C). These findings are consistent with the notion that oligomeric αSyn directly activates eNMDARs or a molecule closely associated with the receptor, but may also affect to a lesser degree AMPARs. Notably, the fact that monomeric αSyn did not affect the current level (Fig. 6A) argues against a contaminant in the αSyn preparations, such as glutamate, that could be responsible for these effects.
Finally, to investigate the effect of αSyn oligomers on neurons in a human context, we recorded from hiPSC-derived cerebrocortical neurons; ∼30% of these cells responded to NMDA application. Among the hiPSC neurons responding to NMDA, oligomeric αSyn generated small currents that were sensitive to APV in all of these cells (t(3) = 13.65, p = 0.0009 by paired t test; Fig. 7), similar to the findings in rat cerebrocortical neurons.
Figure 7.

Oligomeric αSyn induces NMDAR-mediated currents in hiPSC-derived cerebrocortical neurons. A, Whole-cell recording of hiPSC-derived neurons showing that αSyn oligomers evoked inward current in hiPSC neurons, which was largely inhibited by the NMDAR antagonist APV. Bottom, Same recording on a faster timescale. B, Quantification of normalized area under the curve (AUC) showing NMDAR-mediated current in 10 s epochs of recordings induced by αSyn oligomers, determined by addition of APV (n = 4 neurons in 4 independent experiments). Data are mean ± SEM. ***p < 0.001 (Student's t test).
Discussion
In the present study, we investigated the effect of oligomeric αSyn on astrocytes and neurons, and identified a dual pathway for aberrant activation of neurons via excessive glutamatergic input that contributes to synaptic loss in models of PD/LBD. First, we found that αSyn oligomers induce glutamate release from astrocytes, which in turn stimulate neuronal glutamate receptors, including eNMDARs. Second, we discovered a direct effect of αSyn on neurons, through eNMDAR and partially via eAMPARs. Notably, in these experiments, outside-out patches were pulled from neuronal cell bodies, ensuring that the receptors recorded from were extrasynaptic. In both the case of indirect and direct glutamate receptor activation by αSyn oligomers, the aberrant electrical activity could be abrogated with relatively selective eNMDAR antagonists. Most importantly, we also found evidence that αSyn oligomer-induced excessive eNMDAR stimulation can contribute to synaptic spine loss, and this damage could be ameliorated with the new eNMDAR antagonist, NitroSynapsin. Collectively, we propose a dual excitatory pathway converging on eNMDAR activation that contributes to synaptic damage in PD/LBD (Fig. 8).
Figure 8.

Schematic diagram showing dual oligomeric αSyn action on neurons and astrocytes. Right, αSyn directly activates eNMDAR-operated channels. Left, αSyn induces astrocytic glutamate release, which leads to further activation of eNMDARs. This aberrant eNMDAR activity in turn contributes to synaptic damage and loss. Created with www.BioRender.com.
Various species of αSyn have been identified in different synucleinopathies, with varying degrees of neurotoxicity and disease propagation. Interestingly, distinct species are associated with disparate pathologies, and these may correlate to the various neurodegenerative diseases exhibiting αSyn pathology, such as PD, LBD, and multiple system atrophy (Peelaerts et al., 2015; Rodriguez et al., 2015; Lau et al., 2020; Schweighauser et al., 2020). Here, we studied a mixture of αSyn-soluble aggregates, rather than one particular strain, exhibiting a range of sizes and assemblies. While it is not possible to assign a particular conformation to the observed pathologic findings, we can state that these αSyn oligomers/fibrils induce astrocytic glutamate release, excessive extrasynaptic glutamate receptor activity, and synaptic damage. Future work looking at the differential effects of each strain of αSyn oligomer will be needed to mechanistically link conformation to pathology because of glutamate receptor activity. For example, an αSyn strain that releases glutamate from astrocytes or directly interacts with neuronal eNMDARs might be a good target for NitroSynapsin blockade.
Previously, based on both in vitro and in vivo evidence, a mechanism for neuronal damage engendered by aggregated αSyn has been partially ascribed to increasing intracellular nitric oxide (NO) levels with consequent DNA damage and poly(adenosine 5′-diphosphate-ribose) polymerase-1 activation (Kam et al., 2018). However, the upstream trigger for the increase in NO by pathologic αSyn has remained elusive. Our new results offer an explanation for this finding: namely, pathologic αSyn induces astrocytic glutamate release and activation of neuronal eNMDARs, which we have shown leads to a dramatic increase in neuronal NO levels (Talantova et al., 2013; Molokanova et al., 2014). Thus, our data are consistent with the notion that the aberrant eNMDAR activity induced by αSyn oligomers contributes to a rise in neuronal NO, which has been reported to trigger pathologic αSyn-related neurotoxicity (Kam et al., 2018). Importantly, we observed effects of oligomeric αSyn on glutamate-mediated responses both in vitro and ex vivo, supporting the pathophysiological relevance of our findings.
Prior reports have also suggested that aggregated αSyn might injure synapses directly, and effects on NMDARs have been demonstrated, but whether these changes were direct or indirect remained unknown. For example, misfolded αSyn has been shown to increase NMDAR-mediated currents (Diogenes et al., 2012). Moreover, overexpression of αSyn in transgenic mice or in culture systems was shown to increase phosphorylation of NMDAR subunits GluN1and GluN2B (Yang et al., 2016), promote endocytosis of GluN1 (Cheng et al., 2011), and enhance GluN2B-mediated excitotoxicity (Navarria et al., 2015); interactions with the GluN2D subunit have also been reported (Tozzi et al., 2016). Interestingly, αSyn can also form a complex with prion protein, which can interact with NMDARs (Ferreira et al., 2017). To our knowledge, the findings presented here based on patch-clamp recordings from outside-out patches are the first to support a direct functional interaction of oligomeric αSyn with the NMDAR or a closely related molecule that results in an increase in channel activity. We show that blocking the effect of aberrant eNMDAR stimulation with NitroSynapsin can block this effect and protect synaptic spines from αSyn oligomer-mediated damage. Critically, we also show that αSyn monomers cannot result in such damage, suggesting a pathologic role for misfolded protein aggregates on synaptic form and function that involves an excitotoxic component.
Whereas most data to date on pathologic αSyn have been obtained in rodent models, our use of hiPSC-derived neurons allowed us to study the mechanism of action of the misfolded protein in a human context. Accordingly, we used hiPSC neurons and found a similar effect of αSyn oligomers on eNMDAR-mediated currents in these human neurons as in our rodent model systems. While this is an in vitro system, it provides further evidence for the effect of oligomerized αSyn in a human context.
In conclusion, we show that oligomeric/fibrillar αSyn can trigger the release of astrocytic glutamate, resulting in aberrant eNMDAR activity on contiguous neurons. This in turn contributes to synaptic loss because the eNMDAR-selective antagonist NitroSynapsin protected synaptic spines under these conditions. Additionally, we mount evidence that pathologic αSyn oligomers may directly activate glutamate receptors on neurons, principally eNMDARs, which may further contribute to the damage. Importantly, our findings suggest that eNMDAR antagonists, such as NitroSynapsin, might prove useful in ameliorating the synaptic damage observed in PD/LBD and possibly other synucleinopathies.
Footnotes
S.A.L. is an inventor on worldwide patents for the use of memantine and NitroSynapsin for neurodegenerative and neurodevelopmental disorders. Per Harvard University guidelines, S.A.L. participates in a royalty-sharing agreement with his former institution Boston Children's Hospital/Harvard Medical School, which licensed the drug memantine (Namenda) to Forest Laboratories, Inc./Actavis/Allergan/AbbVie, Inc. NitroSynapsin is licensed to EuMentis Therapeutics, Inc. K.B. is currently (but not during the time of the study) an employee of and holds stock/stock options in Biogen, Inc., which is involved in the research, development, and commercialization of therapies for neurological disorders. The remaining authors declare no competing financial interests.
This work was supported in part by National Institutes of Health Grants DP1 DA041722, R01 DA048882, R01 NS086890, R01 AG056259, and RF1 AG057409 to S.A.L. Y.S.E. was supported by National Institutes of Health, National Institute on Aging K99/R00 Pathway to Independence Award K99/R00AG050764. We thank Loren L. Looger and Jonathan S. Marvin for kindly providing the iGluSnFR probe; and Michael S. Sailor (University of California, San Diego) for the use of his dynamic light scattering instrument.
References
- Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, Cox TO, Riddle DM, Zhang B, Trojanowski JQ, Lee VM (2020) Amyloid-β (Aβ) plaques promote seeding and spreading of α-synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology. Neuron 105:260–275. 10.1016/j.neuron.2019.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HD, Hirst WD, Wade-Martins R (2017) The role of astrocyte dysfunction in Parkinson's disease pathogenesis. Trends Neurosci 40:358–370. 10.1016/j.tins.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bras IC, Dominguez-Meijide A, Gerhardt E, Koss D, Lazaro DF, Santos PI, Vasili E, Xylaki M, Outeiro TF (2020) Synucleinopathies: where we are and where we need to go. J Neurochem 153:433–454. 10.1111/jnc.14965 [DOI] [PubMed] [Google Scholar]
- Cheng F, Li X, Li Y, Wang C, Wang T, Liu G, Baskys A, Ueda K, Chan P, Yu S (2011) α-Synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. J Neurochem 119:815–825. 10.1111/j.1471-4159.2011.07460.x [DOI] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M (2007) Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci 27:9220–9232. 10.1523/JNEUROSCI.2617-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delenclos M, Burgess JD, Lamprokostopoulou A, Outeiro TF, Vekrellis K, McLean PJ (2019) Cellular models of α-synuclein toxicity and aggregation. J Neurochem 150:566–576. 10.1111/jnc.14806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diogenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Nasstrom T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergstrom J, Ingelsson M, Quintas A, Sebastiao AM, Lopes LV, Outeiro TF (2012) Extracellular α-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci 32:11750–11762. 10.1523/JNEUROSCI.0234-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira DG, Temido-Ferreira M, Vicente Miranda H, Batalha VL, Coelho JE, Szego EM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, Outeiro TF (2017) α-Synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci 20:1569–1579. 10.1038/nn.4648 [DOI] [PubMed] [Google Scholar]
- Ghatak S, Dolatabadi N, Trudler D, Zhang X, Wu Y, Mohata M, Ambasudhan R, Talantova M, Lipton SA (2019) Mechanisms of hyperexcitability in Alzheimer's disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife 8:e50333. 10.7554/eLife.50333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglieri V, Calabrese V, Calabresi P (2018) α-Synuclein: from early synaptic dysfunction to neurodegeneration. Front Neurol 9:295. 10.3389/fneur.2018.00295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M (2001) α-Synuclein and neurodegenerative diseases. Nat Rev Neurosci 2:492–501. 10.1038/35081564 [DOI] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S (2014) α-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34:249–259. 10.1523/JNEUROSCI.2507-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey LA, Coleman-Jackson R (2019) Pharmacological management of dementia with Lewy bodies. Drugs Aging 36:309–319. 10.1007/s40266-018-00636-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hires SA, Zhu Y, Tsien RY (2008) Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc Natl Acad Sci USA 105:4411–4416. 10.1073/pnas.0712008105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huls S, Hogen T, Vassallo N, Danzer KM, Hengerer B, Giese A, Herms J (2011) AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced α-synuclein oligomers. J Neurochem 117:868–878. 10.1111/j.1471-4159.2011.07254.x [DOI] [PubMed] [Google Scholar]
- Irwin DJ, Hurtig HI (2018) The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. J Alzheimers Dis Parkinsonism 8:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A, Claessens M (2019) Disruptive membrane interactions of α-synuclein aggregates. Biochim Biophys Acta Proteins Proteom 1867:468–482. 10.1016/j.bbapap.2018.10.006 [DOI] [PubMed] [Google Scholar]
- Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, Andrabi SA, Qi C, Poirier GG, Pletnikova O, Troncoso JC, Bekris LM, Leverenz JB, Pantelyat A, Ko HS, Rosenthal LS, et al. (2018) Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson's disease. Science 362:eaat8407. 10.1126/science.aat8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Cho MK, Kumar A, Maier E, Siebenhaar C, Becker S, Fernandez CO, Lashuel HA, Benz R, Lange A, Zweckstetter M (2009) Structural properties of pore-forming oligomers of α-synuclein. J Am Chem Soc 131:17482–17489. 10.1021/ja9077599 [DOI] [PubMed] [Google Scholar]
- Lau A, So RW, Lau HH, Sang JC, Ruiz-Riquelme A, Fleck SC, Stuart E, Menon S, Visanji NP, Meisl G, Faidi R, Marano MM, Schmitt-Ulms C, Wang Z, Fraser PE, Tandon A, Hyman BT, Wille H, Ingelsson M, Klenerman D, et al. (2020) α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci 23:21–31. 10.1038/s41593-019-0541-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801. 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638. 10.1523/JNEUROSCI.0203-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA (2006) NMDA receptors, glial cells, and clinical medicine. Neuron 50:9–11. 10.1016/j.neuron.2006.03.026 [DOI] [PubMed] [Google Scholar]
- Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ (2006) Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54:343–357. 10.1002/glia.20400 [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA, Looger LL (2013) An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods 10:162–170. 10.1038/nmeth.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos M, Augusto E, Oliveira CR, Agostinho P (2008) Amyloid-β peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 156:898–910. 10.1016/j.neuroscience.2008.08.022 [DOI] [PubMed] [Google Scholar]
- McMahon HT, Foran P, Dolly JO, Verhage M, Wiegant VM, Nicholls DG (1992) Tetanus toxin and botulinum toxins type A and B inhibit glutamate, γ-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes: clues to the locus of action. J Biol Chem 267:21338–21343. 10.1016/S0021-9258(19)36614-1 [DOI] [PubMed] [Google Scholar]
- Molokanova E, Akhtar MW, Sanz-Blasco S, Tu S, Pina-Crespo JC, McKercher SR, Lipton SA (2014) Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J Neurosci 34:5023–5028. 10.1523/JNEUROSCI.2907-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarria L, Zaltieri M, Longhena F, Spillantini MG, Missale C, Spano P, Bellucci A (2015) α-Synuclein modulates NR2B-containing NMDA receptors and decreases their levels after rotenone exposure. Neurochem Int 85-86:14–23. 10.1016/j.neuint.2015.03.008 [DOI] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA (2009) Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15:1407–1413. 10.1038/nm.2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G (2001) Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci 21:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, Dineley KT, Parri HR (2013) α7 nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer's mouse model. PLoS One 8:e81828. 10.1371/journal.pone.0081828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Sims RE, Saunders G, Antonio SA, Codadu NK, Parri HR (2017) Astrocyte-mediated neuronal synchronization properties revealed by false gliotransmitter release. J Neurosci 37:9859–9870. 10.1523/JNEUROSCI.2761-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E (2002) Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res 68:568–578. 10.1002/jnr.10231 [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Jiang L, Arbing MA, Nannenga BL, Hattne J, Whitelegge J, Brewster AS, Messerschmidt M, Boutet S, Sauter NK, Gonen T, et al. (2015) Structure of the toxic core of α-synuclein from invisible crystals. Nature 525:486–490. 10.1038/nature15368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000) Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101:657–669. 10.1016/s0092-8674(00)80877-6 [DOI] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ (2010) The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol 120:131–143. 10.1007/s00401-010-0711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, Murayama S, Yoshida M, Hasegawa M, Scheres SH, Goedert M (2020) Structures of α-synuclein filaments from multiple system atrophy. Nature 585:464–469. 10.1038/s41586-020-2317-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388:839–840. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Sulzer D, Edwards RH (2019) The physiological role of α-synuclein and its relationship to Parkinson's disease. J Neurochem 150:475–486. 10.1111/jnc.14810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Xia P, Cui J, Talantova M, Bodhinathan K, Li W, Saleem S, Holland EA, Tong G, Pina-Crespo J, Zhang D, Nakanishi N, Larrick JW, McKercher SR, Nakamura T, Wang Y, Lipton SA (2015) Pharmacologically targeted NMDA receptor antagonism by NitroMemantine for cerebrovascular disease. Sci Rep 5:14781. 10.1038/srep14781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, et al. (2013) Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA 110:E2518–E2527. 10.1073/pnas.1306832110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosatto L, Andrighetti AO, Plotegher N, Antonini V, Tessari I, Ricci L, Bubacco L, Dalla Serra M (2012) α-Synuclein pore forming activity upon membrane association. Biochim Biophys Acta 1818:2876–2883. 10.1016/j.bbamem.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Bagetta V, Tantucci M, Durante V, Quiroga-Varela A, Costa C, Di Filippo M, Ghiglieri V, Latagliata EC, Wegrzynowicz M, Decressac M, Giampà C, Dalley JW, Xia J, Gardoni F, Mellone M, El-Agnaf OM, Ardah MT, Puglisi-Allegra S, et al. (2016) α-Synuclein produces early behavioral alterations via striatal cholinergic synaptic dysfunction by interacting with GluN2D N-methyl-D-aspartate receptor subunit. Biol Psychiatry 79:402–414. 10.1016/j.biopsych.2015.08.013 [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous α-Synuclein pore forming activity upon membrane association: synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA (2010) Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 30:11246–11250. 10.1523/JNEUROSCI.2488-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Hertz E, Zhang X, Leinartaite L, Lundius EG, Li J, Svenningsson P (2016) Overexpression of α-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucocerebrosidase activity. Neurosci Lett 611:51–58. 10.1016/j.neulet.2015.11.023 [DOI] [PubMed] [Google Scholar]